

Abstract
Genomic instability is a characteristic of cancer cells. In order to maintain genomic integrity, cells have evolved a complex DNA repair system to detect, signal and repair a diversity of DNA lesions. Homologous recombination (HR)-mediated DNA repair represents an error-free repair mechanism to maintain genomic integrity and ensure high-fidelity transmission of genetic information. Deficiencies in HR repair are of tremendous importance in the etiology of human cancers and at the same time offer great opportunities for designing targeted therapeutic strategies. The increase in the number of proteins identified as being involved in HR repair has dramatically shifted our concept of the proteins involved in this process: traditionally viewed as existing in a linear and simple pathway, today they are viewed as existing in a dynamic and interconnected network. Moreover, exploration of the targets within this network that can be modulated by small molecule drugs has led to the discovery of many effective kinase inhibitors, such as ATM, ATR, DNA-PK, CHK1, and CHK2 inhibitors. In preclinical studies, these inhibitors have been shown to sensitize cancer cells to chemotherapy and radiation therapy. The most exciting discovery in the field of HR repair is the identification of the synthetic lethality relationship between poly (ADP-ribose) polymerase (PARP) inhibitors and HR deficiency. The promises of clinical applications of PARP inhibitors and the concept of synthetic lethality also bring challenges into focus. Future research directions in the area of HR repair include determining how to identify the patients most likely to benefit from PARP inhibitors and developing strategies to overcome resistance to PARP inhibitors.
Keywords: DNA repair, Genomic instability, Homologous-recombination-mediated DNA repair, Poly polymerase inhibitors, Synthetic lethality
INTRODUCTION
DNA within our cells, the “central store” of our genetic information, is constantly being exposed to DNA-damaging factors. There are three major causes of DNA lesions. First, exogenous DNA damaging agents, such as the ultraviolet (UV) light from sunlight, ionizing radiation and numerous genotoxic chemicals causing DNA structural alterations. Second, by-products of endogenous cellular processes such as reactive oxygen species, and stalled or collapsed replication forks. Third, spontaneous disintegration of DNA chemical bonds, such as deamination of cytosine and hydrolysis of nucleotide residues causing abasic sites. In order to cope with diverse DNA lesions, cells utilize five major, partly overlapping pathways to detect, signal, and repair damaged DNA and maintain genomic stability (Table 1), including single strand break (SSB) repair mediated by nucleotide-excision repair (NER) or base-excision repair (BER); double strand break (DSB) repair mediated by homologous recombination (HR) and non-homologous end joining (NHEJ); and mismatch repair. Classic genes involved in these pathways are shown in the Table 1. Genetic defects of these genes lead to cancer predisposition, which highlights the key role of DNA repair systems in tumor suppression[1].
Table 1.
An overview of DNA damage lesions and repair mechanisms in cancer suppression
| DNA repair pathway | NER | BER | HR | NHEJ | MMR |
| DNA lesions | (6-4) PP | Uracil | Interstrand cross-link | Interstrand | A-G mismatch |
| Bulky adduct | Abasic site | DSB | Cross-link | T-C mismatch | |
| CPD | 8-oxoguanine | DSB | Insertion | ||
| SSB | SSB | Deletion | |||
| Key molecules | XPA-XPG, RPA, ERCC1 | DNA glycosylase, APE1, DNA poly β/δ/ε, XRCC1, DNA ligase 1/3 | ATM, RAD50/MRE11/NBS1 RPA, BRCA1, BRCA2, RAD51 | KU70-KU80, DNAPK, XRCC4 | hMSH2/6 hMLH1, FEN1, Exonuclease 1 |
| Cancer linkage | Xeroderma pigmentosum (skin cancer) | XRCC1 (lung cancer) | Ataxia telangiectasia (lymphomas), nijmegen breakage syndrom (lymphomas), BRCA1/BRCA2 (breast and ovarian cancers), werner syndrome (various cancers), bloom syndrome (leukaemia, lymphoma, others), rothmund-thomson syndrome (osteosarcoma) | Ligase IV deficiency (leukemia) | HNPCC (colorectal cancer) |
NER: Nucleotide-excision repair; BER: Base-excision repair; HR: Homologous recombination; NHEJ: Non-homologous end joining; DSB: Double strand break; MMR: Mismatch repair; (6-4) PP: Pyrimidine (6-4) pyrimidone; CPD: Cyclobutane pyrimidine dimers; HNPCC: Herditary non-ployposis colorectal cancer; SSB: Single strand break; RPA: DNA replication protein A; ERCC1: Excision repair cross-complementation group 1; RAD51: DNA repair protein RAD51 homolog 1; MRE11: Meiotic recombination 11 homolog 1; NBS1: Nijmegen B breakage syndrome 1; BRCA: Breast cancer susceptibility protein; DNAPK: DNA-dependent protein kinase catalytic subunit.
