

Abstract
Inherited mutations in the tumor suppressor genes BRCA1 and BRCA2 cause increased risk of developing various cancers, especially breast and ovarian cancers. Tumors that develop in patients with inherited BRCA1/2 mutations are generally believed to be BRCA1/2 deficient. Cancer cells with BRCA1/2 deficiency are defective in DNA repair by homologous recombination and sensitive to interstrand DNA crosslinking agents, such as cisplatin and carboplatin, and poly(ADP-ribose) polymerase (PARP) inhibitors. Therefore, these agents are logical choices for the treatment for BRCA1/2-deficient tumors and have shown to be clinically effective. However, BRCA1/2-mutated tumors often develop resistance to these drugs. Restoration of BRCA1/2 functions due to secondary BRCA1/2 mutations has been recognized as a mechanism of acquired resistance to cisplatin and PARP inhibitors in BRCA1/2-mutated cancer cells. This indicates that even disease-causing inherited mutations of tumor suppressor genes can be genetically reverted in cancer cells, if the genetic reversion is advantageous for the cells' survival. In this review, we will discuss this drug resistance mechanism.
Drug resistance is a serious obstacle to effective cancer therapy. Though a number of drug resistance mechanisms have been identified(1, 2), in this review, we mainly discuss resistance resulting from restoration of DNA repair. Specifically, we will focus on a recently recognized mechanism of resistance to cisplatin and poly(ADP-ribose) polymerase (PARP) inhibitors: restoration of BRCA1/2 functions due to secondary mutations of BRCA1/2 in BRCA1/2-mutated tumors(3–6). Defects in DNA repair lead to cellular sensitivity to DNA damaging drugs and restoration of DNA repair results in acquired resistance to those drugs. This unexpected drug resistance mechanism has attracted attention from many researchers because of its clinical significance. In this review, we will discuss identification of BRCA1/2 secondary mutation as a drug resistance mechanism, its clinical implications and future directions.
BRCA1, BRCA2 and Sensitivity of Cancer Cells to Cisplatin and PARP Inhibitor
Since DNA repair mechanisms play an important role in determining cellular sensitivity to DNA damaging agents, it is reasonable to speculate that DNA repair plays a critical role in development of resistance to anti-cancer DNA damaging agents (Fig. 1). Cells are equipped with multiple DNA repair mechanisms to deal with DNA damage caused by normal metabolic processes and exogenous agents. Loss of integrity of DNA repair promotes genetic instability, which can lead to tumorigenesis. An additional consequence of defective DNA repair is cellular hypersensitivity to DNA damaging agents, which has been exploited in the widespread use of DNA damaging anti-cancer agents in chemotherapy(7). DNA damaging agents work especially well in individuals with known DNA repair defects in their cancer cells. A widely studied group of such are individuals with mutations in the BRCA1 and BRCA2 tumor suppressor genes.
Fig. 1. Implications of DNA repair defects in cancer.

DNA repair defects (for example, defect of homologous recombination due to BRCA1/2 deficiency) can contribute to genomic instability, which promotes malignant transformation of cells. They also lead to cellular sensitivity to certain DNA damaging agents. Restoration of DNA repair (for example, restoration of homologous recombination DNA repair due to functional restoration of BRCA1/2) can lead to acquired resistance to the DNA damaging agents.
Heterozygous germline mutations in BRCA1/2 are responsible for a large fraction of hereditary breast and ovarian cancers. Additionally, BRCA2 mutation carriers have increased risk of other types of cancer, such as pancreatic, and prostate cancers(8, 9). The risk for developing breast cancer by age 70 is 50–70% for BRCA1/2 mutation carriers, while the risk of developing ovarian cancer by age 70 is 40–50% for BRCA1 mutation carriers and 10–20% for BRCA2 mutation carriers(10, 11). Though individuals are heterozygous for BRCA1/2 mutations in normal cells, neoplastic cells show loss of the wildtype allele and retention of the mutated BRCA1/2 allele; therefore, these tumors are considered to be BRCA1/2 deficient(12–14). In the case of pancreatic carcinomas in BRCA2 mutation carriers, sometimes tumors can be BRCA2 proficient(15).
Biallelic germline mutations of BRCA2 cause a severe form of a rare recessive genetic disease, Fanconi anemia (FA-D1 subgroup)(16). Therefore, BRCA2 is also called FANCD1. Fanconi anemia is characterized by bone marrow failure, cancer/leukemia susceptibility, multiple congenital abnormalities and cellular hypersensitivity to interstrand DNA crosslinking agents, such as cisplatin and mitomycin C(17). Fanconi anemia can be divided into at least 13 complementation groups and all of the responsible genes have been identified. FA-D1 subtype patients are especially cancer prone, and the cumulative probability of any malignancy of FA-D1 subgroup is 97% by age 5.2 years(18).
BRCA1 and BRCA2 proteins play important roles in DNA double-strand break repair by homologous recombination (HR)(19, 20). BRCA1 directly binds to phosphorylated CtIP protein and localizes CtIP to DNA double-strand breaks (DSBs), which facilitates resection of DSB ends to create 3' overhangs of single-stranded DNA (ssDNA)(21, 22). The ssDNA is then coated by RPA, which is subsequently displaced by the recombinase protein RAD51. BRCA2 plays a critical role in this step: BRCA2 directly binds to RAD51 and facilitates loading of RAD51 on ssDNA(23). RAD51 forms a nucleoprotein filament on ssDNA and catalyses the search for a homologous sequence and promotes strand invasion, which is followed by formation and resolution of intermediate recombination structures. BRCA1 forms a protein complex with PALB2 and BRCA2, and regulates BRCA2(24). In addition, BRCA1 has also been reported to be involved in checkpoint function, transcriptional regulation, chromatin remodeling, and ubiquitination(25).
