

Abstract
Background
Quantitative real-time PCR (qRT-PCR) is currently the most accurate method for detecting differential gene expression. Such an approach depends on the identification of uniformly expressed 'housekeeping genes' (HKGs). Extensive transcriptomic data mining and experimental validation in different model plants have shown that the reliability of these endogenous controls can be influenced by the plant species, growth conditions and organs/tissues examined. It is therefore important to identify the best reference genes to use in each biological system before using qRT-PCR to investigate differential gene expression. In this paper we evaluate different candidate HKGs for developmental transcriptomic studies in the economically-important flax fiber- and oil-crop (Linum usitatissimum L).
Results
Specific primers were designed in order to quantify the expression levels of 20 different potential housekeeping genes in flax roots, internal- and external-stem tissues, leaves and flowers at different developmental stages. After calculations of PCR efficiencies, 13 HKGs were retained and their expression stabilities evaluated by the computer algorithms geNorm and NormFinder. According to geNorm, 2 Transcriptional Elongation Factors (TEFs) and 1 Ubiquitin gene are necessary for normalizing gene expression when all studied samples are considered. However, only 2 TEFs are required for normalizing expression in stem tissues. In contrast, NormFinder identified glyceraldehyde-3-phosphate dehydrogenase (GADPH) as the most stably expressed gene when all samples were grouped together, as well as when samples were classed into different sub-groups.
qRT-PCR was then used to investigate the relative expression levels of two splice variants of the flax LuMYB1 gene (homologue of AtMYB59). LuMYB1-1 and LuMYB1-2 were highly expressed in the internal stem tissues as compared to outer stem tissues and other samples. This result was confirmed with both geNorm-designated- and NormFinder-designated-reference genes.
Conclusions
The use of 2 different statistical algorithms results in the identification of different combinations of flax HKGs for expression data normalization. Despite such differences, the use of geNorm-designated- and NormFinder-designated-reference genes enabled us to accurately compare the expression levels of a flax MYB gene in different organs and tissues. Our identification and validation of suitable flax HKGs will facilitate future developmental transcriptomic studies in this economically-important plant.
Background
Flax is one of the earliest cultivated plants known to man and the remains of seeds found in different archeological sites suggest that it was also used before domestication, most probably for textiles [1]. Cultivated flax (Linum usitatissimum L.) is an annual, diploid, self-pollinated crop used as a source of both high-quality, cellulose-rich bast fibers and oil. The main areas of fiber production are currently found in France (77,000 ha) with an annual turnover of over 200 million euros followed by Russia and China (80,000 ha). In contrast, oil varieties (linseed) are mainly grown in India (930,000 ha), Canada (811,000 ha) and China (570,000 ha) [2].
Although flax fibers have now been largely replaced in textiles by cotton or synthetic fibers, they still are used in the fabrication of high-quality linen material and are also incorporated in the polymeric matrix during the fabrication of biocomposites in order to improve their mechanical properties [3].
In the flax stem, fibers are derived from the procambial cells of the protophloem [4] and are referred to as bast fibers. Individual bast fibers are extremely-long (≤ 77 mm) single cells [5] characterized by the presence of a very thick secondary cell-wall containing high amounts of cellulose and very low amounts of lignins as compared to the shorter fibers found in the xylem [6]. Bast fibers provide mechanical support to the plant and are organized in bundles that occupy the great majority of the external tissues between the epidermis and the vascular cambium. Flax fiber extraction is initiated during the retting process when stems are left on the ground in order to promote microbiological-mediated separation of the fiber bundles from the surrounding tissues [7].
Secondary cell-wall formation is most often studied in woody species such as poplar, pine and eucalyptus, or else in model species such as Arabidopsis and tobacco. More recently, growing interest in the unusual structure of the secondary cell wall of bast fibers (high crystalline cellulose, low lignin) is stimulating transcriptomic and genomic studies in flax and other bast fiber species [8-10]. A better understanding of the molecular regulation controlling fiber cell-wall metabolism should increase our knowledge of the very complex coordination necessary for the production and assembly of different polymers within the cell wall.
Flax is a suitable model for genetic and functional genomics as it is autogamous, easy to grow, has a relatively short vegetative stage, and can be genetically transformed [11-13]. The first studies on flax DNA structure were undertaken almost thirty years ago [14,15] and the first cDNA sequence was deposited in Genbank in 1993 [16]. Previous analyses of the flax transcriptome have mainly involved studies of single gene expression by Northern blot [17-23] or multiple gene expression by classical RT-PCR [24]. Only relatively few studies [10,25] have reported the use of qRT-PCR in flax. Quantitative real-time RT-PCR (qRT-PCR) is currently the most accurate method for detecting low abundant mRNAs and is capable of detecting slight variations in gene expression in different tissues of the same plant [26]. When compared to traditional methods used to evaluate transcript accumulation, the main advantages of qRT-PCR are its very high sensitivity and specificity. However, in order to obtain reliable and reproducible results, it is necessary to include suitable internal controls. Indeed, the high sensitivity of qRT-PCR can lead to misinterpretation of expression data [27], especially when biological samples show similar gene expression levels. Slight variations in the RNA integrity or quantity, in the reverse transcription yield, or more generally in the transcriptional activity of the studied organ can all have a major impact on quantification. It is therefore absolutely necessary to develop a normalisation strategy before undertaking gene expression studies. The identification of suitable control genes is thus an important challenge in transcriptome analyses and the major aim is to identify genes ubiquitously expressed in every tissue independently of the experimental context [28].
In the past a number of different genes were commonly used for normalising expression, especially in classical RT-PCR approaches. Most of these genes proved to be suitable for such studies at the time, mainly because of the low sensibility of RT-PCR. More recently, the same reference genes have also been used for qRT-PCR and it has been assumed that their expression is perfectly stable even though this has not necessarily been experimentally verified. Examples of such commonly-used genes include GAPDH (glyceraldehyde-3-phosphate dehydrogenase) [29], cyclophilin and 18S rRNA [30], actin [31], EF1alpha [32], ubiquitin [33] and beta tubulin [34].
Surprisingly, there are relatively few reports concerning the study of stably-expressed genes, referred to as housekeeping genes (HKGs), in different plant species, even though qRT-PCR is routinely used in many research projects. Suitable HKGs have been identified in model species with sequenced genomes such as rice [35], Arabidopsis [36], grapevine [37] and poplar [38], as well as in economically-important crops such as wheat [39], barley [40], Coffee [41], tomato [42], potato [43] and soybean [44]. In most cases, gene expression stability was determined by using statistical approaches such as geNorm [45] and NormFinder [46]. The geNorm algorithm allows the identification of the most suitable reference gene(s) and the optimal number of genes that should be used. It relies on a pairwise comparison and evaluates the variation of relative quantity ratios for each gene pair in a set of expression data. The NormFinder algorithm depends on a statistical and mathematical model that not only estimates the overall expression variation of a candidate gene, but also considers the variation between the chosen subgroups.
In the present study, we have evaluated the expression profiles of 13 putative HKGs during the development of flax plants with a special focus on the stem which contains two major cell-wall tissue types: 1) inner tissues (lignin-rich secondary cell walls of xylem) and 2) outer tissues (lignin-poor, cellulose-rich secondary cell walls of bast fibers). The two algorithms geNorm and NormFinder were used in order to determine the best reference genes needed for normalisation. The selected HKGs were then used to normalise gene expression in flax for an investigation of the expression of LuMYB1 - a flax MYB transcription factor highly similar to AtMYB59 [47].
