

Abstract
Generation of reactive oxygen species (ROS) causes cellular oxidative damage and has been implicated in the etiology of Alzheimer's disease (AD). In contrast, multiple lines of evidence indicate that ROS can normally modulate long-term potentiation (LTP), a cellular model for memory formation. We recently showed that decreasing the level of superoxide through the overexpression of mitochondrial superoxide dismutase (SOD-2) prevents memory deficits in the Tg2576 mouse model of AD. In the current study, we explored whether AD-related LTP impairments could be prevented when ROS generation from mitochondria was diminished either pharmacologically or via genetic manipulation. In wild-type hippocampal slices treated with exogenous amyloid β peptide (Aβ1-42) and in slices from APP/PS1 mutant mice that model AD, LTP was impaired. The LTP impairments were prevented by MitoQ, a mitochondria-targeted antioxidant, and EUK134, an SOD and catalase mimetic. In contrast, inhibition of NADPH oxidase either by diphenyliodonium (DPI) or by genetically deleting gp91phox, the key enzymatic component of NADPH oxidase, had no effect on Aβ-induced LTP blockade. Moreover, live staining with MitoSOX Red, a mitochondrial superoxide indicator, combined with confocal microscopy, revealed that Aβ-induced superoxide production could be blunted by MitoQ, but not DPI, in agreement with our electrophysiological findings. Finally, in transgenic mice overexpressing SOD-2, Aβ-induced LTP impairments and superoxide generation were prevented. Our data suggest a causal relationship between mitochondrial ROS imbalance and Aβ-induced impairments in hippocampal synaptic plasticity.
Introduction
Lack of mechanistic understanding and effective therapies characterize the current state of Alzheimer's disease (AD), a devastating neurodegenerative disease that accounts for the majority of dementia and is an emerging global threat to public health (Selkoe, 2004; Querfurth and LaFerla, 2010). The abnormal brain accumulation of amyloid β (Aβ), a small peptide derived from amyloid precursor protein (APP), presents as a pathologic hallmark of AD (Haass and Selkoe, 2007). Many lines of experimental evidence have demonstrated that high levels of Aβ cause synaptic dysfunction and memory deficits (Haass and Selkoe, 2007; Shankar et al., 2008; Ma et al., 2010; Querfurth and LaFerla, 2010). However, the molecular mechanisms underlying the synaptic toxicity of Aβ are still poorly understood.
There is strong evidence to link reactive oxygen species (ROS) with neurodegenerative diseases, including AD (Lin and Beal, 2006). As cellular energy machinery, mitochondria produce the largest amount of ROS in mammalian cells, with superoxide (O2−̣) being the predominant species leading to the formation of other ROS (Balaban et al., 2005; Murphy, 2009). Oxidative damage by superoxide itself is usually thought to be primarily prevented by mitochondrial superoxide dismutase (SOD-2), which rapidly catalyzes dismutation of superoxide to H2O2 (Murphy, 2009). AD mutant mice with decreased SOD-2 expression exhibit increased levels of brain Aβ and accelerated behavioral abnormalities, including cognitive dysfunction (Li et al., 2004; Esposito et al., 2006). Conversely, we and others recently showed that overexpression of SOD-2 in two different AD mouse models reduced Aβ deposition and prevented memory deficits (Dumont et al., 2009; Massaad et al., 2009). However, whether the decrease in superoxide that results in the aforementioned behavioral improvements in AD model mice is correlated with improvements in synaptic plasticity has not been explored. Given the notion that AD is essentially a disease of synaptic failure (Selkoe, 2002; Tanzi, 2005), it is important to provide direct evidence between ROS imbalances and synaptic dysfunction to enhance our understanding of AD pathophysiology.