Loss of BRCA1/2 function leads to increased chromosomal instability, which can promote tumorigenesis. Importantly, BRCA1/2 deficiency also results in cellular sensitivity to interstrand DNA crosslinking agents such as cisplatin(26, 27), and a promising new class of anti-cancer agents called PARP inhibitors(28, 29).
Cisplatin and its derivative, carboplatin, cause interstrand and intrastrand crosslinks which covalently link DNA, preventing transcription and replication(30). Interstrand crosslinks are mainly recognized during S phase as they can cause approaching replication forks to stall(31), although interstrand crosslink repair also occurs in G1(32). Repair of the interstrand crosslink is a multistep processes(33), beginning with endonucleolytic excision on one strand, followed by translesion synthesis across the interstrand crosslink and removal by excision. The resulting DSB must be resected to generate ssDNAs followed by repair by HR. As HR is defective in BRCA1/2-deficient cells, these cells are hypersensitive to interstrand crosslinking agents.
Platinum compounds (cisplatin and carboplatin) are effective chemotherapeutic agents for ovarian cancers(1). Patients with ovarian carcinomas andBRCA1/2 mutations have longer recurrence-free intervals than patients with sporadic ovarian carcinomas, especially when treated with platinum-based therapy(34–36). However, recurrence does occur in the majority of women with BRCA1/2-associated ovarian carcinoma and the recurrent carcinomas eventually acquire platinum resistance. Studies have suggested several mechanisms that can lead to cisplatin resistance including changes in transport of the drug via altered expression of copper transporters and increased glutathione expression(2).
BRCA1/2-deficient cells are highly sensitive to PARP inhibitors(28, 29), and many clinical trials of PARP inhibitors for the treatment of patients with BRCA1/2-mutated cancers are ongoing(37). PARP-1 attaches ADP-ribose to itself and other targets and facilitates repair of single strand breaks (SSBs). PARP inhibition results in the accumulation and persistence of SSBs, which then can be converted into DSBs during S phase. HR plays a critical role in the repair of DSBs in S phase. Therefore, inhibition of PARP in HR-deficient cells, such as BRCA1/2-deficient cells, results in synthetic lethality(28, 29) and promising results of clinical trials in BRCA-associated carcinomas have been reported(38–41). However, not all of the patients with BRCA1/2-mutated cancer respond to the PARP inhibitor, and even for patients who respond to the therapy, response duration is finite. Therefore, PARP inhibitor resistance is becoming a clinically important issue. The mechanism of clinical PARP inhibitor resistance is not clear, but in BRCA1/2-mutated ovarian carcinomas treated with a PARP inhibitor, there is an association between the clinical benefit rate and platinum-free interval, suggesting that there may be a common mechanism of resistance to cisplatin and PARP inhibitors(40). Data from a murine Brca1/p53-deficient mammary tumor model has shown that resistance to PARP inhibitors can result from upregulation of P-glycoprotein efflux pumps(42).
A Lesson from a Rare Genetic Disease, Fanconi Anemia
It is well known among researchers working on rare genetic diseases, such as Fanconi anemia, that spontaneous genetic alterations that compensate for inherited disease-causing mutations (genetic reversion) sometimes occur(43). These genetic alterations in Fanconi anemia include secondary genetic changes such as back-mutations to wildtype, compensatory mutations in cis, intragenic crossovers and gene conversion(44, 45). This phenomenon is sometimes called “natural gene therapy”(45) because patients with genetic reversion in their hematopoietic cells tend to have milder clinical phenotype. Cells derived from Fanconi anemia patients are usually hypersensitive to interstrand crosslinking agents, but cells with genetic reversion are resistant. Furthermore, secondary mutation of BRCA2/FANCD1 in a mitomycin C-resistant acute myelogenous leukemia cell line from a Fanconi anemia patient with biallelic mutations in BRCA2/FANCD1 has been reported(46). Similarly, a Chinese hamster cell line with biallelic Brca2 mutations, V-C8, is highly sensitive to mitomycin C, and in vitro mitomycin C selection of V-C8 led to generation of mitomycin C-resistant clones that acquired an additional Brca2 mutation leading to the restoration of the open reading frame of one of the Brca2 alleles(47).
This knowledge led us to hypothesize that secondary BRCA1/2 mutations that cancel the effect of the inherited BRCA1/2 mutations are a mechanism of acquired resistance to cisplatin and PARP inhibitor in BRCA1/2-mutated cancer.
BRCA2 Secondary Mutations Occur In Vitro in BRCA2-mutated Cancer Cell Line Models
This hypothesis is now supported by several lines of experimental and clinical evidence. Our group(3, 5) and Ashworth's group(6) have independently demonstrated that cisplatin/PARP inhibitor-resistant clones of BRCA2-mutated cancer cell lines can be generated in vitro and that restoration of functional BRCA2 protein expression due to secondary BRCA2 mutations is a major mechanism for the observed resistance. To date, in vitro models of secondary mutation have only been published for BRCA2-mutated cancer cell lines, not for BRCA1-mutated cancer cell lines.
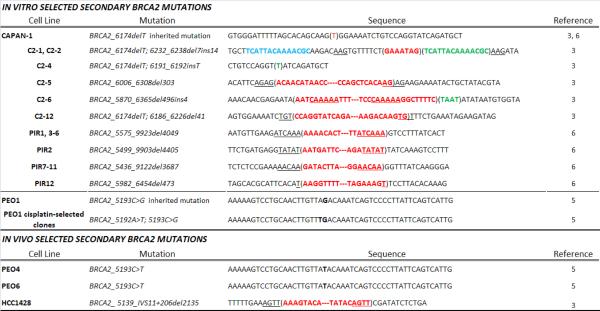
In vitro models of drug resistance have been generated in two BRCA2-mutated cancer cell lines: a pancreatic cancer cell line, Capan-1(3, 6), and an ovarian cancer cell line, PEO1(5). Capan-1 harbors the mutant allele BRCA2.6174delT, but no wildtype BRCA2 allele(48). PEO1 harbors the mutant allele BRCA2.5193C>G (Y1655X), but no wildtype BRCA2 allele(5). Therefore, these two cancer cell lines are BRCA2-deficient and sensitive to cisplatin and PARP inhibitors(3, 5, 6).