Of the various forms of DNA lesions that can result from such exposure, DSBs are probably the most dangerous. DSBs can be generated in response to exogenous factors, most commonly ionizing radiation and chemotherapeutic drugs, and can also be caused by endogenous factors, such as reactive oxygen species and collapsed replication forks[1]. DSBs arise when the two complementary strands of the DNA double helix are broken simultaneously, and thus DSBs pose a major threat to genomic integrity. Specifically, inaccurate repair or lack of repair of a DSB can lead to deleterious genome changes, including mutations, amplifications, deletions, and translocations, and such chromosomal aberrations lead to genomic instability, which is the hallmark of cancer cells[2].
To maintain genomic integrity and prevent tumorigenesis, cells have evolved two main pathways to repair DSBs: NHEJ and HR (Figure 1). NHEJ involves a direct ligation of broken ends. In contrast, HR involves repair of DSBs using the genetic information contained in the homologous sequence. Compared to NHEJ, which is often error-prone and introduces small sequence deletions, HR repair represents an error-free repair mechanism. The choice of cells to use either NHEJ or HR is largely dependent on the phases of the cell cycle. NHEJ is present throughout the cell cycle but is particularly common in the G0 and G1 phases whereas HR predominates in the S and G2 phases implying that HR is a major mechanism to ensure the high-fidelity transmission of genetic information[3]. These distinct features of HR give it a central role in maintaining genomic stability. Deficiency in HR repair has been well documented in the development of human cancer. In this review, we discuss the constantly expanding network of proteins identified as being involved in HR repair and how this network can be exploited in targeted cancer therapy.
Figure 1.

DNA double strand break repair pathways. A: Homologous recombination (HR) repair. MRE11-RAD50-NBS1 (MRN) complex recognizes and senses double strand breaks (DSBs). This complex activates ATM kinase, which in turn initiates the full DNA damage response, particularly in the heterochromatin regions of chromosomes. CtIP-mediated nuclease activity is required for the end resection from 5' to 3', which leads to the formation of single-strand DNA (ssDNA). The exposed ssDNA is coated with DNA replication protein A (RPA) and activates the Ataxia Telangiectasia and Rad3-related protein (ATR) response to facilitate HR repair. Then RAD51 nucleoprotein filament is assembled, which replaces RPA-coated ssDNA, performs homology sequence searching, and mediates strand invasion. DSBs are restored by branch migration of this joint DNA molecule, DNA synthesis, ligation, and resolution of Holliday junctions; B: Non-homologous end joining (NHEJ) repair. The two broken ends are processed and ligated directly by the action of the end-binding KU70/80 complex and DNA-PKcs followed by XRCC4-ligase4.
A NETWORK VIEW OF HR PROTEINS
HR repair involves a variety of proteins, which function in a hierarchically ordered, mutually coordinated manner to detect, signal, and repair DSBs[4]. DSBs are detected by “sensors”-DNA-damage-recognizing proteins. These sensors then trigger the activation of a “transducer” system-a protein kinase cascade-that amplifies and diversifies the DSB signal to activate a series of downstream “effectors” to execute HR repair. The key steps in HR repair include 5’ to 3’ resection of broken ends, search for homologous sequence, and strand invasion, recombination, and ligation.