Herein we investigated the effects of decreasing ROS on Aβ-induced impairments in hippocampal long-term potentiation (LTP), a well established form of synaptic plasticity and the most intensely studied cellular model for memory (Malenka and Nicoll, 1999; Malenka, 2003). By taking advantage of novel, specific pharmacological ROS scavengers and inhibitors, as well as genetic manipulations, we reduced ROS production from either mitochondria or NADPH oxidase, another major source of ROS that plays a key role in synaptic plasticity (Kishida et al., 2006; Kishida and Klann, 2007). We found that hippocampal LTP impairments, either induced by exogenous Aβ or in slices from APP/PS1 AD mutant mice, could be rescued with mitochondrial antioxidants. In contrast, inhibition of NADPH oxidase either pharmacologically or by genetic deletion of gp91phox had no effect on Aβ-induced LTP impairments. Furthermore, in transgenic mice overexpressing SOD-2, Aβ-induced LTP deficits as well as Aβ-induced increases in superoxide levels were prevented. Our data suggest a causal relationship between mitochondrial ROS imbalance and Aβ toxicity in hippocampal synaptic plasticity.
Materials and Methods
Transgenic mouse lines.
All mice were housed in the Transgenic Mouse Facility of New York University, compliant with the NIH Guide for Care and Use of Laboratory Animals. The facility is kept on a 12 h light/dark cycle, with regular feeding and cage cleaning schedule.
APP/PS1 transgenic mice were purchased from the Jackson Laboratory. These mice were created via incorporation of human/murine APP construct bearing the Swedish double mutation and the exon-9-deleted PSEN1 mutation (APPswe + PSEN1/ΔE9) (Jankowsky et al., 2001). Wild-type littermates were used as controls for all experiments with APP/PS mutant mice. gp91phox knock-out mice were generated as described previously (Pollock et al., 1995). These mice lack the gp91phox subunit of NADPH oxidase; age-matched C57BL/6J mice were used as controls for experiments with these mice. SOD-2 transgenic mice overexpressing the human mitochondrial SOD were generated as described previously (Ho et al., 1998). Wild-type littermates were used as controls for all experiments with SOD-2 transgenic mice.
Hippocampal slice preparation and electrophysiology.
Hippocampal slices (400 μm) were prepared from mice 2–4 months of age (unless specified otherwise) using a vibratome as described previously (Hu et al., 2006). The slices were maintained at room temperature in a submersion chamber with artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 24 NaHCO3, and 15 glucose, bubbled with 95% O2/5% CO2. Slices were incubated for at least 2 h before removal for experiments. For electrophysiology experiments, slices were transferred to recording chambers (preheated to 32°C) where they were superfused with oxygenated ACSF. Monophasic, constant-current stimuli (100 μs) were delivered with a bipolar silver electrode placed in the stratum radiatum of area CA3, and the field EPSPs (fEPSPs) were recorded in the stratum radiatum of area CA1 with electrodes filled with ACSF (resistance, 2–4 MΩ). Baseline fEPSPs were monitored by delivering stimuli at 0.033 Hz. fEPSPs were acquired, and amplitudes and maximum initial slopes measured, using pClamp 10 (Molecular Devices). LTP was induced with a high-frequency stimulation (HFS) protocol consisting of two 1 s long 100 Hz trains, separated by 60 s, delivered at 70–80% of the intensity that evoked spiked fEPSPs (Tsokas et al., 2007).
Drug treatment.
Drugs were prepared as stock solutions and were diluted to the final concentration before use. Incubation of hippocampal slices with drugs was performed in either recording chambers or maintenance chambers as needed. The final concentrations and sources of the drugs were as follows: MitoQ 10 Methanesuflonate (MitoQ, 500 nm; Antipodean Pharmaceuticals), EUK134 (250 nm; Cayman Chemical), diphenyliodonium (DPI, 10 nm; Sigma). Aβ1-42 stock (100 μm; Bachem) was prepared in water and stored at −20°C for at least 24 h before use at a final concentration of 500 nm. This preparation yields ample Aβ oligomers (Ma et al., 2010). Scrambled Aβ1-42 peptide (500 nm; Anaspec) was prepared in the same way as Aβ(1-42).
Detection of mitochondrial superoxide in hippocampal slices.
Upon completion of either drug (or vehicle) treatment, hippocampal slices were incubated with 5 μm MitoSOX Red, a mitochondrial superoxide indicator (prepared as 5 mm stock solution in DMSO immediately before the experiments) (Invitrogen) for 10 min. Slices then were fixed with ice-cold 4% paraformaldehyde in PBS overnight at 4°C. Slices were further cut into 40 μm sections with a vibratome and mounted onto presubbed slides with Vectashield mounting medium with DAPI (Vector Laboratories). The sections were imaged using a Leica TCS SP5 confocal microscope (Leica) at 630×. All parameters (pinhole, contrast, gain, offset) were held constant for all sections from the same experiment.