We selected Capan-1 cells in the presence of cisplatin in vitro and obtained 14 cisplatin-resistant clones(3). Interestingly, seven of the 14 cisplatin-resistant subclones harbored secondary BRCA2 mutations that restored the reading frame, resulting in expression of almost full-length BRCA2 protein (see Table 1 for mutation spectrum). The resulting BRCA2 proteins all had intact C-terminal regions containing a DNA binding domain, a nuclear localization signal and a functionally important C-terminal RAD51 binding domain(49).
Table 1. Secondary BRCA2 mutations identified in BRCA2-mutated cancer cell line models.
Deleted sequences are represented in red text and inserted sequences are represented by green text. Point mutations are shown in bold-faced black text. Blue text for clone C2-1 is a templated nucleotide sequence that is inserted downstream after deletion of a 7bp segment. Short sequences of homo logy that may contribute to deletion events via microhomology-mediated end-joining are underlined. (Data of PIR1-12 are adapted with permission from Edwards et al, Nature 2008;451(7182): 1111–5, Table 2 (reference 6).)
|
Ashworth's group selected Capan-1 cells in the presence of PARP inhibitor in vitro and obtained 12 PARP inhibitor-resistant clones(6). All 12 of the PARP inhibitor-resistant clones had secondary mutations that remove the original 6174delT mutation and restored BRCA2 protein with intact C-terminal nuclear localization signal and the RAD51 binding domain (Table 1). Intriguingly, some of the restored BRCA2 proteins still lack the DNA binding domain, suggesting that the DNA binding domain may be dispensable for some functions of BRCA2.
We selected PEO1 cells in the presence of cisplatin or a PARP inhibitor in vitro and obtained 11 cisplatin-selected clones and 3 PARP inhibitor-selected clones(5). Eight of 11 cisplatin- selected clones and all of the 3 PARP inhibitor-selected clones harbored the same secondary BRCA2 mutation: a single-base substitution that altered the original nonsense mutation (Y1655X) to a missense mutation (Y1665L) and resulted in expression of full-length BRCA2 (Table 1).
The BRCA2 proteins resulting from secondary mutations have restored HR functions. First, most of Capan-1 and PEO1 clones with secondary BRCA2 mutations restored ionizing radiation (IR)-induced RAD51 foci formation(3, 5, 6), which is a marker of functional BRCA2(26). Second, the function of these BRCA2 proteins resulting from secondary mutations was confirmed using an I-SceI-dependent DR-GFP recombination reporter assay(3, 5, 6). Finally, siRNA-mediated depletion of BRCA2 in BRCA2-restored cells sensitized the cells to cisplatin and PARP inhibitors, indicating that restored BRCA2 is functional and responsible for the acquired drug resistance(5, 6).
An unfortunate byproduct of resistance to cisplatin or PARP inhibitors due to secondary BRCA2 mutation is cross-resistance to other DNA damaging drugs. Data from the in vitro models show that resistance to cisplatin mediated by BRCA2 restoration also results in resistance to PARP inhibitors and vice versa(3, 5, 6). Not surprisingly, PARP inhibitor-resistant cells respond to docetaxel, an agent that inhibits mitosis by stabilizing microtubules, in a similar manner to parental Capan-1 cells, suggesting that resistance due to BRCA2 restoration modulates response to DNA damaging agents specifically and not chemotherapeutic agents that work via alternative mechanisms(6). Intriguingly, cisplatin-resistant Capan-1 clones with BRCA2 secondary mutations are still sensitive to 6-thioguanine(50), suggesting that 6-thioguanine may be effective for the treatment of cisplatin/PARP inhibitor-resistant BRCA2-restored cancer. These findings indicate that restoration of BRCA2 by secondary BRCA2 mutation can be a mechanism of cisplatin/PARP inhibitor resistance at least in cell lines selected in the presence of cisplatin/PARP inhibitor in vitro. It is important to note that restoration of BRCA2 did not explain cisplatin resistance in all clones selected in cisplatin. Some of the cisplatin-resistant clones without secondary BRCA2 mutation remained sensitive to PARP inhibitor(3, 5), indicating that platinum-resistant carcinomas without secondary BRCA2 mutations may still respond to PARP inhibitor therapy.
BRCA2 Secondary Mutations Can Occur In Vivo – Evidence in Cancer Cell Lines
Analysis of a BRCA2-mutated breast cancer cell line, HCC1428, provided evidence of secondary mutations occurring in vivo(3). HCC1428 was isolated from the pleural effusion of a woman heterozygous for BRCA2_6174delT with breast carcinoma after she had undergone chemotherapy(3, 51). Interestingly, the BRCA2 gene in HCC1428 has a 2135bp internal deletion that spanned the original 6174delT mutation and the exon 11/intron 11 junction (Table 1). This deletion leads to expression of two BRCA2 transcripts encoding intact DNA binding domain and the C-terminal nuclear localization signals. Consistently, HCC1428 is resistant to cisplatin and could be sensitized to cisplatin by depleting BRCA2(3). Since HCC1428 was derived from the patient after chemotherapy, the internal deletion mutant may be a result of in vivo selection in the patient.
