

Abstract
This study reports the synthesis, chromatographic separation and pharmacological evaluation of the two enantiors of the neuropeptide S receptor (NPSR) antagonist (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68). The (9R)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (compound 10) and (9S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (compound 10a) were synthesized and their purity assessed by chiral chromatography. The absolute configuration of the enantiomer 10 has been assigned from the crystal structure of the corresponding (S)-phenyl ethyl amine derivative 8. Calcium mobilization studies performed on cells expressing the recombinant NPSR demonstrated that compound 10 is the active enantiomer while the contribution of 10a to the NPSR antagonist properties of the racemic mixture is negligible.
Introduction
Neuropeptide S (NPS) is the last neuropeptide identified via the reverse pharmacology approach.1 NPS selectively binds and activates a previously orphan GPCR receptor now referred to as NPSR.1 NPSR is widely distributed in the brain while the expression of the NPS peptide precursor is limited to few discrete brain areas.1, 2 The supraspinal administration of NPS in rodents produces a rather unique pattern of actions: stimulation of wakefulness associated with anxiolytic-like effects.1 In addition, NPS has been reported to inhibit food intake, facilitate memory, elicit antinociceptive effects, and recent evidence suggests an involvement of the NPS/NPSR system in drug addiction (see for a review 3).
Potent and NPSR selective antagonists are now required for understanding the biological functions controlled by the NPS/NPSR system. As far as peptide antagonists are concerned, these molecules were recently discovered by replacing Gly5 in the natural peptide sequence with a D-amino acid. Examples of such compounds are [D-Cys(tBu)5]NPS4, [D-Val5]NPS5 and more recently [tBu-D-Gly5]NPS.6 The first example of non-peptide molecules able to interact with the NPSR was reported in the patent literature by Takeda researchers.7 Among the different molecules described in the patent, the compound (9R/S)-3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68; compound 1) has been pharmacologically characterized. In vitro, compound 1 behaves as a selective, potent (pA2 ≅ 8) and competitive antagonist at human8 and murine9 NPSR. In vivo, compound 1 has been reported to prevent the arousal promoting and anxiolytic-like effects elicited by NPS in mice and rats.8, 9 In addition, compound 1 reversed the protective effect of NPS on the NMDA receptor antagonist MK-801-induced neurotoxicity in rats.10 Finally recent findings indicate that in the rat intracerebroventricular injection of NPS increased conditioned reinstatement of cocaine seeking, whereas peripheral administration of compound 1 reduced it.11 These pharmacological investigations were performed using the racemic compound 1. Molecular modelling studies investigated non-peptide ligand binding to NPSR.12 In the frame of these studies, docking analyses were performed and a defined NPSR binding pocked was proposed; of note, only the (S) enantiomer of compound 1 was used in such simulations. The importance of ligand chirality for NPSR interaction has been recently supported by the identification of two novel classes of non-peptide NPSR antagonists: the quinoline13 and the tricyclic imidazole14 based compounds. In both cases, a single bioactive enantiomer was obtained by chiral chromatography separation from the corresponding racemic mixture.
In the present study, we report the synthesis, chiral HPLC analysis, X-ray crystallographic assignment and in vitro pharmacological evaluation of the two compound 1 enantiomers.
Results and Discussion
The reference compound 1 was synthesized following the procedures reported by Okamura et al.8 In Scheme 1 is described the synthetic approach adopted for the synthesis of 10 and 10a starting from (S)-phenyl ethyl amine. As reported in literature15, 16 the use of phenyl ethyl amine was expected to induce the stereochemistry of C9 of the tetrahydro-oxazolo[3,4-a]pyrazine nucleus. Unfortunately, we obtained only a slight chiral induction corresponding approximately to a 60 / 40% ratio determined by NMR spectroscopy. Similar results were obtained using (R)-phenyl ethyl amine as chiral auxiliary. Nevertheless, diastereomers 8 and 8a were successfully separated in good yield by flash chromatography. The removal of the chiral auxiliary to obtain 9 and 9a and the acylation of N7 with p-fluoro-benzylisocyanate to obtain final compounds were achieved using the procedure reported by Okamura et al.8 The purity grade and enantiomeric excess of 10 and 10a were determined by chiral HPLC analysis. The top panel of Figure 1 shows the chromatogram for the single enantiomer 10a (first eluted component), the middle panel that of 10 (second eluted species) and the bottom panel of the figure displays the chromatogram for the racemate. As it is evident from this figure, there is no trace of 10 in the chromatogram corresponding to the elution of 10a, nor of 10a in that of 10. In order to define the absolute configuration of the C9 chiral centre, we explored different crystallization conditions for compounds 8, 8a, 9, 9a and for the final products. Only with compound 8 we were able to obtain crystals suitable for further X-ray investigation. In particular, compound 8 was crystallized from ethanol/ ethyl acetate and its X-ray analysis demonstrated the absolute configuration R at the chiral center C9. The absolute C9 configuration of compound 8 has been assigned by reference to the unchanged chiral centre C10 in configuration S (Figure 2). On the basis of the absolute configuration of 8, we were able to assign the absolute C9 configuration to compounds 8a, 9, 9a and that to the final products 10 and 10a.
Scheme 1. Synthesis of compound 10 and 10a.
