

Abstract
In recent years, there has been a growing interest in using porous microbeads such as agarose beads as solid supports to bind target molecules from complex fluid samples. Porous beads have large surface area to volume ratios and high receptor concentrations, and they facilitate relatively high sensitivity detection and multiplexing. Unfortunately, to take full advantage of the porous beads' attributes, long incubation times are needed due to the relatively slow mass transfer of target molecules from the exterior solution into the beads' interior. To accelerate the mass transfer process, we propose a novel assay in which functionalized porous beads are periodically compressed and expanded. Preliminary experiments were carried out to compare the performance of the pulsating beads with that of conventional, non-pulsating beads. These experiments indicate that the pulsating beads significantly accelerate binding rates with minimal increase in non-specific binding. Thus, pulsing has the potential of significantly reducing assay time.
Keywords: biosensors, microbead, pulsation, microfluidics, uniaxial compression
In recent years, there has been a growing interest in using microbeads as solid support for capturing target molecules in both benchtop apparatuses and in microfluidic systems1-8. Typically, the beads are polymeric (e.g. polystyrene or agarose), range in size from a few micrometers to a few hundred micrometers, and can be readily purchased from many vendors with various surface functionalizations such as oligonucleotides, antibodies, and antigens.
Several recent studies have demonstrated the utility of microbead-based detection in complex biological samples. For example, beads were distributed among wells etched in fiber-optic substrates to detect DNA and inflammatory cytokines in saliva9-11; in silicon wafers for measurement of C-reactive protein in saliva12 and large-scale, rapid blood group DNA typing13; and in PDMS substrates containing an array of dome-shaped structures with interstitial wells to detect antirabbit immunoglobin G14. Beads integrated in microfluidic systems offer reduced assay times compared to their macroscopic counterparts as demonstrated with antibody-coated microbeads used to detect human secretory immunoglobulin A15, carcinoembryonic antigen16, and interferon-gamma17. While some bead-based assays rely solely on binding at the bead's periphery, others employ porous beads. The large surface area to volume ratio and the accessibility to mass transfer of the three-dimensional internal microstructure of porous beads offer a large density of binding sites, which translates to improved sensitivity18. Porous beads commonly suffer, however, from relatively low mass transfer rates of target analytes from the bulk of the solution into the bead interior. Thus, relatively long incubation times are needed to take full advantage of the porous bead's high binding capacity.
Here, we demonstrate, for the first time, that alternating compression and expansion of porous beads significantly enhances the mass transfer of analytes to interior binding sites and, thus, the binding rate. The pliable, sponge-like nature of agarose enables significant bead compression. During bead compression, the nanopores in the polymer matrix collapse and expel fluid from the bead's interior. When the compressive force is relaxed, due to the bead's elasticity19-25, the bead expands and resumes its shape prior to the compression. During the expansion stroke, solution laden with target analyte flows into the bead's interior, allowing target molecules to bind to the immobilized ligands.
To illustrate the concept, we constructed a simple experiment. We inserted both functionalized and unfunctionalized (control) agarose beads in a microfluidic chamber (Figure 1). A programmable micromanipulator was used to manipulate a rod, which periodically pushed the chamber's ceiling down. This process resulted in alternating compression and expansion of the bead. During the compression stroke, the pores in the bead collapsed and liquid was expelled out of the bead. During the expansion stroke, solution laden with target molecules permeated into the bead. The imbibition of the solution resulted in significantly enhanced mass transfer compared to diffusion alone as well as greater binding rates.
Figure 1.
