

Abstract
Although recent publications have linked the molecular events driving facioscapulohumeral muscular dystrophy (FSHD) to expression of the double homeobox transcription factor DUX4, overexpression of FRG1 has been proposed as one alternative causal agent as mice overexpressing FRG1 present with muscular dystrophy. Here, we characterize proliferative defects in two independent myoblast lines overexpressing FRG1. Myoblasts isolated from thigh muscle of FRG1 transgenic mice, an affected dystrophic muscle, exhibit delayed proliferation as measured by decreased clone size, whereas myoblasts isolated from the unaffected diaphragm muscle proliferated normally. To confirm the observation that overexpression of FRG1 could impair myoblast proliferation, we examined C2C12 myoblasts with inducible overexpression of FRG1, finding increased doubling time and G1-phase cells in mass culture after induction of FRG1 and decreased levels of pRb phosphorylation. We propose that depressed myoblast proliferation may contribute to the pathology of mice overexpressing FRG1 and may play a part in FSHD.
Introduction
Facioscapulohumeral muscular dystrophy, or FSHD, primarily affects muscles of the face, shoulders and upper arms. It is the third most common muscular dystrophy, following Duchenne muscular dystrophy and myotonic muscular dystrophy, affecting 1 in 20,000 individuals [1]. Onset of muscle weakness in FSHD patients most commonly occurs between puberty and the second decade of life, ultimately leading to patients becoming wheelchair-bound [2], [3], [4]. Compared to the majority of muscular dystrophies, FSHD is unique in its very low rate of any respiratory or cardiac muscle involvement, which is often the eventual cause of death for patients with other forms of muscular dystrophy [5]. As such, patients with FSHD typically live a normal lifespan, but suffer a severely decreased quality of life.
The molecular basis of FSHD is still under debate, although the genetic event linked with FSHD has been identified to be in the subtelomeric region on the long arm of chromosome 4 [6], [7]. This region, denoted as 4q35, contains a series of 3.3 kb tandem repeat elements, which have been termed D4Z4 repeats [8]. Unaffected individuals have 11 to 150 D4Z4 repeats, but patients with FSHD have had this region truncated to 10 or less [9]. Efforts to identify the molecular basis of this disease have been hampered, however, because the truncation associated with FSHD is not within a well-characterized gene coding or promoter region.
Multiple models have been proposed to explain how a D4Z4 repeat truncation is linked to FSHD, reviewed in [10]. The primary model is that the loss of D4Z4 repeats increases expression of a double homeobox transcription factor DUX4c, a putative gene centromeric to the D4Z4 repeats and highly homologous to DUX4 [11], [12], [13]. DUX4c has been shown to be up-regulated in FSHD biopsies and primary myoblasts, possibly leading to induction of the MYF5 myogenic regulator, which serves to inhibit differentiation and activate proliferation [14], [15]. In addition, overexpression of DUX4 in other cell lines has been shown to cause apoptosis and impair myogenesis in both cell culture models and zebrafish development [16], [17], [18]. A recent chromosomal analysis of affected and unaffected 4q35 alleles has determined that FSHD is linked to a single nucleotide polymorphism located distal to the last D4Z4 repeat [19], which stabilizes the DUX4 transcript through polyadenylation and may result in elevated protein levels and cytotoxicity via still unknown mechanisms.
A second model proposes that the loss of D4Z4 repeats may increase the available pool of a repressive complex comprised of YY1, HMG2B and nucleolin that is normally bound to D4Z4 repeats. YY1 interacts with Ezh2, a histone lysine methyltransferase, playing a key role in expression of muscle genes during embryonic development [20], [21] and MeCP2, a methyl CpG binding protein involved in Rett syndrome [22]. In addition, YY1 may also be able to interact with the chromatin insulator CTCF [23]. HMGB2 may affect the maintenance of heterochromatic regions by interacting with SP100B and subsequently HP1, establishing higher-order chromatin structures [24], [25]. In contrast, nucleolin may have an opposite effect on heterochromatin formation as it serves to decondense chromatin through displacement of histone H1 [26]. Perturbations in any of these proteins due to loss of D4Z4 repeats resulting in increased chromatin accessibility may cause gene deregulation in trans and play a role in the pathogenesis of FSHD.
A third model suggests that D4Z4 may serve as nucleating sites for local transcriptional repression involving the previously mentioned YY1 complex. Loss of D4Z4 may lift repression in cis of the 4q35 region and thus the nearby genes FRG1, FRG2 and ANT1 [27], [28]. Additionally, the identification of a nuclear matrix attachment site (S/MAR) and its disassociation from the nuclear matrix in FSHD patients may change the arrangement of DNA loop domains, causing increased transcription of FRG1 and FRG2 [29], [30], [31]. Presently, it is unclear as to which or how many of these many non-exclusive mechanisms play a causal role in the pathogenesis of FSHD.
Previously, it has been observed that there may be increased transcriptional activation of FRG1, FRG2 and ANT1, the three genes upstream of the D4Z4 tandem repeat elements, in muscle of human FSHD patients [28]; however, these results were not observed in another patient study [32]. Unfortunately, generating a relevant mouse model to study FSHD has been exceptionally difficult because mice do not have D4Z4 repeats in an analogous chromosomal setting. Acting under the assumption that overexpression of FRG1, FRG2 or ANT1 plays a causative role in the development of FSHD, transgenic mice were generated expressing each of these individual genes under the human skeletal actin promoter, specific for expression in muscle, which resulted in the identification of a potential mouse model for FSHD. In contrast to transgenic mice overexpressing FRG2 or ANT1, only transgenic mice overexpressing FRG1 appear to have symptoms characteristic of muscular dystrophy [33]. It should be noted, however, that the FRG1 transgenic mouse model that resulted in dystrophic phenotypes had FRG1 skeletal muscle protein levels considerably higher than that observed in FSHD patients.
Facioscapulohumeral muscular dystrophy region gene-1 (FRG1) is an actin-bundling protein associated with muscle-attachment sites, specifically located to the Z-disc in mature muscle tissue [34], [35]. FRG1 has been shown to play a crucial and specific role in muscle development of Xenopus laevis, further implicating its importance in muscle development and maintenance [36], [37]. Recently, Bodega et al. showed that the FRG1 gene was prematurely expressed during FSHD myoblast differentiation, thereby suggesting that the number of D4Z4 repeats in the array may affect the correct timing of FRG1 expression [27].
Based on this work, we hypothesized that the dystrophic phenotype in FRG1 transgenic mice is caused, at least in part, by decreased proliferation in the muscle satellite cell population, which are the cells responsible for maintaining proper muscle regenerative potential. Satellite cells are often thought of as muscle stem cells, proliferating when there is a need for either muscle repair or growth and then differentiating into skeletal muscle. We have observed that increased levels of FRG1 impair normal satellite cell proliferation and may contribute to disease progression by limiting the pool of cells to repair damaged muscle and/or delaying the kinetics of repair.
Results
Expression of FRG1 in skeletal muscle of mice causes dystrophic characteristics
Mice overexpressing FRG1 have been previously described [33]. In that study, three lines of transgenic mice overexpressing FRG1 specifically in muscle were generated by using the human skeletal actin promoter to drive transcription of the human FRG1 cDNA. These mice develop spinal kyphosis and characteristics of muscular dystrophy, including increased fibrosis in muscles, lower body weight, lower muscle weight and decreased cross-sectional area, reduced tolerance to exercise and mis-splicing of specific transcripts associated with myotonic dystrophy. Mice used in the Gabellini et al. study [33] were kindly provided by Dr. Rossella Tupler. After establishing an independent colony with continual back-crossing to C57BL/6 mice, we performed a limited characterization of the in vivo phenotypes in their highest FRG1 expressing line (H-FRG1 TG) as a confirmation of their reported findings. As expected, these mice begin to show mild spinal kyphosis and reduced body weight by 4 weeks of age, both of which become progressively more severe over time.