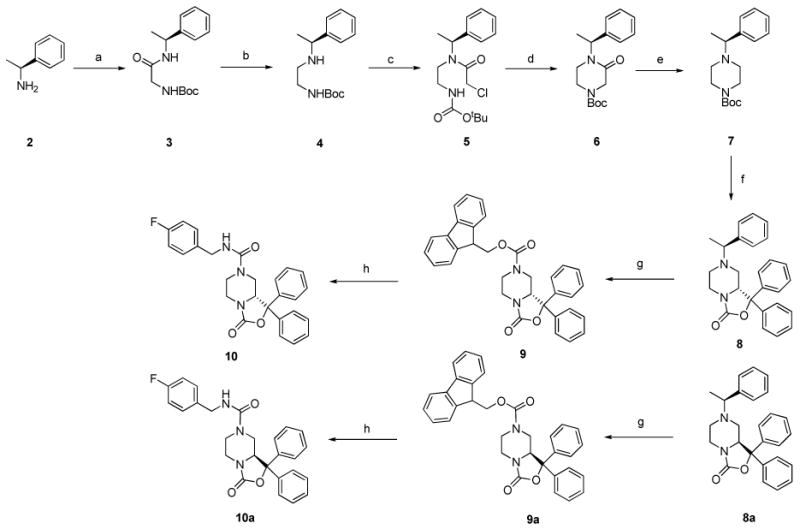
Reagents and conditions: (a) CH2CI2, WSC, Boc-Gly-OH, room temp, 12h; (b) LiAIH4, THF, 0°C, 1h; (c) Chloroacetyl-chloride, EtOAc, NaHCO3, 0°C to room temp., 24h; (d) THF/DMF 1/1, NaH, 0°C to room temp, 24h; (e) THF, LiAIH4, room temp, 4h; (f) THF, Benzophenone, sec-BuLi, TMEDA, -78°C to -30°C to room temp, 24h; (g) CH3CN, Fmoc-CI, reflux, 12h; (h) THF, DBU, p-fluoro-benzylisocyanate, room temp, 12h.
Figure 1.
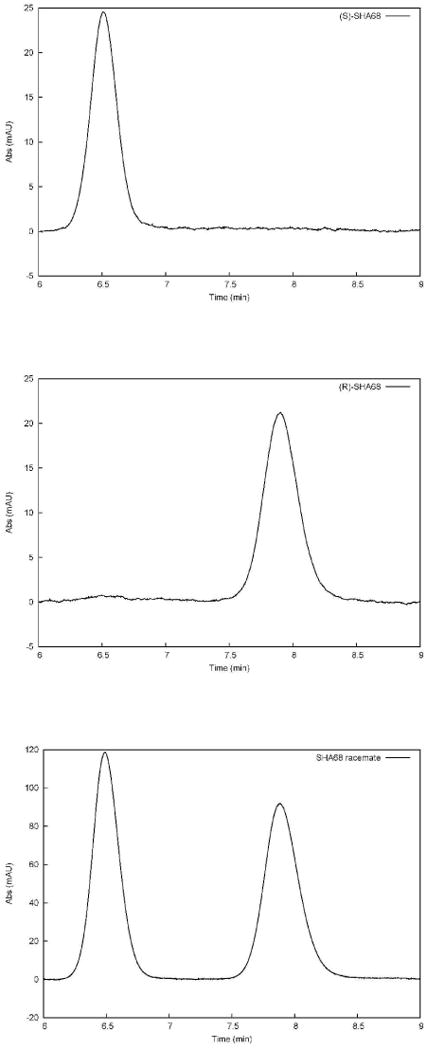
Chromatograms of compound 10a (top panel) and 10 (middle panel) in comparison with the racemate (bottom panel).
Figure 2.
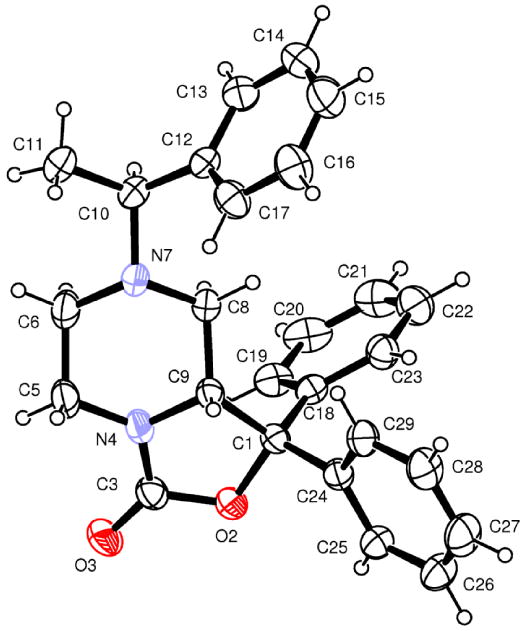
ORTEP view of compound 8. The thermal ellipsoids are drawn at 30% probability level.
In parallel, we performed a series of NMR experiments. In Figure 3 the enlarged [1H]NMR spectra of the C9 proton region of the 10a and 10 isomers are depicted. The coupling constant analysis between Hx and Ha/Hb C8 protons are very similar (11.3/3.7 Hz for 10a, panel A and 11.2/3.7 Hz for 10, panel B) in both compounds. This result together with X-ray data of 8 (see Figure 2), confirms the axial position of C9 proton in both enantiomers.
Figure 3.