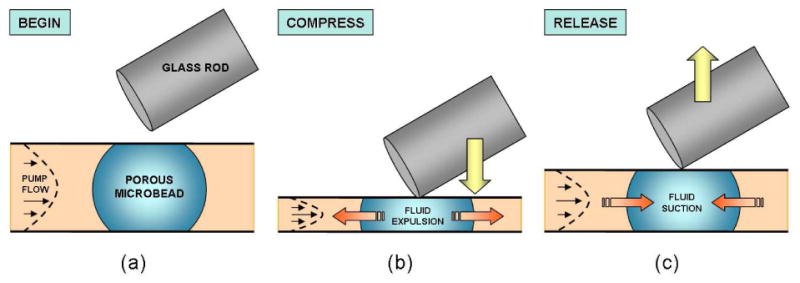
A schematic illustration of the pulsing process used in our proof of concept experiment. (a) Initially a porous, streptavidin-coated, agarose microbead is immobilized in a microconduit and subjected to the continuous flow of biotin-coated quantum dot solution. (b) The bead is compressed with a glass rod. (c) Once the rod is lifted, the conduit and bead return to their initial configuration. The effect of pulsing is monitored by measuring the fluorescent intensity of the bead as a function of time.
The experimental flow cell consisted of three layers: a bottom rigid glass substrate (a 1 mm thick glass slide); a central double-sided adhesive tape with a conduit cut in its center with a laser machine; and a top, flexible, plastic cover made with 100 μm thick cyclic olefin copolymer (COC, Plitek, Des Plaines, IL). The adhesive tape acted as a spacer, dictating the height of the conduit, as well as a sealing material. The COC cover contained inlet and outlet ports.
Streptavidin-agarose “test” beads (6% agarose mass fraction, Pierce Biotechnology, Rockford, IL) and unfunctionalized “control” beads (6% agarose mass fraction, Sepharose CL-6B, Sigma-Aldrich, St. Louis, MO) were inserted into the reaction chamber and allowed to dry at room temperature prior to attaching the COC cover. In the experiments in the 75 and 125 μm tall conduits, we focused, respectively, on beads with dry diameters greater than 30 and 50 μm. After attaching the COC cover, the chip was mounted on an epifluorescence inverted microscope (IX71, Olympus Corporation, Melville, NY) equipped with a CCD camera (Orca-ER, Hamamatsu, Bridgewater, NJ), 100 W mercury discharge lamp, and programmable Eppendorf TransferMan NK2 micromanipulator (Eppendorf North America, Hauppauge, NY). A syringe pump (PHD 2000, Harvard Apparatus, Holliston, MA) was connected to the chip's inlet with a tube and a small PDMS connection port.
Deionized water was initially pumped through the conduit to hydrate the beads. As the beads absorbed water, they swelled considerably. In the absence of the confining conduit's ceiling, the bead's diameter would swell to about 250% of its original size (e.g. a dry bead of 40 μm diameter would expand to 100 μm upon hydration). In our conduit, the bead's expansion in the vertical direction was restricted by the conduit's height, which was smaller than the diameter of the hydrated bead. As a result, the bead lodged against the conduit's floor and ceiling, assumed the shape of a flattened ellipsoid, and remained fixed in place (Figure 1).
After hydrating the beads with water, we pumped a Phosphate Buffered Saline solution (1X PBS, HyClone Laboratories, Inc., Logan, UT) laden with a specified concentration of biotinfunctionalized quantum dots (biotin-QDot605, Invitrogen, Carlsbad, CA) through the conduit. The QDots had an approximate diameter of 11±1 nm and were able to migrate through the ∼50 nm diameter26,27 pores of the (6% mass fraction) agarose beads. The diffusion coefficient of the QDots inside the beads was about an order of magnitude lower than in the bulk solution28. In our experiments, the QDots simulated the target analyte. The QDot solution was pumped through the conduit continuously at a flow rate of 0.1-1 μL min-1. A sufficiently high flow rate was chosen to approximate well-mixed conditions next to the surface of the beads as judged by the uniformity of the QDots' emission intensity outside the beads. We did not observe any significant reduction in QDot concentration next to the beads' surface.