Data analysis.
Data are presented as mean ± SEM. Statistics were performed using Origin software (OriginLab). For comparison between two groups, a two-tailed independent Student's t test was used. For comparisons between multiple groups, a one-way ANOVA was used followed by individual post hoc tests when applicable. Error probabilities of p < 0.05 were considered statistically significant.
Results
Aβ-induced LTP impairments are rescued by EUK134 and MitoQ
We first confirmed that mouse hippocampal slices treated with exogenous Aβ1-42 (500 nm) resulted in impaired LTP in our recording conditions (Fig. 1A,C), consistent with previous reports (Shankar et al., 2008; Ma et al., 2010). To explore the relationship between mitochondrial ROS (mainly superoxide) with Aβ-induced LTP failure, we applied two structurally distinct ROS scavengers: EUK134, a synthetic SOD and catalase mimetic (Rong et al., 1999), and MitoQ, a novel mitochondria-targeted antioxidant (Smith and Murphy, 2010). In slices treated with either EUK134 (250 nm) or MitoQ (500 nm), HFS was capable of inducing LTP in the presence of Aβ (Fig. 1A,C). We further observed that neither EUK134 nor MitoQ alone significantly enhanced HFS-induced LTP (Fig. 1B,C). To ensure that the LTP blockade by Aβ1-42 was not due to nonspecific peptide effects, we applied a scrambled Aβ1-42 peptide to hippocampal slices. As shown in Figure 1D, HFS-induced LTP was not inhibited by the scrambled peptide. Together, these findings suggest that Aβ-induced LTP impairments can be rescued by decreasing mitochondrial ROS levels.
Figure 1.

Aβ-induced LTP impairments are rescued by EUK134 and MitoQ. A, HFS induced LTP in vehicle-treated slices (black triangles, n = 7), but not in slices pretreated with 500 nm Aβ1-42 (gray triangles, n = 9). In contrast, Aβ1-42 treatment did not inhibit LTP in the presence of either EUK134 (250 nm, dark gray circles, n = 6) or MitoQ (500 nm, light gray hexagons, n = 8). The dashed line represents the normalized baseline value of 100%. B, Compared with vehicle-treated slices (gray diamonds, n = 10), either 500 nm MitoQ (black triangles, n = 7), or 250 nm EUK134 (gray circles, n = 7) alone did not have an effect on LTP. In addition, treatment of slices with either EUK134 (gray hexagons, n = 10) or MitoQ (black hexagons, n = 9) did not affect baseline fEPSPs. C, Cumulative data showing mean fEPSP slopes 80 min post-HFS based on the LTP experiments in A and B. D, HFS-induced LTP was expressed normally in slices treated with scrambled Aβ1-42 peptide (500 nm, n = 5). sc, Scrambled.
Decreasing NADPH oxidase activity either with DPI or by deleting the gp91phox subunit does not prevent Aβ-induced LTP impairments
In addition to mitochondria, another source of superoxide in the hippocampus is NADPH oxidase (Kishida et al., 2006; Kishida and Klann, 2007). NADPH oxidase is a membrane-bound enzyme complex composed of six subunits, with gp91phox the subunit that encodes the oxidase component (Kishida and Klann, 2007). To determine whether NADPH oxidase-mediated ROS generation is involved in the impairments in synaptic plasticity induced by Aβ, we first used the NADPH oxidase inhibitor DPI, which has been demonstrated to have neuroprotective effects at low concentrations (Qian et al., 2007). HFS-induced LTP was sustained when induced in the presence of DPI (10 nm) alone, but declined to baseline within 1 h after HFS when Aβ was present (Fig. 2A). In addition, in hippocampal slices from mice in which the gp91phox subunit was deleted, normal LTP was observed following HFS (Fig. 2B). However, in slices treated with Aβ, LTP was impaired (Fig. 2B). These findings indicate that neither pharmacologically nor genetically decreasing of NADPH oxidase activity impacts Aβ-induced LTP impairments in the hippocampus.
Figure 2.