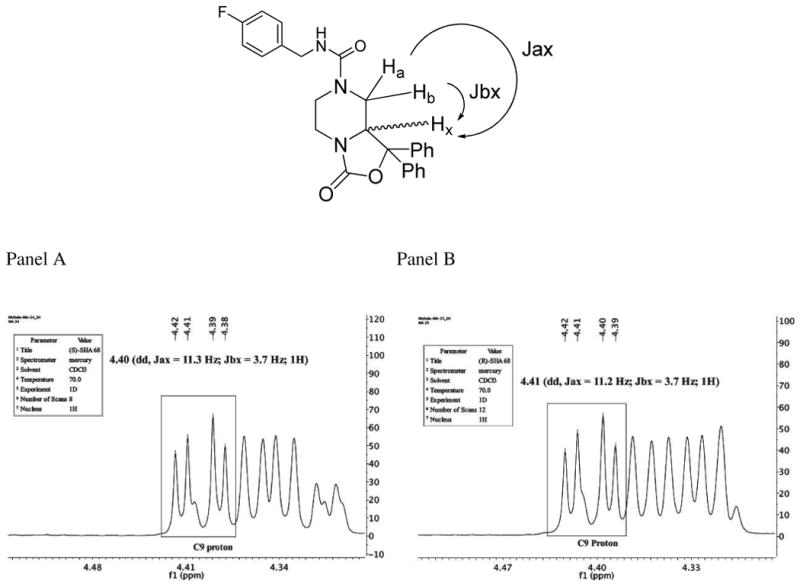
Enlarged [1H]NMR spectra of the C9 proton region of the 10a (panel A) and 10 (panel B) isomers
Next, we evaluated and compared the in vitro NPSR antagonist properties of compound 1, compound 10, and compound 10a. The three samples were tested in calcium mobilization studies performed on HEK293 cells expressing the murine NPSR or the two isoforms of the human receptor (hNPSRAsn107 and hNPSRIle107).17
The natural peptide NPS was able to induce calcium mobilization in a concentration dependent manner in HEK 293 mNPSR (pEC50 8.97 ± 0.11; Emax 250 ± 11%), hNPSRAsn107 (pEC50: 9.07 ± 0.11; Emax 316 ± 13%) and hNPSRIle107 (pEC50: 9.17 ± 0.15; Emax 333 ± 17%). The three samples were challenged against the stimulatory effect of 10 nM NPS in inhibition response curves (Figure 4). Compound 1, compound 10, and compound 10a did not stimulated per se calcium mobilization up to 10 μM. Compound 1 inhibited in a concentration dependent manner the stimulatory effect of NPS showing similar high values of potency (pKB ≅ 8). These values of potency are superimposable to those previously published.8, 9 Compound 10 was also able to antagonize in a concentration dependent manner the stimulatory effect of NPS displaying values of potency similar or slightly higher than the racemic mixture. By contrast, compound 10a showed a slight inhibitory effect only at micromolar concentrations. The values of potency of the three compounds in the three cells lines are summarized in Table 1. Collectively, these results demonstrated that compound 10 is the active enantiomer while the contribution of compound 10a to the biological activity of the racemic mixture is negligible. This information can be extremely useful for the refinement of the recently proposed molecular models of NPSR and its binding pocket.12 As already mentioned in the introduction, the relevance of ligand chirality for NPSR binding is also corroborated by the fact that the biological activity of chemically different molecules, such as the quinoline13 and the tricyclic imidazole14 compounds, could be attributed to a single bioactive enantiomer.
Figure 4.

Inhibition response curves to compound 1 (SHA 68), compound 10 and compound 10a in HEK293 cells expressing the murine NPSR and the human NPSR isoforms. Data are mean ± s.e.m. of four separate experiments made in duplicate.
Table 1. Potencies (pKB) of compound 1 (SHA 68), compound 10 and compound 10a in HEK293 cells expressing the murine NPSR and the human NPSR isoforms.
| Compound | mNPSR | hNPSR Ile107 | hNPSR Asn107 |
|---|---|---|---|
| pKB | pKB | pKB | |
| 1 | 8.16 (7.79 - 8.53) |
8.03 (7.77 - 8.37) |
7.99 (7.73 - 8.25) |
| 10 | 8.29 (7.93 - 8.65) |
8.18 (7.90 - 8.46) |
8.28 (7.72 - 8.84) |
| 10a | <6 | <6 | <6 |
In conclusion, the present study described the synthesis and separation of the two compound 1 enantiomers. The synthetic scheme we used can be easily scaled up to multi-grams. Compound 10 was demonstrated to be the bioactive enantiomer. Nowadays this molecule represents the standard non peptide NPSR antagonist that has been, and surely will be, used to investigate the biological functions controlled by the NPS / NPSR system and to evaluate the therapeutic potential of innovative drugs acting as NPSR selective ligands.
Experimental Section
Materials
HPLC grade solvent were purchased from Sigma Aldrich (Steinheim, Germany). The purity of the tested compound 1, compound 10 and compound 10a has been assessed by RP-HPLC. All compounds showed >95% purity. One-dimensional and two dimensional NMR spectra were recorded on a VARIAN 400 MHz instrument. Chemical shifts are given in ppm (δ) relative to TMS and coupling constants are in Hz. MS analyses were performed on a ESI-Micromass ZMD 2000. Optical rotation data were recorded on a Perkin-Elmer polarimeter 241. Flash chromatography was carried out on a silica gel (Merck, 230–400 Mesh). Silica gel (Polygram SIL G/UV254) was used for thin layer chromatography.