The QDots were imaged with a long pass filter (ex: 470 nm, em: > 515 nm, filter set 11001, Chroma Technology Corporation, Rockingham, VT). The concentration of the QDots in and around selected beads was monitored with a 10× or 20× objective. Fluorescent images were acquired every 5 minutes and processed with Hamamatsu HCImage software.
Bead pulsation was initiated either concurrently with the introduction of the QDot solution or after a predetermined amount of time (to compare the performance of the same bead in the absence and the presence of pulsation). A relatively simple, but effective, means was devised to control bead compression. A 1 mm diameter glass rod was mounted to the arm of the manipulator and was pushed against the COC chip surface (Figure 1) next to the bead(s) of interest. The manipulator's motion was automated with a string of motion commands sent via the com port. We estimate that the height of the beads decreased to about 25 or 30 μm in their most compressed state. The force needed to effect the pulsation was mostly dictated by the mechanical properties of the COC cover and was estimated to be about 225±50 mN. The force needed to compress individual beads is much smaller. We did not measure the pulsating force directly.
We did not observe any leakage during the experiment due to material fatigue or cracking of the COC cover, and expect the single-use (disposable) chip to remain viable for the duration of a typical test. If necessary, materials with better mechanical properties than COC can be selected.
Pulsing was briefly paused (for less than 15% of the experiment's duration) to acquire each fluorescent image. Following the experiment, bead intensity measurements were performed with Wright Cell Imaging Facility (WCIF) ImageJ version 1.37a (National Institutes of Health, Bethesda, MD).
The nanopores of our agarose bead matrix facilitated the migration and subsequent binding of QDots. This process was expedited when the beads were subjected to periodic pulsing. During compression, there was no observable change in the lateral diameter of the beads. The compression-induced reduction in the beads' volume coincided with a decrease in the pores' volume and the interstitial expulsion of fluid from within the beads' pores. During the release, fluid laden with target molecules refilled the pores as the beads regained their initial shape. Figure 2 depicts the detected emission intensity (which is proportional to the concentration of the target analyte) as a function of time for neighboring test and control (unfunctionalized) beads in the absence and in the presence of pulsing. There was no pulsing for the first 30 min of the experiment. At 30 min, pulsing was initiated at 1 Hz and continued for the remainder of the experiment. Inset micrographs for each bead are included in the figure at 25 and 53 min. It is evident that as pulsing started, the rate of increase of emission from the test bead was much greater than in the absence of pulsing, indicating a significant increase in the binding rate. For example, at 25 min (pulsing off), the ratio of test bead intensity to control bead intensity was ∼2, whereas at 53 min (pulsing on) the ratio nearly doubled to ∼4. Although the fluorescent intensity emitted by the test bead was much greater than that emitted by the control bead, we also observed a small increase in the fluorescence of the control bead during pulsing. Post-experiment, confocal imaging suggested that this was likely due to QDot non-specific trapping at two locations: (1) at the interfaces between the control bead and the floor and ceiling of the conduit (resulting in an out-of-focus spot appearing at the bead's center when one is focusing the microscope objective at the bead's midheight plane); and (2) near the center of the control bead due to the great reduction in pore size during compression preventing QDot outward migration (the same effect was not observed in the test beads because unbound QDots did not reach the center of the test bead). The experiments with the control bead indicate that the effect of pulsing on non-specific binding was minimal. For instance, in Figure 2 the test bead intensity at 53 min was 124% above its value at 25 min, whereas the control bead intensity at 53 min was 15% above its value at 25 min.
Figure 2.