Although the proteins involved in HR repair were traditionally viewed as existing in a simple pathway, a more accurate view is that these proteins exist in a complex protein network. In this network, many cellular responses are well known to be utilized in coordination of the HR repair process (Figure 2). For example, cell cycle checkpoints are activated to slow down cell cycle progression and allow sufficient time for DNA repair before cells enter the next cell cycle. Two kinases, CHK1 and CHK2, activate checkpoints. They function as transducers to relay and amplify DNA damage signaling through a phosphorylation cascade initiated by ATM/ATR, two major kinases sensing DNA damage lesions. Transcriptional regulation is activated to induce specific gene expression to facilitate efficient DNA repair. Recently, increasing evidence has shown that epigenetic changes affecting chromatin structure play a critical role in regulating HR repair. Various histone modifications and chromatin remodeling factors appear to function in HR repair to ensure that cells can overcome the highly condensed chromatin structure and allow DNA lesions to be accessed by DNA-damage-signaling and DNA-damage-repairing proteins. In addition, a diversity of posttranslational modifications-including phosphorylation, ubiquitination, sumoylation, acetylation, and methylation-adds a complex regulatory layer to the HR repair network. These modifications have a broad impact on many aspects of HR repair, such as the recognition of DNA lesions, the transduction of DNA damage signals, and the loading of DNA repair proteins[5,6]. For example, recent studies have shown molecular details that ubiquitination of H2AX and H2A mediated by RNF8 provides an additional histone marker other than H2AX phosphorylation for recruiting BRCA1 complex to DNA damage sites[4]. Using RNA interference screening technology, researchers recently performed a genome-wide survey of HR proteins in mammalian cells and discovered novel genes involved in HR[7]. In summary, HR repair involves a complex protein network, and the number of proteins known to be involved in this network is constantly expanding.
Figure 2.
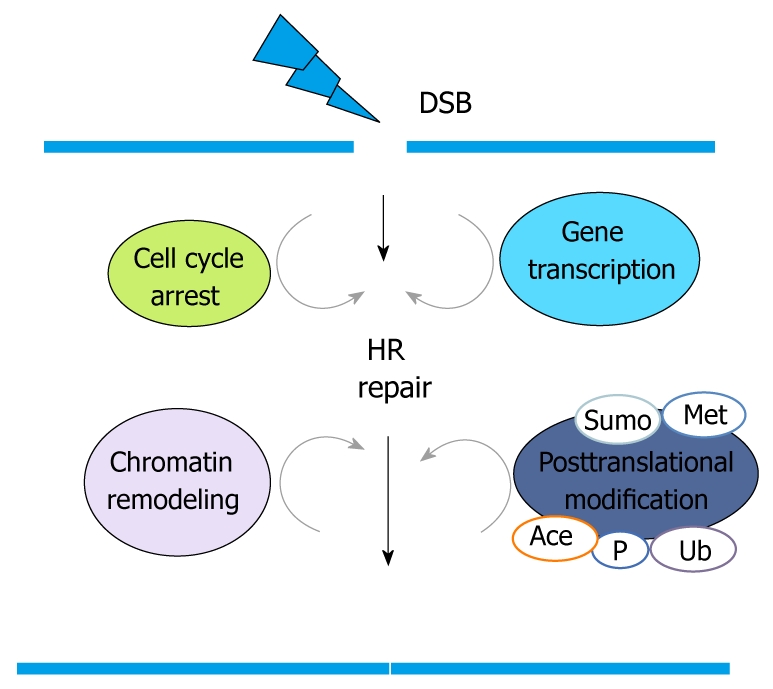
The complex protein network involved in homologous recombination repair. HR: Homologous recombination; DSB: Double strand break.