Typical Procedures for the Synthesis of 10 and 10a
[(1-Phenyl-ethylcarbamoyl)-methyl]-carbamic acid tert-butyl ester (3)
To a stirred solution of Boc-Gly-OH (5 g, 28.5 mmol) in CH2Cl2 (50 mL), WSC (3.64 g, 19 mmol) and (S)-phenylethyl amine (3.45 g, 28.5 mmol) were added. After 24 h at room temperature the reaction was monitored by TLC (EtOAc/light petroleum 2:1). The organic layer was washed with 10% citric acid (20 mL), 5% NaHCO3 (20 mL) and brine (20 mL). The organic phase was dried, concentrated in vacuo and purified by flash chromatography (EtOAc/light petroleum 2:1) to obtain 3 in 60% yield.1H NMR (400MHz, CDCl3): δ 7.31-7.24 (m, 5H, Ar); 6.62 (bs, 1H, NH-CO); 5.31 (bs, 1H, NH-Boc); 5.11 (m, 1H, CH-CH3); 3.75 (m, 2H, CH2-NH-Boc); 1.47 (d, 3H, CH3-CH-Ar, J=6.8 Hz); 1.42 (s, 9H, tbu-); 13C NMR (100MHz, CDCl3): δ 168.6, 156.2, 143.0, 128.7, 127.4, 126.1, 80.2, 48.7, 44.6, 28.3, 21.9; MS (ESI): [M+H]+ =279; [α]D20 = -41 (c = 0.121g/100 ml, chloroform).
[2-(1-Phenyl-ethylamino)-ethyl]-carbamic acid tert-butyl ester (4)
To a stirred suspension of LiAlH4 (0.85g, 22.38mmol) at 0 °C in anhydrous THF, a solution of 3 (3.11 g, 11.19 mmol) was added drop wise. The reaction was monitored by TLC (EtOAc/light petroleum 3:1) and after 24 hours the excess of hydride was quenched with water and the salts were filtered trough a celite pad. The solvent was evaporated in vacuo to yield 4 (2.66 g, 10.07 mmol) in 90% yield. 1H NMR (400MHz, CDCl3): δ 7.32-7.27 (m, 5H, Ar); 4.96 (bs, 1H, NH-Boc); 3.77-3.73 (q, 1H, CH3-CH-Ar, J=6.6 Hz); 3.16-3.13 (m, 2H, NH-CH2-CH2); 2.59-2.51 (m, 2H, NH- CH2-CH2); 1.75 (bs, 1H, -NH); 1.42 (s, 9H, tbu-); 1.36-1.33 (d, 3H, CH3-CH-Ar, J=6.6 Hz);. MS (ESI): [M+H]+ = 265; [α]D20 = -29° (c = 0.11 g/100 mL, chloroform).
{2-[(2-Chloro-acetyl)-(1-phenyl-ethyl)-amino]-ethyl}-carbamic acid tert-butyl ester (5)
To a stirred solution of 4 (1.94 g, 7.34 mmol) in EtOAc (50 mL) at 0 °C, satured solution of NaHCO3 (5 mL) was added. After 10 min, chloroacetyl chloride (1.17 mL, 14.68 mmol) was added drop wise. The reaction was monitored by TLC (EtOAc/light petroleum 3:1) and after 24 h at room temperature, NaHCO3 (2 mL) was added to the organic phase. The organic layer was separated and the aqueous phase was extracted twice with EtOAc (50 mL). The combined organic phases were concentrate to dryness to obtain 5 in quantitative yield. 1H NMR (400MHz, CDCl3): δ 7.22-6.98 (m, 5H, Ar); 5.59-5.43 (q, 1H, CH3-CH-Ar, J=8 Hz); 4.85 (bs, 1H, NH-Boc); 4.10-3.97 (m, 2H, NH- CH2-CH2); 3.76 (s, 2H, C=O-CH2-Cl); 3.18-3.15 (m, 2H, NH- CH2-CH2); 1.40-1.25 (d, 3H, CH3-CH-Ar, J=8 Hz); 1.06 (s, 9H, tbu). 13C NMR (100MHz, CDCl3): δ 170.12, 155.85, 139.47, 128.45, 128.20, 128.03, 80.69, 59.44, 42.86, 41.14, 38.24, 27.48, 19.98. MS (ESI): [M+H]+ = 341.
3-oxo-4-(1-phenyl-ethyl)-piperazine-1-carboxylic acid tert-butyl ester (6)
To a stirred suspension of 60% NaH (1.14 g, 28.61 mmol) in a mixture of THF/DMF 1/1 (20 mL) at 0 °C, a solution of 5 (3.25 g, 9.54 mmol) in THF/DMF 1/1 (10 mL) was added. After 24 h the reaction was quenched by adding NH4Cl satured solution (15 mL) and the solvent was removed in vacuo. The residue was dissolved in EtOAc (50 mL) and washed twice with water (20 mL). The organic layer was dried, evaporated under reduced pressure and the crude product purified by flash chromatography (eluent: EtOAc/light petroleum 1:1) to obtain 6 in 45% yield. 1H NMR (400MHz, CDCl3): δ 7.36-7.28 (m, 5H, Ar); 6.08 (q, 1H, CH3-CH-Ar, J=8Hz); 4.23-4.18 (d, 1H, N-CHeHa-C=O, J=20Hz); 4.10 (d, 1H, N-CHeHa-C=O, J=20Hz); 3.62 (bs, 1H, CH2 Piperazine); 3.27 (bs, 1H, CH2 Piperazine); 3.19 (bs, 1H, CH2 Piperazine); 2.83 (bs, 1H, CH2 Piperazine); 1.53-1.51 (d, 3H, CH3-CH-Ar, J=8Hz); 1.45 (s, 3H, tbu-). 13C NMR (100MHz, CDCl3): δ 165.44, 153.78, 139.41, 128.63, 127.68, 127.36, 80.69, 50.08, 47.98, 40.24, 28.32, 15.34. MS (ESI): [M+H]+ = 305; [α]D20 = -116.0 ° (c = 0.318 g/100 mL, chloroform).