To verify that FRG1 expression was increased specifically in skeletal muscle, we performed Western blot analysis on lysates generated from a variety of tissues from 10-week old mice using a specific α-FRG1 antibody. Equivalent amounts of total protein were loaded and increased FRG1 protein was detected in all tested skeletal muscles of H-FRG1 TG mice compared to wild-type littermate controls ( Figure 1A ). We observed a specific increase in the expression of FRG1 in skeletal muscles such as quadriceps, gastrocnemius and diaphragm muscle, and no increase of FRG1 in cardiac muscle or other tissues, confirming the skeletal muscle-specific expression of FRG1. Endogenous expression of FRG1 was detected in the lung, but there was no significant difference in expression between H-FRG1 TG mice and wild-type littermate controls.
Figure 1. FRG1 expression and dystrophic phenotype of mice.

A) Muscles and tissue from either H-FRG1 TG transgenic mice (FRG1) or wild-type littermate control (C57BL/6) were collected from 10-week old mice. 300 µg of total protein from tissue lysates isolated from quadriceps muscle (Q), gastrocnemius muscle (G), diaphragm muscle (D), whole heart (H), lung tissue (Lu), liver tissue (Li) and brain tissue (B) were probed with α-FRG1 antibody after SDS-PAGE. B) Diaphragm and quadriceps muscle from 13-week old H-FRG1 TG mouse (FRG1) or wild-type littermate control (C57BL/6) stained with hemotoxylin/eosin and viewed under 100× magnification. Arrows note location of centrally located nuclei present in FRG1 cross-section. Scale bar notes 50 µm.
Muscle weights of collected tissues were measured at different ages and normalized to body weight, to account for runted phenotype of H-FRG1 TG mice. Quadriceps and gastrocnemius wet muscle weight comprised a smaller percentage of total body weight in H-FRG1 TG mice compared to wild-type mice, but diaphragm weight showed no significant change (data not shown). This observation agrees with previous observations demonstrating that despite FRG1 expression in all skeletal muscle tissue, only specific muscles may exhibit a dystrophic phenotype [33], [38].
To examine the histology of specific muscles, we collected and cryosectioned muscle tissue from 13-week old H-FRG1 TG mice and wild-type littermate controls. Hemotoxylin and eosin staining of these sections showed an increased incidence of centrally located nuclei and increased fiber size variability in affected muscle tissues, namely the quadriceps, but this was not noted in internal muscles such as the diaphragm ( Figure 1B ). From these experiments we find that overexpression of FRG1 only causes a dystrophic phenotype in a subset of skeletal muscles. We also noted that H-FRG1 TG mice were able to survive past 1 year of age (data not shown) despite the worsening of their muscular dystrophy and other associated phenotypes. The apparent sparing of the diaphragm muscle of any measurable defect may possibly replicate the muscle specificity of FSHD in human patients, which similarly does not appear to affect internal muscles. All of these observations confirm the muscular dystrophy phenotype in H-FRG1 TG mice as originally characterized.
Effects of FRG1 expression in mouse-derived myoblasts
In principle, dystrophic phenotypes in muscle can arise from enhanced degeneration, defective regeneration, or both. For instance, myoblasts from mice lacking A-type lamins that exhibit signs of muscular dystrophy plate with high viability and proliferate normally, but ultimately have impaired differentiation [39]. Thus, muscular dystrophies associated with mutations in the A-type lamin gene, LMNA, may be associated with decreased satellite cell differentiation, causing depressed regeneration in addition to reduced myofiber stability. As a test for potential regeneration defects in satellite cells of H-FRG1 TG mice, we sought to determine whether myoblasts from these mice had altered proliferation or differentiation potential in cell culture. Myoblast cultures were isolated and cultured by standard techniques [40], which, if treated appropriately, are comprised predominantly of proliferating myoblasts and can provide an accurate readout of the regenerative potential of muscle from the H-FRG1 TG mice. We speculated that myoblasts from H-FRG1 TG mice may have similar defects to those from Lmna−/− mice.
We isolated and cultured the satellite cell population from affected quadriceps and unaffected diaphragm muscle of 18-week old H-FRG1 TG mice and wild-type littermate controls. Since the quadriceps muscle, but not the diaphragm, exhibited characteristics of muscular dystrophy, we speculated that there is a specific defect in satellite cells derived from the thigh. We thus performed a clonal assay on primary isolated myoblasts in order to measure the proliferation rates of the isolated satellite cells.
We quantified transcript levels of FRG1 in our isolated satellite cells by qPCR, and observed increased levels of FRG1 transcript in both proliferating and differentiated cultures from H-FRG1 TG mice (Figure S1A). This was somewhat surprising as the promoter driving FRG1 expression, the human skeletal actin promoter, is reported to be active only after differentiation. We also performed Western analysis on cell lysates generated from the same satellite cells to observe protein levels of FRG1. Levels of FRG1 protein are increased in both proliferating and differentiated cultures of satellite cells from of H-FRG1 TG mice (Figure S1B). In addition, FRG1 is detected in satellite cell cultures derived from either diaphragm or thigh muscle (Figure S1C).
For clonal analysis, the tissue-derived satellite cells were plated at a very low density, 1000 cells per 10 cm dish, and allowed to grow for a predefined amount of time. At regular intervals, plates were fixed and nuclei were visualized by staining with methylene blue. We also stained for myosin heavy chain (MyHC) as a control to verify that cells did not prematurely differentiate over the course of the clonal assay, in which bFGF and high serum levels were maintained. After fixation and staining, the total number of cells per clone was determined and binned for comparison in a histogram format. Thigh-derived satellite cells from an 18-week old H-FRG1 TG mouse show a marked decrease in average clone size compared to those derived from a wild-type littermate control ( Figure 2A ). A significant fraction of these cells show arrest in a 2-cell clone size skewing the distribution compared to the wild-type thigh-derived satellite cells. This effect may be even more dramatic considering that single cell clones were not scored in this assay, as we consider single cell clones may potentially be new clones arising from detached satellite cells floating away from the original clone during mitosis. We replicated these observations with an independent satellite cell culture isolated from 20-week old mouse limbs obtained directly from Dr. Rossella Tupler which were comparable to our 18-week old thigh-derived satellite cells at a similar time point (Figure S2). The proliferative defect was not replicated in the diaphragm-derived satellite cells, which show a very similar clone size distribution between the H-FRG1 TG and wild-type C57BL/6 littermates. These findings indicate that FRG1 overexpression leads to a muscle-type specific defect in proliferation, and correlates with the dystrophic phenotype.
Figure 2. Clonal analysis of mouse-derived myoblasts.

A) Myoblasts isolated from diaphragm (D) or thigh (T) of 18-week old H-FRG1 TG mice (FRG1) or wild-type littermate controls (WT) were cultured and plated at low density. Cells were fixed at regular time intervals and stained for myosin heavy chain. Total number of nuclei per clone was counted and a representative graph of data from 96-hours post-plating is shown (n = 100). B) Myoblasts isolated from diaphragm (D) or thigh (T) of 4-week old H-FRG1 TG mice (FRG1) or wild-type littermate controls (WT) were scored for proliferation as above at 72-hours post-plating (50<n<80).