Another example of in vivo selection is seen with the PEO1/PEO4/PEO6 series of ovarian cancer cell lines derived from a woman heterozygous for BRCA2.5193C>G (Y1655X), with stage III ovarian adenocarcinoma(5, 52). PEO1 was derived from ascites at the time of the first relapse when the disease was still sensitive to cisplatin. PEO4 was derived at time of second relapse when the disease was refractory to cisplatin and PEO6 was derived from ascites at the terminal phase. PEO1 is hemizygous for the original nonsense mutation (BRCA2.5193C>G) (Y1655X) and lacks full-length BRCA2 protein expression, and PEO4 and PEO6 had a secondary mutation at the same position (BRCA2.5193C>T) (Y1655Y) that restored BRCA2 protein expression (Table 1). Importantly, we confirmed the same secondary mutation in the clinical samples of the patient(5). These data indicate that the secondary BRCA2 mutation occurred in vivo and may have contributed to the cisplatin resistance observed in the patient.
BRCA1/2 Secondary Mutations in Clinical Samples of Ovarian Cancer
Although cellular models to date have only shown evidence of a role for BRCA2 secondary mutation in drug resistance, clinical data indicate that secondary mutations of both BRCA1(4) and BRCA2(3, 5, 6) can occur in platinum-resistant ovarian carcinomas.
Our group and Ashworth's group reported analyses of a small number of platinum-sensitive and -resistant recurrent ovarian carcinomas from BRCA1/2 mutation carriers (Table 2)(3–6). Interestingly, 7 of 10 platinum-resistant carcinomas had secondary BRCA1/2 mutations (3/5 BRCA1; 4/5 BRCA2) and 1 BRCA1-mutated primary platinum-resistant carcinoma had a secondary BRCA1 mutation, while none of the 5 platinum-sensitive recurrent ovarian carcinomas (3 BRCA1; 2 BRCA2) showed secondary BRCA1/2 mutation. These studies suggest that BRCA1/2 restoration is an important mechanism in clinical cases of platinum resistance in BRCA1/2-associated carcinomas. We are currently analyzing a larger number of cases in order to correlate secondary BRCA1/2 mutations and clinical platinum resistance.
Table 2.
Secondary BRCA1/2 mutations in clinical samples of recurrent ovarian carcinomas.
| Patient | Mutated gene | Inherited mutation | Secondary genetic change | Platinum sensitivity at time of recurrence | Reference |
|---|---|---|---|---|---|
| UW91 | BRCA1 | 185delAG | Yes (185insAG) | refractory | 4 |
| UW208 | BRCA1 | E1250X | No | sensitive | |
| UW-F21 | BRCA1 | 2594delC | Yes (2606_2628del23) | refractory | |
| UW9238 | BRCA1 | 185delAG | Yes (185insAG) | refractory | |
| UW-LS317 | BRCA1 | 3171insTGAGA | No | refractory | |
| UW-F27 | BRCA1 | 185delAG | Yes (185insAG) | refractory | |
| CS1 | BRCA1 | 185delAG | No | sensitive | |
| CS4 | BRCA1 | 185delAG | No | sensitive | |
| CS14 | BRCA1 | 185delAG | No | refractory | |
|
| |||||
| CS2 | BRCA1 | L598X | No | refractory | 3 |
| BRCA2 | 8765delAG | Lost mutated allele | |||
| UW174 | BRCA2 | 5578delAA | No | refractory | |
| UW3548 | BRCA2 | 6174delT | Yes (6174insT) | refractory | |
| CS9 | BRCA2 | 6174delT | No | sensitive | |
| CS15 | BRCA2 | 2699delT | No | sensitive | |
|
| |||||
| UK1999 | BRCA2 | 6174delT | Yes (137bp deletion) | refractory | 6 |
| UK2223 | BRCA2 | 6174delT | Yes (8bp deletion) | refractory | |
We also found a case with primary platinum resistance that showed a secondary mutation (back mutation) of BRCA1 in the primary ovarian carcinoma(4). This patient had a history of chemotherapy for breast cancer many years before she developed ovarian carcinoma. This anecdotal case suggests an interesting possibility that chemotherapy for prior malignancy may be involved in the occurrence of secondary mutation of BRCA1/2 in subsequent cancers, but clearly we need a larger study to test this possibility.
Several reports have shown that the PARP inhibitor Olaparib shows effectiveness in ovarian and breast carcinomas in BRCA1/2-mutation carriers, though disease progression eventually occurs in most patients(38–41). A recent clinical study evaluating efficacy of PARP inhibitor therapy for ovarian carcinomas in BRCA1/2-mutation carriers showed that best results were obtained in individuals that were still responsive to platinum therapy(40). Secondary BRCA1/2 mutation is possibly an important mechanism of clinical resistance to PARP inhibitor and should be tested in future studies.
Animal Models of Brca1/2-mutated Cancer
Heterozygous deletion of Brca1 or Brca2 in mice fails to show a cancer phenotype and constitutional homozygous deletion of Brca1 or Brca2 generally results in embryonic lethality(53). In order to circumvent these problems, several tissue-specific conditional knockout models have been generated to mimic Brca1-(54–57) and Brca2-(58–61) associated breast carcinoma. Though most Brca1/2 mouse models are generated using large deletions that do not result in expression of a transcript, a few do express mutant transcript (e.g. Brca1 exon11 deletion variant), which may explain differences in phenotypes observed in these models(53). To date, murine models have not shown Brca1/2 secondary mutation as a mechanism of drug resistance. However, the large deletions of Brca1/2 used in these models preclude the possibility that secondary mutations can arise that will restore functional Brca1/2 proteins.
Several groups have investigated resistance to cisplatin/carboplatin, PARP inhibitors, doxorubicin and docetaxel in K14CreBrca1flox/flox/p53flox/flox (42, 62) and blg-CreBrca2flox/flox/p53flox/flox (63) mammary tumors by orthotopic transplantation of small tumors into mammary fatpads of wildtype females prior to challenge with drug. Treatment with doxorubicin and docetaxel resulted in variable response of carcinomas, although resistance always occurred to the maximum tolerable dose(62). Doxorubicin-resistant neoplasms upregulated the Mdr1a and Mdr1b genes that encode a P-glycoprotein, suggesting that efficient export of the drug out of cells results in the resistance. Brca1/2- and p53-deficient tumors also responded well to PARP inhibitor, although resistance always occurred due to upregulation of P-glycoprotein pumps(42, 63).