Inhibition of NADPH oxidase activity with either DPI or by genetic deletion of gp91phox does not prevent Aβ-induced LTP impairments. A, LTP persisted in slices treated with DPI alone (10 nm; open circles, n = 5) and Aβ1-42 (500 nm; filled circles, n = 5) treatment resulted in impaired LTP in the presence of DPI. DPI alone (filled triangles, n = 7) did not alter baseline fEPSPs. B, In slices from gp91phox knock-out (KO) mice, HFS elicited persistent LTP (open circles, n = 6) but LTP was impaired in the presence of Aβ1-42 (500 nm; filled circles, n = 7).
MitoQ but not DPI prevents the elevation in mitochondrial superoxide caused by Aβ
To obtain direct evidence that the prevention of the Aβ-induced impairments in synaptic plasticity described above was actually related to a reduction in mitochondrial superoxide levels, we stained live slices with MitoSOX Red, a fluorogenic dye that is selective for the detection of superoxide in mitochondria. The MitoSOX Red reagent is chemically targeted to mitochondria and when oxidized by superoxide exhibits red fluorescence (Hu et al., 2007). Hippocampal slices treated with Aβ demonstrated a significant enhancement in red fluorescence signal in area CA1 (Fig. 3A), indicating an increase in superoxide levels in mitochondria. The Aβ-induced increase in mitochondrial superoxide was prevented when MitoQ was added, as indicated by a blunting of the MitoSOX fluorescent signal (Fig. 3A). In contrast, Aβ elicited a considerable increase in superoxide levels in the presence of DPI (Fig. 3A). To ensure that the increase in MitoSOX staining induced by Aβ was not due to nonspecific peptide effects, we performed experiments in which slices were treated with scrambled Aβ1-42 peptide followed by staining for MitoSOX. As shown in Figure 3B, the scrambled peptide had no impact on MitoSOX staining compared with the control. These results suggest that ROS production in hippocampal slices in response to Aβ is largely derived via mitochondria. Moreover, these findings are in agreement with the results from the electrophysiology experiments in that MitoQ, but not DPI, rescued Aβ-induced impairments in LTP (Figs. 1, 2).
Figure 3.
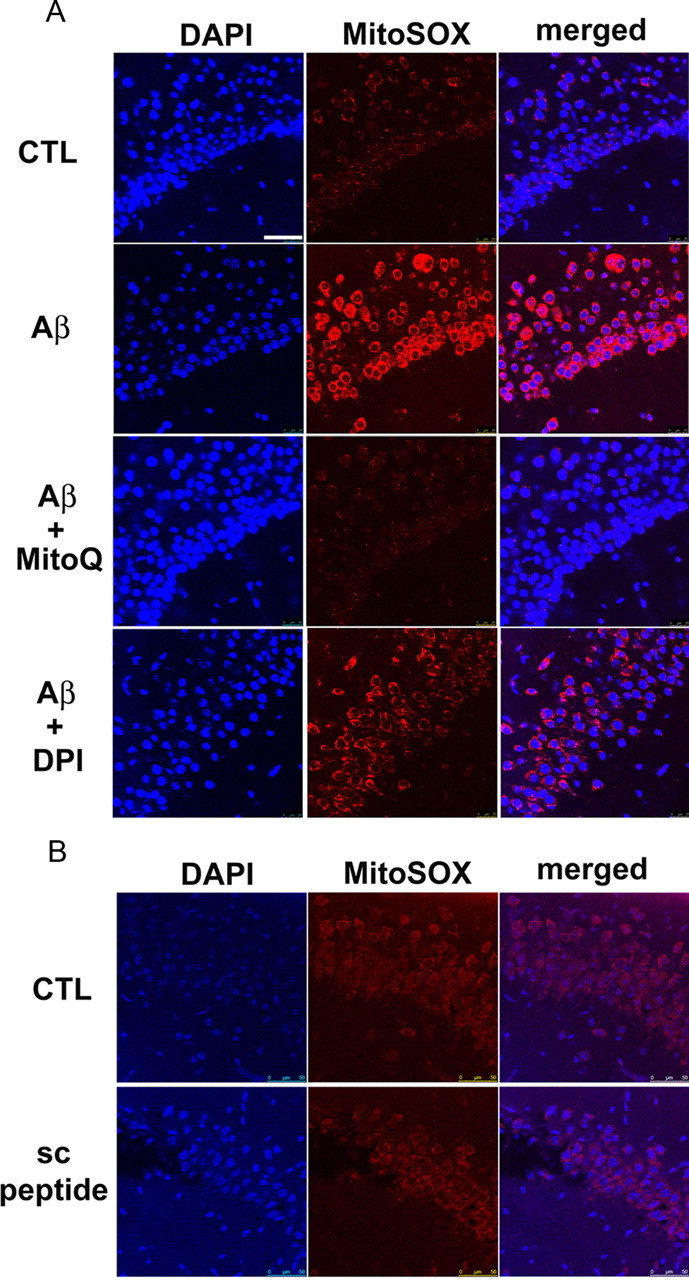
MitoQ, but not DPI, prevents Aβ-induced increases in mitochondrial superoxide. A, Treatment of hippocampal slices with Aβ1-42 (500 nm) for 60 min increased the MitoSOX fluorescent signal (red) in area CA1 compared with control slices. DAPI staining is shown as blue. Pretreatment of slices with MitoQ (500 nm) blunted the Aβ-induced elevation in the MitoSOX fluorescent signal. In slices pretreated with DPI (10 nm), Aβ still caused increases in the MitoSOX fluorescent signal. B, Treatment of slices with a scrambled Aβ1-42 peptide (500 nm) for 60 min did not alter the MitoSOX fluorescent signal. Results are representative of three independent experiments. Scale bar, 50 μm. CTL, Control; sc, scrambled.