HR IN CANCER DEVELOPMENT, TREATMENT, AND DRUG RESISTANCE
The causal link between inappropriate HR repair and tumorigenesis is supported by the fact that mutations of HR repair genes predispose to the development of a wide variety of human cancers. These HR repair genes include BRCA1 and BRCA2 in breast and ovarian cancer; ATM, NBS1, BLM, and WRN in lymphoma and leukemia; RAD54 and CtIP in non-Hodgkin’s lymphoma and colon cancer; RAD51B in lymphoma and uterine leiomyoma; and RECQL4 in skin cancer and osteosarcoma[5]. As the list of proteins known to be involved in the HR network grows, a high number of genes encoding these HR proteins may be silenced or mutated in cancer, which probably compromise or inactivate HR repair.
The mainstream anticancer drugs employed in the clinic are DNA-damaging agents, many of which can directly or indirectly cause toxic DSBs and lead to cell death[8,9]. For example, alkylating agents (e.g. cisplatin), hydroxyurea, and antimetabolites (e.g. 5-fluorouracil) can block replication fork progression, resulting in stalled or collapsed replication forks and replication-associated DSBs. Inhibitors of topoisomerase I (e.g. camptothecin) can also cause replication-associated DSBs. Inhibitors of topoisomerase II (e.g. etoposide) induce DSBs directly during the relaxation of supercoiled DNA. Ionizing radiation and radiomimetic agents (e.g. bleomycin) also cause replication-associated DSBs.
Tumors with HR repair deficiency are sensitive to anticancer drugs that induce DSBs. For instance, primary ovarian cancers are often highly sensitive to platinum-based therapy (e.g. cisplatin), and this sensitivity is closely correlated with compromised HR repair function, such as that due to reduced expression levels of BRCA1 or FANCF or mutations in BRCA1 or BRCA2[10]. In contrast, ovarian tumors with re-expression of FANCF or genetic reversion of BRCA1 or BRCA2 mutations are cisplatin resistant[11-13]. Furthermore, high levels of RAD51 expression have been linked to resistance to etoposide treatment in small cell lung cancer[14,15]. These studies emphasize the impact of HR repair not only in response to therapy but also as a mechanism underlying drug resistance. Ongoing research efforts are focused on discovering targets in the HR repair network that can be modulated by small molecule drugs. Identification of such targets could potentially lead to novel therapeutic strategies.
KINASE INHIBITORS OF THE HR NETWORK
It has been well established that genes encoding kinases are the targets in the human genome most likely to be modifiable with small molecule drugs. Therefore, inhibitors of kinases are at the forefront of targeted therapeutics in cancer treatment. In the HR repair network, phosphorylation mediated by various kinases is of utmost importance in coordinating cellular responses to DSBs. For example, kinases such as ATM, ATR, DNA-PKcs, CHK1, and CHK2 constitute the primary sensors and transducers in the DNA-damage-signaling cascade[4,16,17]. ATM, ATR, and DNA-PKcs belong to the phosphatidylinositol 3-kinase-related protein kinase family. They are located at the top of the signaling cascade. When activated, they phosphorylate a multitude of proteins to initiate a phosphorylation cascade. CHK1 and CHK2 are important transducers that relay DNA damage signals downstream to activate cell cycle checkpoints. CHK1 regulates the S and G2/M checkpoints, while CHK2 elicits an additional G1 arrest. Although historically it was believed that these kinases function in independent pathways in detecting and signaling DNA lesions, emerging evidence now supports cross-talk among the pathways in modulating cellular response to DNA damage[3]. Targeting these kinases by specific inhibitors has been approved to be an effective approach to modulating the HR machinery.
