

Abstract
Efforts toward achieving a practical and diastereoselective intramolecular [4 + 3] cycloaddition of nitrogen-stabilized oxyallyl cations with tethered dienes are described. Epoxidation of N-sulfonyl substituted allenamides with dimethyldioxirane (DMDO) generates nitrogen-stabilized oxyallyl cations that would readily undergo stereoselective [4 + 3] cycloaddition with dienes. Selectivity is found to depend on the tethering length as well as the stability of the oxyallyl cation intermediate, whether generated from N-carbamoyl- or N-sulfonyl-substituted allenamides. The use of chiral N-sulfonyl-substituted allenamide provided minimal diastereoselectivity in the cycloaddition, while high diastereoselectivity can be achieved with a stereocenter present on the tether. These studies provide further support for the synthetic utility of allenamides.
INTRODUCTION
Over the past 30 years, the utility of heteroatom-stabilized oxyallyl cations has risen to the forefront of [4 + 3] cycloadditions.1,2 Heteroatom substituents such as oxygen3 and sulfur,4 as well as halogens5 provide electronically-biased oxyallyl cations which have become attractive intermediates for developing not only highly regioselective, but also stereoselective [4 + 3] cycloadditions.1,2 However, only recently has the utility of nitrogen-substituted oxyallyl cations in these cycloadditions received significant attention.6 Almost ten years ago, we first developed stereoselective [4 + 3] cycloadditions of chiral nitrogen-stabilized7 oxyallyl cations 2b derived from allenamides 1 via epoxidation [Scheme 1].8 These efforts rendered allenamides highly visible as a new organic functional group,9 while allowing us to contribute to the area of [4 + 3] cycloadditions through advancing its regio-10 and stereoselective manifolds11,12 as well as establishing the first asymmetric [4 + 3] cycloaddition using Cu(II)-bisoxazoline catalysts.13,14
Scheme 1.

Intermolecular [4 + 3] Cycloaddition.
The trivalency of the nitrogen atom offers the flexibility to simultaneously tune its electron donating ability toward the oxyallyl cation through various substitutions while tethering a chiral auxiliary [R*] and a coordinating unit [W] to achieve both highly regio- and stereoselective [4 + 3] oxyallyl cycloadditions, which remain a challenge in this field.1,11,12 While we recently reported the first systematic study on the regioselectivity of intermolecular [4 + 3] cycloadditions with unsymmetrical furans,10a we have also been engaging in developing intramolecular variants11d,e of our [4 + 3] cycloaddition via two approaches: I: N-Tethered 4→6, and II: C-Tethered through either the α-position 7→8 or γ-position 9→10 [Scheme 2]. We elected to focus on Approach-I because it underscores a distinct advantage as well as significance of using nitrogen-stabilized oxyallyl cations in constructing complex nitrogen heterocycles for natural or non-natural product synthesis,15 thereby also further accentuating the synthetic power of allenamides.
Scheme 2.

Approaches to Intramolecular [4 + 3].
Our first report on the intramolecular [4 + 3] cycloaddition utilized N-carbamoyl-substituted N-tethered allenamides that could be prepared in three steps from furanyl iodide 11 [Scheme 3].11e Initial attempts using a direct addition of 2–5 equivalents of DMDO led to low yields of the desired cycloadducts 13a and 13b with mostly being oxidative ring opening of the furan. However, it was quickly found that slow addition of DMDO via a syringe pump allowed for selective epoxidation of the allenic double bond in 14 or 15 and slowed the competing oxidation of furan. The ensuing intramolecular [4 + 3] cycloaddition of the corresponding oxyallyl cations with furan led to the desired cycloadducts 16 and 17 in 80% and 75% yields, respectively, as single diastereomers. In addition, a small amount of the epoxidized cycloadduct 18 as also isolated from the reaction of 14. While using the improved syringe pump addition protocol, we were able to show that a few other N-carbamoyl-substituted allenamides could also undergo successful intramolecular [4 + 3] cycloaddition,11e most of these examples in our communication were over-engineered, and the generality as well as practicality remained elusive. We reported here details of our efforts in evolving this intramolecular reaction into a useful and stereoselective cycloaddition manifold for constructing nitrogen heterocycles.
Scheme 3.

Previous Work on [4 + 3] Cycloadditions.
RESULTS AND DISCUSSION
I. N-Carbamoyl Substituted Allenamides
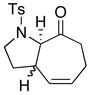
To commence our studies, we re-investigated intramolecular [4 + 3] cycloadditions of N-carbamoyl-substituted allenamides but chose to use those containing a chiral carbamoyl group, which could serve as an auxiliary to provide asymmetric induction in the intramolecular cycloaddition and gain access to optically enriched cycloadducts. Consequently, our first attempt using chiral allenamide 19 with the menthyl auxiliary gave cycloadduct 20 in 78% yield and what appeared to be a single diastereomer based on NMR [Scheme 4]. Using a 3-carbon tether, the yield dropped dramatically in giving cycloadduct 22. The stereoselectivity also dropped significantly with respect to the ring fusion with only a 60:40 diastereomeric ratio, which was also observed with achiral allenamides in our previous work.11e Furthermore, an increase to the 4-carbon tether failed to give the desired cycloadduct 24 regardless of reaction temperature, most likely due to the increase in conformational entropy associated with the longer carbon tether.
Scheme 4.

[4 + 3] Cycloaddition of Chiral Allenamides.
While we were quite pleased with the apparent asymmetric induction achieved in cycloadduct 20 when using the chiral menthyl auxiliary for the 2-carbon tether, the excitement did not last long. Upon examination of the X-ray crystal structure obtained for cycloadduct 20, we were very surprised to find that the X-ray structure contains a 1:1 mixture of diastereomers with respect to the chiral auxiliary [Figure 1]. While it is quite unusual to co-crystallize two diastereomers, this data suggests that the chiral influence of the carbamate auxiliary is too far removed from the rest of the molecule to provide any facial bias during the cycloaddition, and that it exerted very little influence in differentiating the two diastereomers spectroscopically based on proton NMR [Figure 2].16
Figure 1.
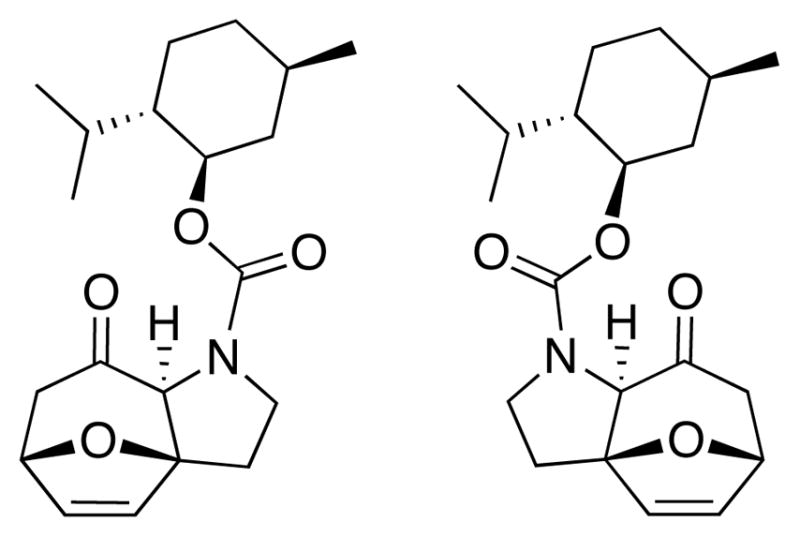
1:1 Mixture of Diastereomeric 20
Figure 2.

1H NMR of the [4 + 3] Cycloadduct 20.
II. N-Sulfonyl Substituted Allenamides
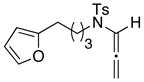
We then decided to shift our efforts from N-carbamoyl-substituted to N-sulfonyl-substituted allenamides to investigate the difference in reactivity of N-sulfonyl oxyallyl cations and perhaps improve overall selectivity in the [4 + 3] cycloaddition. Having recently published a facile route to N-sulfonyl-substituted allenamides17 featuring a Mitsunobu reaction, allenamide 27 could be prepared in two steps from alcohol 25 in excellent yield. [Scheme 5]. Alternatively, γ-substituted allenamide (±)-29a and α-substituted allenamide 32 could be obtained from alcohols 25 and 30, respectively, utilizing a four-step sequence featuring two Mitsunobu reactions.18 It is worth noting that all of the alcohols are commercially available, making it possible to quickly access a variety of substituted allenamides depending on the substituted alcohol.
Scheme 5.

Synthesis of N-Sulfonyl Allenamides.
Our initial examination of N-Ts-substituted allenamide 27 in the [4 + 3] cycloaddition revealed N-sulfonyl allenamides to be much more reactive toward epoxidation as the reaction took place readily at −78 °C giving cycloadduct 34 in almost quantitative yield [Scheme 6]. The more electron-deficient N-p-nitrobenzenesulfonyl-substituted allenamide 33 also afforded cycloadduct 35 in 93% yield requiring only slightly longer reaction time after addition of DMDO. This is in great contrast with the reactivity of N-carbamoyl-substituted allenamides in which DMDO epoxidation is much slower at temperatures below −45 °C and often results in competing oxidative ring-opening of the furan.
Scheme 6.