To determine whether there is an age-dependent increase in severity of the observed proliferation defect, we isolated and performed a clonal assay on both thigh- and diaphragm-derived satellite cells isolated from 4-week old mice, which appear asymptomatic, to compare to the aforementioned data from more severely symptomatic 18-week old mice. For each of these populations, we scored multiple time points of a clonal assay, to more thoroughly assay the proliferative defect. The clone size distributions of myoblasts from asymptomatic 4-week old mice did not show any significant proliferative defect when compared to their 18-week old counterparts at a similar clone size ( Figure 2B ). To more easily compare the data from these clone size distributions, we obtained the average clone size for each of these populations and calculated the average doubling time of each of these lines. The data show that the proliferative defects associated with FRG1 overexpression appear to be age-specific, as myoblasts obtained from quadriceps muscle of 4-week old H-FRG1 TG mice are indistinguishable from those derived from littermate controls, whereas satellite cells from 18-week old mice H-FRG1 TG mice have a severe defect ( Table 1 ). These effects are further reflected in the general health of the mice and severity of muscle dystrophy at later ages. It is interesting to observe that by 52-weeks of age, we were unable to successfully culture satellite cells from the thigh muscle of H-FRG1TG mice, possibly due to either extremely depressed proliferation rates or exhaustion of the satellite cell population. It should also be noted that we never observed any significant differentiation defect in any of our satellite cell cultures (data not shown). We conclude that the overexpression of FRG1 in muscle tissue causes muscle-type-specific and age-dependent impairment in the ability of satellite cells to proliferate when isolated in cell culture.
Table 1. Doubling times derived from clonal assays.
| 4 wk. old | 18 wk. old | ||||
| Hours post-plating | 48 h. | 72 h. | 48 h. | 96 h. | 144 h. |
| BL/6-D | 18 | 17.4 | 19.9 | 22.2 | 25.6 |
| FRG1-D | 17.4 | 17.7 | 25.7 | 21.9 | 24.4 |
| BL/6-T | 18.6 | 18.8 | 23 | 28.1 | 26 |
| FRG1-T | 18.5 | 19.4 | 31.1 | 33.3 | 28.2 |
Average calculated doubling time of myoblasts isolated from 4-week or 18-week old H-FRG1 TG mice (FRG1) or wild-type littermate control (BL/6) at varying times post-plating. Doubling times were calculated from average clone size with the following formula: [T *Ln(2)]/Ln(average clone size).
Inducible FRG1 expression in C2C12 myoblasts
In addition to experiments performed in mouse-derived satellite cell culture, we sought to establish an independent cell culture model system that would be free of any potential artifacts introduced during generation of the H-FRG1TG mice. Initially, we performed viral transduction of FRG1 under the CMV promoter in the pMXIH vector as previously described [41]. Findings in early passages after selection indicated a proliferative defect associated with FRG1 overexpression. Unfortunately, we observed a loss of the proliferative defect over time as measured by BrdU-positive cells (Figure S3). The loss of defects in proliferation was accompanied by an increase in cells staining negative for FRG1, suggesting either that expression was being actively silenced or, more likely, high FRG1 expressing cells were being outcompeted over time by lower expressing cells with a faster rate of proliferation. Given this complication, we adopted another strategy to drive FRG1 overexpression.
We chose to employ an inducible system to control expression of FRG1 that would allow us to turn on expression when needed. We utilized C2C12 myoblasts that have an integrated cassette expressing FLAG-tagged FRG1 under a tetracycline-responsive promoter (iC2C12-FRG1) generated as previously described and kindly provided to us by Dr. Michael Kyba [17]. FRG1 expression is induced in iC2C12-FRG1 myoblasts by the addition of doxycycline and levels of induction can be modulated by adjusting levels of the drug. There is no detectable expression of FRG1 in uninduced cells as assayed by Western blot of whole cell lysates using an α-FLAG antibody. In comparison, robust expression of FRG1 was observed under a variety of induction conditions ranging from 250 to 1000 ng/mL doxycycline ( Figure 3A ).
Figure 3. Expression of FRG1 in iC2C12-FRG1 myoblasts.

A) Western blot of lysates from iC2C12-FRG1 myoblasts before induction with doxycycline or after induction for 24 hours with concentrations ranging from 250 ng/mL to 1000 ng/mL using α-FLAG antibody. β-actin loading control shown below. B) Localization of FRG1 by immunofluorescence in iC2C12-FRG1either uninduced or induced with 500 ng/mL doxycycline for 24 hours. DAPI stain is represented in the blue channel and α-FRG1 antibody staining is represented in the green channel.
FRG1 is believed to play a role in post-transcriptional mRNA processing and localizes to nuclear Cajal bodies [42], therefore we performed immunofluorescence to investigate whether FRG1 was properly localized in iC2C12-FRG1 myoblasts after induction. iC2C12-FRG1 myoblasts were cultured on glass coverslips and cells were grown for 24 hours in the presence or absence of doxycycline followed by fixation and staining. Staining with DAPI and an α-FLAG antibody revealed nuclear localization of FRG1-FLAG following induction ( Figure 3B ). There was some variation in magnitude of expression of FRG1-FLAG on a cell-to-cell basis, but we observed robust partially punctuate staining in the nucleus, consistent with possible localization to Cajal bodies.
Interestingly, we observed in our initial characterization of induced iC2C12-FRG1 myoblasts a similar phenomenon as the virally transduced C2C12 cells in that after numerous passages in the presence of doxycycline, induced cells showed much lower expression levels of FRG1-FLAG (Figure S4) and likely a loss of any proliferative defect. A similar explanation is likely that lower FRG1 expressing cells in this non-clonal population ultimately outcompete higher expressing cells and take over the culture.
Characterizing defects in proliferation following FRG1 expression in iC2C12-FRG1 myoblasts
To investigate the proliferation defect caused by FRG1 overexpression, we conducted a number of assays in iC2C12-FRG1 myoblasts. We first assayed the proliferation rates of cells in mass culture in the presence or absence of doxycycline. In order to determine growth rates, identical numbers of cells were plated in wells of a 24-well plate and at regular time intervals the total number of cells per well were counted using a hemocytometer. We determined that induction of FRG1-FLAG in iC2C12-FRG1 myoblasts has a negative effect on their proliferation rate, as shown by the increased doubling time when grown in doxycycline ( Figure 4A ). Naïve C2C12 myoblasts were not affected by exposure to doxycycline (data not shown), demonstrating that the phenotype is likely specific to FRG1 overexpression.
Figure 4. Characterization of proliferation defect in iC2C12-FRG1 myoblasts.

A) Mass culture doubling times calculated from hemocytometer counts of iC2C12-FRG1 myoblasts with or without induction of expression by doxycycline over 120 hours. Below are the cell cycle profiles of uninduced iC2C12-FRG1 myoblasts as well as after 24 hours and 72 hours in the presence of doxycycline. Cell cycle profiles were calculated from DNA content analysis by flow cytometry on proliferating myoblasts that were fixed and DAPI stained. *denotes statistical significance p<0.05, **p<0.005. B) Cell cycle profiles of synchronized iC2C12-FRG1 myoblasts over 24 hours in the absence or presence of doxycycline showing fraction of cells in G1, S or G2-phase. Comparison of data is plotted in a line graph fashion showing G1 in green, S in blue and G2 in red to demonstrate the generalized lag exhibited by iC2C12-FRG1 myoblasts expressing FRG1. Uninduced iC2C12-FRG1 myoblasts are graphed with a dashed line, while iC2C12-FRG1 myoblasts with doxycycline-induced FRG1 expression are graphed with a solid line. n>20,000 cells analyzed for each time point.