A number of kinase inhibitors have been identified as potentially useful in cancer treatment (Figure 3). UCN-01, a staurosporine analogue, was identified as an inhibitor of CHK1. It disrupts the G2/M checkpoint in response to ionizing radiation in p53-negative cells. This result raises the interesting possibility that UCN-01 might preferentially sensitize p53-mutant cancer cells to DNA-damage-inducing agents by inhibition of the G2/M checkpoint. Three CHK2 inhibitors are in the early phase of clinical development: AZD7762 (AstraZeneca), PF47736 (Pfizer), and XL844 (Exelixis). These drugs cause inhibition of cell-cycle arrest, progressive DNA damage, inhibition of DNA repair, and ultimately apoptosis of tumor cells. An inhibitor of ATM kinase activity, KU55933 (AstraZeneca), is currently in preclinical development. CGK733 was found to be a potent inhibitor of both ATM and ATR. NU7441 has an inhibitory effect on the activation of DNA-PKcs[8,9,18].
Figure 3.

Double strand break repair pathways and the points of action of cancer treatment drugs. HR: Homologous recombination; NHEJ: Non-homologous end joining; DSB: Double strand break; RPA: DNA replication protein A; RAD51: DNA repair protein RAD51 homolog 1; MRE11: Meiotic recombination 11 Homolog 1; NBS1: Nijmegen B breakage syndrome 1; BRCA: Breast cancer susceptibility protein; XRCC: X-ray repair cross-complementing protein.
Because these inhibitors do not selectively target cancer cells, there are considerable concerns about their clinical application because of their potential toxic effects on normal cells. One solution to minimize potential toxic effects on normal cells is to use combination therapy in which kinase inhibitors are combined with DNA-damaging therapy. The rationale for the combination strategy is that kinase inhibitors should improve the therapeutic index by inhibiting DNA repair and thereby hypersensitizing tumor cells to DNA-damaging agents. Recent experimental and preclinical studies support the notion that kinase inhibitors can be used as chemosensitizers and radiosensitizers. In combination with gemcitabine, the CHK2 inhibitor, XL844, was found to be well tolerated in a xenograft model. The DNA-PKcs inhibitor, NU7441, was shown to sensitize cells to topoisomerase II inhibitors and radiation therapy. These results reveal the promise of the therapeutic strategy of combining DNA-damage-inducing agents with inhibitors of HR repair[8,9,18].
Another potential strategy for minimizing potential cytotoxic effects of kinase inhibitors on normal cells is to apply such therapy to tumors with specific genetic defects that render them preferentially sensitive to DNA repair inhibitors. A recent study revealed that two pancreatic tumor lines that exhibited defects in the Fanconi anemia pathway were more sensitive to the ATM inhibitor, KU55933, than were their isogenic control cells[19]. This finding provides a rationale for the application of an ATM inhibitor in the treatment of pancreatic cancer with a defective Fanconi anemia repair pathway. Moreover, loss of expression of FANC genes has been associated with a variety of cancers, including head and neck, lung, ovarian, and cervical cancer[18]. Hence, ATM inhibitors could be more broadly used for targeted therapies in Fanconi anemia pathway-deficient cancers. This study also points to a new direction in the application of DNA repair inhibitors: selective targeting of the intrinsic genetic defects in tumor cells. The most important advance in this area is the discovery of the so-called synthetic lethality interaction between poly (ADP-ribose) polymerase (PARP) inhibitors and HR repair deficiency[20], which is described in the next section.
SYNTHETIC LETHALITY IN HR-DEFICIENT TUMORS
Synthetic lethality was first used to describe the relationship that exists between two genes when a mutation in either gene alone is not lethal, but mutations in both genes cause death of the cell. In the context of discovering anticancer drugs, a synthetic lethality relationship exists between two genes when chemical inhibition of a target gene kills cells that harbor a specific genetic alteration, such as loss of a tumor suppressor or activation of an oncogene. This therapeutic strategy provides a means for selectively targeting cancer cells with genetic alterations and sparing normal cells. The drugs identified from synthetic lethality relationships hold great promise for the development of new therapeutic approaches with very minimal side effects.