Intramolecular [4 + 3] Cycloaddition.
We proceeded to examine the scope of the intramolecular [4 + 3] cycloaddition of N-sulfonyl-substituted allenamides [Table 1]. First, methyl substitution at the α-or γ-allenic position (entries 1–2, respectively) gave cycloadducts 36 and 37a with the major isomer of 37a shown as assigned via NOE experiments. Attempts with γ-phenyl substituted allenamide (±)-29b (entries 3–4) gave mostly hydrolysis of the allenamide and oxidation of furan, perhaps resulting from a more stabilized and less reactive oxyallyl cation. However, a cyclohexyl substitution was well-tolerated (entry 6) giving cycloadduct 39 containing a spirocenter in 90% yield.
Table 1.
Scope of [4 + 3] Cycloadditions.
| entry | allenamidesa | temp [°C] | cycloadductsb | yield [%]c |
|---|---|---|---|---|
| 1 32 |
|
−78 |
 36 |
69 |
| 2 (±)-29a: R = Me |
|
−78 |
 37a 37b |
93d |
| 3 (±)-29b: R = Ph | −78 | <5e | ||
| 4 (±)-29b: R = Ph | 0 | 37b | <5e | |
| 6 38 |
|
 39 |
90 | |
| 7 40a: P = Bn |
|
−78 |
 41a |
81 |
| 8 40b: P = TBS | −78 | 41b | 97 | |
| 9 42 |
|
−78 |
 43 43 |
<5e |
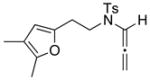
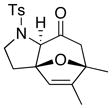
| 10 42 | 0 | <5e | ||
| 11 44: R = H |
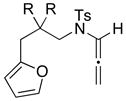
|
−78 |
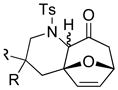 45 45 47 47 |
<5e |
| 12 44: R = H | 0 | 45f | ||
| 13 46: R = Me | −78 | <5e | ||
| 14 46: R = Me | 0 | 64g | ||
| 15 4B |
|
0 |
 49 |
17g |
| 16 50 |
|
−78 |
 51 51 |
<5e |
| 17 50 | 10 | 73f |
All reactions were carried out in CH2Cl2 at 0.05 M with MS 4Å.
Single diastereomers unless otherwise noted.
Isolated yields.
Major isomer shown.
Mostly decomposition by hydrolysis or oxidation of furan.
dr = 75:25
dr = 90:10.
Various furan substitutions were also investigated (entries 7–10). Disubstituted furans with either benzylor silyl-ethers afforded highly functionalized cycloadducts 41a and 41b, respectively, in excellent yields. This suggests that even at low temperatures, epoxidation of the allenic double bond is still favored over oxidation of furan, which again contrasts with the reactivity of N-carbamate allenamides. However, when using a trisubstituted furan (see 42) under the reaction conditions, epoxidation of furan and oxidative ring-opening occurred exclusively regardless of the reaction temperature. These results provide an interesting comparison between the electron-donating ability of the N-sulfonyl into the allenic double bond with that of the electron-rich furan.
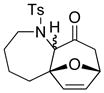
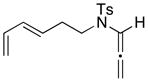
The three-carbon tethered allenamides 44 and 46 showed significantly better results compared with the N-carbamate allenamides (vide supra). Not only were yields much higher with or without a gem-dimethyl group, but the overall diastereoselectivity also increased (entries 12 and 14). Furthermore, despite a large entropic barrier, the four-carbon tethered allenamide 48 did afford the desired cycloadduct 49, albeit in a modest 17% yield. It is worth noting that higher temperatures were required for the longer tethers to overcome the entropic barrier needed for furan to orient itself with the oxyallyl cation before competing oxidation or hydrolysis took place. Finally, the cycloaddition proceeded with a simple butadiene giving cycloadduct 51 in 73% yield (entry 17). Higher temperature was also required in this case as the unreactive S-trans conformation likely dominates at −78 °C.
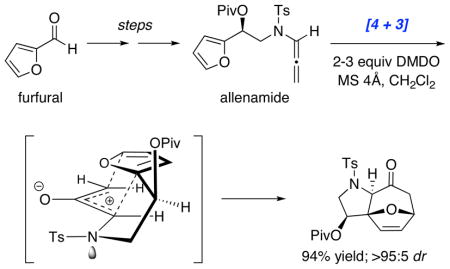
X-ray analysis of cycloadduct 35 unambiguously confirmed the [4 + 3] cycloaddition pathway with N-sulfonyl allenamides and provides a general mechanistic model of the cycloaddition as shown in Figure 3. Although two possible transition structures are at play, 52-endo [or compact]19 and 52-exo [or extended]19, which could both provide the observed stereochemistry in the cycloadduct, the oxyallyl cation 52-endo should experience more A1,3 strain with the planar nitrogen substituents whereas 52-exo possesses a more preferred W-conformation.1,2 DFT calculations of the two possible transition state conformations also shows a tremendous preference for 52-exo.20 Thus, Approach-I in the intramolecular [4 + 3] cycloaddition of nitrogen-stabilized oxyallyl cations likely proceeds in an exo manner.
Figure 3.

A Proposed Model for [4 + 3] Cycloaddition
III. Achieving Diastereoselectivity in the [4 + 3] Cycloaddition
With the improved reactivity of N-sulfonyl-substituted allenamides in the intramolecular [4 + 3] cycloaddition, we decided to once again investigate the effect of a chiral auxiliary in the reaction. From our previous work, we were able to achieve high diastereoselectivities and gain access to optically active cycloadducts containing seven- or eight-membered rings fused to the cycloheptane when using chiral oxazolidinone-substituted allenamides 53 [Scheme 7].11e In the ensuing cycloaddition, the preferred W-conformation and a similar exo approach with chiral oxyallyl 54 would lead to the observed major diastereomer of 55. However, the multi-step synthesis required to make the cyclic allenamides as well as the potential difficulty in cleaving the auxiliary somewhat limited the utility of this approach.
Scheme 7.

Previous Work on [4 + 3] Cycloaddition.
We chose to investigate the chiral camphor-derived auxiliary for our N-sulfonyl-substituted allenamides for two reasons: 1) the possible coordinating effect of the ketone with a Lewis acid during the cycloaddition; 2) previously reported success in other cycloadditions using camphor auxiliaries.21 As shown in Scheme 8, reaction of camphor-derived allenamide 56 in the presence of ZnCl2 gave cycloadduct 57 in excellent yield but with only modest diastereoselectivity. Similar results were obtained upon conversion of the ketone to a silyl-ether on the auxiliary (58→59). Despite the improved reactivity of N-sulfonyl-substituted allenamides in the [4 + 3] cycloaddition, these results further support our previous conclusion that N-substituted auxiliaries are too far removed to provide asymmetric induction during the cycloaddition.
Scheme 8.

Cycloadditions of Camphor-Derived Allenamides.
Knowing that the chiral auxiliary had little effect on the overall conformation during the cycloaddition, we chose to prepare allenamides containing a stereocenter on the tether in order to provide more of a conformational bias and perhaps lead to better selectivity [Scheme 9]. We chose to prepare diol 60 from the commercially available tri-O-acetyl-D-glucal using a previously reported protocol.22 However, using either FeCl3-6H2O or InCl3-3H2O, we found poor overall conversion under the reaction conditions. Selective protection of the primary alcohol with tosyl chloride in the presence of dibutyltin oxide23 followed by protection of the secondary hydroxyl group provided tosylate 61 in good yield.24 Interestingly, in an attempt to displace the tosylate with N-tosyl propargyl amine to obtain propargyl amide 62, we instead obtained the [4 + 2] cycloadduct 64 in 65% yield presumably through our previously reported tandem isomerization-cycloaddition sequence.17 Although the [4 + 2] product was not desired, we were impressed by the overall yield for the three-step tandem sequence. Furthermore, we were encouraged by the diastereoselectivity observed, hoping for similar or better results in the [4 + 3] cycloaddition.
Scheme 9.

Initial Route to Chiral Allenamide 63.
Using a revised route reported by Hashmi25, we were able to obtain the racemic propargyl amide (±)-62 starting from furfural [Scheme 10]. Cyanohydrin formation followed by silyl protection of the resultant alcohol gave (±)-65 in 72% yield. Reduction of the nitrile to a primary amine followed by tosylation then provided sulfonamide (±)-66 in good overall yield. Finally, propargylation and subsequent isomerization using our protocol afforded the desired allenamide (±)-63. It is worth noting that Hashmi’s synthesis was also done asymmetrically using enzyme catalysis for the cyanohydrin formation.25,26 We are also aware of the fact that O’Doherty has documented an array of beautiful and practical asymmetric protocols for accessing diols and amino alcohols related to 60 and 66 that we could adopt in future applications.27
Scheme 10.

A Revised Route to (±)-63: Showing One Enantiomer.
Other derivatives were also prepared in order to investigate their effect on the diastereoselectivity in the [4 + 3] cycloaddition. As shown in Table 2, our first attempt using TIPS-protected allenamide (±)-63 under our standard conditions gave the desired cycloadduct in 85% yield with an 80:20 diastereomeric ratio, similar to that of the [4 + 2] product. The yield remained the same at a higher reaction temperature although a slight decrease in selectivity was observed (entry 2). Interestingly, the smaller TES group gave a slight increase in selectivity and yield (entry 3).
Table 2.
Diastereoselective [4 + 3] Cycloadditions
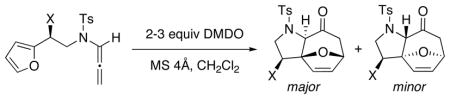 | ||||||
|---|---|---|---|---|---|---|
| entry | allenamidea | X= | temp [°C] | cycloadduct | yield [%]b | major:minor |
| 1 | (±)-63 | -OTIPS | −78 | 67 | 85 | 80:20 |
| 2 | (±)-63 | -OTIPS | 0 | 67 | 86 | 75:25 |
| 3 | (±)-68 | -OTES | −78 | 69 | 97 | 83:17 |
| 4 | (±)-70 | -OPiv | −78 | 71 | 94 | >95:5 |
| 5 | (±)-72 | -NBocp-Ns | −78 | 73 | <5 | - |
| 6 | (±)-72 | -NBocp-Ns | 0 | 73 | 49 | >95:5 |
All reactions were carried out at 0.05 M with 50 mg MS 4Å.
Isolated yields.
While we were pleased with the reasonable selectivity observed using silyl-protecting groups, we reasoned that we might be able to achieve even better diastereoselectivity using a pivalate protecting group because the shorter oxygen-carbon bond could provide a stronger conformational bias during the ensuing cycloaddition. To our delight, the pivalate-protected allenamide (±)-70 not only afforded the desired cycloadduct in excellent yield but also with excellent diastereoselectivity (entry 4). Furthermore, while reaction of nitrogen-substituted allenamide (±)-72 failed at −78 °C giving only oxidative ring-opening and hydrolysis products, the desired cycloadduct 73 could also be obtained with excellent selectivity and reasonable yield at higher temperature (entry 6). We reasoned that the steric bulk of the substituents on the nitrogen atom may actually hinder the ensuing cycloaddition thus allowing the competing oxidative ring-opening pathway to dominate, especially at low temperatures.
IV. Spectroscopic Comparisons of [4 + 2] and [4 + 3] Cycloadducts and Mechanistic Pathways
An interesting comparison can be made between [4 + 2] and [4 + 3] cycloadducts derived from TIPS-protected allenamide (±)-63. As shown in Table 3, the two cycloadducts have similar key 1H NMR resonances with the largest difference obviously being the Ha proton on an sp2 versus sp3 carbon. However, an interesting dichotomy arises when comparing the major isomers of [4 + 2] and [4 + 3] cycloadducts (64-major and 67-major, respectively). The major isomer from the [4 + 2] cycloaddition possesses an anti relationship between the silyl-ether and the oxa-bicyclic bridge while that of the [4 + 3] cycloadduct possesses a syn relationship as assigned via NOE experiments.
Table 3.
Comparison of [4 + 2] and [4 + 3] Cycloadducts.
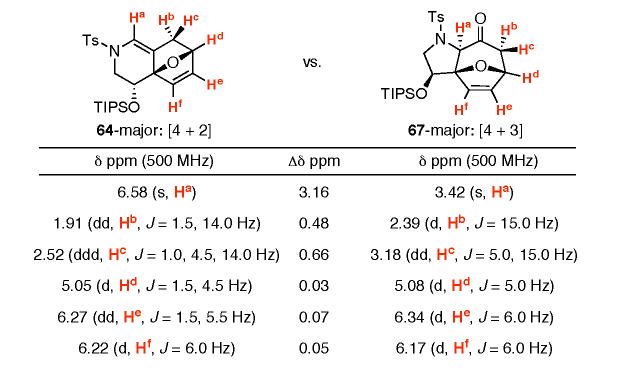
|
A working model was proposed based on the stereochemical assignments and our previous mechanistic understanding of the [4 + 2] and [4 + 3] pathways [Figure 4]. In the [4 + 2] pathway, the most stable conformation would place the large silyl-ether substituent in a pseudo-equatorial position away from the ensuing cycloaddition thus leading to the observed isomer. For the [4 + 3] pathway, if we maintain the preferred W-conformation of the oxyallyl-cation, we can envision two possible exo-approaches. While exo-I would suffer from steric interaction of the large silyl-ether pointing into the ensuing cycloaddition thereby leading to the minor isomer, exo-II places the silyl-ether away from the cycloaddition and would lead to the observed major isomer.
Figure 4.

NOE Experiments and Proposed Model.
The ability to access both [4 + 2] and [4 + 3] cycloadducts through divergent pathways highlights yet another synthetic utility of N-sulfonyl allenamides [Scheme 11]. Not only do N-sulfonyl-substituted allenamides show improved reactivity compared to N-carbamoyl-substituted allenamides, but their facile preparation from a variety of commercially available starting materials also provides a practical approach to accessing a diverse array of complex heterocyclic scaffolds via thermal or electrophilic activation conditions.
Scheme 11.