To further investigate the proliferative defect of iC2C12-FRG1 myoblasts expressing FRG1, we utilized flow cytometry to determine the fraction of cycling cells in specific phases of the cell cycle. Actively proliferating iC2C12-FRG1 myoblasts grown in either the presence or absence of doxycycline were fixed and stained with DAPI. Subsequent flow cytometry analysis of the cells for DNA content indicated an increased fraction of FRG1 overexpressing iC2C12-FRG1 myoblasts in G1-phase, coupled with a corresponding decrease in the fraction of cells in S-phase ( Figure 4A ). These findings suggest that cells overexpressing FRG1 are delayed in transit through G1. We believe that this shift in cell cycle profile may at least in part explain the gross proliferation defect that we have observed.
One potential weakness of just looking at a cell cycle phase distribution is that absolute cell cycle length is indeterminate in a mass culture cell cycle profile. To address this issue, we performed a mitotic shakeoff assay to synchronize proliferating iC2C12-FRG1 myoblasts so that transit through specific cell cycle phases could be measured. Since cells progressing through mitosis are less adherent to culture dishes, this technique provides a way to synchronize cells in the absence of any drugs to mediate cell cycle arrest. After being plated in the presence or absence of doxycycline for a period of 24 hours, mitotic cells were isolated by mild shaking and rocking for a period of 20 minutes to generate synchronized cultures. Fixation at 2-hour intervals followed by subsequent analysis by flow cytometry for DNA content was performed to track cells as they progressed through the cell cycle. We observed that FRG1 overexpressing cells exhibited a consistent 1–2 hour delay in exit from G1 and entry into S-phase ( Figure 4B & Table 2 ). This finding is consistent with the FACS analysis of asynchronous cells, which indicated that a higher percentage of FRG1 overexpressing cells were in G1 phase. It should be noted that it is difficult to monitor progression beyond one cell cycle in this assay since the iC2C12-FRG1 myoblasts rapidly lose synchronicity.
Table 2. Raw cell cycle profile data.
| 2 | 4 | 6 | 8 | 10 | 12 | 14 | 16 | 18 | 20 | 22 | 24 | |
| ICE FRG1 −dox | ||||||||||||
| %G1 | 86.41 | 82.4 | 74.58 | 48.95 | 21.03 | 4.31 | 20.55 | 40.35 | 53.35 | 56.66 | 43.65 | 29.16 |
| %S | 3.11 | 4 | 12.52 | 34.53 | 74.94 | 92.66 | 32.41 | 14.43 | 20.47 | 27.68 | 48.95 | 51.91 |
| %G2 | 10.48 | 13.6 | 12.9 | 16.52 | 4.03 | 3.03 | 47.04 | 45.22 | 26.18 | 15.66 | 7.4 | 18.93 |
| ICE FRG1 +dox | ||||||||||||
| %G1 | 87.83 | 87.67 | 80.81 | 66.74 | 38.25 | 12.04 | 16.5 | 34.75 | 56.41 | 64.96 | 50.97 | 39.37 |
| %S | 2.31 | 1.82 | 7.83 | 21.04 | 49.45 | 87.96 | 52.06 | 18.24 | 11.67 | 19.11 | 35.2 | 46.6 |
| %G2 | 9.86 | 10.51 | 11.36 | 12.22 | 12.3 | 0 | 31.44 | 47.01 | 31.92 | 15.93 | 13.83 | 14.23 |
Values from WinCycle analysis of synchronized ICE-FRG1 myoblasts in the presence or absence of doxycycline-induced FRG1 expression showing fraction of cells in G1, S or G2-phase of the cell cycle plotted in table form to emphasize differences. N>20,000 analyzed for each timepoint.
Cell cycle specific factors are affected by FRG1 expression
The retinoblastoma protein (pRb) is one of the primary regulators of the mammalian cell cycle, inhibiting E2F-dependent transcription and maintaining cells in G1-phase when hypophosphorylated. Phosphorylation by cyclin-dependent kinases interferes with the capacity of pRb to repress E2F-dependent transcription, permitting S-phase entry [43], [44]. Since FRG1 overexpression led to increased G1 phase occupancy, we examined the phosphorylation state of pRb as an indicator of these altered cell cycle kinetics. We observe via Western blot that compared to naïve C1C12 or uninduced iC2C12-FRG1 myoblasts, levels of 807/811 phosphorylated pRb are lower in induced iC2C12-FRG1 myoblasts ( Figure 5A ). Total levels of pRb appear lower, however this is likely an artifact of the antibody used, since it is known to have somewhat greater affinity for hyperphosphorylated pRb. Decreased pRb phosphorylation is consistent with a decreased proliferation rate and increased G1 phase occupancy.
Figure 5. Phosphorylation of pRb is perturbed by FRG1 expression.
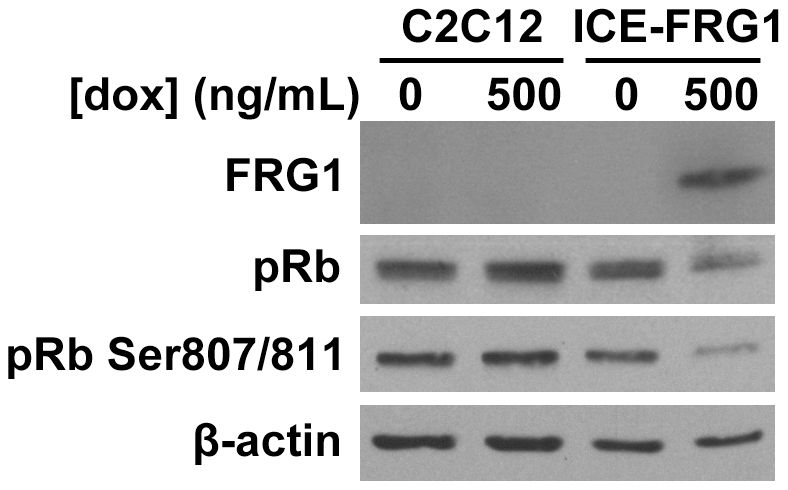
Western blot analysis for pRb, pRb Ser807/811 and FRG1 in iC2C12-FRG1 cells grown in the presence or absence of doxycycline. β-actin loading control shown below.
Discussion
In this study, we demonstrate decreased proliferation rates of myoblasts expressing FRG1, an attribute that could contribute to the long term reduction in muscle regenerative potential and muscular dystrophy observed in transgenic mice overexpressing FRG1. We have verified expression and muscular dystrophy in the H-FRG1 TG mouse and seen that thigh-derived myoblasts, but not diaphragm-derived myoblasts, from these animals demonstrate a proliferative defect by clonal analysis. We also find that induction of FRG1 in myoblasts by a tetracycline-responsive system negatively affects proliferation as determined in cell cycle profiles measured by flow cytometry and hypophosphorylation of pRb.
Reduced myoblast proliferation is not commonly linked to muscular dystrophy, which is more classically attributed to death of muscle fibers, such as in Duchenne muscular dystrophy [45], or a combination of enhanced fiber degeneration and defective differentiation kinetics such as in Lmna −/− mouse models [39], [46]. However, there have been reports of depressed proliferation kinetics in Duchenne muscular dystrophy myoblasts, compounding the existing mechanisms of muscular dystrophy [47]. Since the proliferation defect gets more severe in myoblasts isolated from H-FRG1 TG mice of increasing age, it is difficult to differentiate between two models: (1) that the proliferation defect precedes onset of the dystrophic phenotype or (2) that the defect derives from reduced satellite potential with age resulting from increasing strain on satellite cells to repair damage.
Our findings in C2C12 cells may indicate that a cell cycle defect can occur as a primary result of FRG1 overexpression. However, it ultimately remains unclear precisely how FRG1 impacts cell cycle progression. FRG1 has been shown to localize to Cajal bodies in the nucleus, where it is reported to regulate RNA processing [42], [48]. Misprocessing of RNA transcripts has also been linked to myotonic dystrophy [49], [50]. One possible hypothesis for induction of G1 arrest by FRG1 involves altered splicing of transcripts encoding cell cycle components. For instance, altered processing of the cyclin E RNA produced different isoforms of the protein with different affinities for Cdk2 [51], [52], [53].