The most successful synthetic lethality relationship exploited in HR repair for cancer treatment is the relationship between PARP and BRCA1- or BRCA2-deficient cells (Figure 4). PARP is an enzyme that facilitates repair of SSBs by promoting base-excision repair, a DNA repair pathway recognizing and eliminating damaged DNA bases. In normal cells, in the absence of PARP activity, damaged DNA bases accumulate and arrest DNA replication forks at the damage sites. These collapsed or stalled replication forks transform SSBs to replication associated DSBs, which are predominantly repaired through the HR mechanism in replicating cells. Therefore, in normal cells, DNA damage generated by PARP inhibitors is well tolerated because of functional compensation from the HR repair network. In contrast, cells with deficient HR repair, such as cancer cells with BRCA1 and BRCA2 deficiency, cannot cope with this increased DNA damage and therefore exhibit hypersensitivity to PARP inhibitors[21]. This synthetic lethality relationship is the underlying mechanism proposed by two studies that showed that inhibitors of PARP1 are toxic to cells deficient in BRCA1 or BRCA2, whereas cells with competent BRCA1 or BRCA2 functions were less sensitive to PARP inhibitors by orders of magnitude[22,23]. More exciting, results from a small early-stage clinical trial involving 60 patients were recently published and showed that the PARP inhibitor, Olaparib, has anticancer effects against breast cancers and ovarian cancers with BRCA1 and BRCA2 mutations at safely administrable doses with minimal side effects[24]. Because mutations of BRCA1 or BRCA2 are frequently observed in the triple-negative subtype of breast cancer, effective treatment of which remains a clinical challenge, it has been proposed that PARP inhibitors may provide specific therapeutic benefits to patients with this breast cancer subtype[25].
Figure 4.

Mechanism of synthetic lethality interaction between poly (ADP-ribose) polymerase inhibitors and homologous recombination repair deficiency. DSB: Double strand break; SSB: Single strand break; HR: Homologous recombination; RPA: DNA replication protein A; BRCA: Breast cancer susceptibility protein; PARP: Poly (ADP-ribose) polymerase.
Several screening studies have been performed with the goal of identifying other genes required to maintain cell viability in the absence of PARP1 activity. The screening data support our current understanding of the synthetic lethality mechanism resulting from PARP1 inhibition and loss of BRCA1 or BRCA2 function. Defects in a number of genes required for HR repair were discovered to cause synthetic lethality when combined with PARP inhibition[26,27]. Furthermore, defects in some genes implicated in checkpoint activation were also found to cause synthetic lethality when combined with PARP inhibition, indicating that blocking cell cycle arrest may render cells hypersensitive to PARP inhibitors by reducing the time available for HR repair[28]. Finally, a recent RNAi screening study showed that deficiency of USP11, a novel gene implicated in DSB repair, can sensitize cells to PARP inhibitor treatment[29].
In conclusion, the identification of a synthetic lethality interaction between PARP inhibitors and HR deficiency not only provides a promising therapeutic approach to treat cancers deficient in BRCA1 or BRCA2, but also opens new avenues for developing targeted cancer therapies by utilizing the concept of synthetic lethality.
FUTURE DIRECTIONS
Currently, our understanding of the HR repair network is expanding. Development of HR inhibitors and targeting of tumor cells with specific genetic defects in the HR network represent extremely promising opportunities to optimize current therapeutic strategies and discover new synthetic lethality relationships. Many challenges in the area of synthetic lethality are beginning to come into focus.