MYB transcription factors are of particular interest in studies on secondary cell wall development since they have been shown to regulate the expression of genes coding enzymes of the phenylpropanoid biosynthetic pathway responsible for the production of lignin monomers [48,49]. We decided to investigate the expression profiles of LuMYB1 since initial transcriptome studies in our laboratory (data not shown) had indicated that the gene was most highly expressed in inner stem tissues and therefore potentially associated with secondary cell wall formation in flax xylem tissues. In addition, AtMYB59 has been shown to undergo alternative splicing leading to different transcripts in rice and Arabidopsis, and therefore represents an interesting model for gene regulation studies.
Results
Selection of candidate reference genes and amplification specificity
Many reference genes, assumed to have a stable expression have been used to normalize expression in transcriptomics. In this study, primers were designed for 20 commonly used housekeeping genes (HKGs) representing different functional classes in flax. Sequences were identified after performing BLASTX searches on a collection of ESTs obtained from a flax fiber-rich cDNA library [24] and at the NCBI for the tubulin sequence. Finally, 13 primer pairs were selected on the basis of their amplification efficiency. The candidate reference genes including those encoding actin, cyclophilin, two elongation factors, five different eukaryotic translation initiation factors, GAPDH, ubiquitin, ubiquitin extension protein and tubulin are described in Table 1. The expression stabilities of these potential flax HKGs were assessed by qRT-PCR on a series of two biological repeats of 13 tissue samples obtained from different tissues at 3 developmental stages. Theses samples correspond to: 1) leaves, apical- and medium-stem tissues (vegetative stage), 2) roots, flowers, apical inner- and outer-stem tissues, medium inner- and outer-stem tissues (flowering stage) and 3) apical inner- and outer-stem tissues, medium inner- and outer-stem tissues (green-capsule stage). The different stem samples and developmental stages were chosen since previous studies [6,50] have shown that the main developmental modifications associated with bast fiber formation and cell wall maturation occur in the apical and medium stem tissues between the vegetative and green capsule stages.
Table 1.
Flax candidate HKG description and comparison with Arabidopsis orthologs.
| Gene abbreviation | GenBank accession number | Gene description | Arabidopsis ortholog locus | Arabidopsis BlastX E-value |
|---|---|---|---|---|
| ACT | GR508908 | Actin | NM_121018 | 1e-145 |
| CYC | GR508910 | Cyclophilin | AF020433 | 1e-48 |
| EF1A | GR508911 | Elongation Factor 1-α | AK230352 | 2e-155 |
| EF2 | GR508907 | Elongation Factor 2 | AC009894 | 3e-162 |
| ETIF1 | GR508906 | Eukaryotic translation initiation Factor 1 | NM_122959 | 2e-49 |
| ETIF3E | GR420420 | Eukaryotic translation initiation Factor 3 E | NM_115589 | 2e-54 |
| ETIF3H | GR420421 | Eukaryotic translation initiation Factor 3 H | NM_100960 | 1e-115 |
| ETIF4F | GR420423 | Eukaryotic translation initiation Factor 4 E | ATU62044 | 8e-72 |
| ETIF5A | GR508912 | Eukaryotic translation initiation Factor 5 A | NM_101261 | 4e-78 |
| GAPDH | CV478202 | Glyceraldehyde 3-phosphate dehydrogenase | NM_101214 | 1e-120 |
| UBI | CV478829 | Ubiquitin | DQ793133 | 3e-71 |
| UBI2 | GR508909 | Ubiquitin extension protein | NM_130278 | 5e-66 |
| TUA | CA482867 | α-Tubulin | AK226954 | 8e-103 |
Specific transcript amplification was confirmed by the presence of a single peak in the melting curve obtained after amplification. The amplified products were also further analyzed by agarose gel electrophoresis and ethidium bromide staining (Figure 1). Only a single band with the expected size was detected in each experiment. The amplification efficiencies are indicated in Table 2.
Figure 1.

Real-time quantitative RT-PCR amplification specificity. Amplified fragments obtained after qRT-PCR were separated by agarose gel electrophoresis. Amplification primers were designed for Actin (ACT), GAPDH, Cyclophilin (CYC), Elongation factor 1A (EF1A) and 2 (EF2), Ubiquitin (UBI), Ubiquitin extension protein (UBI 2), Tubulin (TUA), Eucaryotic Translation Initiation Factors 1A (ETIF1A), 3E (ETIF3E), 3H (ETIF3H), 4E (ETIF4E) and 5A (ETIF5A).
Table 2.
Description of candidate reference genes, primers and amplicons for flax housekeeping gene selection.
| Gene abbreviation | Gene description | Primer Sequence (Forward/Reverse Primer) |
Amplicon length (bp) | Efficiencies (%) |
|---|---|---|---|---|
| ACT | Actin | 5'-TCCAGGCCGTTCTTTCTCTA-3' | 153 | 97.9 |
| 5'-CTGTAAGGTCACGACCAGCA-3' | ||||
| CYC | Cyclophilin | 5'-TGATTGCGGTCAGCTGTAAG-3' | 147 | 93.5 |
| 5'-AGGTGAAACGCTAGGCAGAA-3' | ||||
| EF1A | Elongation Factor 1-α | 5'-GCTGCCAACTTCACATCTCA-3' | 140 | 101.9 |
| 5'-GATCGCCTGTCAATCTTGGT-3' | ||||
| EF2 | Elongation Factor 2 | 5'-GTGGTGCTGAGATCACGAAA-3' | 109 | 98.9 |
| 5'-AGACGGTTATGCTTGTTGGG-3' | ||||
| ETIF1 | Eukaryotic translation initiation Factor 1 | 5'-CCTTGTAGGGCTGAGGGATT-3' | 145 | 104.8 |
| 5'-CTCATCAAGACCACCAGCAA-3' | ||||
| ETIF3E | Eukaryotic translation initiation Factor 3 E | 5'-TTACTGTCGCATCCATCAGC-3' | 106 | 101.8 |
| 5'-GGAGTTGCGGATGAGGTTTA-3' | ||||
| ETIF3H | Eukaryotic translation initiation Factor 3 H | 5'-CAGCGTGCTTGAAGTAACCA-3' | 115 | 101.7 |
| 5'-AACCTCCCTCAAGCATCTCA-3' | ||||
| ETIF4E | Eukaryotic translation initiation Factor 4 E | 5'-TGTGGCTCGAAACTCTGATG-3' | 145 | 103.5 |
| 5'-CCCATCTGGACAGCTTCATT-3' | ||||
| ETIF5A | Eukaryotic translation initiation Factor 5 A | 5'-TGCCACATGTGAACCGTACT-3' | 159 | 103.1 |
| 5'-CTTTACCCTCAGCAAATCCG-3' | ||||
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase | 5'-AGGTTCTTCCCGCTCTCAAT-3' | 138 | 101.7 |
| 5'-CCTCCTTGATAGCAGCCTTG-3' | ||||
| UBI | Ubiquitin | 5'-CTCCGTGGAGGTATGCAGAT-3' | 114 | 94.7 |
| 5'-TTCCTTGTCCTGGATCTTCG-3' | ||||
| UBI2 | Ubiquitin extension protein | 5'-CCAAGATCCAGGACAAGGAA-3' | 129 | 98.6 |
| 5'-GAACCAGGTGGAGAGTCGAT-3' | ||||
| TUA | α-Tubulin | 5'-CCTGTTGGGAGCTTTACTGC-3' | 100 | 102.6 |
| 5'-AAGGTGTTGAAGGCATCGTC-3' |
Expression profile of the reference genes
In order to determine the expression rates of the selected genes, all PCR assays were performed in triplicate. The Ct values for the 13 genes were pooled so that the expression levels of each gene could be compared. These values ranged from 19.70 to 29.25 in all tested samples (Figure 2A) and from 20.93 to 29.24 if only stem tissues were considered (Figure 2B). Six genes (TUA, UBI, GAPDH, EF2, EF1A, and ETIF5A) showed the highest expression levels irrespective of whether all tissues or just stem tissues are considered. ETIF5A showed the lowest median and mean Ct values and UBI2 showed the highest values in both total and stem tissues.