One contentious observation regarding overexpression of FRG1 in patients with FSHD is that other research groups have been unable to replicate the results published showing increased FRG1 transcript in affected muscle [28]. Some studies of myoblasts subjected to microarray analysis or measuring RNA transcription of FRG1 have not yielded any results showing an increase of FRG1 transcript [54], [55], while similar experiments with qRT-PCR done in other groups have shown an increased trend [27]. Recently, however, Bodega et al. reported that overexpression of FRG1 in muscle biopsies is not a uniform finding and may depend on the composition and age of the muscle biopsy, as FRG1 is only upregulated during an early state of differentiation into myotubes. Regardless of whether the levels of FRG1 are increased or not in FSHD patients, the H-FRG1 TG mouse is a valuable tool for studying the mechanics of muscular dystrophy, though it is important to note that FRG1 expression in these mice is many fold higher than observed in any human patients. Ultimately, FRG1 remains relatively poorly characterized and our findings may help to further elucidate its function.
It has been reported that myoblasts isolated from human muscle biopsies exhibit a morphological difference upon differentiation [56]. The authors observed that, compared to differentiated wild-type myotubes, FSHD myotubes were thinner, less branched, more disorganized and were comprised of fewer myoblasts as measured by total number of nuclei per myotube. Converse to our findings, they did not note any proliferation defects in their myoblast cultures, which may be attributed to differences in their isolation technique, sample sources and/or assay techniques. Regardless of the caveats, this raises the possibility of a compound defect in both proliferation and differentiation as a mechanism for the development of FSHD.
One disparity in our data may be the difference observed between the proliferative defect described in mass culture (Figure 4A) and that seen by mitotic shakeoff analyzed by flow cytometry (Figure 4B). Although the difference seen by mitotic shakeoff is less severe than that observed by mass culture doubling time, it is worth noting that the cells are kept under different conditions in each of these assays. In mass culture, myoblasts find themselves in much denser conditions with self-conditioned media. In the mitotic shakeoff, myoblasts are in a much less dense environment and exposed to fresh media and growth factors. It is possible that, given enough time, the cell cycle profiles of myoblasts isolated by mitotic shakeoff would exhibit a more severe defect with increased cell density, but it is impossible to determine this within the time frame whereby cells remain synchronized.
In summary, our experiments have demonstrated the expression of FRG1 in mouse muscle causes a tissue-specific and age-dependent proliferative defect in the satellite cell population, possibly playing a part in the development of muscular dystrophies and FSHD in humans.
Materials and Methods
Animal care and genotyping
High-expressing FRG1 transgenic mice (H-FRG1 TG) were kindly provided by Dr. R. Tupler [33]. DNA was extracted from tail samples by digestion with TENS solution (50 mM Tris pH 7.5, 100 mM EDTA pH 8.0, 400 mM NaCl, 0.4% SDS, proteinase K 0.5 mg/mL) followed by ethanol precipitation of genomic DNA. PCR genotyping of FRG1 mice was performed with the following primers: HSA-FRG1-5′ 5′-GAT CTA GCG GCC GCC ATG GCC GAG TAC TCC TAT GTG AAG TCT-3′ and HSA-FRG1-3′ 5′-GCG CGC TTA ATT AAT CAC TTG CAG TAT CTG TCG GCT TTC A-3′. Mice were bred and maintained under specific pathogen-free conditions. All experiments were performed in compliance with the University of Washington Institutional Animal Care and Use Committee protocol #2362-04.
Analysis of FRG1 expression
Tissue lysates were generated from quadriceps, gastrocnemius, diaphragm, heart, liver, lung and brain tissue isolated from H-FRG1 TG mice and wild-type littermate controls. Homogenization was done in 5 mL of ice-cold RIPA buffer supplemented with protease inhibitors (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% deoxycholate, 1 mM Na3VO4, 1 mM NaF, 1 mM PMSF, 1 µg/mL aprotinin, 1 µg/mL leupeptin) per gram of tissue for 30 seconds using the Omni-Tip system (OMNI International). Samples were incubated on ice for 30 minutes, spun down at 16,500 rcf for 15 minutes at 4°C, followed by transferring of supernatant and recentrifugation for an additional 10 minutes to generate tissue lysates.
RNA was isolated from both proliferating and differentiated mouse muscle satellite cell cultures using the RNAqueous kit (Ambion Inc). qPCR was performed using primers as previously described [33]. Cell lysates were also generated from the aforementioned muscle satellite cell cultures by homogenization in RIPA buffer as described above.
Histological analysis of muscle
Quadriceps, gastrocnemius, soleus and diaphragm were isolated from H-FRG1 TG mice and wild-type littermate controls, blotted briefly on filter paper and weighed on an analytical balance. Tissues were mounted in Tissue-Tek OCT compound (Sakura Finetek), frozen in a liquid nitrogen-cooled bath of isopentane and stored at −80°C. Cryosections were collected using a Leica CM1850 for hemotoxylin and eosin staining. Stained sections were viewed and photographed under 100× magnification on a Zeiss Axiovert 200 M. Analysis of images was performed using AxioVision v4.3.
Isolation of mouse muscle satellite cells and cell culture
To isolate satellite cells, 4-week and 18-week old FRG1 or wild-type mouse thigh or diaphragm muscle tissue was dissected away and placed in chilled Growth Media (GM) consisting of Ham's F10C+15% horse serum (Atlanta Biologicals Lot D0195)+50 µg/ml gentamicin with the addition of 0.25 µg/ml Fungizone. Connective tissue and fat were dissected away from muscle tissue under a dissecting microscope and muscle was weighed on an analytical balance. Isolation of the myoblastic population was performed as previously described [40], with the following modifications and details. Diced muscle was minced for 2 minutes using curved-tip scissors and treated with 50 µL of 0.05% trypsin-EDTA (Gibco) diluted in Hank's saline per mg of tissue. The tissue/trypsin mixture was incubated at 37°C and pipetted thoroughly every 5 minutes for 20 minutes total. The trypsinized tissue mixture was then passed through a 70 µm cell strainer (BD Falcon) to reduce fibroblast contamination and the strainer was rinsed with an additional 100 µL of F10C per mg of tissue. The flowthrough was then centrifuged at 800 rpm (Sorvall Legend T) for 5 minutes and plated on gelatin-coated dishes in GM+6 ng/ml basic fibroblast growth factor (bFGF)+0.25 µg/ml Fungizone [40]. The satellite cell cultures were observed carefully and differentially passaged to eliminate any fibroblast contamination over 5 passages. To differential passage, satellite cells were treated to a mild trypsinization and observed under a microscope until the round satellite cells were seen detaching from the plate. Because of their morphological differences, satellite cells will detach from the dish before the flattened and larger fibroblasts. Once the satellite cells were observed detaching, the trypsin solution was immediately removed and plated to isolate the satellite cell population.
The established mouse-derived satellite cell cultures were cultured on 10 cm collagen-coated tissue culture plates in GM with 4 ng/ml bFGF. Cells were supplemented with fresh bFGF every 12 hours to inhibit differentiation of satellite cells into myocytes. Passaging occurred when cells reached 5×105 cells in a 10 cm dish by rinsing with Saline A (10 mM dextrose, 30 mM HEPES, 3 mM KCl, 130 mM NaCl, 1 mM Na2HPO4-7H2O), trypsinization and replating at 5×104 in a new 10 cm dish.
iC2C12-FRG1 myoblasts were generated as described and generously provided by Dr. Michael Kyba at the University of Texas Southwestern [57]. iC2C12-FRG1 myoblasts were maintained in DMEM (Hyclone)+10% FBS (Atlanta Biologicals Lot A0087) taking care to passage before confluency to avoid loss of the myoblastic population. Induction of FRG1-FLAG in iC2C12-FRG1 myoblasts was achieved by incubating cells with 500 ng/mL doxycycline for 24 hours.