Divergent Reactivity of N-Sulfonyl Allenamides.
CONCLUSION
We have described here our efforts toward achieving a practical and diastereoselective intramolecular [4 + 3] cycloaddition of nitrogen-stabilized oxyallyl cations with tethered dienes. Selectivity is found to depend greatly on the tethering length as well as the stability of the oxyallyl cation intermediate, whether generated from N-carbamoyl- or N-sulfonyl-substituted allenamides. Oxyallyl cations derived from the N-sulfonyl-substituted allenamides show overall improved reactivity in the cycloaddition allowing for greater diversity in substrate scope. While the use of chiral auxiliaries provided minimal diastereoselectivity in the cycloaddition, high diastereoselectivity can be achieved with a stereocenter present on the tether. These studies provide further support for the synthetic utility of allenamides and promising potential for future development in intramolecular [4 + 3] cycloadditions of nitrogen-stabilized oxyallyl cations.
EXPERIMENTAL SECTION
General Experimental Methods
All reactions were performed in flame-dried glassware under nitrogen atmosphere. Solvents were distilled prior to use. 1H and 13C NMR spectra were obtained 400 or 500 MHz spectrometers using CDCl3 with TMS or residual solvent as standard unless otherwise noted. Infrared spectra were obtained in CHCl3 or neat. Optical rotations were measured on a digital polarimeter, using a 1 mL cell with a 1 dm path length. For low (MS) and high (HRMS) resolution, m/z ratios are reported as values in atomic mass units. Mass analysis was done in either APCI mode or EI mode. All spectral data obtained for new compounds are reported here.
General Procedure for Isomerizaton of Propargyl Amides to Allenamides
To a flame-dried 25 mL RB-flask filled was added propargyl amide 26 (245 mg, 0.8 mmol) and anhydrous THF (0.3 M) under N2. The solution was cooled to 0 °C and t-BuOK (20 mol% from 1M solution in THF) was added. The resulting mixture was warmed to room temperature or the reported reaction temperature and stirred for 2–6 hr. The reaction was monitored by TLC, and after completion, was filtered through celite or a small bed of silica gel with ethyl acetate and concentrated under reduced pressure. The crude residue was purified via silica gel flash column chromatography (isocratic eluent: EtOAc in hexanes) to provide the desired allenamide 27 (235 mg, 96%) as a colorless oil.
Allenamide 21 (178 mg, 0.51 mmol) was prepared in 85% yield according to the general procedure. Rf = 0.65 [1:9 EtOAc/hexanes]; [α]D23 = −45.4° [c 1.0, CH2Cl2]; pale yellow oil; 1H NMR (500 MHz, CDCl3) (due to rotamers, many of the signals were not well-resolved and/or line-broadened) δ 0.79 (d, 3H, J = 7.0 Hz), 0.87–0.91 (m, 6H), 0.92–0.98 (m, 1H), 1.00–1.08 (m, 1H), 1.10 (t, 1H, J = 7.0 Hz), 1.33–1.40 (m, 1H), 1.44–1.53 (m, 1H), 1.64–1.71 (m, 2H), 1.85–1.96 (m, 2H), 2.24–2.29 (m, 1H), 2.62 (t, 1H, J = 7.5 Hz), 3.26 (brs, 1H), 3.35–3.49 (m, 2H), 4.63 (td, 1H, J = 4.0, 10.5 Hz), [extra resonance due to rotamer, 4.56 (td, J = 4.0, 10.5 Hz)] 5.31 (brs, 2H), 5.99 (brs, 1H), 6.27 (brs, 1H), Due to rotamers, α-H on allenamide split into two signals: 7.01 (t, 0.5H, J = 6.0 Hz) and 7.17 (t, 0.5H, J = 6.0 Hz), 7.29 (br s, 1H); 13C NMR (125 MHz, CDCl3) Not recorded to due to rotamers IR (neat) cm−1 2952s, 2930m, 1703s, 1457m, 1236m; mass spectrum (ESI): m/e (% relative intensity) 369.4 (5) (M+Na+H)+, 369.4 (80) (M+Na)+, 346.4 (5) (M+H)+, 310.4 (100); HRMS (ESI) m/e calcd for C21H31NO3+ (M+Na)+ 368.2197, found 368.2203.
Allenamide 23 (1.01g, 2.8 mmol) was prepared in 89% yield according to the general procedure. Rf = 0.80 [1:4 EtOAc/hexanes]; [α]D23 = −44.4° [c 1.0, CH2Cl2]; pale yellow oil; 1H NMR (400 MHz, CDCl3) (due to rotamers, many of the signals were not well-resolved and/or line-broadened) δ 0.78 (d, 3H, J = 7.2 Hz), 0.90 (dd, 6H, J = 2.4, 7.2 Hz), 1.05 (qd, 2H, J = 3.2, 12.8 Hz), 1.26–1.29 (m, 1H), 1.35–1.40 (m, 1H), 1.44–1.49 (m, 1H), 1.55–1.64 (m, 4H), 1.65–1.72 (m, 2H), 1.88–1.94 (m, 1H), 2.05 (d, 1H, J = 12.8 Hz), 2.63 (t, 2H, J = 7.6 Hz), 3.32–3.46 (m, 2H), 4.63 (td, 1H, J = 4.4, 10.4 Hz), 5.30–5.34 (m, 2H), 5.96 (d, 1H, J = 2.4 Hz), 6.26 (brs, 1H), Due to rotamers, α-H on allenamide split into two signals: 7.01 (t, 0.5H, J = 6.2 Hz) and 7.17 (t, 0.5H, J = 6.2 Hz), 7.28 (brs, 1H); 13C NMR (125 MHz, CDCl3) Not recorded to due to rotamers IR (neat) cm−1 2953m, 1698s, 1457m, 1401s, 1302m; mass spectrum (ESI): m/e (% relative intensity) 383.4 (10) (M+Na+H)+, 382.4 (100) (M+Na)+, 360.4 (20) (M+H)+; HRMS (ESI) m/e calcd for C22H33NO3+ (M+Na)+ 382.2353, found 382.2364.
Allenamide 32 (40 mg, 0.13 mmol) was prepared in 40% yield according to the general procedure at 40 °C. Rf = 0.42 [1:5 EtOAc/hexanes]; colorless oil;1H NMR (400 MHz, CDCl3) δ 1.99 (t, 3H, J = 3.2 Hz), 2.41 (s, 3H), 2.87 (t, 2H, J = 7.2 Hz), 3.39 (t, 2H, J = 7.2 Hz), 4.66 (qt, 2H, J = 3.2 Hz), 6.09 (dd, 1H, J = 0.4, 3.2 Hz), 6.27 (dd, 1H, J = 2.0, 3.2 Hz), 7.26 (brs, 1H), 7.27 (d, 2H, J = 8.4 Hz), 7.64 (d, 2H, J = 8.4 Hz);13C NMR (100 MHz, CDCl3) δ 21.3, 21.7, 48.9, 81.6, 106.6, 107.3, 110.4, 128.0, 129.4, 134.2, 141.5, 143.6, 152.5, 207.1; IR (neat) cm−1 2925m, 1597m, 1348s, 1159s, 1092m; mass spectrum (ESI): m/e (% relative intensity) 340.9 (10) (M+Na)+, 339.8 (100), 318.9 (10) (M+H)+, 317.9 (60) (M)+; HRMS (ESI) m/e calcd for C17H19NO3S+ (M+Na)+ 340.1159, found 340.1163.
Allenamide 40a (90 mg, 0.205 mmol) was prepared in 75% yield according to the general procedure. Rf = 0.30 [1:5 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 2.41 (s, 3H), 2.83 (t, 2H, J = 7.6 Hz), 2.88 (t, 2H, J = 7.2 Hz), 3.36 (t, 2H, J = 7.6 Hz), 3.69 (t, 2H, J = 7.2 Hz), 4.52 (s, 2H), 5.30 (d, 2H, J = 6.0 Hz), 5.93 (s, 2H), 6.83 (t, 1H, J = 6.0 Hz), 7.24–7.32 (m, 7H), 7.68 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.7, 27.3, 29.0, 45.4, 68.5, 73.1, 88.0, 100.2, 106.7, 107.1, 127.3, 127.7, 127.8, 128.5, 129.9, 135.6, 138.4, 143.9, 150.8, 152.0, 201.3; IR (neat) cm−1 2861w, 1597w, 1453m, 1353s, 1161s; mass spectrum (ESI): m/e (% relative intensity) 461.2 (10) (M+Na+H)+, 460.2 (100) (M+Na)+, 438.2 (10) (M+H)+; HRMS (ESI) m/e calcd for C25H27NO4S+ (M+Na)+ 460.1553, found 460.1551.
Allenamide 44 (170 mg, 0.49 mmol) was prepared in 68% yield from the corresponding propargyl amide according to the general procedure. Rf = 0.45 [1:5 EtOAc/hexanes]; white solid; mp = 64–65 °C; 1H NMR (500 MHz, CDCl3) δ 0.96 (s, 6H), 2.42 (s, 3H), 2.61 (s, 2H), 2.98 (s, 2H), 5.17 (d, 2H, J = 6.5 Hz), 6.03 (d, 1H, J = 3.0 Hz), 6.28 (dd, 1H, J = 1.5, 2.5 Hz), 6.74 (t, 1H, J = 6.5 Hz), 7.29 (d, 1H, J = 1.5 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.66 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 25.6, 36.3, 39.1, 56.0, 88.0, 102.5, 107.8, 110.3, 127.7, 129.7, 134.8, 141.2, 143.8, 153.6, 203.1; IR (neat) cm−1 2925m, 2189w, 1659w, 1459m, 1166s; mass spectrum (ESI): m/e (% relative intensity) 341.3 (15) (M+Na+H)+, 340.2 (100) (M+Na)+, 318.3 (5) (M+H)+; HRMS (ESI) m/e calcd for C17H19NO3S+ (M+Na)+ 340.0978, found 340.0980.