MitoQ and EUK134 rescue hippocampal LTP impairments in APP/PS1 mutant mice
To further explore the role of mitochondrial ROS in AD-related impairments in hippocampal synaptic plasticity, we used slices from an APP/PS1 AD transgenic mouse model. Hippocampal LTP in APP/PS1 mice (10–12 months old) was inhibited, compared with littermate controls (Fig. 4A,B), consistent with previously described learning deficits in these mice at this age (Reiserer et al., 2007). Interestingly, when slices from APP/PS1 mice were treated with either MitoQ (500 nm) or EUK134 (250 nm), LTP was significantly enhanced to the level of wild type (Fig. 4A–C). These results are similar to those from experiments in which Aβ-induced impairments in LTP were rescued by MitoQ and EUK134 (Fig. 1A) and are consistent with the notion that Aβ-induced production of ROS from mitochondria impairs synaptic plasticity.
Figure 4.

MitoQ and EUK134 prevent LTP impairments in APP/PS1 mutant mice. A, HFS-induced LTP was impaired in slices from 10 to 12 month old APP/PS1 transgenic mice (gray circles, n = 8), compared with slices from wild-type (WT) mice in which HFS induced normal LTP (black circles, n = 6). In APP/PS1 slices treated with MitoQ (500 nm), HFS induced LTP similar to that observed in wild-type mice (triangles, n = 7). B, Cumulative data showing mean fEPSP slopes 80 min post-HFS. C, Impairment of hippocampal LTP in APP/PS1 mice (gray circles, n = 10) was prevented with EUK134 (250 nm) treatment (open circles, n = 4).
Aβ fails to block hippocampal LTP and increase mitochondrial superoxide levels in SOD-2 transgenic mice
A key antioxidant enzyme that scavenges superoxide in mitochondria is SOD-2 (Kishida and Klann, 2007). We previously showed that SOD-2 overexpression decreased mitochondrial superoxide in cultured neurons and extended the life span of the mice (Hu et al., 2007). Given our findings that indicate that reducing mitochondrial superoxide levels with pharmacological agents (MitoQ and EUK134) could alleviate Aβ-induced LTP impairments, we hypothesized that normal LTP would be expressed in Aβ-treated slices from SOD-2 transgenic mice. As shown in Figure 5, Aβ did not inhibit LTP in hippocampal slices from SOD-2 transgenic mice. In comparison, in slices from age-matched wild-type mice treated with Aβ, LTP declined to baseline levels within 1 h after HFS. These results indicate that increased endogenous expression of SOD-2 prevents Aβ-induced impairments in hippocampal synaptic plasticity.
Figure 5.