4-(1-phenyl-ethyl)-piperazine-carboxylic acid tert-butyl ester (7)
To a stirred suspension of LiAlH4 (453 mg, 18.9 mmol) in anhydrous THF (20 mL) at room temperature, a solution of 6 (1.15 g, 3.78 mmol) in THF (10 mL) was added. After 30 minutes the reaction was completed as showed by TLC analysis (EtOAc/light petroleum 1:2). The reaction was quenched by adding 15% NaOH (1 mL) and Et2O (20 mL). The resulting precipitate was filtered through a Celite pad and the solvent was concentrate to dryness. The crude product was purified by flash chromatography (eluent: EtOAc/light petroleum 1:2) to give 7 (920 mg, 3.17 mmol) in 84% yield. 1H NMR (400MHz, CDCl3): δ 7.31-7.25 (m, 5H, Ar); 3.41-3.35 (m, 5H, CH2-N-Boc, CH-CH3); 2.41-2.33 (m, 4H, CH2-N); 1.43 (s, 9H, tbu-); 1.36 (d, 3H, CH3-CH-Ar). 13C NMR (100MHz, CDCl3): δ 146.80, 134.13, 128.38, 127.74, 127.10, 85.27, 50.36, 29.78, 28.49, 27.48. MS (ESI): [M+H]+ = 291; [α]D20 = -32° (c = 0.0104 g/100 mL, chloroform).
1,1-Diphenyl-7-(1-phenyl-ethyl)-hexahydro-oxazolo[3,4-a] pyrazin-3-one (8 and 8a)
To a stirred solution of 7 (380 mg, 1.31 mmol) in anhydrous THF (5 mL), TMEDA (0.53 mL, 3.54 mmol) was added. The reaction was cooled at -78 °C and sec-BuLi 1.4M in exane (2.53 mL, 3.54 mmol) was added. The reaction was heated at -35 °C and after 2 h a solution of benzophenone (480 mg, 2.62 mmol) in anhydrous THF (7 mL) was added drop wise. The reaction became green and was stirred at room temperature for 24 h. After this time the reaction was monitorated by TLC (EtOAc/light petroleum 1:2) and quenched by adding NH4Cl satured solution (20 mL). The solvent was removed in vacuo and the aqueous phase extracted 3 times with EtOAc (30 mL). The combined organic layer was dried and evaporated to dryness. The crude diastereomers mixture was purified by flash chromatography using EtOAc/light petroleum 1:2 as eluent to obtain the fast running diastereomer 8a in 40% yield and the low running distereomer 8 in 45% yield.
8a: 1H NMR (400 MHz, CDCl3): δ 7.55 - 7.49 (m, 2H), 7.41 - 7.21 (m, 11H), 7.19 - 7.14 (m, 2H), 4.51 (dd, 1H, J = 10.9, 3.6 Hz), 3.74 (ddd, 1H, J = 13.2, 3.5, 1.3 Hz), 3.34 (q, 1H, J = 6.7 Hz), 3.04 (ddd, 1H, J = 13.0, 12.1, 3.6 Hz), 2.70 - 2.61 (m, 2H), 1.86 (td, 1H, J = 11.9, 3.6 Hz), 1.50 - 1.41 (m, 1H), 1.22 (d, 3H, J = 6.7 Hz). 13C NMR (100 MHz, CDCl3): δ 156.17, 142.85, 142.52, 138.91, 128.69, 128.58, 128.50, 128.35, 128.01, 127.51, 127.37, 126.19, 125.95, 85.50, 64.52, 61.56, 52.66, 49.30, 42.07, 19.34. MS ESI [M+H+]= 399; [α]D20= +216 (c = 0.108 g/100 mL, chloroform).
8: 1H NMR (400 MHz, CDCl3): δ 7.50 - 7.45 (m, 2H), 7.39 - 7.20 (m, 11H), 7.18 - 7.14 (m, 2H), 4.44 (dd, 1H, J = 3.56, 10.93 Hz), 3.86 - 3.79 (m, 1H), 3.48 (q, 1H, J = 6.8 Hz), 3.11 (ddd, 1H, J = 13.0, 12.0, 3.81 Hz), 2.80 - 2.73 (m, 1H), 2.44 (ddd, 1H, J = 11.5, 3.5, 1.6 Hz,), 2.07 - 1.97 (m, 1H), 1.50 (m, 1H), 1.27 (d, 3H, J = 6.8 Hz). 13C NMR (100 MHz, CDCl3): δ 156.20, 142.48, 142.28, 138.81, 128.65, 128.51, 128.32, 127.98, 127.59, 127.28, 126.11, 125.92, 125.84, 85.39, 63.86, 61.67, 53.24, 47.58, 42.11, 17.16; MS ESI [M+H+]= 399; [α]D20= -132°(c = 0.11 g/100 mL, chloroform).
3-Oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 9H-fluoren-9-ylmethyl ester (9 and 9a)
To a stirred solution of 8 or 8a (200 mg, 0.52 mmol) in acetonitrile (10 mL) at reflux, Fmoc-Cl (148 mg, 0.57 mmol) dissolved in acetonitrile (7 mL) was added. The reaction, monitored by TLC (EtOAc/light petroleum 1:2), was completed in 12 h. The desired precipitate was filtered off to obtain 9 or 9a in about 67% yield and pure enough to be used in the next reaction.