Allenamide 48 (296 mg, 0.89 mmol) was prepared in 99% yield from the corresponding propargyl amide according to the general procedure. Rf = 0.47 [1:3 EtOAc/hexanes]; pale yellow oil; 1H NMR (500 MHz, CDCl3) δ 1.57 (sept, 2H, J = 7.0 Hz), 1.65 (sept, 2H, J = 7.5 Hz), 2.42 (s, 3H), 2.61 (t, 2H, J = 7.0 Hz), 3.10 (t, 2H, J = 7.0 Hz), 5.25 (d, 2H, J = 6.5 Hz), 5.96 (dd, 1H, J = 0.5, 8.0 Hz), 6.26 (dd, 1H, J = 2.0, 8.0 Hz), 6.81 (t, 1H, J = 6.0 Hz), 7.28 (d, 1H, J = 1.5 Hz), 7.30 (d, 2H, J = 8.0 Hz), 7.67 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 25.1, 27.4, 27.6, 46.4, 87.6, 100.3, 105.1, 110.2, 127.3, 129.9, 135.6, 140.9, 143.8, 155.9, 201.6; IR (thin film) cm−1 3260w, 2926m, 1597w, 1347s, 1160s; mass spectrum (ESI): m/e (% relative intensity) 355.3 (10) (M+Na+H)+, 354.3 (100) (M+Na)+, 332.3 (10) (M+H)+; HRMS (ESI) m/e calcd for C18H21NO3S+ (M+Na)+ 354.1135, found 354.1142.
Allenamide 56 (70 mg, 0.19 mmol) was prepared in 70% yield from the corresponding propargyl amide according to the general procedure. Rf = 0.51 [1:2 EtOAc/hexanes]; [α]D23 = +14.2° [c 0.55, CH2Cl2]; yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.87, (s, 3H), 1.13 (s, 3H), 1.42 (ddd, 1H, J = 4.0, 5.6, 13.2 Hz), 1.65 (ddd, 1H, J = 4.4, 5.2, 14.0 Hz), 1.93 (d, 1H, J = 18.4 Hz), 2.01–2.07 (m, 1H), 2.10 (t, 1H, J = 4.4 Hz), 2.38 (dt, 1H, J = 4.0, 18.4 Hz), 2.45–2.52 (m, 1H), 2.81 (d, 1H, J = 14.8 Hz), 2.95 (t, 2H, J = 7.6 Hz), 3.38 (d, 1H, J = 14.4 Hz), 3.62 (sept, 1H, J = 7.6 Hz), 3.67 (sept, 1H, J = 7.6 Hz), 5.45 (d, 2H, J = 6.0 Hz), 6.10 (d, 1H, J = 3.6 Hz), 6.28 (t, 1H, J = 2.4 Hz), 6.74 (t, 1H, J = 6.4 Hz), 7.31 (dd, 1H, J = 1.2, 2.0 Hz); 13C NMR (100 MHz, CDCl3) δ 19.9, 20.1, 25.4, 27.0, 27.6, 42.7, 43.0, 45.6, 47.9, 48.0, 58.5, 88.2, 99.8, 106.7, 110.4, 141.6, 152.3, 200.8, 214.9; IR (thin film) cm−1 2921m, 1742s, 1350s, 1235m, 1089s; mass spectrum (ESI): m/e (% relative intensity) 386.8 (10) (M+Na)+, 385.9 (100), 363.9 (20) (M)+; HRMS (ESI) m/e calcd for C19H25NO4S+ (M+Na)+ 386.1397, found 386.1394.
Allenamide 58 (107 mg, 0.22 mmol) was prepared in 97% yield from the corresponding propargyl amide according to the general procedure. Rf = 0.43 [1:10 EtOAc/hexanes]; [α]D23 = −22.8° [c 0.54, CH2Cl2]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 0.06 (s, 3H), 0.11 (s, 3H), 0.84 (s, 3H), 0.87 (s, 9H), 0.99 (s, 3H), 1.06–1.12 (m, 1H), 1.39 (ddd, 1H, J = 3.6, 9.6, 11.2 Hz), 1.68–1.75 (m, 4H), 1.98 (td, 1H, J = 3.2, 11.6 Hz), 2.63 (d, 1H, J = 13.6 Hz), 2.90 (t, 2H, J = 7.6 Hz), 3.48 (d, 1H, J = 13.2 Hz), 3.56 (sept, 1H, J = 6.4 Hz), 3.60 (sept, 1H, J = 6.8 Hz), 4.01 (dd, 1H, J = 3.6, 6.8 Hz), 5.42 (d, 2H, J = 6.0 Hz), 6.08 (d, 1H, J = 0.8, 3.2 Hz), 6.28 (dd, 1H, J = 2.0, 3.2 Hz), 6.74 (t, 1H, J = 6.0 Hz), 7.31 (dd, 1H, J = 0.8, 1.6 Hz); 13C NMR (100 MHz, CDCl3) δ −4.8, −3.7, 17.9, 20.3, 20.9, 26.1, 27.4, 27.7, 29.3, 42.5, 44.7, 45.3, 48.7, 49.1, 50.2, 76.3, 88.0, 100.1, 106.6, 110.5, 141.6, 152.5, 200.7; IR (thin film) cm−1 2928m, 1595w, 1358s, 1151s, 1083s; mass spectrum (ESI): m/e (% relative intensity) 503.5 (15) (M+Na+H)+, 502.5 (100) (M+Na)+; HRMS (ESI) m/e calcd for C25H41NO4SSi+ (M+Na)+ 502.2418, found 502.2401.
Allenamide (±)-63 (153 mg, 0.32 mmol) was prepared in 61% yield according to the general procedure. Rf = 0.36 [1:8 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.98 (d, 9H, J = 5.6 Hz), 1.01–1.07 (m, 12H), 2.41 (s, 3H), 3.30 (dd, 1H, J = 7.2, 13.6 Hz), 3.45 (dd, 1H, J = 6.8, 13.6 Hz), 5.08 (t, 2H, J = 7.2 Hz), 5.24 (d, 2H, J = 6.0 Hz), 6.26 (dd, 1H, J = 3.2 Hz), 6.29–6.31 (m, 1H), 6.68 (t, 1H, J = 6.4 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.34 (dd, 1H, J = 0.8, 1.2 Hz), 7.66 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 12.4, 17.9, 18.0, 21.7, 51.7, 67.1, 88.2, 101.0, 107.8, 110.2, 127.4, 129.8, 135.2, 141.9, 143.8, 154.5, 201.4; IR (neat) cm−1 2943m, 2866w, 1462w, 1359s, 1164s; mass spectrum (ESI): m/e (% relative intensity) 499.2 (5) (M+Na+H)+, 498.2 (25) (M+Na)+, 476.2 (100) (M+H)+; HRMS (ESI) m/e calcd for C25H37NO4SSi+ (M+H)+ 476.2286, found 476.2269.
Allenamide (±)-68 (115 mg, 0.26 mmol) was prepared in 82% yield according to the general procedure. Rf = 0.35 [1:8 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.57 (qt, 6H, J = 7.6 Hz), 0.90 (t, 9H, J = 8.0 Hz), 2.41 (s, 3H), 3.37 (AB of ABX, 2H, JAX = JBX = 6.8, JAB = 13.6 Hz), 4.96 (t, 1H, J = 6.8 Hz), 5.27 (d, 2H, J = 6.4 Hz), 6.26 (d, 1H, J = 3.2 Hz), 6.30 (dd, 1H, J = 1.6, 3.2 Hz), 6.73 (t, 1H, J = 6.4 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.37 (d, 1H, J = 1.6 Hz), 7.67 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 4.7, 6.8, 21.7, 51.5, 66.7, 88.1, 100.9, 107.5, 110.3, 127.4, 129.8, 135.2, 142.0, 143.8, 154.4, 201.5; IR (neat) cm−1 2940m, 1460w, 1355s, 1159s, 1090s; mass spectrum (ESI): m/e (% relative intensity) 457.2 (15) (M+Na+H)+, 456.1 (50) (M+Na)+, 434.2 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H31NO4SSi+ (M+Na)+ 456.1636, found 456.1634.
Allenamide (±)-70 (90 mg, 0.22 mmol) was prepared in 52% yield according to the general procedure. Rf = 0.31 [1:8 EtOAc/hexanes]; white solid; 47–49 °C; 1H NMR (400 MHz, CDCl3) δ 1.22 (s, 9H), 2.42 (s, 3H), 3.31(dd, 1H, J = 4.0, 14.0 Hz), 3.84 (dd, 1H, J = 9.2, 14.0 Hz), 5.29 (dd, 1H, J = 6.4, 10.0 Hz), 5.41 (dd, 1H, J = 6.4, 10.4 Hz), 6.05 (dd, 1H, J = 4.4, 9.2 Hz), 6.32 (dd, 1H, J = 2.0, 3.2 Hz), 6.34 (d, 1H, J = 3.6 Hz), 6.76 (t, 1H, J = 6.4 Hz), 7.31 (d, 2H, J = 8.4 Hz), 7.36 (dd, 1H, J = 0.8, 1.6 Hz), 7.68 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 21.7, 27.2, 39.0, 48.2, 65.2, 88.5, 100.4, 109.1, 110.5, 127.4, 129.9, 135.0, 142.8, 144.0, 150.3, 177.6, 201.6; IR (neat) cm−1 2972w, 1728s, 1357s, 1166s, 1146s; mass spectrum (ESI): m/e (% relative intensity) 426.1 (20) (M+Na)+, 421.2 (100), 404.2 (5) (M+H)+; HRMS (ESI) m/e calcd for C21H25NO5S+ (M+Na)+ 426.1346, found 426.1337.
Allenamide (±)-72 (65 mg, 0.107 mmol) was prepared in 59% yield according to the general procedure. Rf = 0.32 [1:8 EtOAc/hexanes]; orange foam solid; mp = 73–75 °C; 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 9H), 2.44 (s, 3H), 3.53 (dd, 1H, J = 4.0, 14.0 Hz), 4.24 (dd, 1H, J = 9.6, 14.0 Hz), 5.35 (dd, 1H, J = 6.0, 10.4 Hz), 5.47 (dd, 1H, J = 6.0, 10.4 Hz), 5.92 (dd, 1H, J = 4.4, 9.2 Hz), 6.36 (s, 2H), 6.90 (t, 1H, J = 6.0 Hz), 7.34 (d, 2H, J = 8.4 Hz), 7.36 (brs, 1H), 7.67 (d, 2H, J = 8.4 Hz), 8.25 (d, 2H, J = 8.8 Hz), 8.37 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ 21.7, 27.9, 46.6, 52.8, 85.8, 89.1, 100.1, 108.1, 110.8, 123.9, 127.4, 130.1, 130.3, 134.5, 142.0, 144.4, 145.6, 149.6, 150.1, 150.5, 201.3; IR (neat) cm−1 3154w, 1730s, 1597m, 1531s, 1148s; mass spectrum (ESI): m/e (% relative intensity) 627.1 (25) (M+Na+H)+, 626.1 (80) (M+Na)+, 621.2 (100), 604.2 (5) (M+H)+; HRMS (ESI) m/e calcd for C27H29N3O9S2 + (M+Na)+ 626.1238, found 626.1208.
Allenamide 38 (110 mg, 0.296 mmol) was prepared in 44% yield (77% brsm) from amide 28 and (2-iodovinylidene)cyclohexane according to our previously reported Cu(I) cross-coupling procedure.28 Rf = 0.56 [1:2 EtOAc/hexanes]; pale yellow oil; 1H NMR (400 MHz, CDCl3) δ 1.47–1.52 (m, 3H), 1.54 (s, 2H), 1.58–1.65 (m, 2H), 2.09 (t, 3H, J = 5.0 Hz), 2.41 (s, 3H), 2.86 (t, 2H, J = 8.0 Hz), 3.33 (t, 2H, J = 8.0 Hz), 6.03 (d, 1H, J = 3.0 Hz), 6.26 (t, 1H, J = 2.5 Hz), 6.58 (brs, 1H), 7.28 (brs, 1H), 7.29 (d, 2H, J = 8.5 Hz), 7.69 (d, 2H, J = 8.5 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6, 25.9, 26.8, 27.3, 32.8, 45.3, 96.8, 106.3, 110.3, 116.1, 127.3, 129.7, 135.6, 141.4, 143.6, 152.6, 189.0; IR (neat) cm−1 2929m, 1447w, 1348m, 1163s; mass spectrum (ESI): m/e (% relative intensity) 394.9 (10) (M+Na)+, 393.9 (100), 372.9 (10) (M+H)+, 371.9 (60) (M)+; HRMS (ESI) m/e calcd for C21H25NO3S+ (M+H)+ 372.1628, found 372.1625.