Aβ failed to block hippocampal LTP in SOD-2 transgenic mice. A, HFS-induced LTP (dark gray triangles, n = 8) was blocked by Aβ1-42 (500 nm) in wild-type (WT) hippocampal slices (light gray triangles, n = 7). In slices from SOD-2 transgenic mice, HFS was able to induce LTP in the presence of Aβ1-42 (500 nm; gray circles, n = 5). B, Cumulative data showing mean fEPSP slopes 80 min post-HFS.
In a parallel series of experiments, we treated slices with Aβ and then stained them with MitoSOX to determine how mitochondrial superoxide levels were regulated in SOD-2 transgenic mice. The basal levels of superoxide in mitochondria were slightly elevated in SOD-2 transgenic mice compared with wild-type littermates (Fig. 6). Importantly, Aβ-induced increases in mitochondrial superoxide levels were prevented in SOD-2 transgenic mice (Fig. 6). These results are consistent with those in which Aβ failed to inhibit LTP in slices from SOD-2 transgenic mice, and are consistent with the idea Aβ-induced increases in mitochondrial superoxide cause impairments in hippocampal synaptic plasticity.
Figure 6.

Aβ-induced increases in mitochondrial superoxide were prevented in SOD-2 transgenic mice. Aβ-induced increases in the MitoSOX fluorescent (red) signal were absent in slices from SOD-2 transgenic mice. MitoSOX fluorescent intensity was slightly elevated in SOD-2 transgenic mice compared with wild-type (WT) littermates. Results are representative of three independent experiments. Scale bar, 50 μm.
Discussion
Many lines of evidence indicate that impairment of synaptic plasticity by abnormal accumulation of soluble Aβ occurs before plaque formation and neurodegeneration in AD, lending support to the notion that Alzheimer's disease is a synaptic failure (Selkoe, 2002; Oddo et al., 2003; Jacobsen et al., 2006; Haass and Selkoe, 2007; Ma et al., 2010; Tomiyama et al., 2010). Developing a mechanistic understanding of and accordingly reversing such synaptic dysfunction caused by Aβ could yield important insights into therapeutic targets for AD. In the present study, we have demonstrated that Aβ-induced inhibition of LTP was reversed by a mitochondria-targeted ROS scavenger (MitoQ), general ROS scavengers (EUK134), and by genetically overexpressing SOD-2, the mitochondrial SOD. The reversal of the synaptic plasticity deficits by the mitochondria-targeted scavengers correlated with their ability to prevent increases in mitochondrial superoxide elicited by Aβ. To our knowledge, our findings are the first to demonstrate a causal relationship between synaptic plasticity dysfunction and mitochondrial ROS production in models of AD.
Previous studies indicated that the role that ROS play in synaptic plasticity and memory is a double-edged sword. On one hand, ROS have a physiological role, in that their production is necessary to maintain normal synaptic plasticity. For example, superoxide levels in hippocampal neurons were increased upon activation of NMDA receptors, a key event for LTP induction (Bindokas et al., 1996). It was further shown that LTP induction was abolished by exogenous SOD and cell-permeable manganese porphyrin compounds that mimic SOD (Klann, 1998; Klann et al., 1998). In addition, the effects of ROS on LTP involve the activation of protein kinase C and mitogen-activated protein kinases, both critical signaling molecules for LTP induction (Knapp and Klann, 2002; Kishida et al., 2005; Huddleston et al., 2008). On the other hand, ROS have been linked to synaptic pathology associated with aging and neurodegenerative diseases, including AD (Balaban et al., 2005; Lin and Beal, 2006). Indeed, various types of ROS-mediated oxidative stress are used consistently as biomarkers of AD brain pathology and antioxidants have been used as a therapeutic treatment for AD in clinical trials (Praticò, 2008).