Clonal analysis of proliferation
To perform clonal assays, proliferating satellite cells were trypsinized and plated at 1000 cells per 10 cm dish in GM with 6 ng/mL bFGF to prevent premature differentiation. Cells were left undisturbed to minimize the formation of satellite clones and fixed with AFA (50% ethanol, 5% formalin, 5% acetic acid) at regular intervals. Myoblast clones were then immunostained for myosin heavy-chain (MyHC) using mouse monoclonal MF-20, biotinylated rabbit anti-mouse IgG, streptavidin and biotinylated horseradish peroxidase (Vector Labs, Inc.), followed by counterstaining with 1% methylene blue as previously described [39]. Clones were then scored by visualization at 25×/50× magnification on a dissecting microscope for both number and percent differentiation. Doubling times were calculated with the formula “Doubling Time = [Time post-plating *Ln(2)]/Ln(average clone size)”.
Viral transduction of C2C12 myoblasts
Expression vectors for FRG1 were generated in the pMXIH vector as described previously [41]. 293T cells were transfected with either pMXIH or pMXIH-FRG1 in combination with the ϕ-ampho packaging plasmid using calcium phosphate. Virus-containing media were filtered through a 0.45 µm filter and applied to C2C12 myoblasts to generate C2C12-pMXIH vector control myoblasts and C2C12-FRG1 myoblasts which should stably express FRG1 protein.
Flow cytometry and other proliferation assays
To assay doubling time of iC2C12-FRG1 myoblasts, proliferating cells were grown in the presence or absence of doxycycline to induce expression of FRG1. 2000 cells were plated per well of a 24-well dish. One well of cells plated in this dish per condition was trypsinized every 24 hours over 120 hours and total cell number was counted in triplicate on an improved Neubauer hemocytometer. Cell number was graphed and exponential curve fit was performed using Microsoft Excel to determine the growth constant and calculate the doubling time.
For BrdU staining of virally-transduced C2C12 myoblasts, after viral transduction, cells were plated on glass coverslips and pulsed with BrdU cell proliferation reagent (Amersham) for 60 minutes. Cells were then fixed in 4% formalin, permeabilized with 0.5% Triton X-100 in PBS and blocked in 5% goat serum, 5% horse serum, 0.2% Tween-20, 0.2% fish skin gelatin in PBS. α-BrdU mouse antibody (BD Biosciences) was diluted 1∶350 in PBS and incubated for 1 hour at 37°C followed by goat α-mouse antibody conjugated to AlexaFluor488 diluted 1∶400. Coverslips were mounted using Vectashield (Vector Labs).
For flow cytometry assays, proliferating iC2C12-FRG1 myoblasts were also plated in 10 cm dishes for cell cycle profile analysis. After trypsinization, cell pellets were resuspended in DAPI solution (2 mM CaCl2, 22 mM MgCl2 0.1 mg/mL BSA, 0.1% Nonidet P-40, 10 µg/mL DAPI, 10% DMSO) and run on an InFlux flow cytometer (Cytopeia). 20,000 cells were run through the flow cytometer measuring DNA content. Analysis was done using WinCycle (Phoenix Flow Systems) to determine cell cycle phase distributions as previously described [58].
For determining initial effects of FRG1 expression in synchronized cells, proliferating iC2C12-FRG1 myoblasts either exposed to doxycycline for 24 hours previously or grown in the absence of doxycycline were synchronized by mitotic shakeoff as described [59]. Three 10-cm dishes were tapped vigorously against a hard surface to shake off mitotic cells, incubated with gentle rocking for 20 minutes to prevent reattachment, then tapped vigorously again to further dislodge mitotic cells, and finally the medium containing mitotic cells was centrifuged and plated in a single 60-mm dish. Twelve 60-mm dishes of synchronized cells were collected and placed either in the presence or absence of doxycycline. Every 2 hours after the initial plating, cells were trypsinized and pellets were fixed and resuspended in the same DAPI solution used for flow cytometry to cover a total period of 24 hours. DNA content was measure as previously mentioned using flow cytometry, with 20,000 cells being analyzed.
Immunofluorescence and immunoblotting
iC2C12-FRG1 myoblasts with or without the induction of FRG1-FLAG were grown on round glass coverslips, fixed in a 4% formalin solution and stained with mouse monoclonal α-FLAG antibody (Sigma) at 1∶10,000 dilution. For cell lysates, proliferating satellite cells or iC2C12-FRG1 myoblasts were lysed in RIPA buffer at 4°C, incubated on ice for 30 minutes, and centrifuged at 16,500 rcf for 10 minutes to remove cell debris. Cell lysates were run on a 10% SDS-PAGE gel, transferred to nitrocellulose overnight at 4°C in a Tris-glycine buffer and incubated overnight at 4°C with one of the following primary antibodies. Monoclonal α-FRG1 at 1∶1000 (Abnova clone 4A5), mouse monoclonal pRb at 1∶1000 (BD Pharmingen clone 14001A), rabbit anti-mouse α-pRb Ser795 at 1∶1000 (Cell Signaling), rabbit anti-mouse α-pRb Ser807/811 at 1∶1000 (Cell Signaling), or mouse monoclonal α-β-actin at 1∶10000 (Abcam). Western blotting was completed the next day using the corresponding secondary antibody of donkey anti-rabbit HRP at 1∶10000 (Amersham Biosciences) or rabbit anti-mouse HRP at 1∶10000 (Amersham Biosciences). Blots were visualized using ECL substrate (PerkinElmer) on Kodak Biomax Light (Sigma) film.
Supporting Information
Examination of FRG1 expression in muscle-derived myoblasts. A) Levels of FRG1 in 20-week old diaphragm-derived myoblasts from either H-FRG1 TG (FRG1) or wild-type littermate control (C57BL/6) as assayed by qPCR normalized to GAPDH. Proliferating cultures were judged to have less than 5% differentiated cells while differentiated cultures exhibited greater than 70% differentiation. *indicates no expression detected. B) Western analysis on proliferating and differentiated diaphragm-derived myoblast cultures from 20-week old mice as described above probing for total FRG1 levels. β-actin loading control shown below. C) Western analysis on proliferating satellite cell cultures from diaphragm or thigh of 4-week old mice probing for total FRG1 protein levels. β-actin loading control shown below.
(TIF)
Additional clonal analysis of mouse-derived myoblasts. Myoblasts isolated from hindlimb of 18-week old H-FRG1 TG (FRG1) or wild-type littermate controls (WT) were cultured and plated at low-density. Total number of nuclei per clone were counted at 120-hours post-plating (n = 100). Similarly myoblasts isolated from dissected hindlimbs of 20-week old H-FRG1 TG mouse (FRG1) or wild-type littermate controls (WT) were subjected to this procedure in the lower figure showing total number of nuclei per clone at 120-hours post-plating (n = 13 for FRG1 line, n = 73 for WT line).
(TIF)
Loss of proliferative defect in virus-transduced C2C12 myoblasts. C2C12 mouse myoblasts transduced with either vector control (pMXIH) or a FRG1-expressing construct (FRG1) were scored for incorporation of BrdU after 60-minute pulse to determine % of S-phase cells. Transduced myoblasts show proliferative defect at early passages (passage 8) but lose the phenotype over time.