General Procedure for the Intramolecular [4 + 3] Cycloaddition of Allenamides
To a solution of allenamide (±)-70 (40 mg, 0.10 mmol) and 50 mg of 4Å molecular sieves in CH2Cl2 (2 mL) was added DMDO (2.8 mL, 0.07 M in acetone, 2 equiv) as a chilled solution (at −78 °C) via syringe pump over ~1–2 hours. The syringe pump was cooled by dry ice at all times during the addition. After the addition, the solution was filtered through celite with ethyl acetate and concentrated under reduced pressure. The resulting crude residue was purified via silica gel flash column chromatography (isocratic eluent: EtOAc in hexanes) to provide the desired cycloadduct 71 (39 mg, 0.093 mmol, 94% yield, >95:5 dr). Rf = 0.11 [1:3 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.98 (s, 9H), 2.41 (s, 3H), 2.42 (d, 1H, J = 16.0 Hz), 3.13 (dd, 1H, J = 5.6, 16.0 Hz), 3.52 (d, 1H, J = 12.8 Hz), 3.56 (s, 1H), 4.02 (dd, 1H, J = 4.0, 12.8 Hz), 5.07 (d, 1H, J = 4.0 Hz), 5.11 (d, 1H, J = 6.4 Hz), 6.05 (dd, 1H, J = 0.4, 6.0 Hz), 6.42 (dd, 1H, J = 1.6, 6.0 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.81 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6, 26.8, 27.1, 38.6, 44.2, 55.6, 69.1, 78.5, 93.6, 128.1, 128.2, 129.8, 134.1, 137.3, 144.0, 176.8, 200.4; IR (thin film) cm−1 2976m, 1733s, 1597m, 1240s, 1140s; mass spectrum (ESI): m/e (% relative intensity) 443.2 (10) (M+Na+H)+, 442.1 (100) (M+Na)+, 420.1 (40) (M+H)+; HRMS (ESI) m/e calcd for C21H25NO6S+ (M+H)+ 420.1476, found 420.1490.
Cycloadduct 20 (40 mg, 0.115 mmol) was isolated in 78% yield from allenamide 19 as a 1:1 mixture of diastereomers according to the general procedure. Rf = 0.15 [1:2 EtOAc/hexanes]; [α]D23 = −469.1° [c 0.13, CH2Cl2]; white solid; 1H NMR (500 MHz, CDCl3) (due to rotamers, many of the signals were not well-resolved and/or line-broadened) δ 0.79 (d, 3H, J = 7.0 Hz), 0.89 (d, 6H, J = 6.5 Hz); 0.80–1.08 (m, 3H), 1.26–1.34 (m, 1H), 1.45–1.50 (m, 1H), 1.65 (d, 2H, J = 10.0 Hz), 1.92–1.96 (m, 1H), 2.10 (dd, 1H, J = 6.0, 13.0 Hz), 2.14–2.20 (m, 1H), 2.41 (ddd, 1H, J = 1.0, 4.0, 16.0 Hz), 2.93 (ddd, 1H, J = 2.5, 9.5, 16.0 Hz), 3.52 (td, 1H, J = 6.0, 11.5 Hz), 3.90 (brs, 1H), 4.05 (brs, 1H), 4.58 (td, 1H, J = 4.0, 11.0 Hz), 5.03 (d, 1H, J = 6.5 Hz), 6.27 (d, 1H, J = 6.0 Hz), 6.45 (dt, 1H, J = 1.5, 6.0 Hz); 13C NMR (125 MHz, CDCl3) Not recorded to due to rotamers. IR (thin film) cm−1 2954s, 2870s, 1731s, 1694s, 1416m; mass spectrum (ESI): m/e (% relative intensity) 371.3 (15) (M+Na+H)+, 370.3 (100) (M+Na)+; HRMS (ESI) m/e calcd for C20H29NO4+ (M+Na)+ 370.1989, found 370.1999.
Cycloadduct 22 (14 mg, 0.038 mmol) was isolated in 26% yield (60:40 mixture of diastereomers with respect to the ring fusion) from allenamide 21 according to the general procedure. Major: Rf = 0.18 [1:2 EtOAc/hexanes]; colorless oil; 1:1 mixture with respect to the auxiliary (due to rotamers, many of the signals were not well-resolved and/or line-broadened). 1H NMR (500 MHz, CDCl3) [extra resonances from rotamers reported in brackets] δ 0.77 (d, 1.5H, J = 7.0 Hz), [0.81, (d, 1.5H, J = 7.0 Hz)], 0.82–0.92 (m, 6H); 0.95–1.08 (m, 2H), 1.24–1.32 (m, 2H), 1.41–1.46 (m, 1H), 1.58–1.68 (m, 3H), 1.76–1.82 (m, 1H), 1.86–1.98 (m, 2H), 2.02–2.18 (m, 2H), 2.45 (dd, 1H, J = 7.5, 16.0 Hz), 2.85 (dd, 1H, J = 5.0, 16.0 Hz), 3.24 (brs, 1H), 3.70–3.86 (m, 2H), 4.52 (m, 1H), 5.01 (d, 1H, J = 5.0 Hz), 6.17 (dd, 1H, J = 6.0, 15.0 Hz), 6.34 (td, 1H, J = 1.5, 6.0 Hz); 13C NMR (125 MHz, CDCl3) Not recorded to due to rotamers. IR (thin film) cm−1 2949s, 1725s, 1705s, 1425m, 1355m; mass spectrum (ESI): m/e (% relative intensity) 385.4 (15) (M+Na+H)+, 384.4 (100) (M+Na)+; HRMS (ESI) m/e calcd for C21H31NO4+ (M+Na)+ 384.2146, found 384.2146. Minor: Rf = 0.24 [1:2 EtOAc/hexanes]; colorless oil; 1:1 mixture with respect to the auxiliary. 1H NMR (400 MHz, CDCl3) (due to rotamers, many of the signals were not well-resolved and/or line-broadened) [extra resonances from rotamers reported in brackets] δ 0.75 (d, 1.5H, J = 6.8 Hz), [0.80 (d, 1.5H, J = 6.8 Hz)], 0.86–0.93 (m, 6H); 0.95–1.08 (m, 2H), 1.31–1.48 (m, 2H), 1.62–1.29 (m, 3H), 1.78 (dd, 1H, J = 4.6, 13.2 Hz), 1.84–1.94 (m, 2H), 2.02–2.10 (m, 2H), 2.38 (dd, 1H, J = 4.6, 14.4 Hz), 2.80 (dq, 1H, J = 3.2, 11.6 Hz), 3.27 (dt, 1H, J = 4.6, 14.4 Hz) 3.46 (d, 1H, J = 5.6 Hz), 4.05–4.18 (m, 1H), 4.55 (td, 0.5H, J = 4.5, 11.2 Hz), [4.48 (td, 0.5H, J = 4.5, 11.2 Hz)], 5.01–5.04 (m, 1H), 5.91 (t, 1H, J = 6.0 Hz), 6.33 (dt, 1H, J = 2.0, 6.0 Hz); 13C NMR (100 MHz, CDCl3) Not recorded to due to rotamers. IR (thin film) cm−1 2946s, 1727s, 1703s, 1424m, 1356m; mass spectrum (ESI): m/e (% relative intensity) 385.4 (15) (M+Na+H)+, 384.4 (100) (M+Na)+, 360.5 (15) (M+H)+, 360.5 (20) (M−H)+; HRMS (ESI) m/e calcd for C21H31NO4 + (M+Na)+ 384.2146, found 384.2140.
Cycloadduct 34 (41 mg, 0.128 mmol) was obtained in 98% yield from allenamide 27 according to the general procedure. Rf = 0.17 [1:2 EtOAc/hexanes]; white solid; mp=157–158 °C; 1H NMR (500 MHz, CDCl3) δ 1.89–1.96 (m, 1H), 2.08 (ddd, 1H, J = 2.0, 5.5, 13.5 Hz), 2.40 (d, 1H, J = 16.0 Hz), 2.43 (s, 3H), 3.03 (dd, 1H, J = 6.5, 16.0), 3.61 (s, 1H), 3.62–3.67 (m, 2H), 5.04 (d, 1H, J = 6.5 Hz), 6.09 (d, 1H, J = 6.0 Hz), 6.40 (dd, 1H, J = 1.5, 6.0 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.83 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 31.9, 44.2, 48.9, 71.4, 77.6, 92.8, 128.0, 129.8, 131.7, 134.9, 137.0, 143.8, 201.4; IR (neat) cm−1 2950m, 1705s, 1465w, 1325m, 1155s; mass spectrum (ESI): m/e (% relative intensity) 342.8 (10) (M+Na)+, 341.9 (100), 319.9 (20) (M)+; HRMS (ESI) m/e calcd for C16H17NO4S+ (M+Na)+ 342.0771, found 342.0764.
Cycloadduct 35 (33 mg, 0.084 mmol) was obtained in 93% yield from allenamide 33 according to the general procedure. Rf = 0.14 [1:2 EtOAc/hexanes]; white solid; mp=194–195 °C; 1H NMR (400 MHz, CDCl3) δ 2.17–2.21 (m, 2H), 2.41 (dd, 1H, J = 0.8, 16.4 Hz), 2.96 (ddd, 1H, J = 0.8, 6.8, 16.8 Hz), 3.61 (td, 1H, J = 7.2, 10.0 Hz), 3.85–3.90 (m, 1H), 3.94 (s, 1H), 5.06 (dd, 1H, J = 0.8, 6.8 Hz), 6.21 (d, 1H, J = 6.0 Hz), 6.47 (dd, 1H, J = 2.4, 6.0 Hz), 8.16 (d, 2H, J = 8.8 Hz), 8.37 (d, 2H, J = 8.8 Hz); 13C NMR (100 MHz, CDCl3) δ 32.9, 44.0, 49.1, 71.6, 77.3, 92.8, 124.3, 129.0, 131.9, 137.4, 145.2, 150.2, 201.7; IR (neat) cm−1 2961m, 2923w, 1599m, 1258m, 1013s; mass spectrum (ESI): m/e (% relative intensity) 373.8 (10) (M+Na)+, 372.8 (100), 350.8 (5) (M)+; HRMS (ESI) m/e calcd for C15H14N2O6S+ (M+Na)+ 373.0465, found 373.0451.