Brca1-deleted neoplasms treated with cisplatin showed a significant reduction in size after treatment and eventually grew back(62). However, cisplatin-treated recurrent tumors were still sensitive to the drug(62). Moreover, PARP inhibitor-resistant Brca1-deleted tumors also had reduction in tumor volume after cisplatin treatment(42), consistent with the common notion that P-glycoprotein is not important for transport of cisplatin(64). The fact that cisplatin-treated tumors recurred and were subsequently sensitive to another round of treatment with cisplatin suggests that there is a subset of the tumor population that evades killing by cisplatin(62). In contrast, another report showed that cisplatin resistance in mammary tumors in mice with Brca1flox/flox/p53flox/flox (Brca1 exon11 deletion variant) does occur(65). The difference of the two reports may be due to difference of Brca1 alleles (null(62) vs. exon 11 deletion(65)) or difference of cisplatin treatment regimens.
Murine models of Brca1/2-deficiency with large deletions of Brca1/2 do not mimic the most common BRCA1/2 mutations observed in patients and it is imperative to generate models that contain point mutations or smaller deletions that are more commonly observed in women with inherited BRCA1/2 mutations. Nevertheless, findings from the current murine models provide insight into resistance observed in the clinic. As clinical trials testing efficacy of PARP inhibitors in BRCA1/2-deficient tumors are maturing, analysis of recurrent resistant tumors may yield both BRCA1/2-reexpression and increased export of the drug by P-glycoprotein as resistance mechanisms.
Mechanism of Secondary BRCA1/2 Mutation
The work reviewed here demonstrates that secondary BRCA1/2 mutations are an important mechanism of platinum resistance. How do these secondary mutations arise (Fig. 2)? We can easily imagine that at least 3 factors can contribute to this phenomenon. First, treatment with DNA damaging agents such as cisplatin or carboplatin could increase the mutation rate. Second, BRCA1/2 deficiency could increase the mutation rate, since error-free HR DNA repair cannot work efficiently in the absence of BRCA1/2 and cells rely on more error-prone mechanisms of DNA repair(66). Increased mutation rates could contribute to the occurrence of secondary mutations in BRCA1/2 themselves. Third, treatment with DNA damaging agents or with PARP inhibitors could serve as a selective pressure for BRCA1/2-restored cells.
Fig. 2. Model for involvement of BRCA1/2 restoration in acquired resistance to platinum chemotherapy.

(adapted with modifications from Sakai et al, Nature 2008;451(7182): 1116–20, supplemental material). Initially tumors are BRCA1/2 deficient and sensitive to platinum compounds (cisplatin and carboplatin). Rare cells restore functional BRCA1/2 due to secondary BRCA1/2 mutation. The secondary mutation may occur during the course of chemotherapy or may pre-exist prior to chemotherapy. BRCA1/2 deficiency and treatment with DNA damaging agents can lead to increased mutation rate, which contribute to occurrence of secondary mutations in BRCA1 and BRCA2 themselves. Treatment with platinum compounds can also serve as a selective pressure for BRCA1/2-restored cells. BRCA1/2-restored cells survive the treatment and proliferate to form a BRCA1/2-proficient tumor, which would be resistant to both platinum compounds and PARP inhibitors.
Secondary BRCA1/2 mutations may be acquired during the course of chemotherapy. This can be a result of the increased amount of damaged DNA lesions caused by chemotherapy. Repair of these lesions would be error prone in the HR-defective, BRCA1/2-deficient cell background, which would increase the chance of occurrence of secondary BRCA1/2 mutations. In this model, the BRCA1/2-deficient neoplasm would shrink upon treatment with chemotherapy, leaving cells with acquired BRCA1/2 secondary mutations to expand to form a drug resistant malignancy.
Alternatively, resistance may result from the presence of a subset of BRCA1/2-expressing cells that may exist in the tumor prior to chemotherapy. Tumors can be very genetically heterogeneous in nature. It is possible that a BRCA1/2-deficient tumor comprises a small subset of cells that express functional BRCA1/2 protein due to a reversion event that occurred during the expansion of the tumor population. A precedent for this explanation exists. Lung cancers may express the EGFR mutation T790M that confers resistance to EGFR tyrosine kinase inhibitors in rare cells prior to drug exposure if sensitive detection methods are used(67). Similarly, in some cases of chronic myelogenous leukemia, BCR-ABL mutations that confer resistance to imatinib therapy are detectable by PCR in rare neoplastic cells prior to imatinib exposure(68). Treatment of such a neoplasm with a platinum agent, PARP inhibitor, or other drug that selectively kills BRCA1/2-deficient cells would result initially in a decrease of tumor size. The few BRCA1/2-expressing cells in the tumor population would survive the treatment. These cells can proliferate to form a BRCA1/2-proficient tumor, which would be resistant to further treatment with cisplatin or PARP inhibitor. In this model, drug treatment simply selects for the survival of the population of BRCA1/2-expressing, drug-resistant cells. Further work is needed to distinguish between these two models.
At the moment, we do not know which error-prone DNA repair mechanisms are involved in generating secondary mutations, but microhomology-mediated end-joining(69) and translesion synthesis(70) are obvious candidates. Some of the secondary BRCA1/2 mutations are point mutations or back mutations to wildtype (Tables 1 and 2). Error-prone translesion synthesis polymerases may be involved for the generation of these types of mutations(70). Microhomology-mediated end-joining may be a mechanism that plays a major role in occurrence of secondary BRCA2 mutations of BRCA2-deficient cells. Sequences surrounding the secondary BRCA2 deletions in some of the drug-resistant Capan-1 clones and HCC1428 breast cancer cell line have short regions of sequence identity associated with the ends of the deleted region (Table 1)(3, 6), suggesting involvement of microhomology-mediated end-joining. However, microhomology-mediated end-joining may not play a major role in secondary BRCA1 mutations of BRCA1-deficient cancer cells, since BRCA1 is reported to facilitate microhomology-mediated end-joining(71).