First, it is important to determine how to identify patients with HR-deficient tumors, who are most likely to benefit from treatment with PARP inhibitors. Given the high number of proteins in the HR repair network, it is possible that many cancers have lost or compromised HR repair capacity. The mechanism underlying the synthetic lethality relationship between PARP inhibitors and HR deficiency indicates that PARP inhibitors can be expanded beyond use against breast and ovarian tumors with mutations in BRCA1 or BRCA2 to use against all tumors with HR deficiency. How to identify patients with such tumors? One possible approach is to develop molecular markers that can indicate the activity of the HR repair pathway. For example, RAD51 foci formation could be assayed by immunofluorescent staining. RAD51 is the key molecule involved in HR repair and has recombinase activity, which allows it to strand search and invade. Upon DSB formation, RAD51 is recruited to the DNA lesion and forms discrete nuclear foci. Abolishment of RAD51 foci formation has been widely used as a functional assay to indicate HR repair deficiency. Another potential approach to assessing the status of HR repair is to phosphorylated forms of proteins involved in DNA damage response and repair. We are currently devoting our research efforts to developing and characterizing a clinically applicable molecular tool that can identify cancer cells with HR deficiency. We believe that our studies to address this critical question will have a significant impact on broadening the applications of PARP inhibitors and identifying patients most likely to benefit from this therapy.
Second, methods need to be developed to overcome resistance to PARP inhibitors. The early clinical studies indicated that 35% of patients with BRCA1or BRCA2 mutations were not responsive to PARP inhibitors[24]. Understanding the mechanisms underlying resistance to PARP inhibitors and developing methods to overcome such resistance is an important research area. More and more research evidence indicates that secondary mutations in BRCA1 or BRCA2-deficient tumors may cause resistance to PARP inhibitors. Resistance to cisplatin and related compounds in ovarian cancer with BRCA1 or BRCA2 mutations has been found to arise from reversion mutations in BRCA1 or BRCA2 alleles, which restore HR function[11-13]. Notably, two recent studies reported that loss of 53BP1 rescues BRCA1 deficiency, and reduced 53BP1 expression was observed in subsets of sporadic triple-negative and BRCA1-associated breast cancers[30,31]. Optimal combination treatment may provide a means by which we can genetically or chemically avert the emergence of resistance.
A third area of research is to seek combination treatments that can potentiate PARP inhibitors in HR-deficient cancer cells. Recent data showed that PARP1 is involved in HR repair at replication forks. This combined role of PARP1 in repair of SSBs and HR repair may provide another explanation for the extremely strong synthetic lethality interaction between PARP1 inhibitors and BRCA1 or BRCA2 mutations. It may be reasonable to combine PARP inhibitors with selective HR inhibitors or SSB-inducing agents such as camptothecin to enhance the killing effects of PARP inhibitors. As NHEJ is the other pathway involved in repair of DSBs, deficiency in HR repair may be functionally compensated for by activation of the NHEJ repair activity. Thereby, the combination of PARP inhibitors and inhibitors of the NHEJ repair pathway may have synergistic effects on killing cancer cells. A promising approach to inhibiting the NHEJ repair pathway is to use DNA-PKcs inhibitors to target the activation of DNA-PKcs, which is required for proper NHEJ repair.
Fourth, it will be important to identify new synthetic lethality relationships so that these can be exploited for targeting cancer cells deficient in a variety of types of DNA repair. For example, a new synthetic lethality relationship has recently been identified between the mismatch repair pathway and proofreading DNA polymerases[32]. The therapeutic exploration of this relationship is particularly pertinent to the treatment of colorectal cancer, which has a strong correlation with mismatch repair pathway deficiency.
Finally, posttranslational modifications have recently been revealed to be involved in the HR repair network, including ubiquitination, which is implicated in establishing DNA damage signaling to recruit DNA repair factors and also in promoting protein-protein interactions in this process[33]. Exploring the potential targets of E3 ligase activity in HR repair may lead to identification of a new collection of inhibitors.