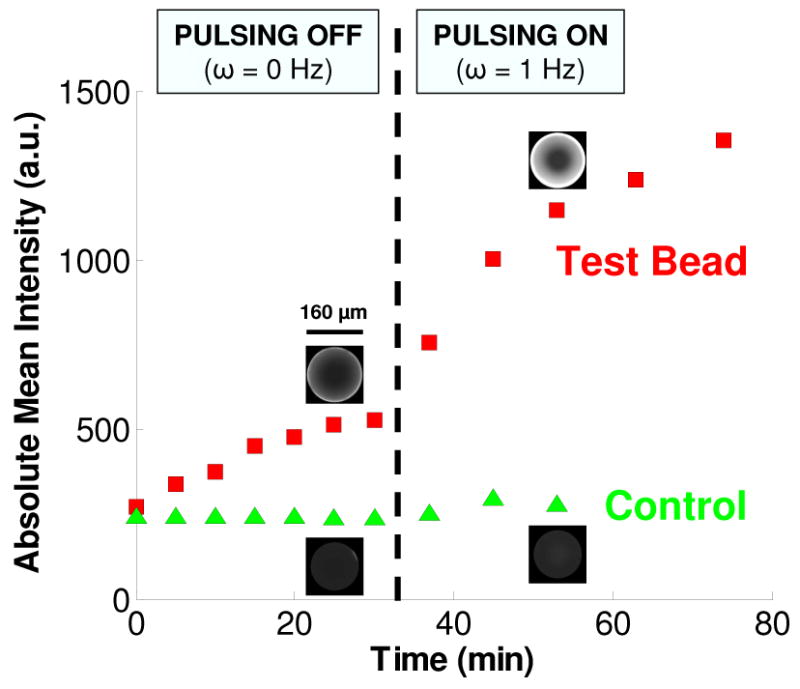
The emission intensities of a streptavidin-coated, test bead and a control (unfunctionalized) bead as functions of time. During the first 30 min, the beads are not pulsed. Pulsing at 1 Hz commences at 30 min and is maintained for the duration of the experiment. The micrographs for each bead are at t = 25 min and t = 53 min. The concentration of QDots in the buffer is 100 nM. Images are taken with a 20× objective at 2 ms exposure. The conduit is 125 μm tall.
Figure 3 depicts the signal intensities of a test bead subjected to pulsing at 2 Hz and a test bead in the absence of pulsing as functions of time. Micrographs for each bead are included at 27 and 46 min. Witness that the pulsed bead's signal intensity increased more rapidly than that of the non-pulsed bead. To achieve, for example, a signal to noise (S/N) ratio of 3, it takes approximately 15 min for the pulsed bead and 40 min for the non-pulsed bead (background noise is assumed to be equal to the bead's mean intensity at t = 0 min). One hour after the start of the experiment, the intensity of the pulsed bead is more than twice that of the non-pulsed bead. These results demonstrate the enhancement in mass transfer and subsequent binding due to pulsation. For the pulsation frequencies tested between 0.5 and 2 Hz (data not shown here), with the precision of our experiments, we did not see a significant dependence of bead intensity on pulsing frequency. Limitations of our experimental apparatus prevented us from testing higher frequencies. However, we would expect that for a given assay, an optimal pulsing frequency exists that depends on factors such as the bead pore size, target size, diffusion coefficients, and interaction kinetics.
Figure 3.
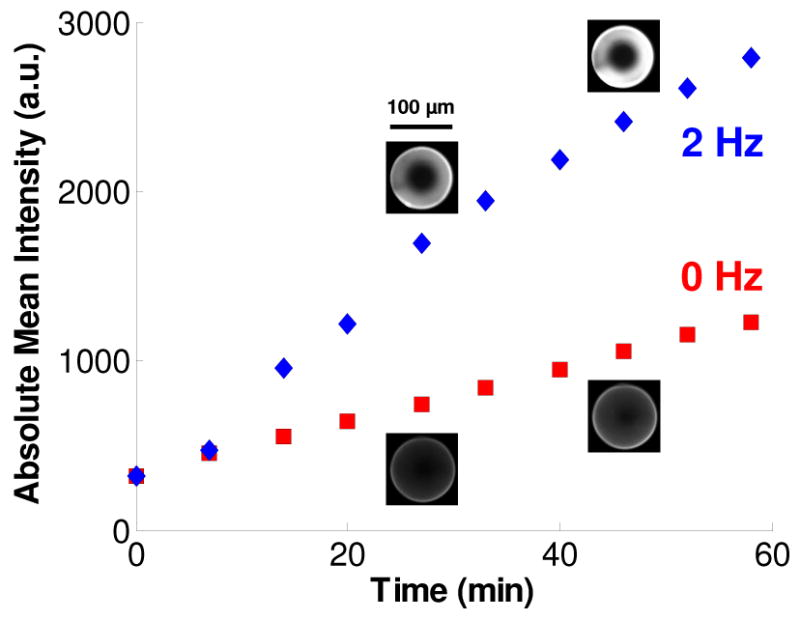
The signal intensity emitted by functionalized agarose beads in the presence of pulsing (2 Hz) and in the absence of pulsing as functions of time. The micrographs for each bead are at times t = 27 min and t = 46 min. 75 μm tall conduit. 10 nM QDot solution. 10× objective and 30 ms exposure.