3-Oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (10 and 10a)
To a stirred solution of 9 (59 mg, 0.11 mmol) in anhydrous THF (15 mL) p-fluoro-benzylisocianate (34.4 mg, 0.228 mmol) and DBU (19.2 mg, 0.126 mmol) were added. The reaction was monitored by TLC (EtOAc/light petroleum 1:2) and by mass spectrometry. After 24 h the reaction was treated as for 8 and 8a. The organic phase was dried and evaporate to dryness to give 10 in 76% yield after column chromatography using EtOAc/light petroleum 1/1 as eluent. 1H NMR (400 MHz, CDCl3): δ 7.51 - 7.47 (m, 2H), 7.41 - 7.18 (m, 10H), 7.03 - 6.94 (m, 2H), 4.95 (t, 1H, J = 5.5 Hz), 4.45 - 4.27 (m, 3H), 4.03 (ddd, 1H, J = 13.5, 3.5, 1.2 Hz), 3.81 (dd, 1H, J = 13.1, 2.7 Hz), 3.69 - 3.60 (m, 1H), 3.05 (td, 1H, J = 12.7, 3.7 Hz), 2.93 - 2.82 (m, 1H), 2.14 (dd, 1H, J = 13.3, 11.3 Hz). 13C NMR (100 MHz, CDCl3): δ 157.20, 156.11, 141.81, 138.30, 134.91, 129.48, 129.41, 129.17, 129.09, 128.82, 128.72, 128.37, 125.99, 125.85, 115.70, 115.48, 85.90, 60.55, 46.58, 44.47, 43.76, 41.37. MS ESI [M+H+]= 445.9; [α]D20= +92 (c= 0.1 g/100 mL, MeOH). Compound 10a was obtained in the same manner, starting from 9a. Analytical data: Yield: 83%; 1H NMR (400 MHz, CDCl3): δ 7.51 - 7.47 (m, 2H), 7.41 - 7.18 (m, 10H), 7.03 - 6.94 (m, 2H), 4.95 (t, 1H, J = 5.5 Hz), 4.45 - 4.27 (m, 3H), 4.03 (ddd, 1H, J = 13.3, 3.6, 1.3 Hz), 3.81 (dd, 1H, J = 13.1, 2.7 Hz), 3.69 - 3.60 (m, 1H), 3.05 (td, 1H, J = 12.7, 3.7 Hz), 2.93 - 2.82 (m, 1H), 2.14 (dd, 1H, J = 13.3, 11.3 Hz). 13C NMR (100 MHz, CDCl3): δ 157.20, 156.11, 141.81, 138.30, 134.91, 129.48, 129.41, 129.17, 129.09, 128.82, 128.72, 128.37, 125.99, 125.85, 115.70, 115.48, 85.90, 60.55, 46.58, 44.47, 43.76, 41.37. MS ESI [M+H+]= 445.9; [α]D20= -91 (c= 0.12 g/100 mL, MeOH).
Chiral chromatography analysis
A micro HPLC (Agilent 1100 micro series, Agilent Technologies) equipped with a micro diode array detector was employed. A 150mm × 2mm stainless steel column packed with Lux Cellulose-1 (cellulose tris 3,5-dimethylphenylcarbamate from Phenomenex) was used for all the measurements. The average size of the packing material was 3 μm. The mobile phase was a binary mixture of hexane/isopropyl alcohol (80/20 v/v). Flow rate was 200uL/min. Injection volume was 3μL. Analyte solutions were filtered with PFTE filters (0.45μm, Supelco, Bellefonte, PA, USA) before injection. All chromatograms were recorded at 230 nm. The retention times for the first (10a) and second (10) eluted enantiomers were 6.5 and 7.9 min, respectively.
Crystal structure determination of compound 8
The crystal data of compound 8 were collected at room temperature using a Nonius Kappa CCD diffractometer with graphite monochromated Mo-Kα radiation. The data sets were corrected for Lorentz and polarization effects. The structure was solved by direct methods18 and refined using full-matrix least-squares with all non-hydrogen atoms anisotropically and hydrogens included on calculated positions, riding on their carrier atoms. All calculations were performed using SHELXL-9719 and PARST20 implemented in WINGX21 system of programs.
Crystal Data
C26H26N2O2, orthorhombic, space group P212121, a = 11.2339(2), b = 11.6808(3), c = 16.4783(5) Å, V = 2162.30(9) Å3, Z = 4, Dc = 1.224 g cm-3, intensity data collected with θ ≤ 26°, 4215 independent reflections measured, 3460 observed reflections [I > 2σ(I)], final R index = 0.0365 (observed reflections), Rw = 0.0904 (all reflections), S = 1.048. The absolute configuration has not been established by anomalous dispersion effects in diffraction measurements on the crystal. The enantiomer has been assigned by reference to an unchanging chiral centre in the synthetic procedure. ORTEP22 view of compound 8 is shown in Figure 2.
CCDC deposition number: 810351.