A plausible explanation for these seemingly contradicting data is that ROS act both as signaling molecules required for normal synaptic function and as oxidative stressors causing harmful effects that impede synaptic plasticity. Usually endogenous antioxidants (SOD, vitamin E, C, etc) are capable of clearing sufficient levels of ROS, preventing neurons from being damaged. However, it is likely that with aging and in pathological conditions such as AD, unusually high levels of ROS are produced consistently, overwhelming the ability of the endogenous antioxidants to remove them, resulting in impairment of synaptic function. For example, we observed a robust increase of mitochondrial superoxide caused by Aβ in hippocampal slices (Figs. 3, 6), which was not observed in control, healthy slices. Moreover, ROS include a variety of species produced from different sources (Balaban et al., 2005; Murphy, 2009). It may be simplistic to label ROS as either “good” or “bad” molecules; they appear to play different roles in synaptic plasticity, depending on their subcellular localization and on the age of the animals being studied. For instance, three SOD isozymes have been identified in mammals and they all catalyze the same chemical reaction through which superoxide is dismutated and detoxified. Although hippocampal LTP was impaired in transgenic mice overexpressing Cu/Zn-SOD (SOD-1), which is found mainly in intracellular compartments (Kamsler and Segal, 2003), in SOD-2 (located in mitochondria) overepxressing transgenic mice, LTP was not inhibited (Hu et al., 2007), which was consistent with our observations in the current study (Fig. 5). In addition, in mice that overexpress extracellular SOD, the expression of LTP was age-dependent in that young mice (3–4 months of age) exhibited impaired LTP (Thiels et al., 2000; Hu et al., 2006) whereas old mice (>20 months of age) exhibited enhanced LTP (Hu et al., 2006). Interestingly, in the present study, we induced LTP with two trains of HFS, a milder LTP-inducing stimulation (Tsokas et al., 2007), and observed that there was a trend toward enhanced LTP in wild-type slices treated with MitoQ (Fig. 1) and in slices from SOD-2 transgenic mice (Fig. 5). The molecular mechanisms that underlie enhanced synaptic plasticity and cognitive function recently have emerged as an important topic (Lee and Silva, 2009). Exploration of the signaling mechanisms linking the reduction of mitochondrial superoxide and enhanced LTP may yield important insights on this issue.
In view of the complexity of ROS function in the nervous system and their diverse effects on synaptic function, it is not surprising that current clinical trials with antioxidants such as vitamin E resulted in either a marginally positive effect or no effect on the cognitive function of Alzheimer patients (Praticò, 2008). Our findings suggest that the specificity and subcellular targeting of antioxidants play an important role in the modulation of synaptic plasticity by ROS, and thus may determine their effectiveness in treating cognitive dysfunction. From this point of view, generating more specific mitochondrial antioxidants might provide a way to improve antioxidant therapy for the treatment of Alzheimer's disease.
Footnotes
This work was supported by National Institutes of Health Grants NS034007 and NS047384 (E.K.) and AG029977 (R.G.P.), and the Alzheimer's Association (E.K.).
M.P.M. holds stock in Antipodean Pharamaceuticals, which is commercializing MitoQ as a pharmaceutical.
References
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont M, Wille E, Stack C, Calingasan NY, Beal MF, Lin MT. Reduction of oxidative stress, amyloid deposition, and memory deficit by manganese superoxide dismutase overexpression in a transgenic mouse model of Alzheimer's disease. FASEB J. 2009;23:2459–2466. doi: 10.1096/fj.09-132928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer's disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5167–5179. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Ho YS, Vincent R, Dey MS, Slot JW, Crapo JD. Transgenic models for the study of lung antioxidant defense: enhanced manganese-containing superoxide dismutase activity gives partial protection to B6C3 hybrid mice exposed to hyperoxia. Am J Respir Cell Mol Biol. 1998;18:538–547. doi: 10.1165/ajrcmb.18.4.2959. [DOI] [PubMed] [Google Scholar]
- Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. J Neurosci. 2006;26:3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu D, Cao P, Thiels E, Chu CT, Wu GY, Oury TD, Klann E. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol Learn Mem. 2007;87:372–384. doi: 10.1016/j.nlm.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huddleston AT, Tang W, Takeshima H, Hamilton SL, Klann E. Superoxide-induced potentiation in the hippocampus requires activation of ryanodine receptor type 3 and ERK. J Neurophysiol. 2008;99:1565–1571. doi: 10.1152/jn.00659.2007. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5161–5166. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Kamsler A, Segal M. Paradoxical actions of hydrogen peroxide on long-term potentiation in transgenic superoxide dismutase-1 mice. J Neurosci. 2003;23:10359–10367. doi: 10.1523/JNEUROSCI.23-32-10359.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Pao M, Holland SM, Klann E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J Neurochem. 2005;94:299–306. doi: 10.1111/j.1471-4159.2005.03189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klann E. Cell-permeable scavengers of superoxide prevent long-term potentiation in hippocampal area CA1. J Neurophysiol. 1998;80:452–457. doi: 10.1152/jn.1998.80.1.452. [DOI] [PubMed] [Google Scholar]
- Klann E, Roberson ED, Knapp LT, Sweatt JD. A role for superoxide in protein kinase C activation and induction of long-term potentiation. J Biol Chem. 1998;273:4516–4522. doi: 10.1074/jbc.273.8.4516. [DOI] [PubMed] [Google Scholar]
- Knapp LT, Klann E. Potentiation of hippocampal synaptic transmission by superoxide requires the oxidative activation of protein kinase C. J Neurosci. 2002;22:674–683. doi: 10.1523/JNEUROSCI.22-03-00674.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Calingasan NY, Yu F, Mauck WM, Toidze M, Almeida CG, Takahashi RH, Carlson GA, Flint Beal M, Lin MT, Gouras GK. Increased plaque burden in brains of APP mutant MnSOD heterozygous knockout mice. J Neurochem. 2004;89:1308–1312. doi: 10.1111/j.1471-4159.2004.02455.x. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, Tampellini D, Klann E, Blitzer RD, Gouras GK. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e12845. doi: 10.1371/journal.pone.0012845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC. The long-term potential of LTP. Nat Rev Neurosci. 2003;4:923–926. doi: 10.1038/nrn1258. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA. Long-term potentiation: a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- Massaad CA, Washington TM, Pautler RG, Klann E. Overexpression of SOD-2 reduces hippocampal superoxide and prevents memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:13576–13581. doi: 10.1073/pnas.0902714106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. 1995;9:202–209. doi: 10.1038/ng0295-202. [DOI] [PubMed] [Google Scholar]
- Praticò D. Oxidative stress hypothesis in Alzheimer's disease: a reappraisal. Trends Pharmacol Sci. 2008;29:609–615. doi: 10.1016/j.tips.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Qian L, Gao X, Pei Z, Wu X, Block M, Wilson B, Hong JS, Flood PM. NADPH oxidase inhibitor DPI is neuroprotective at femtomolar concentrations through inhibition of microglia over-activation. Parkinsonism Relat Disord. 2007;13:S316–S320. doi: 10.1016/S1353-8020(08)70023-3. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Reiserer RS, Harrison FE, Syverud DC, McDonald MP. Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer's disease. Genes Brain Behav. 2007;6:54–65. doi: 10.1111/j.1601-183X.2006.00221.x. [DOI] [PubMed] [Google Scholar]
- Rong Y, Doctrow SR, Tocco G, Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci U S A. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer disease: mechanistic understanding predicts novel therapies. Ann Intern Med. 2004;140:627–638. doi: 10.7326/0003-4819-140-8-200404200-00047. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE. The synaptic Abeta hypothesis of Alzheimer disease. Nat Neurosci. 2005;8:977–979. doi: 10.1038/nn0805-977. [DOI] [PubMed] [Google Scholar]
- Thiels E, Urban NN, Gonzalez-Burgos GR, Kanterewicz BI, Barrionuevo G, Chu CT, Oury TD, Klann E. Impairment of long-term potentiation and associative memory in mice that overexpress extracellular superoxide dismutas. J Neurosci. 2000;20:7631–7639. doi: 10.1523/JNEUROSCI.20-20-07631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T, Matsuyama S, Iso H, Umeda T, Takuma H, Ohnishi K, Ishibashi K, Teraoka R, Sakama N, Yamashita T, Nishitsuji K, Ito K, Shimada H, Lambert MP, Klein WL, Mori H. A mouse model of amyloid beta oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsokas P, Ma T, Iyengar R, Landau EM, Blitzer RD. Mitogen-activated protein kinase upregulates the dendritic translation machinery in long-term potentiation by controlling the mammalian target of rapamycin pathway. J Neurosci. 2007;27:5885–5894. doi: 10.1523/JNEUROSCI.4548-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]