Figure 2.

Expression levels of candidate reference genes in flax tissue samples. Box-plot graphs of Ct values for each reference gene in 13 flax tissue samples (A) or only in stem tissues (B). The black lines show the median values surrounded by lower and upper boxes indicating the first and third quartile. The vertical lines (whiskers) indicate value ranges. The gene abbreviations are the same as in Figure 1.
With regards to gene expression variation, TUA showed the highest value (6.98 cycles) and ETIF4E showed the lowest (3.67 cycles) when all tissues are considered. As might be expected, expression variation was lower when only stem samples are considered. Once again TUA showed the highest value (6.1 cycles) whereas the lowest value was observed for EF1A (3.2 cycles). These results indicated that none of the selected genes showed a near constant expression level and it was therefore necessary to evaluate the best candidates for gene expression normalization.
Expression stability analysis
Since the 13 candidate HKGs show wide variations in expression levels in different flax tissues, it is necessary to use statistical approaches to rank the expression levels and determine the number of housekeeping genes necessary for accurate gene-expression profiling in the selected tissues. We decided to use the two most widely-used algorithms, geNorm and NormFinder.
The geNorm program can be used to identify (in a given set of candidate genes) those genes that are the most stably expressed in different organs or tissues [45]. In this program, an average expression stability value M is calculated for all candidate genes and those genes with the lowest M values are considered to be the most stable. We used geNorm to analyse candidate gene expression levels in 1) total tissues and 2) stem tissues only (Figure 3). When all samples were analysed together, the M value was lowest for EF1A and ETIF5A indicating that they are the most stably expressed gene pair out of the 13 candidates. The alpha-Tubulin gene was the least stably expressed. When only stem tissues were considered, ETIF1 and ETIF4F proved to be the best candidates for normalization (low M value) while EF2 was the worst (high M value). However, in all cases the M values were less than 1 indicating that the selected genes all show relatively acceptable expression stabilities. Nevertheless, our results highlight the fact that it is probably better to choose different reference genes depending on the biological samples to be studied. For example, when only flax stem tissues are considered, the five Eukaryotic translation initiation factors are found amongst the six most stably-expressed genes. In contrast, when all tissues are considered the same five genes show less overall stability and ETIF3H, for example, is the third least-stable gene. Similarly, some genes are stably-expressed when all tissues are considered but appear to be less stable when only stem tissues are taken into account. For example, the ubiquitin gene is the third most-stably expressed gene in total tissues but is only the eighth most stably expressed gene in stems tissues. Finally, the alpha tubulin and elongation factor 2 genes are the two least stable genes in both total tissues and in stem tissues.
Figure 3.

Average expression stability values (M) of flax candidate reference genes. M values of the reference genes were calculated with the geNorm algorithm. Ranking of the stability was performed on 13 different flax tissues (A) or on stem tissues (B). The lowest average expression stability value indicates the most stably expressed gene. The gene abbreviations are the same as in Figure 1.
We also used the geNorm program to calculate the optimal number of HKGs required for accurate normalization in flax (Figure 4). The software calculates the pairwise variation Vn/Vn+1 between two sequential normalization factors NFn and NFn+1 in order to determine the necessity of including an additional control gene for normalization. As long as the V value remains higher than 0.15, an additional reference gene should be added. Our results (Figure 4) showed that the pairwise variation values changed depending upon the samples analyzed. For example, in all tissues, normalization requires the use of three reference genes since only the V3/4 value (0.127) is less than 0.15. However, gene normalization in stem tissues requires the use of only two reference genes since the V2/3 value (0.119) is inferior to the 0.15 cut-off level.
Figure 4.

Determination of the optimal number of reference genes. Pairwise variation (V) values were calculated with the geNorm algorithm. Values under 0.15 indicate that no additional genes are required for the normalization of expression in the selected tissues.
We then used the NormFinder algorithm [46] to identify the most stable gene among our candidates and test whether the use of only one HKG would be suitable for gene expression studies. The NormFinder algorithm requires the analysis of a minimum of 3 genes and a minimum of 2 samples per group. It can analyze the expression data obtained from quantitative methods and choose the best normalization gene from a set of candidates. The software calculates and ranks the stability values for candidate genes within the sample set investigated according to their expression profile. The lowest stability value indicates the most stably expressed gene.
The results of our NormFinder analysis in flax are shown in Table 3. The samples were assembled in one main group or several sub-groups. If all the samples were combined in one group, the best reference gene was GAPDH (stability value 0.069). This gene was also detected as the most stably expressed gene (stability values 0.04 - 0.061) when the samples were distributed into either 2 or 3 sub-groups. However, if the analysis was performed on one main group containing only stem tissues, the most stable gene proved to be ETIF3H (stability value 0.056). Nevertheless, in this case GAPDH proved to be the second-most stably expressed gene (stability value 0.064).
Table 3.
Ranking of candidate genes according to NormFinder.