Calcium mobilization experiments
HEK293 cells stably expressing the murine NPSR or the human receptor isosforms NPSRIle107 and NPSRAsn107 were generated as previously described.17 HEK293mNPSR and HEK293hNPSRIle107 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum, 2mM l-glutamine, hygromycin B (100 mg/L). HEK293hNPSRAsn107 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum, 2 mM glutamine, zeocin (100 mg/L). Cells were cultured at 37 °C in 5% CO2 humidified air. Cells were seeded at a density of 50,000 cells/well into poly-D-lysine coated 96-well black, clear-bottom plates. The following day, the cells were incubated with medium supplemented with 2.5 mM probenecid, 3 μM of the calcium sensitive fluorescent dye Fluo-4 AM and 0.01% pluronic acid, for 30 min at 37 °C. After that time the loading solution was aspirated and 100 μL/well of assay buffer (Hank's Balanced Salt Solution; HBSS) supplemented with 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2.5 mM probenecid and 500 μM Brilliant Black (Aldrich) was added. Concentrated solutions (1 mM) of NPS were made in bidistilled water and kept at −20 °C. Compound 1, compound 10 and compound 10a were dissolved DMSO at a final concentration of 10 mM and stock solutions were kept at −20 °C until use. The successive dilutions were carried out in HBSS/HEPES (20mM) buffer (containing 0.02% bovine serum albumin fraction V). After placing both plates (cell culture and master plate) into the fluorometric imaging plate reader FlexStation II (Molecular Devices, Sunnyvale, CA), fluorescence changes were measured. On-line additions were carried out in a volume of 50 μL/well. To facilitate drug diffusion into the wells in antagonist type experiments, the present studies were performed at 37 °C and three cycles of mixing (25 μL from each well moved up and down 3 times) were performed immediately after antagonist injection to the wells. Inhibition response curves were determined against the stimulatory effect of 10 nM NPS. Compound 1, compound 10 and compound 10a were injected into the wells 24 min before adding NPS.
Data analysis and terminology
The pharmacological terminology adopted in this paper is consistent with IUPHAR recommendations. Data were expressed as mean ± sem of at least four independent experiments made in duplicate. Maximum change in fluorescence, expressed in percent of baseline fluorescence, was used to determine agonist response. Non-linear regression analysis using GraphPad Prism software (v.4.0) allowed logistic iterative fitting of the resultant responses and the calculation of agonist potencies and maximal effects. Agonist potencies are given as pEC50 (the negative logarithm to base 10 of the molar concentration of an agonist that produces 50% of the maximal possible effect). Compound 1, compound 10 and compound 10a antagonist properties were evaluated in inhibition response curve experiments; the antagonist potency, expressed as pKB, was derived from the following equation:
where IC50 is the concentration of antagonist that produces 50% inhibition of the agonist response, [A] is the concentration of agonist, EC50 is the concentration of agonist producing a 50% maximal response and n is the Hill coefficient of the concentration response curve to the agonist.
Supplementary Material
Acknowledgments
We are grateful to Dr. Alberto Casolari and Dr Elisa Durini for the NMR analysis and Professor Vinicio Zanirato for the helpful discussion about NMR spectra of compound 8. This work was supported by funds from the University of Ferrara (FAR grants to GC and SS), the Italian Ministry of the University (PRIN grant to GC and SS and CHEM-PROFARMA-NET grant to AC), the Compagnia di S. Paolo Foundation (NPSNP grant to GC), and the National Institute of Mental Health (MH-71313 grant to RKR).
Abbreviations used
- DBU
1,8-Diazabicyclo[5.4.0]undec-7-ene
- DMEM
Dulbecco's modified Eagle's medium
- DMF
N,N-Dimethylformamide
- Fmoc-Cl
9-Fluorenylmethyl chloroformate
- HBSS
Hank's Balanced Salt Solution
- HEK
Human Embryonic Kidney
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- MS-ESI
electron spray ionization mass spectrometry
- NMDA
N-Methyl-D-aspartic acid
- PFTE
Polytetrafluoroethylene
- RP-HPLC
reversed-phase high-performance liquid chromatography
- THF
tetrahydrofuran
- TMEDA
tetramethylethylenediamine
- WSC
1-Ethyl-3-(3′-dimethylaminopropyl)carbodiimide
Footnotes
Supporting Information Available: monodimensional and bidimensional NMR spectra of compounds 8, 8a, and final products and crystal data of compound 8 (cif file) are available free of charge via the internet at http://pubs.acs.org
References
- 1.Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–497. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 2.Xu YL, Gall CM, Jackson VR, Civelli O, Reinscheid RK. Distribution of neuropeptide S receptor mRNA and neurochemical characteristics of neuropeptide S-expressing neurons in the rat brain. J Comp Neurol. 2007;500:84–102. doi: 10.1002/cne.21159. [DOI] [PubMed] [Google Scholar]
- 3.Guerrini R, Salvadori S, Rizzi A, Regoli D, Calo G. Neurobiology, pharmacology, and medicinal chemistry of neuropeptide S and its receptor. Med Res Rev. 2010;30:751–777. doi: 10.1002/med.20180. [DOI] [PubMed] [Google Scholar]