Modification of Response to Chemotherapy in BRCA1-deficient Cells by 53BP1 Deficiency
Chemotherapeutic response of tumors with BRCA1/2 deficiency may be affected by other genetic or epigenetic modifications. It is possible that suppressor mutations of other genes in BRCA1/2-deficient cells may be able to reverse sensitivity of BRCA1/2-deficient cells to cisplatin and PARP inhibitors, without restoring BRCA1/2 themselves. Two recent reports have shown that loss of the DNA-damage response protein 53bp1 in mice with Brca1-null or Brca1 exon11 deletion mutation can rescue the embryonic lethality of Brca1-deficient mice(72, 73). Brca1/53bp1-deficient cells have increased proliferative potential, chromosomal stability and HR efficiency relative to Brca1-deficient mouse cells. Importantly, 53bp1 deficiency in Brca1-mutated cells also restored resistance to cisplatin and PARP inhibitor. It has also been shown that some estrogen receptor-negative, progesterone receptor-negative and HER2-non-overexpressing (triple negative) and BRCA1/2-associated breast cancers have reduced 53BP1 expression(72). These data suggest that suppression of BRCA1 deficiency by the loss of 53BP1 may be a modifier of clinical progression of BRCA-associated neoplasms. Further studies are needed to determine if 53BP1 loss in BRCA1-deficient tumors can lead to clinical cisplatin/PARP inhibitor resistance.
Clinical Implications
The most profound implication of the work reviewed here on the clinical management of BRCA1/2-associated malignancies is that secondary BRCA1/2 mutation may predict whether a patient will respond to platinum based chemotherapy. This may also be true for patients treated with PARP inhibitors, although there is the additional complication of upregulation of P-glycoprotein pumps contributing to resistance to this class of drugs.
This work also suggests that restoration of DNA repair plays an important role in acquired resistance to DNA damaging drugs. Therefore, drug-resistant cancers with secondary BRCA1/2 mutations may be re-sensitized to those drugs if HR can be inhibited by another drug. For example, proteasome inhibitors(74), CDK inhibitors(75) and HSP90 inhibitors(76) have been reported to inhibit RAD51 foci formation. These drugs may be good candidates for the treatment of platinum/PARP inhibitor-resistant malignancies in combination with cisplatin or PARP inhibitors.
Future Directions
Here we have reviewed evidence for the contribution of secondary BRCA1/2 mutations to resistance to cisplatin and PARP inhibitors in BRCA1/2-mutated cancers. There still remain many unanswered important questions related to this mechanism. First, we need to perform larger clinical studies in order to correlate secondary BRCA1/2 mutations with clinical outcomes, such as resistance to platinum agents, PARP inhibitors and other chemotherapeutics. Second, we need to clarify which error-prone DNA repair mechanisms (for example, microhomology-mediated end-joining(69) and translesion synthesis(70)) are involved in the occurrence of secondary mutations. Third, it is unclear whether secondary mutations arise as a result of drug treatment or are pre-existing in the tumor cell population. Fourth, we want to know whether secondary mutations of other DNA repair genes or DNA damage response genes, such as ATM, NBS1, CHEK2, PALB2/FANCN, BRIP1/FANCJ, RAD51C/FANCO, and other Fanconi anemia genes, are involved in resistance to DNA damaging agents. Fifth, we want to know whether re-expression of BRCA1/2 mediates therapeutic resistance in sporadic carcinomas with nonhereditary BRCA1/2 deficiency due to epigenetic silencing and other mechanisms. For example, in platinum-resistant recurrent ovarian cancer with BRCA1 promoter methylation, re-expression of BRCA1 could result from partial demethylation of the promoter. Sixth, secondary BRCA1/2 mutation is not the only mechanism of drug resistance, and we need to clarify other mechanisms, which may include a suppressor of BRCA1/2 deficiency that restores DNA repair even in the absence of functional BRCA1/2 protein. Efforts to understand the role of DNA repair in chemoresistance may eventually lead to novel strategies to prevent or overcome drug resistance in the future and improve patient survival.
Acknowledgments
We thank all the members of Taniguchi lab for discussion and comments. This work was supported by NIH/NCI grants (R01CA125636 and Pacific Ovarian Cancer Research Consortium, (P50 CA083636)), and Howard Hughes Medical Institute.
Footnotes
Authors declare no conflict of interest.