Our preliminary data demonstrates that alternating compression and expansion of porous beads can significantly increase mass transfer into the bead's interior and shorten assay time. We repeated similar experiments 16 times and observed similar results. The method used in our experiments to compress the beads and prove the concept is somewhat primitive and may not be appropriate for all circumstances. There are, however, many more elegant alternatives to induce bead deformation. The chip may be fitted with a small actuator, such as a membrane that is deformed with hydrostatic pressure induced by an external pressure or heat source; a membrane driven by electrostatic forces; a cell phone vibration motor; or a piezoelectric element, to name just a few options. Other alternatives include embedding magnetic particles in the bead and applying non-uniform, alternating magnetic fields or embedding hydrogels that undergo phase transition upon temperature variations29,30. To the best of our knowledge, this is a first report on using a pulsating bead to enhance mass transfer. The technique has the potential of shortening assay times and improving detection sensitivity within given time constraints.
Acknowledgments
This work was supported, in part, by funding from a Department of Education Graduate Assistance in Areas of National Need (GAANN) fellowship in Lab-on-Chip Technologies (JAT) and NIH/NIDCR Grant U01-DE-017855 (HHB).
References
- 1.Derveaux S, Stubbe BG, Braeckmans K, Roelant C, Sato K, Demeester J, De Smedt SC. Anal Bioanal Chem. 2008;391:2453–2467. doi: 10.1007/s00216-008-2062-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Qiu X, Thompson JA, Chen Z, Liu C, Chen D, Ramprasad S, Mauk MG, Ongagna S, Barber C, Abrams WR, Malamud D, Corstjens PLAM, Bau HH. Biomed Microdevices. 2009;11:1175–1186. doi: 10.1007/s10544-009-9334-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson JA, Du X, Grogan JM, Schrlau MG, Bau HH. J Micromech Microengineering. 2010;20:115017. [Google Scholar]
- 4.Thompson JA, Bau HH. J Chromatogr B. 2010;878:228–236. doi: 10.1016/j.jchromb.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ng JK, Selamat ES, Liu W. Biosens Bioelectron. 2008;23:803–810. doi: 10.1016/j.bios.2007.08.026. [DOI] [PubMed] [Google Scholar]
- 6.Lim CT, Zhang Y. Biosens Bioelectron. 2007;22:1197–1204. doi: 10.1016/j.bios.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Verpoorte E. Lab Chip. 2003;3:60N–68N. doi: 10.1039/b313217j. [DOI] [PubMed] [Google Scholar]