| 1 Group | 2 Groups | 3 Groups | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Total | Stems | Stems/Others | Apical/Medium/Others | Ext/Int/Others | ||||||
| Rank | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability | Gene | Stability |
| 1 | GAPDH | 0,069 | ETIF3H | 0,056 | GAPDH | 0,061 | GAPDH | 0,048 | GAPDH | 0,040 |
| 2 | UBI2 | 0,099 | GAPDH | 0,064 | ETIF4F | 0,072 | UBI2 | 0,071 | UBI2 | 0,066 |
| 3 | UBI | 0,103 | ETIF3E | 0,080 | UBI2 | 0,081 | ETIF3E | 0,079 | TUA | 0,070 |
| 4 | ETIF3E | 0,108 | ETIF5A | 0,080 | ETIF3E | 0,082 | ETIF4F | 0,081 | UBI | 0,075 |
| 5 | EF1A | 0,112 | EF1A | 0,087 | UBI | 0,104 | ACT | 0,091 | ACT | 0,076 |
| 6 | ETIF5A | 0,118 | CYC | 0,100 | ACT | 0,106 | UBI | 0,094 | ETIF5A | 0,076 |
| 7 | ETIF4F | 0,138 | EF2 | 0,103 | EF1A | 0,129 | CYC | 0,113 | EF1A | 0,076 |
| 8 | CYC | 0,145 | UBI | 0,105 | CYC | 0,130 | EF1A | 0,113 | ETIF1 | 0,078 |
| 9 | EF2 | 0,145 | ACT | 0,107 | ETIF5A | 0,135 | ETIF5A | 0,118 | ETIF4F | 0,082 |
| 10 | ETIF1 | 0,157 | ETIF1 | 0,131 | EF2 | 0,142 | EF2 | 0,126 | ETIF3E | 0,084 |
| 11 | ACT | 0,174 | UBI2 | 0,133 | ETIF1 | 0,149 | ETIF1 | 0,127 | CYC | 0,095 |
| 12 | ETIF3H | 0,189 | ETIF4F | 0,137 | TUA | 0,180 | TUA | 0,166 | EF2 | 0,100 |
| 13 | TUA | 0,206 | TUA | 0,176 | ETIF3H | 0,208 | ETIF3H | 0,182 | ETIF3H | 0,124 |
| Best combination of two genes | EF1A EF2 |
0.028 | EF1A ETIF5A |
0,03 | ETIF3E UBI |
0,03 | ||||
LuMYB1 expression
The use of the geNorm and NormFinder algorithms allowed the identification of different potential HKGs for normalization of gene expression levels in flax. In order to see whether the normalization by these different HKGs modified the qRT-PCR-determined expression levels of a gene of interest, we decided to analyze the developmental expression of a flax MYB gene, LuMYB1. We had previously isolated two different LuMYB1 transcripts, LuMYB1-1 and LuMYB1-2 during a flax EST sequencing project [24]. The alignment of the two sequences showed that they are perfectly identical except for the upstream region of LuMYB1-2 that lacks a 68 bp fragment. LuMYB1 is an ortholog of AtMYB59 which is found to undergo alternative splicing in Arabidopsis [47]. AtMYB59 gives rise to four spliced transcripts (Figure 5A), three of which encode proteins with either one or two MYB domains. A similar pattern of alternative splicing also occurs in two homologous genes (OsMYBAS1, OsMYBAS2) in rice. In this species, three of the four transcripts have been detected and can be aligned with AtMYB59 (Figure 5A). In flax, the LuMYB1-1 and LuMYB1-2 transcripts can also be aligned with the Arabidopsis and rice type 1 and 2 transcripts.
Figure 5.
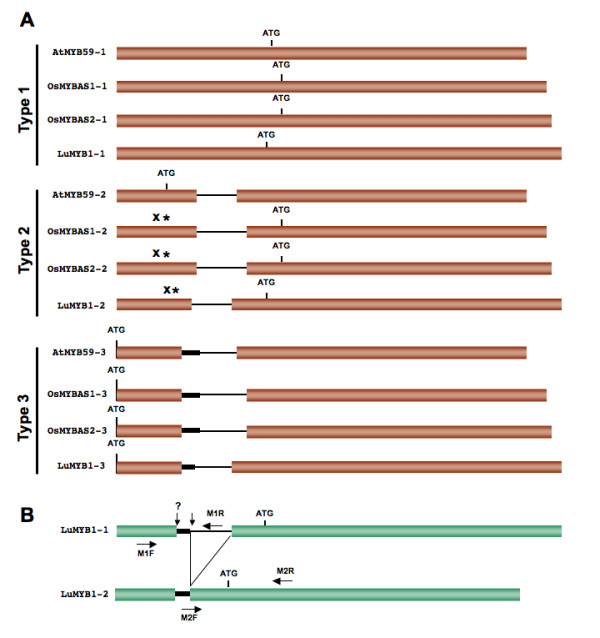
Schematic representation of the differently spliced transcripts of MYB homologues. A: The transcribed sequences are shown in red boxes while the spliced fragments are represented as thin lines. B: Representation of LuMYB1 splice variants. The spliced positions are indicated with vertical arrows. The primer positions used for qRT-PCR are shown with horizontal arrows. X indicates conserved but non-functional start codons and the asterisks indicate stop codons. ATG indicates functional start codons. The hypothetical LuMYB1-3 transcript should be formed by splicing at the position indicated by ?.
Interestingly, both the rice and flax (but not Arabidopsis) type 2 transcripts contain a stop codon close to (and in frame with) the corresponding ATG start codon in Arabidopsis. As a result in both rice and flax, translation is initiated at the next ATG that corresponds to the functional ATG in type 1 transcripts. In consequence, the flax type 1 and 2 ORFs, as in rice, are identical. We have not yet detected a flax type 3 transcript (LuMYB1-3) in our ESTs even though sequence data predicts the existence of such a transcript. Nevertheless, we designed qRT-PCR primers to specifically amplify the two longest flax transcripts. LuMYB1-1 was detected with the M1R primer located in the intron, and LuMYB1-2 with the M2F primer overlapping the 3' splicing site (Figure 5B).
The relative expression levels of LuMYB1 were estimated in the same 13 flax samples that were used previously for the normalization studies (Figure 6). In order to see whether the use of different HKGs (chosen according to geNorm or NormFinder) modified the determined expression profiles of the 2 LuMYB1 splice variants, we compared different HKGs combinations in 1) all flax tissues and 2) in just stem tissues (Figure 6). For all tissues, normalization was accomplished by using the three most stable genes EF1A, ETIF5A and UBI (determined by the geNorm analysis), or by GAPDH, the most stable gene determined by NormFinder when all the samples were assembled into one main group (Table 3, Figure 6A, B). When only stem tissues were analyzed, normalization was accomplished by using the 2 most stable genes ETIF1 and ETIF4F (determined by geNorm), or ETIF3H, the most stable gene determined by NormFinder when only stem tissues are considered (Table 3, Figure 6C, D). Our results indicate that LuMYB1-1 is expressed in all tissues except for the flowers, and that the highest expression levels are found in inner stem tissues, regardless of whether HKGs identified by geNorm or NormFinder were used for normalization. However, since only 1 HKG (GAPDH or ETIF3H) is identified for all samples in the NormFinder algorithm when no sub-groups are defined, we decided to define different sub-groups so as to identify the best HKG couples for normalizing expression data. Different sub-groups were identified (Table 3, Figure 6B) in order to see whether this influenced the choice of HKG couples and subsequent expression normalization in different samples. When all samples were divided into 2 sub-groups (stems/other samples), NormFinder identified EF1A and EF2 as the most stable couple (stability value 0.028). When 3 sub-groups were identified, the best HKG couple identified by NormFinder depended upon the sub-groups chosen. For example, EF1A and ETIF5A (stability value 0.033) represented the best HKG couple for the sub-groups 'apical/medium/other samples', while ETIF3E and UBI (stability value 0.026) represented the best HKG couple for the sub-groups 'external/internal/other samples'. However, despite the fact that NormFinder identified different HKG couples depending upon the sub-groups defined, the use of these different HKG couples to normalize LuMYB1-1 expression data largely confirmed the expression profile previously identified by geNorm and NormFinder (no sub-group, 1 HKG). Determination of the LuMYB1-2 expression profile indicated that the gene was expressed in all tissues (including the flowers) and that, as previously observed for LuMYB1-1, the highest expression levels could be observed in the inner stem tissues (Figure 6E, F). This general expression pattern (highest expression in stem inner tissues) was obtained with both geNorm- and NormFinder-designated HKGs. As for LuMYB1-1, the definition of different sub-groups (and hence different HKG couples) did not fundamentally change the determined LuMYB1-2 expression profile. Nevertheless, our results underline the fact that the use of a particular algorithm and choice of sample set can influence the determined expression value of the gene of interest when the values are relatively close. For example, the use of geNorm for the determination of LuMYB1-2 expression in the stem group (Figure 6G) would suggest that there is little difference between expression levels in stem medium inner tissues at the flowering stage and stem apical inner tissues at the green capsule stage, whereas the use of other HKGs (Figure 6E, F, H) would suggest that LuMYB1-2 expression is higher in stem inner tissues at the green capsule stage.