- 4.Camarda V, Rizzi A, Ruzza C, Zucchini S, Marzola G, Marzola E, Guerrini R, Salvadori S, Reinscheid RK, Regoli D, Calo G. In vitro and in vivo pharmacological characterization of the neuropeptide s receptor antagonist [D-Cys(tBu)5]neuropeptide S. J Pharmacol Exp Ther. 2009;328:549–555. doi: 10.1124/jpet.108.143867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guerrini R, Camarda V, Trapella C, Calo G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Synthesis and biological activity of human neuropeptide S analogues modified in position 5: identification of potent and pure neuropeptide S receptor antagonists. J Med Chem. 2009;52:524–529. doi: 10.1021/jm8012294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guerrini R, Camarda V, Trapella C, Calo G, Rizzi A, Ruzza C, Fiorini S, Marzola E, Reinscheid RK, Regoli D, Salvadori S. Further studies at neuropeptide s position 5: discovery of novel neuropeptide S receptor antagonists. J Med Chem. 2009;52:4068–4071. doi: 10.1021/jm900604g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fukatsu K, Nakayama Y, Tarui N, Mori M, Matsumoto H, Kurasawa O, Banno H. Bicyclic piperazine compound and use thereof. PCT Int Appl. WO2005021555. [Google Scholar]
- 8.Okamura N, Habay SA, Zeng J, Chamberlin AR, Reinscheid RK. Synthesis and pharmacological in vitro and in vivo profile of 3-oxo-1,1-diphenyl-tetrahydro-oxazolo[3,4-a]pyrazine-7-carboxylic acid 4-fluoro-benzylamide (SHA 68), a selective antagonist of the neuropeptide S receptor. J Pharmacol Exp Ther. 2008;325:893–901. doi: 10.1124/jpet.107.135103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruzza C, Rizzi A, Trapella C, Pela M, Camarda V, Ruggieri V, Filaferro M, Cifani C, Reinscheid RK, Vitale G, Ciccocioppo R, Salvadori S, Guerrini R, Calo G. Further studies on the pharmacological profile of the neuropeptide S receptor antagonist SHA 68. Peptides. 2010;31:915–925. doi: 10.1016/j.peptides.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 10.Okamura N, Reinscheid RK, Ohgake S, Iyo M, Hashimoto K. Neuropeptide S attenuates neuropathological, neurochemical and behavioral changes induced by the NMDA receptor antagonist MK-801. Neuropharmacology. 2010;58:166–172. doi: 10.1016/j.neuropharm.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kallupi M, Cannella N, Economidou D, Ubaldi M, Ruggeri B, Weiss F, Massi M, Marugan J, Heilig M, Bonnavion P, de Lecea L, Ciccocioppo R. Neuropeptide S facilitates cue-induced relapse to cocaine seeking through activation of the hypothalamic hypocretin system. Proc Natl Acad Sci U S A. 2010;107:19567–19572. doi: 10.1073/pnas.1004100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dal Ben D, Antonini I, Buccioni M, Lambertucci C, Marucci G, Vittori S, Volpini R, Cristalli G. Molecular modeling studies on the human neuropeptide S receptor and its antagonists. ChemMedChem. 2010;5:371–383. doi: 10.1002/cmdc.200900467. [DOI] [PubMed] [Google Scholar]
- 13.Melamed JY, Zartman AE, Kett NR, Gotter AL, Uebele VN, Reiss DR, Condra CL, Fandozzi C, Lubbers LS, Rowe BA, McGaughey GB, Henault M, Stocco R, Renger JJ, Hartman GD, Bilodeau MT, Trotter BW. Synthesis and evaluation of a new series of Neuropeptide S receptor antagonists. Bioorg Med Chem Lett. 2010;20:4700–4703. doi: 10.1016/j.bmcl.2010.04.143. [DOI] [PubMed] [Google Scholar]
- 14.Trotter BW, Nanda KK, Manley PJ, Uebele VN, Condra CL, Gotter AL, Menzel K, Henault M, Stocco R, Renger JJ, Hartman GD, Bilodeau MT. Tricyclic imidazole antagonists of the Neuropeptide S Receptor. Bioorg Med Chem Lett. 2010;20:4704–4708. doi: 10.1016/j.bmcl.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 15.Juaristi E, Leon-Romo JL, Reyes A, Escalante J. Recent applications of alpha-phenylethylamine (alpha-PEA) in the preparation of enantiopure compounds. Part 3: alpha-PEA as chiral auxiliary. Part 4: alpha-PEA as chiral reagent in the stereodifferentiation of prochiral substrates. Tetrahedron Asymmetry. 1999;10:2441–2495. [Google Scholar]
- 16.Guizzetti S, Benaglia M, Rossi S. Highly stereoselective metal-free catalytic reduction of imines: an easy entry to enantiomerically pure amines and natural and unnatural alpha-amino esters. Org Lett. 2009;11:2928–2931. doi: 10.1021/ol900945h. [DOI] [PubMed] [Google Scholar]
- 17.Reinscheid RK, Xu YL, Okamura N, Zeng J, Chung S, Pai R, Wang Z, Civelli O. Pharmacological Characterization of Human and Murine Neuropeptide S Receptor Variants. J Pharmacol Exp Ther. 2005;315:1338–1345. doi: 10.1124/jpet.105.093427. [DOI] [PubMed] [Google Scholar]
- 18.Altomare A, Burla MC, Camalli M, Cascarano GL, Giacovazzo C, Guagliardi A, Moliterni AG, Polidori G, Spagna R. SIR97: a new tool for crystal structure determination and refinement. J Appl Crystallogr. 1999;32:115–119. [Google Scholar]
- 19.Sheldrich GM. Program for the crystal structure refinement. University of Gottingen, Germany. 1997 http://shelx.uni-ac.gwdg.de/SHELX/
- 20.Nardelli M. PARST95- an updata to PARST: a system of Fortran routines for calculating molecular structure parameters from the results of crystal structure analyses. J Appl Crystallogr. 1995;28:659. [Google Scholar]
- 21.Farrugia LJ. WinGX suite for small-molecule single crystal crystallography. J Appl Crystallogr. 1999;32:837–838. [Google Scholar]
- 22.Farrugia LJ. ORTEP-3 for Windows – a version of ORTEP-III with a Graphical User Interface (GUI) J Appl Crystallogr. 1997;30:565. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.