References
- 1.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–16. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 2.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakai W, Swisher EM, Karlan BY, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–6. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sakai W, Swisher EM, Jacquemont C, et al. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009;69:6381–6. doi: 10.1158/0008-5472.CAN-09-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards SL, Brough R, Lord CJ, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 7.Evers B, Helleday T, Jonkers J. Targeting homologous recombination repair defects in cancer. Trends Pharmacol Sci. 2010;31:372–80. doi: 10.1016/j.tips.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 8.Kadouri L, Hubert A, Rotenberg Y, et al. Cancer risks in carriers of the BRCA1/2 Ashkenazi founder mutations. J Med Genet. 2007;44:467–71. doi: 10.1136/jmg.2006.048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thompson D, Easton DF. Cancer Incidence in BRCA1 Mutation Carriers. J Natl Cancer Inst. 2002;94:1358–65. doi: 10.1093/jnci/94.18.1358. [DOI] [PubMed] [Google Scholar]
- 10.King MC, Marks JH, Mandell JB. Breast and Ovarian Cancer Risks Due to Inherited Mutations in BRCA1 and BRCA2. Science. 2003;302:643–6. doi: 10.1126/science.1088759. [DOI] [PubMed] [Google Scholar]
- 11.Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25:1329–33. doi: 10.1200/JCO.2006.09.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neuhausen SL, Marshall CJ. Loss of heterozygosity in familial tumors from three BRCA1-linked kindreds. Cancer Res. 1994;54:6069–72. [PubMed] [Google Scholar]
- 13.Collins N, McManus R, Wooster R, et al. Consistent loss of the wild type allele in breast cancers from a family linked to the BRCA2 gene on chromosome 13q12–13. Oncogene. 1995;10:1673–5. [PubMed] [Google Scholar]
- 14.Gudmundsson J, Johannesdottir G, Bergthorsson JT, et al. Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12-q13. Cancer Res. 1995;55:4830–2. [PubMed] [Google Scholar]
- 15.Skoulidis F, Cassidy LD, Pisupati V, et al. Germline Brca2 Heterozygosity Promotes Kras(G12D) -Driven Carcinogenesis in a Murine Model of Familial Pancreatic Cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 16.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–9. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 17.D'Andrea AD. Susceptibility pathways in Fanconi's anemia and breast cancer. N Engl J Med. 2010;362:1909–19. doi: 10.1056/NEJMra0809889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999;4:511–8. doi: 10.1016/s1097-2765(00)80202-6. [DOI] [PubMed] [Google Scholar]
- 20.Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell. 2001;7:263–72. doi: 10.1016/s1097-2765(01)00174-5. [DOI] [PubMed] [Google Scholar]
- 21.Yu X, Wu LC, Bowcock AM, Aronheim A, Baer R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J Biol Chem. 1998;273:25388–92. doi: 10.1074/jbc.273.39.25388. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008;283:7713–20. doi: 10.1074/jbc.M710245200. [DOI] [PubMed] [Google Scholar]
- 23.Jensen RB, Carreira A, Kowalczykowski SC. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature. 2010;467:678–83. doi: 10.1038/nature09399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia. Cancer Res. 2010;70:7353–9. doi: 10.1158/0008-5472.CAN-10-1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy CG, Moynahan ME. BRCA gene structure and function in tumor suppression: a repair-centric perspective. Cancer J. 2010;16:39–47. doi: 10.1097/PPO.0b013e3181cf0204. [DOI] [PubMed] [Google Scholar]
- 26.Yuan SS, Lee SY, Chen G, Song M, Tomlinson GE, Lee EY. BRCA2 is required for ionizing radiation-induced assembly of Rad51 complex in vivo. Cancer Res. 1999;59:3547–51. [PubMed] [Google Scholar]
- 27.Bhattacharyya A, Ear US, Koller BH, Weichselbaum RR, Bishop DK. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J Biol Chem. 2000;275:23899–903. doi: 10.1074/jbc.C000276200. [DOI] [PubMed] [Google Scholar]
- 28.Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 29.Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 30.McCabe KM, Olson SB, Moses RE. DNA interstrand crosslink repair in mammalian cells. J Cell Physiol. 2009;220:569–73. doi: 10.1002/jcp.21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol Cell Biol. 2000;20:8283–9. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar S, Davies AA, Ulrich HD, McHugh PJ. DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J. 2006;25:1285–94. doi: 10.1038/sj.emboj.7600993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knipscheer P, Raschle M, Smogorzewska A, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science. 2009;326:1698–701. doi: 10.1126/science.1182372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyd J, Sonoda Y, Federici MG, et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA. 2000;283:2260–5. doi: 10.1001/jama.283.17.2260. [DOI] [PubMed] [Google Scholar]
- 35.Cass I, Baldwin RL, Varkey T, Moslehi R, Narod SA, Karlan BY. Improved survival in women with BRCA-associated ovarian carcinoma. Cancer. 2003;97:2187–95. doi: 10.1002/cncr.11310. [DOI] [PubMed] [Google Scholar]
- 36.Chetrit A, Hirsh-Yechezkel G, Ben-David Y, Lubin F, Friedman E, Sadetzki S. Effect of BRCA1/2 mutations on long-term survival of patients with invasive ovarian cancer: the national Israeli study of ovarian cancer. J Clin Oncol. 2008;26:20–5. doi: 10.1200/JCO.2007.11.6905. [DOI] [PubMed] [Google Scholar]
- 37.Rouleau M, Patel A, Hendzel MJ, Kaufmann SH, Poirier GG. PARP inhibition: PARP1 and beyond. Nat Rev Cancer. 2010;10:293–301. doi: 10.1038/nrc2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 39.Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376:245–51. doi: 10.1016/S0140-6736(10)60893-8. [DOI] [PubMed] [Google Scholar]
- 40.Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2512–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 41.Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 42.Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105:17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40:721–8. doi: 10.1136/jmg.40.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hamanoue S, Yagasaki H, Tsuruta T, et al. Myeloid lineage-selective growth of revertant cells in Fanconi anaemia. Br J Haematol. 2006;132:630–5. doi: 10.1111/j.1365-2141.2005.05916.x. [DOI] [PubMed] [Google Scholar]