Figure 6.

Relative quantification of two LuMYB1 splice forms. The relative expression levels of LuMYB1-1 (A, B, C and D) and LuMYB1-2 (E, F, G and H) were determined after normalization. The total tissue (A and E) or stem (C and G) sample sets were normalized by geNorm with EF1A, ETIF5A and UBI or with ETIF1 and ETIF4F respectively. The NormFinder algorithm was also used with different combinations of HKG. The total tissues (B and F) were divided in 1, 2 or 3 groups and normalized with GAPDH alone (1 group, yellow bar), EF1A and EF2 (stem/others, pink bar), EF1A and ETIF5A (apical/medium/others, green bar) ETIF3E and UBI (external/internal/others, blue bar). When the expression was measured only in one group containing the stem tissues (D and H), ETIF3H was used for normalization. V: vegetative F: flowering and C: green capsule stages; A: apical and B: basal parts of the stem; I: internal and E: external stem tissues; LEA: leaves; FLO: flowers. Values = relative expression levels +/- SD.
Discussion
Over the last years several very powerful techniques have been developed to detect differences in gene expression levels between different cell types, tissues and organs. Among these, qRT-PCR is now commonly used in many laboratories to undertake accurate expression profiling of different candidate genes that have been previously identified, either because they are known to be involved in a specific biological process, or else because they have shown an interesting expression profile in global expression analyses (microarrays).
However, reliable relative quantification can only be performed if an accurate normalization is performed following the choice and verification of suitable (i.e. stable) HKGs. Although microarray data have been analyzed to determine the most stably-expressed genes in Arabidopsis [36] and rice [51], it is not always possible to directly transpose suitable HKGs identified in one species to another species. For example UBQ10 is very stable in Arabidopsis [36] but not in rice [52], nor in soybean [44] nor in Brachypodium [53]. Therefore, since there are no universally-suitable reference genes, it is necessary to verify the expression levels of the candidate reference genes under the same experimental conditions as those used for the gene of interest.
In this study, we have measured the expression levels and stability of candidate genes in different tissues at three developmental stages in flax plants. Our major research interest in this species concerns the cell wall formation and development of the phloem (bast) fibers located in the external tissues of the stem. The polymer composition of the secondary cell walls in these long fiber cells (as well as that of the shorter xylem cells located in the inner tissues) has been previously analyzed [6] and we have shown that the abundance and structure of the phenolic polymer lignin varies according to the age and physiological state of the plant. In order to further investigate cell wall formation (including lignification) in flax, we are currently undertaking gene expression profiling by qRT-PCR on different tissues. However, due to important differences between the structure and the metabolic state of cells in flax inner and outer tissues, it is necessary to identify the most relevant combination of reference genes. Normalization of gene expression in flax has previously been done with either a homolog to an Arabidopsis NADH Ubiquinone oxidoreductase determined by cDNA-AFLP [25], or with an Elongation Factor EF1A homolog [10].
We originally selected 20 candidate HKGs from the literature and after determination of amplification efficiencies for the selected primers we retained 13 candidates. We then evaluated their normalization potential using 2 commonly-used algorithms (geNorm [45] and NormFinder [46]) in a systematic study of their expression stability in different flax tissues including stems, flowers, roots and leaves. The NormFinder algorithm [46] generally ranked GAPDH as the most stably expressed gene in all the flax samples irrespective of whether the samples were assembled into 1 main group or divided into several sub-groups. In only 1 case did NormFinder rank GAPDH as the second most stable gene. These results are consistent with those observed recently in Brachypodium distachyon [53] during cold/heat stress and in Coffea arabica [41] in different organs and tissues. In contrast to NormFinder, the geNorm algorithm indicated that we should use 3 HKGs (EFIA, ETIF5A, Ubiquitin) for studies involving a mix of root, leaf, stem and flower tissues. However, the same program indicated that only 2 genes (ETIF1, ETIF4F) should be used when just stem tissues were considered. While EF1A is often described as a stable gene and has been used as a reference in many species [37,43,52,54], there are only relatively few reports of Eucaryotic Translation Initiation Factor genes being used as reference genes. For example in soybean, ETIF1A and ETIF1B genes were ranked as the most stable genes tested when 21 samples were pooled, or when photoperiodic treatments were modified [44]. In contrast, expression patterns of several translation initiation factors were shown to be unstable in wheat [55], and have been excluded as good candidate reference genes in poplar [38] and Darnel ryegrass [54].
We also evaluated the stability of tubulin and actin genes that are very often used to normalize expression data. Although actin has been shown to be a suitable normalization gene in developmental studies [44], it also appears to be unstable in many biological processes [27]. In the same way, tubulin has been shown to be stable during development in orobranche [56], but is apparently unstable (geNorm analyses) during development [53] and abiotic stress [54]. Our results would also suggest that these 2 genes are among the most instable for expression profiling during development in flax.
Altogether, our results showed that the geNorm and NormFinder algorithms came to different conclusions concerning the best candidate HKG(s) to use for expression normalization in flax. In order to see whether the use of these different HKGs modified the determined expression profiles (and hence possible interpretations of biological role) of a gene of interest, we analyzed the expression of LuMYB1 in different tissues and organs of Flax. LuMYB1 is a homolog of AtMYB59, a gene that has been shown to undergo alternative splicing in Arabidopsis and rice [47]. We decided to investigate the expression profile of LuMYB1 since our team is interested in gaining a better understanding of the regulation of cell wall formation in flax and different MYB transcription factors have been previously shown to play an important role in controlling this process [48]. In addition, preliminary transcriptome data in our lab had indicated that LuMYB1 was highly expressed in stem inner tissues. Such an observation could suggest that this gene is involved in the transcriptional regulation of cell-wall genes during the formation of xylem.
Our results showed that the 2 different flax spliceforms (LuMYB1-1 and LuMYB1-2) are strongly expressed in the inner stem tissues, but only expressed at low levels in external stem tissues, leaves and roots. In addition, only LuMYB1-2 is expressed in flowers. Such a result is interesting since previous work [47] has shown that the corresponding Arabidopsis gene is mainly expressed in roots, leaves and seedlings, but only poorly expressed in stem tissues. More recently [57], AtMYB59 has been shown to be specifically expressed during the S phase of the cell cycle in Arabidopsis cell suspensions, suggesting that this gene plays a key role in cell cycle regulation. Our observation that the corresponding flax gene (both spliceforms) are highly expressed in inner stem tissues could also suggest a role for LuMYB1 in cell cycle events since these flax tissues are the site of intense mitotic activity associated with the division of vascular cambium initials to form new xylem cells. However, further work is obviously necessary to confirm this hypothesis.