Footnotes
Peer reviewers: Melanie H Kucherlapati, PhD, Instructor, Associate Geneticist, Department of Medicine, Division of Genetics, Harvard Medical School, Brigham and Women’s Hospital, 77 Avenue Louis Pasteur, NRB 160B, Boston, MA 02115, United States; Godefridus J Peters, PhD, Professor of Pharmacology, Department of Medical Oncology, VU University Medical Center, PO Box 7057, 1007 MB Amsterdam, The Netherlands
S- Editor Cheng JX L- Editor Webster JR E- Editor Ma WH
References
- 1.Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11:196–207. doi: 10.1038/nrm2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 3.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mutat Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 4.Huen MS, Chen J. The DNA damage response pathways: at the crossroad of protein modifications. Cell Res. 2008;18:8–16. doi: 10.1038/cr.2007.109. [DOI] [PubMed] [Google Scholar]
- 5.Helleday T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis. 2010;31:955–960. doi: 10.1093/carcin/bgq064. [DOI] [PubMed] [Google Scholar]
- 6.Peng G, Lin SY. The linkage of chromatin remodeling to genome maintenance: contribution from a human disease gene BRIT1/MCPH1. Epigenetics. 2009;4:457–461. doi: 10.4161/epi.4.7.10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Słabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, Junqueira M, Paszkowski-Rogacz M, Teyra J, Heninger AK, Poser I, et al. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol. 2010;8:e1000408. doi: 10.1371/journal.pbio.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 9.Martin SA, Lord CJ, Ashworth A. DNA repair deficiency as a therapeutic target in cancer. Curr Opin Genet Dev. 2008;18:80–86. doi: 10.1016/j.gde.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 10.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med. 2003;9:568–574. doi: 10.1038/nm852. [DOI] [PubMed] [Google Scholar]
- 11.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, Boyd J, Reis-Filho JS, Ashworth A. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451:1111–1115. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 13.Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res. 2008;68:2581–2586. doi: 10.1158/0008-5472.CAN-08-0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen LT, Lundin C, Helleday T, Poulsen HS, Sørensen CS, Petersen LN, Spang-Thomsen M. DNA repair rate and etoposide (VP16) resistance of tumor cell subpopulations derived from a single human small cell lung cancer. Lung Cancer. 2003;40:157–164. doi: 10.1016/s0169-5002(03)00026-6. [DOI] [PubMed] [Google Scholar]
- 15.Hansen LT, Lundin C, Spang-Thomsen M, Petersen LN, Helleday T. The role of RAD51 in etoposide (VP16) resistance in small cell lung cancer. Int J Cancer. 2003;105:472–479. doi: 10.1002/ijc.11106. [DOI] [PubMed] [Google Scholar]
- 16.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 17.Chen Y, Poon RY. The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci. 2008;13:5016–5029. doi: 10.2741/3060. [DOI] [PubMed] [Google Scholar]
- 18.O'Connor MJ, Martin NM, Smith GC. Targeted cancer therapies based on the inhibition of DNA strand break repair. Oncogene. 2007;26:7816–7824. doi: 10.1038/sj.onc.1210879. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy RD, Chen CC, Stuckert P, Archila EM, De la Vega MA, Moreau LA, Shimamura A, D'Andrea AD. Fanconi anemia pathway-deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117:1440–1449. doi: 10.1172/JCI31245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 21.Moeller BJ, Pasqualini R, Arap W. Targeting cancer-specific synthetic lethality in double-strand DNA break repair. Cell Cycle. 2009;8:1872–1876. doi: 10.4161/cc.8.12.8743. [DOI] [PubMed] [Google Scholar]
- 22.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 23.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 24.Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O'Connor MJ, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- 25.Iglehart JD, Silver DP. Synthetic lethality--a new direction in cancer-drug development. N Engl J Med. 2009;361:189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 26.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66:8109–8115. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 27.Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2008;7:2010–2019. doi: 10.1016/j.dnarep.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, Rayter S, Tutt AN, Ashworth A. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–1377. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wiltshire TD, Lovejoy CA, Wang T, Xia F, O'Connor MJ, Cortez D. Sensitivity to poly(ADP-ribose) polymerase (PARP) inhibition identifies ubiquitin-specific peptidase 11 (USP11) as a regulator of DNA double-strand break repair. J Biol Chem. 2010;285:14565–14571. doi: 10.1074/jbc.M110.104745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, Oka S, Kay E, Lord CJ, Ashworth A. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17:235–248. doi: 10.1016/j.ccr.2009.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nat Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]