Cycloadduct 36 (23mg, 0.069 mmol) was obtained in 69% yield from allenamide 32 according to the general procedure. Rf = 0.15 [1:2 EtOAc/hexanes]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 0.97 (s, 3H), 2.17 (ddd, 1H, J = 2.0, 8.0, 13.5 Hz), 2.31 (d, 1H, J = 16.0 Hz), 2.30–2.35 (m, 1H), 2.41 (s, 3H), 2.83 (dd, 1H, J = 5.5, 15.5 Hz), 3.68 (td, 1H, J = 2.0, 9.5 Hz), 3.99 (qt, 1H, J = 9.0 Hz), 4.94 (d, 1H, J = 5.5 Hz), 6.04 (d, 1H, J = 6.0 Hz), 6.33 (dd, 1H, J = 1.5, 6.0 Hz), 7.28 (d, 2H, J = 8.0 Hz), 7.70 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 18.0, 21.7, 29.3, 43.6, 47.6, 74.4, 77.5, 95.2, 127.5, 129.7, 130.5, 137.1, 138.2, 143.4, 201.8; IR (thin film) cm−1 2925m, 1718s, 1460w, 1334m, 1158s; mass spectrum (ESI): m/e (% relative intensity) 356.8 (15) (M+Na)+, 355.8 (100), 333.9 (10) (M)+; HRMS (ESI) m/e calcd for C17H19NO4S+ (M+Na)+ 356.0927, found 356.0934.
Cycloadduct 37a (15 mg, 0.045 mmol) was obtained in 95% yield as ~80:20 inseparable mixture from allenamide (±)-29a according to the general procedure. Rf = 0.10 [1:2 EtOAc/hexanes]; colorless oil; 1H NMR (500 MHz, CDCl3) Major δ 1.0 (d, 3H, J = 7.0 Hz), 1.91 (q, 1H, J = 10.0 Hz), 2.13 (ddd, 1H, J = 2.0, 7.0, 13.5 Hz), 2.43 (s, 3H), 3.10 (s, 1H), 3.23 (quint, 1H, J = 7.0 Hz), 3.44 (td, 1H, J = 2.0, 10.0 Hz), 3.86 (qd, 1H, J = 1.5, 10.0 Hz), 4.88 (d, 1H, J = 5.0 Hz), 6.05 (d, 1H, J = 6.0 Hz), 6.36 (dd, 1H, J = 1.5, 6.0 Hz), 7.35 (d, 2H, J = 8.5 Hz), 7.81 (d, 2H, J = 8.5 Hz); Select resonances for minor δ 1.37 (d, 3H, J = 7.0 Hz), 2.13 (dd, 1H, J = 6.0, 13.5 Hz), 2.45 (s, 3H), 3.14 (s, 1H), 3.55 (td, 1H, J = 6.0, 11.0 Hz), 3.68 (t, 1H, J = 10.0 Hz), 4.22 (ddd, 1H, J = 5.5, 8.0, 13.5 Hz), 4.65 (brs, 1H), 6.11 (d, 1H, J = 6.0 Hz), 6.46 (dd, 1H, J = 1.5, 6.0 Hz), 7.87 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 10.3, 21.7, 27.8, 30.7, 48.7, 70.1, 82.7, 93.3, 128.2, 129.9, 132.1, 133.3, 135.8, 144.0, 203.7; IR (thin film) cm−1 2916s, 2848s, 1717s, 1349m, 1163s; mass spectrum (ESI): m/e (% relative intensity) 356.8 (10) (M+Na)+, 355.8 (100), 333.9 (20) (M)+; HRMS (ESI) m/e calcd for C17H19NO4S+ (M+Na)+ 356.0927, found 356.0937.
Cycloadduct 39 (28mg, 0.072 mmol) was obtained in 90% yield from allenamide 38 according to the general procedure. Rf = 0.25 [1:2 EtOAc/hexanes]; light brown solid; mp=134–135 °C; 1H NMR (400 MHz, CDCl3) δ 1.16–1.20 (m, 1H), 1.36–1.48 (m, 3H), 1.56–1.66 (m, 2H), 1.69–1.86 (m, 3H), 1.89–2.10 (m, 2H), 2.15 (ddd, 1H, J = 4.4, 8.4, 13.2 Hz), 2.43 (s, 3H), 3.19 (s, 1H), 3.50 (td, 1H, J = 1.2, 10.4 Hz), 3.80 (td, 1H, J = 7.2, 10.4 Hz), 4.82 (s, 1H), 6.03 (d, 1H, J = 6.0 Hz), 6.40 (dd, 1H, J = 0.8, 6.0 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.84 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 21.7, 21.8, 22.1, 25.6, 29.8, 31.1, 33.0, 49.2, 56.2, 69.6, 84.3, 93.1, 128.3, 129.8, 131.9, 133.3, 136.2, 143.9, 206.3; IR (neat) cm−1 2921m, 1721s, 1464m, 1333m, 1165m; mass spectrum (ESI): m/e (% relative intensity) 410.9 (10) (M+Na)+, 409.8 (100), 387.9 (20) (M)+; HRMS (ESI) m/e calcd for C21H25NO4S+ (M+Na)+ 410.1397, found 410.1403.
Cycloadduct 41a (25 mg, 0.055 mmol) was obtained in 81% yield from allenamide 40a according to the general procedure. Rf = 0.22 [1:2 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.91 (q, 1H, J = 9.6 Hz), 2.04–2.08 (m, 1H), 2.09 (q, 2H, J = 5.6 Hz), 2.43 (s, 3H), 2.52 (d, 1H, J = 16.0 Hz), 2.82 (d, 1H, J = 16.0 Hz), 3.55–3.64 (m, 5H), 4.48 (s, 2H), 5.97 (d, 1H, J = 6.0 Hz), 6.31 (d, 1H, J = 6.0 Hz), 7.28–7.36 (m, 7H), 7.84 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 21.7, 32.2, 37.1, 48.9, 49.3, 65.6, 70.1, 73.3, 85.9, 92.7, 127.8, 127.9, 128.1, 128.6, 129.8, 130.9, 135.0, 138.3, 139.7, 143.8, 202.0; IR (thin film) cm−1 2925m, 1719s, 1352m, 1267m, 1165s; mass spectrum (ESI): m/e (% relative intensity) 477.2 (10) (M+Na)+, 476.2 (100), 454.2 (10) (M)+; HRMS (ESI) m/e calcd for C25H27NO5S+ (M+H)+ 454.1683, found 454.1684.
Cycloadduct 41b (33 mg, 0.068 mmol) was obtained in 97% yield from allenamide 40b according to the general procedure. Rf = 0.10 [1:6 EtOAc/hexanes]; white solid; mp=106–107 °C; 1H NMR (400 MHz, CDCl3) δ 0.03 (s, 3H), 0.04 (s, 3H), 0.87 (s, 9H), 1.87–1.95 (m, 1H), 2.00 (td, 2H, J = 3.6, 6.4 Hz), 2.08 (ddd, 1H, J = 2.4, 5.2, 13.2 Hz), 2.44 (s, 3H), 2.54 (d, 1H, J = 16.0 Hz), 2.82 (dd, 1H, J = 0.4, 16.0 Hz), 3.61–3.65 (m, 2H), 3.74 (t, 2H, J = 6.4 Hz), 5.96 (d, 1H, J = 5.6 Hz), 6.33 (d, 1H, J = 6.0 Hz), 7.33 (dd, 2H, J = 0.8, 8.8 Hz), 7.84 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ −5.2, −5.1, 18.4, 21.8, 26.1, 32.3, 40.0, 48.9, 49.4, 58.6, 70.2, 86.0, 92.6, 128.1, 129.8, 130.7, 135.2, 140.0, 143.8, 202.1; IR (neat) cm−1 2883m, 1735s, 1507w, 1365m, 1216m; mass spectrum (ESI): m/e (% relative intensity) 500.8 (20) (M+Na)+, 499.8 (100), 477.9 (5) (M)+; HRMS (ESI) m/e calcd for C24H35NO5SSi+ (M+Na)+ 500.1898, found 500.1906.
Cycloadduct 45 (14 mg, 0.042 mmol) was obtained in 45% yield as a 3:1 mixture of diastereomers from allenamide 44 according to the general procedure. Minor isomer was found to epimerize upon column chromatography. Rf = 0.15 [1:2 EtOAc/hexanes]; white solid; mp=184–185 °C; 1H NMR (500 MHz, CDCl3) δ 1.51 (td, 1H, J = 5.0, 14.0 Hz), 1.68 (dquint, 1H, J = 2.5, 13.0 Hz), 1.99 (d, 1H, J = 14.0 Hz), 2.09 (qd, 1H, J = 2.5, 13.0 Hz), 2.17 (quartet of triplets, 1H, J = 3.5, 13.5 Hz), 2.42 (d, 1H, J = 16.0 Hz), 2.43 (s, 3H), 2.72 (s, 1H), 3.54 (dd, 1H, J = 5.0, 15.5 Hz), 3.93 (dd, 1H, J = 3.0, 11.5 Hz), 5.11 (d, 1H, J = 4.5 Hz), 5.80 (d, 1H, J = 6.0 Hz), 6.34 (dd, 1H, J = 2.0, 6.0 Hz), 7.35 (d, 2H, J = 8.0 Hz), 7.70 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 20.2, 21.7, 30.6, 46.3, 47.4, 71.4, 79.9, 85.1, 128.6, 129.8, 131.9, 132.9, 137.2, 144.1, 202.1; IR (neat) cm−1 2971m, 1738s, 1337m, 1158s, 1098m; mass spectrum (ESI): m/e (% relative intensity) 356.1 (100) (M+Na+H)+, 334.1 (95) (M+H)+; HRMS (ESI) m/e calcd for C17H19NO4S+ (M+H)+ 334.1108, found 334.1115.
Cycloadduct 47 (28 mg, 0.072 mmol) was obtained in 90% yield from allenamide 46 according to the general procedure. Rf = 0.25 [1:2 EtOAc/hexanes]; white solid; mp=155–156 °C; 1H NMR (500 MHz, CDCl3) δ 0.91 (s, 3H), 1.36 (s, 3H), 1.42 (d, 1H, J = 14.5 Hz), 1.82 (dd, 1H, J = 2.0, 14.5 Hz), 1.89 (d, 1H, J = 11.5 Hz), 2.43 (s, 3H), 2.44 (d, 1H, J = 13.5 Hz), 2.71 (s, 1H), 3.52 (dd, 1H, J = 2.0, 12.0 Hz), 3.55 (dd, 1H, J = 4.5, 14.0 Hz), 5.11 (d, 1H, J = 4.5 Hz), 5.72 (d, 1H, J = 6.0 Hz), 6.30 (dd, 1H, J = 1.5, 6.0 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.70 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 25.8, 29.8, 30.8, 43.2, 46.7, 59.1, 71.5, 80.1, 86.1, 128.4, 129.8, 133.0, 133.8, 136.7, 143.9, 202.2; IR (neat) cm−1 2923m, 1709s, 1327m, 1162s, 1095s; mass spectrum (ESI): m/e (% relative intensity) 385.2 (10) (M+Na+H)+, 384.1 (100) (M+Na)+; HRMS (ESI) m/e calcd for C19H23NO4S+ (M+H)+ 362.1421, found 362.1408.
Cycloadduct 49 (7 mg, 0.020 mmol) was obtained in 17% yield from allenamide 48 according to the general procedure. Rf = 0.17 [1:3 EtOAc/hexanes]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.71 (br d, 2H, J = 14.0 Hz), 1.84–1.95 (m, 2H), 2.02 (dd, 1H, J = 6.0, 14.5 Hz), 2.22 (t, 1H, J = 14.0 Hz), 2.38 (d, 1H, J = 18.0 Hz), 2.41 (s, 3H), 2.75–2.80 (m, 1H), 3.58 (d, 1H, J = 15.0 Hz), 4.38 (s, 1H), 4.98 (d, 1H, J = 6.5 Hz), 6.26 (d, 1H, J = 5.5 Hz), 6.33 (d, 1H, J = 6.0 Hz), 7.27 (d, 2H, J = 8.0 Hz), 7.73 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 21.8, 30.6, 33.5, 46.5, 47.3, 70.3, 76.7, 89.8, 128.1, 129.3, 133.7, 137.3, 137.5, 143.3, 205.1; IR (thin film) cm−1 2926m, 1723s, 1330s, 1265m, 1153s; mass spectrum (ESI): m/e (% relative intensity) 371.3 (10) (M+Na+H)+, 370.3 (100) (M+Na)+; HRMS (ESI) m/e calcd for C18H21NO4S+ (M+Na)+ 370.1084, found 370.1101.