In addition, our results also revealed that both the choice of HKGs and the decision or not to analyze samples together or in sub-groups modified the determined expression profiles for the gene of interest LuMYB1. Although such choices did not affect the identity of the 4 (stem) tissues showing the highest expression levels, they did affect the ranking of these different tissues. Such an observation suggests that care should be taken when interpreting the biological significance of small differences in gene expression, and that in this case, the researchers should probably consider using different normalizations to verify their data. In addition, it is clear that functional approaches are also necessary to fully understand the role of a given gene in a particular biological process. Nevertheless, our results showing important differences in LuMYB1 expression levels (regardless of the normalization type) would suggest that this gene is potentially associated with the formation of xylem tissue in flax stems.
Conclusions
A considerable quantity of transcriptional data is currently available for major model plant species and in silico analyses can therefore be used to identify suitable HKGs for gene expression normalization in these species. However, for most other plant species, the suitability of potential HKGs identified in the literature or in heterologous databases must be verified by qRT-PCR. In this study, we have identified several suitable reference genes for studying developmental gene expression in flax, with a special focus on stem tissues. The use of HKGs identified by both geNorm and NormFinder algorithms allowed us to determine the expression profiles of a gene of interest (LuMYB1) in a large range of different flax tissues. Our results also showed that certain classically-used reference genes such as actin and tubulin are not necessarily the most suitable for studies of quantitative gene expression in flax. In conclusion, our study has identified suitable HKGs for future developmental transcriptome studies in flax. These genes also represent potentially interesting targets for normalizing gene expression in flax under stress conditions, although it would obviously be necessary to verify their stabilities in this case.
Methods
Plant material
Flax plants (Linum usitatissinum L. cv Barbara were grown in a greenhouse under 16 h/20°C day and 8 h/18°C night conditions. The plants were harvested at three developmental stages: (1) vegetative (56 days after sowing), (2) flowering (131 days after sowing, 50% open flowers) and (3) green capsule stage (159 days after sowing). Following harvest, organs were dissected and the stems were divided into three equal parts: - apical, medium and basal (the latter was discarded because the secondary cell walls of bast fibers have obtained their maximum thickness in this part of the stem and residual metabolism is low and unrelated to fiber maturation events).
For the flowering and green capsule stages, the outer fiber-bearing tissues were separated from the inner woody tissues. The tissues were immediately frozen in liquid nitrogen and stored at -80°C.
RNA isolation, quality control and cDNA synthesis
Frozen tissues were ground in liquid nitrogen using a mortar and a pestle. Total RNA was extracted using the RNeasy Plant Mini Kit (Qiagen) according to the manufacturer's instructions. RNA purity was assessed on a biophotometer (Eppendorf) by determining the OD260/OD280 and OD260/OD230 ratios, which were between 1.8 and 2. Potentially-contaminating DNA was eliminated by treatment with DNAse I using the DNA-free kit (Ambion). RNA concentration and quality were determined by capillary electrophoresis on an Experion labchip electrophoresis system (Bio-Rad) One microgram of total RNA was reverse-transcribed using the Iscript cDNA synthesis kit (Bio-Rad) according to the manufacturer's instructions. The cDNAs were diluted 1:256 with nuclease free water.
Design and validation of reference gene primers
Specific primer pairs were first designed for 20 commonly used housekeeping genes representing distinct functional classes and gene families. These include ubiquitins, actin, tubulin, elongation factors, cytosolic cylophilins and translational initiation factors (Table 2). The sequence of these genes were identified by BLAST searches (Table 1) in a flax EST database obtained from a cDNA library derived from the outer fiber-bearing tissues of flax [24]. For primer design, Primer3 software [58] was used (160 bp maximum length, optimal Tm at 60°C, GC % between 20% and 80%). The absence of secondary structure in the primer annealing fragments was verified using mfold [59]. Five-point standard curves of a 4-fold dilution series (1:16 to 1:4096) were used to calculate the PCR efficiency (E) of each primer pair. The PCR efficiency is given by the equation E = (10(-1/m) -1) × 100 where m is the slope of the linear regression model fitted over log transformed data of the input cDNA concentration versus Ct values according to the linear equation y = m log(x) + b. After evaluation of the 20 primer pairs, 13 were retained on the basis of their amplification efficiency value (93-105%).
qRT-PCR conditions and analyses
The qRT-PCRs were carried out in 96-wells plates with a MyIQ real time PCR detection system (Bio-Rad) using Quantitect SYBR Green PCR Kit (Qiagen) in a reaction volume of 20 μL (5 μL diluted cDNAs, 10 μL of 2× SYBR Green mix and primer pairs at 0.4 μM). Aliquots from the same cDNA solutions were used with all primer sets in each experiment. All PCR reactions were performed under the following conditions: 95°C for 15 min, 40 cycles of 10 s at 95 °C and 30 s at 60°C. For each primer pair, a melting curve was generated in order to confirm the specificity of the amplification and the PCR products were checked on a 4% agarose gel. Data were analysed using Bio-Rad iQ5 software. The efficiency (E) value of each reaction was between 1.93 and 2.04 with R2 values higher than 0.998. Each experiment was repeated three times on two biological replicates, each one represented by three technical repetitions. PCR reactions on samples lacking the cDNA template or the reverse transcriptase during the cDNA synthesis were also performed as negative controls for each primer pair.
Statistical analyses of gene expression stability
The stability of the candidate reference genes was evaluated by two statistical approaches. In both approaches, expression levels were expressed relative to the sample with the highest expression. Ct values were converted into relative quantities and imported into the geNorm v3,5 software http://medgen.ugent.be/~jvdesomp/genorm/ and into the NormFinder software http://www.mdl.dk/publicationsnormfinder.htm.
The geNorm algorithm first calculates an expression stability value (M) for each gene and then the pairwise variation (V) of this gene with the others. All the tested genes are ranked according to their stability in the tested tissues and the number of HKGs necessary for an optimal normalization is indicated. NormFinder also ranks the stability of the tested genes, but independently of each other.
Expression of LuMYB1
In order to validate the selected HKG, the relative expression level of LuMYB1 was calculated for each cDNA sample. Two primer pairs were designed in order to discriminate the two splice variants LuMYB1-1 [genbank:GQ374577] (M1F: GAGGACATCCTCCTGGTCAA; M1R: AGACCAACCTTCCCCAGATT) and LuMYB1-2 [genbank:GQ374578] (M2F: GGAGACGCGTAATAGGTTTG; M2R: GCGAGCAATTCTTGACCATCTG). For this study, two biological repeats of three technical measures were made in the same conditions as described above. The expression levels of the two forms were calculated according to the equation:
Authors' contributions
RH performed the sample preparations, experimental procedures and data analysis. SH supervised the study and critically revised the manuscript. GN participated in the qPCR experiments, designed this work and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Rudy Huis, Email: rudy_huis@hotmail.com.
Simon Hawkins, Email: simon.hawkins@univ-lille1.fr.
Godfrey Neutelings, Email: godfrey.neutelings@univ-lille1.fr.
Acknowledgements
The authors gratefully acknowledge S. Fénart for his helpful advice on statistics. RH is supported by the French Ministère de la Recherche. This work was financed by the French Nord-Pas de Calais regional project ARCir Plant Teq 4: 'Identification des determinants moléculaires de la qualité des fibres de lin'.