- 8.Jokerst JV, Raamanathan A, Christodoulides N, Floriano PN, Pollard AA, Simmons GW, Wong J, Gage C, Furmaga WB, Redding SW, McDevitt JT. Biosens Bioelectron. 2009;24:3622–3629. doi: 10.1016/j.bios.2009.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blicharz TM, Siqueira WL, Helmerhorst EJ, Oppenheim FG, Wexler PJ, Little FF, Walt DR. Anal Chem. 2009;81:2106–2114. doi: 10.1021/ac802181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bowden M, Song LN, Walt DR. Anal Chem. 2005;77:5583–5588. doi: 10.1021/ac050503t. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson JA, Steemers FJ, Walt DR. Anal Chem. 2000;72:5618–5624. doi: 10.1021/ac0008284. [DOI] [PubMed] [Google Scholar]
- 12.Christodoulides N, Mohanty S, Miller CS, Langub MC, Floriano PN, Dharshan P, Ali MF, Bernard B, Romanovicz D, Anslyn E, Fox PC, McDevitt JT. Lab Chip. 2005;5:261–269. doi: 10.1039/b414194f. [DOI] [PubMed] [Google Scholar]
- 13.Hashmi G, Shariff T, Seul M, Vissavajjhala P, Hue-Roye K, Charles-Pierre D, Lomas-Francis C, Chaudhuri A, Reid ME. Transfusion. 2005;45:680–688. doi: 10.1111/j.1537-2995.2005.04362.x. [DOI] [PubMed] [Google Scholar]
- 14.Lim CT, Zhang Y. Small. 2007;3:573–579. doi: 10.1002/smll.200600435. [DOI] [PubMed] [Google Scholar]
- 15.Sato K, Tokeshi M, Odake T, Kimura H, Ooi T, Nakao M, Kitamori T. Anal Chem. 2000;72:1144–1147. doi: 10.1021/ac991151r. [DOI] [PubMed] [Google Scholar]
- 16.Sato K, Tokeshi M, Kimura H, Kitamori T. Anal Chem. 2001;73:1213–1218. doi: 10.1021/ac000991z. [DOI] [PubMed] [Google Scholar]
- 17.Sato K, Yamanaka M, Takahashi H, Tokeshi M, Kimura H, Kitamori T. Electrophoresis. 2002;23:734–739. doi: 10.1002/1522-2683(200203)23:5<734::AID-ELPS734>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 18.Ali MF, Kirby R, Goodey AP, Rodriguez MD, Ellington AD, Neikirk DP, McDevitt JT. Anal Chem. 2003;75:4732–4739. doi: 10.1021/ac034106z. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Wang D, Lu W, Lin Y, Tung K. Chem Eng Sci. 2008;63:195–203. [Google Scholar]
- 20.Egholm RD, Christensen SF, Szabo P. J Appl Polym Sci. 2006;102:3037–3047. [Google Scholar]
- 21.Liu KK. J Phys D-Appl Phys. 2006;39:R189–R199. [Google Scholar]
- 22.He JY, Zhang ZL, Kristiansen H. J Appl Polym Sci. 2009;113:1398–1405. [Google Scholar]
- 23.Knaebel A, Lequeux F. Polym Gels Networks. 1997;5:577–584. [Google Scholar]
- 24.Andrei DC, Briscoe BJ, Luckham PF, Williams DR. J Chim Phys Phys-Chim Biol. 1996;93:960–976. [Google Scholar]
- 25.Yan Y, Zhang Z, Stokes JR, Zhou Q, Ma G, Adams MJ. Powder Technol. 2009;192:122–130. [Google Scholar]
- 26.Hagel L, Ostberg M, Andersson T. J Chromatogr A. 1996;743:33–42. [Google Scholar]
- 27.Jokerst JV, Chou J, Camp JP, Wong J, Lennart A, Pollard AA, Floriano PN, Christodoulides N, Simmons GW, Zhou Y, Ali MF, McDevitt JT. Small. 2011;7:613–624. doi: 10.1002/smll.201002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thompson JA, Bau HH. 2011 In Preparation for Submission. [Google Scholar]
- 29.Gijs MAM. Microfluid Nanofluid. 2004;1:22–40. [Google Scholar]
- 30.Richter A, Kuckling D, Howitz S, Gehring T, Arndt KF. J Microelectromech Syst. 2003;12:748–753. [Google Scholar]