Cycloadduct 51 (24 mg, 0.078 mmol) was obtained in 73% yield as a 3:1 mixture of diastereomers from allenamide 50 according to the general procedure. Major: Rf = 0.20 [1:3 EtOAc/hexanes]; white solid; mp=83–84 °C; 1H NMR (500 MHz, CDCl3) δ 1.67–1.72 (m, 1H), 1.99 (quintet of doublets, 1H, J = 3.0, 7.0 Hz), 2.31–2.36 (m, 1H), 2.43 (s, 3H), 2.45 (d, 1H, J = 15.0 Hz), 2.54–2.59 (m, 1H), 2.68 (d, 1H, J = 5.0 Hz) 2.70 (dd, 1H, J = 2.0, 5.0 Hz), 2.97–3.00 (m, 1H), 3.27 (q, 1H, J = 9.0 Hz), 3.44 (td, 1H, J = 3.0, 8.0 Hz), 4.91 (d, 1H, J = 9.0 Hz), 5.48 (ddd, 1H, J = 1.0, 4.5, 11.0 Hz), 5.84 (ddd, 1H, J = 1.5, 2.5, 7.5 Hz), 7.31 (d, 2H, J = 8.5 Hz), 7.75 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 21.4, 21.7, 31.6, 42.4, 43.3, 46.3, 67.5, 127.8, 129.6, 129.8, 129.9, 135.5, 143.7, 206.4; IR (neat) cm−1 2917s, 1726s, 1340m, 1154s, 1105m; mass spectrum (ESI): m/e (% relative intensity) 329.1 (10) (M+Na+H)+, 328.1 (55) (M+Na)+, 306.1 (100) (M+H)+; HRMS (ESI) m/e calcd for C16H19NO3S+ (M+H)+ 306.1159, found 306.1147. Minor: Rf = 0.16 [1:3 EtOAc/hexanes]; colorless oil; 1H NMR (500 MHz, CDCl3) δ 1.63–1.70 (m, 2H), 2.08 (dt, 1H, J = 5.5, 11.5 Hz), 2.13–2.16 (m, 1H), 2.43 (s, 3H), 2.53–2.62 (m, 4H), 3.15 (ddd, 1H, J = 5.5, 6.0, 7.0 Hz), 3.73 (dd, 1H, J = 8.0, 10.5 Hz), 4.43 (d, 1H, J = 11.5 Hz), 5.84 (ddd, 1H, J = 1.5, 3.0, 11.0 Hz), 6.02 (quintet of doublets, 1H, J = 2.5, 6.0 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.88 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 21.7, 23.9, 32.3, 40.7, 43.2, 48.3, 72.0, 127.9, 129.7, 131.0, 132.1, 136.9, 143.6, 205.8; IR (thin film) cm−1 2849m, 1723s, 1335m, 1140s, 1098m; mass spectrum (ESI): m/e (% relative intensity) 328.1 (10) (M+Na)+, 306.1 (100) (M+H)+; HRMS (ESI) m/e calcd for C16H19NO3S+ (M+H)+ 306.1159, found 306.1151.
Cycloadduct 57 (25 mg, 0.065 mmol) was obtained in 80% yield as a 65:35 inseparable mixture of diastereomers from allenamide 56 according to the general procedure. Rf = 0.24 [1:1 EtOAc/hexanes]; colorless oil; 1H NMR (500 MHz, CDCl3) Major δ 0.91 (s, 3H), 1.13 (s, 3H), 1.42 (td, 1H, J = 4.0, 9.5 Hz), 1.67–1.75 (m, 1H), 1.93 (d, 1H, J = 18.0 Hz), 2.01–2.07 (m, 1H), 2.09 (t, 1H, J = 5.0 Hz), 2.11–2.15 (m, 1H), 2.28 (td, 1H, J = 8.5, 13.0 Hz), 2.36–2.42 (m, 1H), 2.41 (dd, 1H, J = 5.0, 17.5 Hz), 2.45–2.54 (m, 1H), 2.96 (dd, 1H, J = 7.5, 17.5 Hz), 3.17 (d, 1H, J = 15.0 Hz), 3.48–3.56 (m, 1H), 3.60 (s, 1H), 3.90 (d, 1H, J = 15.0 Hz), 4.10 (dd, 1H, J = 8.5, 10.5 Hz), 4.26 (s, 1H), 5.05 (d, 1H, J = 7.5 Hz), 6.28 (d, 1H, J = 6.0 Hz), 6.48 (dd, 1H, J = 1.5, 6.0 Hz); Select resonances for minor δ 1.11 (s, 3H), 1.91 (d, 1H, J = 18.0 Hz), 2.99 (dd, 1H, J = 7.0, 17.0 Hz), 4.03 (dd, 1H, J = 8.5, 10.5 Hz), 4.22 (s, 1H), 6.27 (d, 1H, J = 6.5 Hz), 6.46 (dd, 1H, J = 2.0, 6.5 Hz); 13C NMR (125 MHz, CDCl3): Not obtained for inseparable mixture. IR (thin film) cm−1 2959m, 1739s, 1415w, 1332s, 1144s; mass spectrum (ESI): m/e (% relative intensity) 403.1 (10) (M+Na+H)+, 402.2 (100) (M+Na)+, 380.2 (10) (M+H)+; HRMS (ESI) m/e calcd for C19H25NO5S+ (M+Na)+ 402.1346, found 402.1351.
Cycloadduct 59 (25 mg, 0.065 mmol) was obtained in 97% yield as a 60:40 inseparable mixture of diastereomers from allenamide 58 according to the general procedure. Rf = 0.50 [1:5 EtOAc/hexanes]; pale yellow oil; 1H NMR (500 MHz, CDCl3) Major δ 0.07 (s, 3H), 0.19 (s, 3H), 0.89 (s, 9H), 0.90 (s, 3H), 1.03 (s, 3H), 1.05–1.10 (m, 1H), 1.35–1.41 (m, 1H), 1.67–1.78 (m, 4H), 1.92–1.94 (m, 1H), 2.00–2.10 (m, 2H), 2.24 (td, 1H, J = 8.0, 12.8 Hz), 2.40 (d, 1H, J = 17.2 Hz), 2.95 (dd, 1H, J = 7.6, 17.2 Hz), 3.35–3.42 (m, 1H), 3.48 (d, 1H, J = 13.6 Hz), 3.59 (d, 1H, J = 13.6 Hz), 4.00–4.07 (m, 2H), 4.32 (s, 1H), 5.04 (d, 1H, J = 7.2 Hz), 6.29 (d, 1H, J = 5.6 Hz), 6.46–6.48 (m, 1H); Select resonances for minor δ 0.05 (s, 3H), 0.90 (s, 3H), 0.87 (s, 9H), 0.91 (s, 3H), 1.25 (s, 3H), 2.39 (d, 1H, J = 17.6 Hz), 2.94 (dd, 1H, J = 7.6, 17.6 Hz), 3.45 (d, 1H, J = 14.0 Hz), 3.73 (d, 1H, J = 14.4 Hz), 4.17 (dd, 1H, J = 8.0, 11.6 Hz), 4.27 (s, 1H), 6.27 (d, 1H, J = 5.6 Hz); 13C NMR (125 MHz, CDCl3): Not obtained for inseparable mixture. IR (thin film) cm−1 2955m, 1737s, 1329s, 1248m, 1099m; mass spectrum (ESI): m/e (% relative intensity) 519.5 (20) (M+Na+H)+, 518.5 (100) (M+Na)+; HRMS (ESI) m/e calcd for C25H41NO5SSi+ (M+Na)+ 518.2367, found 518.2383.
Cycloadduct 67 (35 mg, 0.071 mmol) was obtained in 85% yield as a 4:1 mixture of diastereomers from allenamide (±)-63 according to the general procedure. Major: Rf = 0.19 [1:4 EtOAc/hexanes]; white solid; mp=97–98 °C; 1H NMR (500 MHz, CDCl3) δ 0.91–0.95 (m, 3H), 0.90 (d, 18H, J = 8.0 Hz), 2.39 (d, 1H, , J = 15.0 Hz), 2.40 (s, 3H), 3.18 (dd, 1H, J = 5.0, 15.0 Hz), 3.33 (d, 1H, J = 11.0 Hz), 3.42 (s, 1H), 4.04 (dd, 1H, J = 4.0, 11.5 Hz), 4.30 (d, 1H, J = 4.0 Hz), 5.08 (d, 1H, J = 5.0 Hz), 6.17 (d, 1H, J = 6.0 Hz), 6.34 (d, 1H, J = 6.0 Hz), 7.31 (d, 2H, J = 8.0 Hz), 7.80 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 21.1, 17.9, 18.0, 21.6, 44.4, 59.3, 68.4, 72.5, 78.8, 95.7, 128.1, 128.9, 129.8, 133.5, 136.5, 143.8, 200.9; IR (neat) cm−1 2944m, 1721s, 1462m, 1352s, 1165s; mass spectrum (ESI): m/e (% relative intensity) 515.2 (5) (M+Na+H)+, 509.2 (100), 492.2 (75) (M+H)+; HRMS (ESI) m/e calcd for C25H37NO5SSi+ (M+H)+ 492.2235, found 492.2236. Minor: Rf = 0.09 [1:4 EtOAc/hexanes]; white solid; mp=126–128 °C; 1H NMR (500 MHz, CDCl3) δ 0.89–0.97 (m, 3H), 1.00 (s, 18H), 2.40 (d, 1H, J = 17.0 Hz), 2.43 (s, 3H), 3.01 (dd, 1H, J = 6.5, 17.0 Hz), 3.40 (t, 1H, J = 10.5 Hz), 3.81 (dd, 1H, J = 7.5, 10.5 Hz), 3.83 (s, 1H), 4.19 (dd, 1H, J = 7.5, 9.5 Hz), 5.13 (d, 1H, J = 6.5 Hz), 6.10 (d, 1H, J = 6.0 Hz), 6.47 (dd, 1H, J = 1.5, 6.0 Hz), 7.33 (d, 2H, J = 8.0 Hz), 7.83 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 12.3, 18.0, 18.1, 21.7, 44.4, 53.5, 69.2, 70.4, 77.3, 91.8, 127.9, 129.8, 131.0, 135.8, 138.0, 144.0, 201.4; IR (neat) cm−1 2918s, 1724s, 1460m, 1347s, 1116s; mass spectrum (ESI): m/e (% relative intensity) 514.2 (20) (M+Na)+, 509.2 (100), 492.2 (75) (M+H)+; HRMS (ESI) m/e calcd for C25H37NO5− SSi+ (M+H)+ 492.2235, found 492.2231.