References
- Zohary D, Hopf M. Domestication of plants in the old world. Third. Oxford: Oxford University Press; 2004. [Google Scholar]
- ITL. http://www.lin-itl.com/
- Bodros E, Pillin I, Montrley N, Baley C. Could biopolymers reinforced by randomly scattered flax fibre be used in structural applications? Composites Science and Technology. 2007;67:462–470. doi: 10.1016/j.compscitech.2006.08.024. [DOI] [Google Scholar]
- Gorshkova TA, Wyatt SE, Salnikov VV, Gibeaut DM, Ibragimov MR, Lozovaya VV, Carpita NC. Cell-Wall Polysaccharides of Developing Flax Plants. Plant Physiol. 1996;110:721–729. doi: 10.1104/pp.110.3.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy N, Yang Y. Biofibers from agricultural byproducts for industrial applications. Trends in Biotechnology. 2005;23:22–27. doi: 10.1016/j.tibtech.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Day A, Ruel K, Neutelings G, Cronier D, David H, Hawkins S, Chabbert B. Lignification in the flax stem: evidence for an unusual lignin in bast fibers. Planta. 2005;222:234–245. doi: 10.1007/s00425-005-1537-1. [DOI] [PubMed] [Google Scholar]
- Akin DE, Morrison WH, Rigsby LL, Dodd RB. Plant factors influencing enzyme retting of fiber and seed flax. J Agric Food Chem. 2001;49:5778–5784. doi: 10.1021/jf010804d. [DOI] [PubMed] [Google Scholar]
- Wrobel-Kwiatkowska M, Starzycki M, Zebrowski J, Oszmianski J, Szopa J. Lignin deficiency in transgenic flax resulted in plants with improved mechanical properties. J Biotechnol. 2007;128:919–934. doi: 10.1016/j.jbiotec.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Ebskamp MJ. Engineering flax and hemp for an alternative to cotton. Trends Biotechnol. 2002;20:229–230. doi: 10.1016/S0167-7799(02)01953-4. [DOI] [PubMed] [Google Scholar]
- Roach MJ, Deyholos MK. Microarray analysis of flax (Linum usitatissimum L.) stems identifies transcripts enriched in fibre-bearing phloem tissues. Mol Genet Genomics. 2007;278:149–165. doi: 10.1007/s00438-007-0241-1. [DOI] [PubMed] [Google Scholar]
- Lamblin F, Saladin G, Dehorter B, Cronier D, Grenier E, Lacoux J, Bruyant P, Laine E, Chabbert B, Girault F. et al. Overexpression of a heterologous sam gene encoding S-adenosylmethionine synthetase in flax (Linum usitatissimum) cells: Consequences on methylation of lignin precursors and pectins. Physiol Plant. 2001;112:223–232. doi: 10.1034/j.1399-3054.2001.1120211.x. [DOI] [PubMed] [Google Scholar]
- Vrinten P, Hu Z, Munchinsky MA, Rowland G, Qiu X. Two FAD3 desaturase genes control the level of linolenic acid in flax seed. Plant Physiol. 2005;139:79–87. doi: 10.1104/pp.105.064451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillot S, Rosiau E, Laplace C, Thomasset B. Influence of light intensity and selection scheme on regeneration time of transgenic flax plants. Plant Cell Rep. 2009;28:359–371. doi: 10.1007/s00299-008-0638-2. [DOI] [PubMed] [Google Scholar]
- Cullis CA. DNA sequence organization in the flax genome. Biochim Biophys Acta. 1981;652:1–15. doi: 10.1016/0005-2787(81)90203-3. [DOI] [PubMed] [Google Scholar]
- Goldsbrough PB, Cullis CA. Characterisation of the genes for ribosomal RNA in flax. Nucleic Acids Res. 1981;9:1301–1309. doi: 10.1093/nar/9.6.1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Funk C, Brash A. Molecular cloning of an allene oxide synthase: a cytochrome P450 specialized for the metabolism of fatty acid hydroperoxides. Proc Natl Acad Sci USA. 1993;90:8519–8523. doi: 10.1073/pnas.90.18.8519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson MJ, Zhang R, Pryor A. Nucleotide sequence relationships of double-stranded RNAs in flax rust, Melampsora lini. Curr Genet. 1993;24:428–432. doi: 10.1007/BF00351852. [DOI] [PubMed] [Google Scholar]
- Omann F, Beaulieu N, Tyson H. cDNA sequence and tissue-specific expression of an anionic flax peroxidase. Genome. 1994;37:137–147. doi: 10.1139/g94-018. [DOI] [PubMed] [Google Scholar]
- Roberts JK, Pryor A. Isolation of a flax (Linum usitatissimum) gene induced during susceptible infection by flax rust (Melampsora lini) Plant J. 1995;8:1–8. doi: 10.1046/j.1365-313X.1995.08010001.x. [DOI] [PubMed] [Google Scholar]
- Day A, Dehorter B, Neutelings G, Czeszak X, Chabbert B, Belingheri L, David H. Caffeoyl-coenzyme A 3-O-methyltransferase enzyme activity, protein and transcript accumulation in flax (Linum usitatissimum) stem during development. Physiol Plant. 2001;113:275–284. doi: 10.1034/j.1399-3054.2001.1130216.x. [DOI] [PubMed] [Google Scholar]
- Wrobel M, Zebrowski J, Szopa J. Polyhydroxybutyrate synthesis in transgenic flax. J Biotechnol. 2004;107:41–54. doi: 10.1016/j.jbiotec.2003.10.005. [DOI] [PubMed] [Google Scholar]
- Harms K, Atzorn R, Brash A, Kuhn H, Wasternack C, Willmitzer L, Pena-Cortes H. Expression of a Flax Allene Oxide Synthase cDNA Leads to Increased Endogenous Jasmonic Acid (JA) Levels in Transgenic Potato Plants but Not to a Corresponding Activation of JA-Responding Genes. Plant Cell. 1995;7:1645–1654. doi: 10.1105/tpc.7.10.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S, Abdel-Reheem M, Bhella R, McCracken C, Hildebrand D. Characteristics of high alpha-linolenic acid accumulation in seed oils. Lipids. 2008;43:749–755. doi: 10.1007/s11745-008-3207-0. [DOI] [PubMed] [Google Scholar]
- Day A, Addi M, Kim W, David H, Bert F, Mesnage P, Rolando C, Chabbert B, Neutelings G, Hawkins S. ESTs from the fibre-bearing stem tissues of flax (Linum usitatissimum L.): expression analyses of sequences related to cell wall development. Plant Biol (Stuttg) 2005;7:23–32. doi: 10.1055/s-2004-830462. [DOI] [PubMed] [Google Scholar]
- Gutierrez L, Conejero G, Castelain M, Guenin S, Verdeil JL, Thomasset B, Van Wuytswinkel O. Identification of new gene expression regulators specifically expressed during plant seed maturation. J Exp Bot. 2006;57:1919–1932. doi: 10.1093/jxb/erj138. [DOI] [PubMed] [Google Scholar]
- Gachon C, Mingam A, Charrier B. Real-time PCR: what relevance to plant studies? J Exp Bot. 2004;55:1445–1454. doi: 10.1093/jxb/erh181. [DOI] [PubMed] [Google Scholar]
- Guenin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L. Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J Exp Bot. 2009;60:487–493. doi: 10.1093/jxb/ern305. [DOI] [PubMed] [Google Scholar]
- Sturzenbaum SR, Kille P. Control genes in quantitative molecular biological techniques: the variability of invariance. Comp Biochem Physiol B Biochem Mol Biol. 2001;130:281–289. doi: 10.1016/S1096-4959(01)00440-7. [DOI] [PubMed] [Google Scholar]