(TIF)
Loss of FRG1 expression in iC2C12-FRG1 myoblasts over time. iC2C12-FRG1 myoblasts were cultured and maintained with or without the presence of 500 ng/mL doxycycline to induce FRG1 expression. Immunofluorescence with an α-FLAG antibody (green) and DAPI staining (blue) reveals loss of FRG1 expression after ∼20 passages under induction conditions, but robust expression with an acute induction of 24 hours.
(TIF)
Acknowledgments
The authors would like to thank Rubysue Mangalindan, Ashot Safarli and other members of the Ladiges lab in the Department of Comparative Medicine for handling and maintenance of mouse colonies. Flow cytometry and assistance with analysis was graciously provided by Donna Prunkard in the Rabinovitch lab, Department of Pathology, UW.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by a grant from the Pacific Northwest Friends of FSHD. S.C.C. is supported by the cardiovascular pathology training grant NIH T32 HL007312. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.van der Maarel SM, Frants RR, Padberg GW. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta. 2007;1772:186–194. doi: 10.1016/j.bbadis.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, et al. Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35–facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet. 1995;4:951–958. doi: 10.1093/hmg/4.5.951. [DOI] [PubMed] [Google Scholar]
- 3.Tawil R, Forrester J, Griggs RC, Mendell J, Kissel J, et al. Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. The FSH-DY Group. Ann Neurol. 1996;39:744–748. doi: 10.1002/ana.410390610. [DOI] [PubMed] [Google Scholar]
- 4.Ricci E, Galluzzi G, Deidda G, Cacurri S, Colantoni L, et al. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol. 1999;45:751–757. doi: 10.1002/1531-8249(199906)45:6<751::aid-ana9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 5.Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34:1–15. doi: 10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 6.Sarfarazi M, Wijmenga C, Upadhyaya M, Weiffenbach B, Hyser C, et al. Regional mapping of facioscapulohumeral muscular dystrophy gene on 4q35: combined analysis of an international consortium. Am J Hum Genet. 1992;51:396–403. [PMC free article] [PubMed] [Google Scholar]
- 7.Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, et al. Location of facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet. 1990;336:651–653. doi: 10.1016/0140-6736(90)92148-b. [DOI] [PubMed] [Google Scholar]
- 8.Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 9.van Deutekom JC, Wijmenga C, van Tienhoven EA, Gruter AM, Hewitt JE, et al. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum Mol Genet. 1993;2:2037–2042. doi: 10.1093/hmg/2.12.2037. [DOI] [PubMed] [Google Scholar]
- 10.de Greef JC, Frants RR, van der Maarel SM. Epigenetic mechanisms of facioscapulohumeral muscular dystrophy. Mutat Res. 2008;647:94–102. doi: 10.1016/j.mrfmmm.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gabriels J, Beckers MC, Ding H, De Vriese A, Plaisance S, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene. 1999;236:25–32. doi: 10.1016/s0378-1119(99)00267-x. [DOI] [PubMed] [Google Scholar]
- 12.Beckers M, Gabriels J, van der Maarel S, De Vriese A, Frants RR, et al. Active genes in junk DNA? Characterization of DUX genes embedded within 3.3 kb repeated elements. Gene. 2001;264:51–57. doi: 10.1016/s0378-1119(00)00602-8. [DOI] [PubMed] [Google Scholar]
- 13.Clapp J, Mitchell LM, Bolland DJ, Fantes J, Corcoran AE, et al. Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:264–279. doi: 10.1086/519311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansseau E, Laoudj-Chenivesse D, Marcowycz A, Tassin A, Vanderplanck C, et al. DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS One. 2009;4:e7482. doi: 10.1371/journal.pone.0007482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord. 2007;17:611–623. doi: 10.1016/j.nmd.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. Embo J. 2008;27:2766–2779. doi: 10.1038/emboj.2008.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Snider L, Asawachaicharn A, Tyler AE, Geng LN, Petek LM, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet. 2009;18:2414–2430. doi: 10.1093/hmg/ddp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camano P, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 329:1650–1653. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caretti G, Di Padova M, Micales B, Lyons GE, Sartorelli V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev. 2004;18:2627–2638. doi: 10.1101/gad.1241904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laible G, Wolf A, Dorn R, Reuter G, Nislow C, et al. Mammalian homologues of the Polycomb-group gene Enhancer of zeste mediate gene silencing in Drosophila heterochromatin and at S. cerevisiae telomeres. Embo J. 1997;16:3219–3232. doi: 10.1093/emboj/16.11.3219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forlani G, Giarda E, Ala U, Di Cunto F, Salani M, et al. The MeCP2/YY1 interaction regulates ANT1 expression at 4q35: novel hints for Rett syndrome pathogenesis. Hum Mol Genet. 19:3114–3123. doi: 10.1093/hmg/ddq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Donohoe ME, Zhang LF, Xu N, Shi Y, Lee JT. Identification of a Ctcf cofactor, Yy1, for the X chromosome binary switch. Mol Cell. 2007;25:43–56. doi: 10.1016/j.molcel.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 24.Lehming N, Le Saux A, Schuller J, Ptashne M. Chromatin components as part of a putative transcriptional repressing complex. Proc Natl Acad Sci U S A. 1998;95:7322–7326. doi: 10.1073/pnas.95.13.7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lomberk G, Wallrath L, Urrutia R. The Heterochromatin Protein 1 family. Genome Biol. 2006;7:228. doi: 10.1186/gb-2006-7-7-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erard MS, Belenguer P, Caizergues-Ferrer M, Pantaloni A, Amalric F. A major nucleolar protein, nucleolin, induces chromatin decondensation by binding to histone H1. Eur J Biochem. 1988;175:525–530. doi: 10.1111/j.1432-1033.1988.tb14224.x. [DOI] [PubMed] [Google Scholar]
- 27.Bodega B, Ramirez GD, Grasser F, Cheli S, Brunelli S, et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol. 2009;7:41. doi: 10.1186/1741-7007-7-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gabellini D, Green MR, Tupler R. Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell. 2002;110:339–348. doi: 10.1016/s0092-8674(02)00826-7. [DOI] [PubMed] [Google Scholar]
- 29.Petrov A, Allinne J, Pirozhkova I, Laoudj D, Lipinski M, et al. A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for the facio-scapulo-humeral dystrophy. Genome Res. 2008;18:39–45. doi: 10.1101/gr.6620908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petrov A, Pirozhkova I, Carnac G, Laoudj D, Lipinski M, et al. Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc Natl Acad Sci U S A. 2006;103:6982–6987. doi: 10.1073/pnas.0511235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pirozhkova I, Petrov A, Dmitriev P, Laoudj D, Lipinski M, et al. A functional role for 4qA/B in the structural rearrangement of the 4q35 region and in the regulation of FRG1 and ANT1 in facioscapulohumeral dystrophy. PLoS One. 2008;3:e3389. doi: 10.1371/journal.pone.0003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R. Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology. 2007;68:569–577. doi: 10.1212/01.wnl.0000251269.31442.d9. [DOI] [PubMed] [Google Scholar]
- 33.Gabellini D, D'Antona G, Moggio M, Prelle A, Zecca C, et al. Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature. 2006;439:973–977. doi: 10.1038/nature04422. [DOI] [PubMed] [Google Scholar]