- 45.Mankad A, Taniguchi T, Cox B, et al. Natural gene therapy in monozygotic twins with Fanconi anemia. Blood. 2006;107:3084–90. doi: 10.1182/blood-2005-07-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ikeda H, Matsushita M, Waisfisz Q, et al. Genetic reversion in an acute myelogenous leukemia cell line from a Fanconi anemia patient with biallelic mutations in BRCA2. Cancer Res. 2003;63:2688–94. [PubMed] [Google Scholar]
- 47.Wiegant WW, Overmeer RM, Godthelp BC, van Buul PP, Zdzienicka MZ. Chinese hamster cell mutant, V-C8, a model for analysis of Brca2 function. Mutat Res. 2006;600:79–88. doi: 10.1016/j.mrfmmm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 48.Abbott DW, Freeman ML, Holt JT. Double-strand break repair deficiency and radiation sensitivity in BRCA2 mutant cancer cells. J Natl Cancer Inst. 1998;90:978–85. doi: 10.1093/jnci/90.13.978. [DOI] [PubMed] [Google Scholar]
- 49.Esashi F, Galkin VE, Yu X, Egelman EH, West SC. Stabilization of RAD51 nucleoprotein filaments by the C-terminal region of BRCA2. Nat Struct Mol Biol. 2007;14:468–74. doi: 10.1038/nsmb1245. [DOI] [PubMed] [Google Scholar]
- 50.Issaeva N, Thomas HD, Djureinovic T, et al. 6-thioguanine selectively kills BRCA2-defective tumors and overcomes PARP inhibitor resistance. Cancer Res. 2010;70:6268–76. doi: 10.1158/0008-5472.CAN-09-3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gazdar AF, Kurvari V, Virmani A, et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int J Cancer. 1998;78:766–74. doi: 10.1002/(sici)1097-0215(19981209)78:6<766::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 52.Langdon SP, Lawrie SS, Hay FG, et al. Characterization and properties of nine human ovarian adenocarcinoma cell lines. Cancer Res. 1988;48:6166–72. [PubMed] [Google Scholar]
- 53.Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–97. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- 54.Mak TW, Hakem A, McPherson JP, et al. Brca1 required for T cell lineage development but not TCR loci rearrangement. Nat Immunol. 2000;1:77–82. doi: 10.1038/76950. [DOI] [PubMed] [Google Scholar]
- 55.Liu X, Holstege H, van der Gulden H, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci U S A. 2007;104:12111–6. doi: 10.1073/pnas.0702969104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu X, Wagner KU, Larson D, et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation. Nat Genet. 1999;22:37–43. doi: 10.1038/8743. [DOI] [PubMed] [Google Scholar]
- 57.Xu X, Qiao W, Linke SP, et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001;28:266–71. doi: 10.1038/90108. [DOI] [PubMed] [Google Scholar]
- 58.Ludwig T, Fisher P, Murty V, Efstratiadis A. Development of mammary adenocarcinomas by tissue-specific knockout of Brca2 in mice. Oncogene. 2001;20:3937–48. doi: 10.1038/sj.onc.1204512. [DOI] [PubMed] [Google Scholar]
- 59.Cheung AM, Hande MP, Jalali F, et al. Loss of Brca2 and p53 synergistically promotes genomic instability and deregulation of T-cell apoptosis. Cancer Res. 2002;62:6194–204. [PubMed] [Google Scholar]
- 60.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–25. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 61.McAllister KA, Bennett LM, Houle CD, et al. Cancer susceptibility of mice with a homozygous deletion in the COOH-terminal domain of the Brca2 gene. Cancer Res. 2002;62:990–4. [PubMed] [Google Scholar]
- 62.Rottenberg S, Nygren AO, Pajic M, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci U S A. 2007;104:12117–22. doi: 10.1073/pnas.0702955104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hay T, Matthews JR, Pietzka L, et al. Poly(ADP-ribose) polymerase-1 inhibitor treatment regresses autochthonous Brca2/p53-mutant mammary tumors in vivo and delays tumor relapse in combination with carboplatin. Cancer Res. 2009;69:3850–5. doi: 10.1158/0008-5472.CAN-08-2388. [DOI] [PubMed] [Google Scholar]
- 64.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–34. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 65.Shafee N, Smith CR, Wei S, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res. 2008;68:3243–50. doi: 10.1158/0008-5472.CAN-07-5480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tutt A, Bertwistle D, Valentine J, et al. Mutation in Brca2 stimulates error-prone homology-directed repair of DNA double-strand breaks occurring between repeated sequences. Embo J. 2001;20:4704–16. doi: 10.1093/emboj/20.17.4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maheswaran S, Sequist LV, Nagrath S, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008;359:366–77. doi: 10.1056/NEJMoa0800668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roche-Lestienne C, Soenen-Cornu V, Grardel-Duflos N, et al. Several types of mutations of the Abl gene can be found in chronic myeloid leukemia patients resistant to STI571, and they can pre-exist to the onset of treatment. Blood. 2002;100:1014–8. doi: 10.1182/blood.v100.3.1014. [DOI] [PubMed] [Google Scholar]
- 69.McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet. 2008;24:529–38. doi: 10.1016/j.tig.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xie K, Doles J, Hemann MT, Walker GC. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1011412107. doi:10.1073/pnas.1011412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhong Q, Chen CF, Chen PL, Lee WH. BRCA1 facilitates microhomology-mediated end joining of DNA double strand breaks. J Biol Chem. 2002;277:28641–7. doi: 10.1074/jbc.M200748200. [DOI] [PubMed] [Google Scholar]
- 72.Bouwman P, Aly A, Escandell JM, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–95. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bunting SF, Callen E, Wong N, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res. 2007;67:7395–405. doi: 10.1158/0008-5472.CAN-07-1015. [DOI] [PubMed] [Google Scholar]
- 75.Deans AJ, Khanna KK, McNees CJ, Mercurio C, Heierhorst J, McArthur GA. Cyclin-dependent kinase 2 functions in normal DNA repair and is a therapeutic target in BRCA1-deficient cancers. Cancer Res. 2006;66:8219–26. doi: 10.1158/0008-5472.CAN-05-3945. [DOI] [PubMed] [Google Scholar]
- 76.Dungey FA, Caldecott KW, Chalmers AJ. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol Cancer Ther. 2009;8:2243–54. doi: 10.1158/1535-7163.MCT-09-0201. [DOI] [PMC free article] [PubMed] [Google Scholar]