- Magneschi L, Kudahettige RL, Alpi A, Perata P. Expansin gene expression and anoxic coleoptile elongation in rice cultivars. J Plant Physiol. 2009;166(14):1576–80. doi: 10.1016/j.jplph.2009.03.008. [DOI] [PubMed] [Google Scholar]
- Brentner LB, Mukherji ST, Merchie KM, Yoon JM, Schnoor JL, Van Aken B. Expression of glutathione S-transferases in poplar trees (Populus trichocarpa) exposed to 2,4,6-trinitrotoluene (TNT) Chemosphere. 2008;73:657–662. doi: 10.1016/j.chemosphere.2008.07.059. [DOI] [PubMed] [Google Scholar]
- Bracha-Drori K, Shichrur K, Lubetzky TC, Yalovsky S. Functional analysis of Arabidopsis postprenylation CaaX processing enzymes and their function in subcellular protein targeting. Plant Physiol. 2008;148:119–131. doi: 10.1104/pp.108.120477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomal C, Bedon F, Caron S, Mansfield SD, Levasseur C, Cooke JE, Blais S, Tremblay L, Morency MJ, Pavy N. et al. Involvement of Pinus taeda MYB1 and MYB8 in phenylpropanoid metabolism and secondary cell wall biogenesis: a comparative in planta analysis. J Exp Bot. 2008;59:3925–3939. doi: 10.1093/jxb/ern234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquea F, Gutierrez F, Medina C, Arce-Johnson P. A novel Otubain-like cysteine protease gene is preferentially expressed during somatic embryogenesis in Pinus radiata. Mol Biol Rep. 2008;35:567–573. doi: 10.1007/s11033-007-9124-0. [DOI] [PubMed] [Google Scholar]
- Diretto G, Welsch R, Tavazza R, Mourgues F, Pizzichini D, Beyer P, Giuliano G. Silencing of beta-carotene hydroxylase increases total carotenoid and beta-carotene levels in potato tubers. BMC Plant Biol. 2007;7:11. doi: 10.1186/1471-2229-7-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BR, Nam HY, Kim SU, Kim SI, Chang YJ. Normalization of reverse transcription quantitative-PCR with housekeeping genes in rice. Biotechnol Lett. 2003;25:1869–1872. doi: 10.1023/A:1026298032009. [DOI] [PubMed] [Google Scholar]
- Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005;139:5–17. doi: 10.1104/pp.105.063743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid KE, Olsson N, Schlosser J, Peng F, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006;6:27. doi: 10.1186/1471-2229-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner AM, Yakovlev IA, Strauss SH. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004;4:14. doi: 10.1186/1471-2229-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolacci AR, Tanzarella OA, Porceddu E, Ciaffi M. Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol. 2009;10:11. doi: 10.1186/1471-2199-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faccioli P, Ciceri GP, Provero P, Stanca AM, Morcia C, Terzi V. A combined strategy of "in silico" transcriptome analysis and web search engine optimization allows an agile identification of reference genes suitable for normalization in gene expression studies. Plant Mol Biol. 2007;63:679–688. doi: 10.1007/s11103-006-9116-9. [DOI] [PubMed] [Google Scholar]
- Barsalobres-Cavallari CF, Severino FE, Maluf MP, Maia IG. Identification of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions. BMC Mol Biol. 2009;10:1. doi: 10.1186/1471-2199-10-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Exposito-Rodriguez M, Borges AA, Borges-Perez A, Perez JA. Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 2008;8:131. doi: 10.1186/1471-2229-8-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot N, Hausman JF, Hoffmann L, Evers D. Housekeeping gene selection for real-time RT-PCR normalization in potato during biotic and abiotic stress. J Exp Bot. 2005;56:2907–2914. doi: 10.1093/jxb/eri285. [DOI] [PubMed] [Google Scholar]
- Jian B, Liu B, Bi Y, Hou W, Wu C, Han T. Validation of internal control for gene expression study in soybean by quantitative real-time PCR. BMC Mol Biol. 2008;9:59. doi: 10.1186/1471-2199-9-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64:5245–5250. doi: 10.1158/0008-5472.CAN-04-0496. [DOI] [PubMed] [Google Scholar]
- Li J, Li X, Guo L, Lu F, Feng X, He K, Wei L, Chen Z, Qu LJ, Gu H. A subgroup of MYB transcription factor genes undergoes highly conserved alternative splicing in Arabidopsis and rice. J Exp Bot. 2006;57:1263–1273. doi: 10.1093/jxb/erj094. [DOI] [PubMed] [Google Scholar]
- Zhong R, Ye ZH. Regulation of cell wall biosynthesis. Curr Opin Plant Biol. 2007;10:564–572. doi: 10.1016/j.pbi.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Zhou J, Lee C, Zhong R, Ye ZH. MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. Plant Cell. 2009;21:248–266. doi: 10.1105/tpc.108.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorshkova TA, Sal'nikov VV, Chemikosova SB, Ageeva MV, Pavlencheva NV, van Dam J. The snap point: a transition point in Linum usitatissimum bast fiber development. Industrial Crops and Products. 2003;18:213–221. doi: 10.1016/S0926-6690(03)00043-8. [DOI] [Google Scholar]
- Jain M, Khurana JP. Transcript profiling reveals diverse roles of auxin-responsive genes during reproductive development and abiotic stress in rice. FEBS J. 2009;276:3148–3162. doi: 10.1111/j.1742-4658.2009.07033.x. [DOI] [PubMed] [Google Scholar]
- Jain M, Nijhawan A, Tyagi AK, Khurana JP. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun. 2006;345:646–651. doi: 10.1016/j.bbrc.2006.04.140. [DOI] [PubMed] [Google Scholar]
- Hong SY, Seo PJ, Yang MS, Xiang F, Park CM. Exploring valid reference genes for gene expression studies in Brachypodium distachyon by real-time PCR. BMC Plant Biol. 2008;8:112. doi: 10.1186/1471-2229-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R, Hollenbeck V, Dombrowski J. Evaluation of Reference Genes for Quantitative RT-PCR in Lolium perenne. Crop Science. 2008;48:1881–1887. doi: 10.2135/cropsci2007.10.0597. [DOI] [Google Scholar]
- Gallie DR, Le H, Caldwell C, Browning KS. Analysis of translation elongation factors from wheat during development and following heat shock. Biochem Biophys Res Commun. 1998;245:295–300. doi: 10.1006/bbrc.1998.8427. [DOI] [PubMed] [Google Scholar]
- Gonzaìlez-Verdejo C, Die J, Nadal S, Jimeìnez-Mariìn A, Moreno M, Romaìn B. Selection of housekeeping genes for normalization by real-time RT-PCR: Analysis of Or-MYB1 gene expression in Orobanche ramosa development. Analytical Biochemistry. 2008;379:38–43. doi: 10.1016/j.ab.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Mu RL, Cao YR, Liu YF, Lei G, Zou HF, Liao Y, Wang HW, Zhang WK, Ma B, Du JZ. et al. An R2R3-type transcription factor gene AtMYB59 regulates root growth and cell cycle progression in Arabidopsis. Cell Res. 2009;19:1291–1304. doi: 10.1038/cr.2009.83. [DOI] [PubMed] [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]