- 34.Liu Q, Jones TI, Tang VW, Brieher WM, Jones PL. Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J Cell Sci. 123:1116–1123. doi: 10.1242/jcs.058958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hanel ML, Sun CY, Jones TI, Long SW, Zanotti S, et al. Facioscapulohumeral muscular dystrophy (FSHD) region gene 1 (FRG1) is a dynamic nuclear and sarcomeric protein. Differentiation. doi: 10.1016/j.diff.2010.09.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hanel ML, Wuebbles RD, Jones PL. Muscular dystrophy candidate gene FRG1 is critical for muscle development. Dev Dyn. 2009;238:1502–1512. doi: 10.1002/dvdy.21830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wuebbles RD, Long SW, Hanel ML, Jones PL. Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol. 2010;3:386–400. [PMC free article] [PubMed] [Google Scholar]
- 38.D'Antona G, Brocca L, Pansarasa O, Rinaldi C, Tupler R, et al. Structural and functional alterations of muscle fibres in the novel mouse model of facioscapulohumeral muscular dystrophy. J Physiol. 2007;584:997–1009. doi: 10.1113/jphysiol.2007.141481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frock RL, Kudlow BA, Evans AM, Jameson SA, Hauschka SD, et al. Lamin A/C and emerin are critical for skeletal muscle satellite cell differentiation. Genes Dev. 2006;20:486–500. doi: 10.1101/gad.1364906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Neville C, Rosenthal N, McGrew M, Bogdanova N, Hauschka S. Skeletal muscle cultures. Methods Cell Biol. 1997;52:85–116. [PubMed] [Google Scholar]
- 41.Kudlow BA, Jameson SA, Kennedy BK. HIV protease inhibitors block adipocyte differentiation independently of lamin A/C. Aids. 2005;19:1565–1573. doi: 10.1097/01.aids.0000186827.91408.90. [DOI] [PubMed] [Google Scholar]
- 42.van Koningsbruggen S, Dirks RW, Mommaas AM, Onderwater JJ, Deidda G, et al. FRG1P is localised in the nucleolus, Cajal bodies, and speckles. J Med Genet. 2004;41:e46. doi: 10.1136/jmg2003.012781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frolov MV, Dyson NJ. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J Cell Sci. 2004;117:2173–2181. doi: 10.1242/jcs.01227. [DOI] [PubMed] [Google Scholar]
- 44.Trimarchi JM, Lees JA. Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol. 2002;3:11–20. doi: 10.1038/nrm714. [DOI] [PubMed] [Google Scholar]
- 45.Watchko JF, O'Day TL, Hoffman EP. Functional characteristics of dystrophic skeletal muscle: insights from animal models. J Appl Physiol. 2002;93:407–417. doi: 10.1152/japplphysiol.01242.2001. [DOI] [PubMed] [Google Scholar]
- 46.Mittelbronn M, Sullivan T, Stewart CL, Bornemann A. Myonuclear degeneration in LMNA null mice. Brain Pathol. 2008;18:338–343. doi: 10.1111/j.1750-3639.2008.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Melone MA, Peluso G, Petillo O, Galderisi U, Cotrufo R. Defective growth in vitro of Duchenne Muscular Dystrophy myoblasts: the molecular and biochemical basis. J Cell Biochem. 1999;76:118–132. doi: 10.1002/(sici)1097-4644(20000101)76:1<118::aid-jcb12>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 48.van Koningsbruggen S, Straasheijm KR, Sterrenburg E, de Graaf N, Dauwerse HG, et al. FRG1P-mediated aggregation of proteins involved in pre-mRNA processing. Chromosoma. 2007;116:53–64. doi: 10.1007/s00412-006-0083-3. [DOI] [PubMed] [Google Scholar]
- 49.Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 50.Ranum LP, Day JW. Myotonic dystrophy: RNA pathogenesis comes into focus. Am J Hum Genet. 2004;74:793–804. doi: 10.1086/383590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mumberg D, Wick M, Burger C, Haas K, Funk M, et al. Cyclin ET, a new splice variant of human cyclin E with a unique expression pattern during cell cycle progression and differentiation. Nucleic Acids Res. 1997;25:2098–2105. doi: 10.1093/nar/25.11.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sewing A, Ronicke V, Burger C, Funk M, Muller R. Alternative splicing of human cyclin E. J Cell Sci. 1994;107(Pt 2):581–588. doi: 10.1242/jcs.107.2.581. [DOI] [PubMed] [Google Scholar]
- 53.Porter DC, Keyomarsi K. Novel splice variants of cyclin E with altered substrate specificity. Nucleic Acids Res. 2000;28:E101. doi: 10.1093/nar/28.23.e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klooster R, Straasheijm K, Shah B, Sowden J, Frants R, et al. Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur J Hum Genet. 2009;17:1615–1624. doi: 10.1038/ejhg.2009.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masny PS, Chan OY, de Greef JC, Bengtsson U, Ehrlich M, et al. Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur J Hum Genet. 2009 doi: 10.1038/ejhg.2009.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barro M, Carnac G, Flavier S, Mercier J, Vassetzky Y, et al. Myoblasts from affected and non affected FSHD muscles exhibit morphological differentiation defects. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kyba M, Perlingeiro RC, Daley GQ. HoxB4 confers definitive lymphoid-myeloid engraftment potential on embryonic stem cell and yolk sac hematopoietic progenitors. Cell. 2002;109:29–37. doi: 10.1016/s0092-8674(02)00680-3. [DOI] [PubMed] [Google Scholar]
- 58.Rabinovitch PS. DNA content histogram and cell-cycle analysis. Methods Cell Biol. 1994;41:263–296. doi: 10.1016/s0091-679x(08)61723-9. [DOI] [PubMed] [Google Scholar]
- 59.Schorl C, Sedivy JM. Analysis of cell cycle phases and progression in cultured mammalian cells. Methods. 2007;41:143–150. doi: 10.1016/j.ymeth.2006.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Examination of FRG1 expression in muscle-derived myoblasts. A) Levels of FRG1 in 20-week old diaphragm-derived myoblasts from either H-FRG1 TG (FRG1) or wild-type littermate control (C57BL/6) as assayed by qPCR normalized to GAPDH. Proliferating cultures were judged to have less than 5% differentiated cells while differentiated cultures exhibited greater than 70% differentiation. *indicates no expression detected. B) Western analysis on proliferating and differentiated diaphragm-derived myoblast cultures from 20-week old mice as described above probing for total FRG1 levels. β-actin loading control shown below. C) Western analysis on proliferating satellite cell cultures from diaphragm or thigh of 4-week old mice probing for total FRG1 protein levels. β-actin loading control shown below.
(TIF)
Additional clonal analysis of mouse-derived myoblasts. Myoblasts isolated from hindlimb of 18-week old H-FRG1 TG (FRG1) or wild-type littermate controls (WT) were cultured and plated at low-density. Total number of nuclei per clone were counted at 120-hours post-plating (n = 100). Similarly myoblasts isolated from dissected hindlimbs of 20-week old H-FRG1 TG mouse (FRG1) or wild-type littermate controls (WT) were subjected to this procedure in the lower figure showing total number of nuclei per clone at 120-hours post-plating (n = 13 for FRG1 line, n = 73 for WT line).
(TIF)
Loss of proliferative defect in virus-transduced C2C12 myoblasts. C2C12 mouse myoblasts transduced with either vector control (pMXIH) or a FRG1-expressing construct (FRG1) were scored for incorporation of BrdU after 60-minute pulse to determine % of S-phase cells. Transduced myoblasts show proliferative defect at early passages (passage 8) but lose the phenotype over time.
(TIF)
Loss of FRG1 expression in iC2C12-FRG1 myoblasts over time. iC2C12-FRG1 myoblasts were cultured and maintained with or without the presence of 500 ng/mL doxycycline to induce FRG1 expression. Immunofluorescence with an α-FLAG antibody (green) and DAPI staining (blue) reveals loss of FRG1 expression after ∼20 passages under induction conditions, but robust expression with an acute induction of 24 hours.
(TIF)