Cycloadduct 69 (35 mg, 0.071 mmol) was obtained in 85% yield as a 4:1 mixture of diastereomers from allenamide (±)-68 according to the general procedure. Major: Rf = 0.24 [1:3 EtOAc/hexanes]; white solid; mp=73–74 °C; 1H NMR (500 MHz, CDCl3) δ 0.44 (qd, 6H, J = 3.5, 7.5 Hz), 0.79 (t, 9H, J = 7.5 Hz), 2.39 (d, 1H, J = 15.5 Hz), 2.41 (s, 3H), 3.14 (dd, 1H, J = 5.0, 15.5 Hz), 3.32 (d, 1H, J = 11.5 Hz), 3.42 (s, 1H), 3.99 (dd, 1H, J = 4.0, 11.5 Hz), 4.12 (d, 1H, J = 3.5 Hz), 5.06 (d, 1H, J = 5.5 Hz), 6.11 (d, 1H, J = 6.0 Hz), 6.34 (dd, 1H, J = 1.5, 6.0 Hz), 7.32 (d, 2H, J = 8.0 Hz), 7.79 (d, 2H, J = 8.5 Hz); 13C NMR (100 MHz, CDCl3) δ 4.6, 6.7, 21.6, 44.4, 59.2, 68.3, 71.7, 78.7, 95.5, 128.1, 128.8, 129.6, 133.5, 136.7, 143.7, 200.9; IR (neat) cm−1 2866m, 1719s, 1415w, 1350s, 1166s; mass spectrum (ESI): m/e (% relative intensity) 473.2 (10) (M+Na+H)+, 467.2 (80), 450.2 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H31NO5SSi+ (M+H)+ 450.1765, found 450.1750. Minor: Rf = 0.12 [1:3 EtOAc/hexanes]; white solid; mp=129–131 °C; 1H NMR (500 MHz, CDCl3) δ 0.56 (q, 6H, J = 8.0 Hz), 0.90 (t, 9H, J = 8.0 Hz), 2.40 (d, 1H, J = 17.0 Hz), 2.44 (s, 3H), 3.00 (dd, 1H, J = 6.5, 17.0 Hz), 3.38 (t, 1H, J = 10.5 Hz), 3.77 (dd, 1H, J = 7.5, 10.5 Hz), 3.85 (s, 1H), 4.07 (dd, 1H, J = 7.5, 9.5 Hz), 5.14 (d, 1H, J = 6.5 Hz), 6.08 (d, 1H, J = 6.0 Hz), 6.48 (dd, 1H, J = 1.5, 6.0 Hz), 7.34 (d, 2H, J = 8.0 Hz), 7.83 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3) δ 0.2, 4.7, 6.7, 21.8, 44.4, 53.3, 69.1, 70.1, 91.7, 127.9, 129.8, 130.8, 135.9, 138.3, 144.0, 201.3; IR (neat) cm−1 2849m, 1725s, 1462m, 1352s, 1163s; mass spectrum (ESI): m/e (% relative intensity) 472.2 (30) (M+Na)+, 467.2 (80), 451.2 (20) (M+2H)+, 450.2 (100) (M+H)+; HRMS (ESI) m/e calcd for C22H31NO5SSi+ (M+H)+ 450.1765, found 450.1756.
Cycloadduct 73 (15 mg, 0.024 mmol) was obtained in 49% yield with >95:5 dr from allenamide (±)-72 according to the general procedure. Rf = 0.11 [1:3 EtOAc/hexanes]; yellow solid; mp=104–106 °C; 1H NMR (500 MHz, CDCl3) δ 1.35 (s, 9H), 2.46 (s, 3H), 2.47 (d, 1H, J = 16.5 Hz), 3.07 (dd, 1H, J = 6.5, 16.5 Hz), 3.72 (dd, 1H, J = 9.0, 10.5 Hz), 3.82 (s, 1H), 4.43 (dd, 1H, J = 9.0, 10.5 Hz), 4.93 (t, 1H J = 9.0 Hz), 5.10 (d, 1H, J = 6.5 Hz), 6.20 (d, 1H, J = 6.0 Hz), 6.48 (dd, 1H, J = 2.0, 6.0 Hz), 7.38 (d, 2H, J = 8.0 Hz), 7.89 (d, 2H, J = 8.5 Hz), 8.08 (d, 2H, J = 8.5 Hz), 8.32 (d, 2H, J = 8.5 Hz); 13C NMR (125 MHz, CDCl3) δ 0.2, 21.8, 27.9, 44.4, 49.3, 56.2, 69.5, 77.7, 86.6, 91.7, 124.0, 128.3, 129.5, 130.0, 131.7, 134.5, 137.8, 144.5, 146.0, 150.4, 200.1; IR (neat) cm−1 2928w, 1732s, 1717s, 1350s, 1265m, 1147s; mass spectrum (ESI): m/e (% relative intensity) 644.4 (5) (M+Na+2H)+, 643.4 (20) (M+Na+H)+, 642.4 (100) (M+Na)+; HRMS (ESI) m/e calcd for C27H29N3O10S2 + (M+Na)+ 642.1187, found 642.1213.
A solution of tosylate 61 (50 mg, 0.11 mmol), N-tosyl propargyl amine (36 mg, 0.17 mmol), and Cs2CO3 (45 mg, 0.13 mmol) in DMF (0.11 mL) was heated to 80 °C overnight. The solution was then filtered through celite with ethyl acetate and concentrated under reduced pressure. The resulting crude residue was purified via silica gel flash column chromatography (isocratic eluent: EtOAc in hexanes) to provide the desired cycloadduct 64 (35 mg, 0.074 mmol) in 65% yield as ~80:20 mixture of diastereomers. Rf = 0.20 [1:10 EtOAc/hexanes]; colorless oil; Major isomer reported. 1H NMR (500 MHz, CDCl3) δ 0.91–0.96 (m, 3H), 0.97 (s, 18H), 1.91 (dd, 1H, J = 1.5, 14.0 Hz), 2.42 (s, 3H), 2.52 (ddd, 1H, J = 1.0, 4.5, 14.0 Hz), 2.85–2.90 (m, 1H), 3.79–3.96 (m, 2H), 5.05 (d, 1H, J = 1.5, 4.5 Hz), 6.22 (d, 1H, J = 6.0 Hz), 6.27 (dd, 1H, J = 1.5, 5.5 Hz), 6.58 (s, 1H), 7.30 (d, 2H, J = 8.0 Hz), 7.67 (d, 2H, J = 8.0 Hz); 13C NMR (125 MHz, CDCl3): δ 12.3, 18.0, 18.1, 21.7, 31.0, 48.0, 65.9, 78.3, 89.4, 117.3, 119.2, 127.3, 130.0, 133.0, 134.9, 135.2, 144.0; 1H NMR (500 MHz, C6D6) δ 1.02 (sept, 3H, J = 6.5 Hz), 1.05 (d, 18H, J = 6.5 Hz), 1.40 (dd, 1H, J = 1.0, 14.0 Hz), 1.80 (s, 3H), 2.12 (ddd, 1H, J = 1.0, 4.0, 14.0 Hz), 2.93 (dd, 1H, J = 11.5, 12.0 Hz), 4.17 (dd, 1H, J = 5.0, 11.0 Hz), 4.26 (ddd, 1H, J = 0.5, 5.0, 12.0 Hz), 4.54 (dd, 1H, J = 1.5, 4.5 Hz), 5.72 (d, 1H, J = 6.0 Hz), 6.01 (d, 1H, J = 5.5 Hz), 6.68 (d, 2H, J = 8.0 Hz), 6.74 (s, 1H), 7.69 (d, 2H, J = 8.0 Hz); IR (thin film) cm−1 2943m, 2866m, 1735w, 1357m, 1165s; mass spectrum (ESI): m/e (% relative intensity) 498.2 (10) (M+Na)+, 477.2 (20) (M+2H)+, 476.2 (100) (M+H)+; HRMS (ESI) m/e calcd for C25H37NO4SSi+ (M+H)+ 476.2286, found 476.2266.
Compound (±)-65 (12 g, 42.94 mmol) was prepared in 72% yield over two steps according to the procedure reported by Hashmi.25 Rf = 0.63 [1:4 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 1.07 (d, 9H, J = 6.8 Hz), 1.09 (d, 9H, J = 6.8 Hz), 1.13–1.22 (m, 3H), 5.63 (s, 1H), 6.38 (dd, 1H, J = 2.0, 3.6 Hz), 6.52 (dd, 1H, J = 0.8, 3.6 Hz), 7.42 (dd, 1H, J = 0.8, 2.0 Hz); 13C NMR (100 MHz, CDCl3) δ 12.1, 17.7, 17.8, 58.4, 109.3, 110.8, 117.4, 143.7, 148.0; IR (neat) cm−1 2945s, 2868m, 1719w, 1463s, 1091s; mass spectrum (ESI): m/e (% relative intensity) 302.3 (100) (M+Na)+, 253 (20) (M−CN)+; HRMS (ESI) m/e calcd for C15H25NO2Si+ (M+Na)+ 302.1547, found 302.1557.
(±-66). Compound (±)-66 (3.09 g, 7.06 mmol) was prepared in 66% yield over two steps according to the procedure reported by Hashmi.25 Rf = 0.47 [1:4 EtOAc/hexanes]; white solid; mp = 41–42 °C; 1H NMR (400 MHz, CDCl3) δ 0.93 (d, 9H, J = 5.6 Hz), 0.95–1.03 (m, 12H), 2.42 (s, 3H), 3.22 (dt, 1H, J = 6.0, 12.0 Hz), 3.30 (dt, 1H, J = 6.0, 12.0 Hz), 4.63 (t, 1H, J = 6.0 Hz), 4.85 (t, 1H, J = 6.0 Hz), 6.22 (d, 1H, J = 3.2 Hz), 6.29 (dd, 1H, J = 1.2, 2.4 Hz), 7.29 (d, 2H, J = 8.0 Hz), 7.30 (brs, 1H), 7.70 (d, 2H, J = 8.0 Hz); 13C NMR (100 MHz, CDCl3) δ 12.3, 17.9, 18.0, 21.7, 48.2, 67.3, 108.0, 110.4, 127.3, 129.9, 137.0, 142.2, 143.6, 153.8; IR (neat) cm−1 3289m, 2944m, 2865m, 1738w, 1326s, 1160s; mass spectrum (ESI): m/e (% relative intensity) 461.4 (15) (M+Na+H)+, 460.4 (100) (M+Na)+; HRMS (ESI) m/e calcd for C22H35NO4SSi+ (M+Na)+ 460.1949, found 460.1934.
Amide (±)-62 (2.8 g, 5.88 mmol) was prepared in 95% yield according to the procedure reported by Hashmi.25 Rf = 0.51 [1:4 EtOAc/hexanes]; colorless oil; 1H NMR (400 MHz, CDCl3) δ 0.97 (d, 9H, J = 6.0 Hz), 1.02–1.05 (m, 3H), 1.03 (d, 9H, J = 6.0 Hz), 1.98 (t, 1H, J = 2.4 Hz), 2.41 (s, 3H), 3.40 (dd, 1H, J = 7.2, 14.4 Hz), 3.52 (ddd, 1H, J = 0.8, 6.4, 14.4 Hz), 3.62 (dd, 1H, J = 2.4, 18.4 Hz), 4.19 (ddd, 1H, J = 0.8, 2.4, 18.4 Hz), 5.08 (t, 1H, J = 6.8 Hz), 6.30 (dd, 1H, J = 0.8, 3.2 Hz), 6.32 (dd, 1H, J = 2.0, 3.2 Hz), 7.27 (d, 2H, J = 8.0 Hz), 7.37 (dd, 1H, J = 0.8, 2.0 Hz), 7.71 (d, 2H, J = 8.4 Hz); 13C NMR (100 MHz, CDCl3) δ 12.4, 17.9, 18.1, 21.7, 38.5, 51.5, 68.4, 73.6, 77.4, 108.3, 110.5, 128.0, 129.6, 136.2, 142.1, 143.6, 154.5; IR (neat) cm−1 3285m, 2928m, 2854m, 1348s, 1159s; mass spectrum (ESI): m/e (% relative intensity) 499.2 (5) (M+Na+H)+, 498.2 (30) (M+Na)+, 493.3 (100); HRMS (ESI) m/e calcd for C25H37NO4SSi+ (M+Na)+ 498.2105, found 498.2124.
Supplementary Material
Acknowledgments
Authors thank NIH [GM066055] for financial support and Dr. Victor Young [University of Minnesota] for X-ray structural analysis.
Footnotes
SUPPORTING INFORMATION AVAILABLE. 1 H NMR, 13C NMR spectra, and X-ray structural data are available free of charge via Internet at http://pubs.acs.org.
References
- 1.For excellent reviews on heteroatom-substituted oxyallyl cations, see: Harmata M. Adv Synth Catal. 2006;348:2297.Harmata M. Recent Res Devel In Organic Chem. 1997;1:523.Also see: Katritzky AR, Dennis N. Chem Rev. 1989;89:827.
- 2.For general reviews on [4 + 3] cycloadditions, see: Harmata M. Chem Commun. ASAP; 2010. Battiste MA, Pelphrey PM, Wright DL. Chem Eur J. 2006;12:3438. doi: 10.1002/chem.200501083.Antoline JE, Hsung RP. ChemTracts. 2005;18:562.Hartung IV, Hoffmann HMR. Angew Chem Int Ed. 2004;43:1934. doi: 10.1002/anie.200300622.Harmata M, Rashatasakhon P. Tetrahedron. 2003;59:2371.Harmata M. Acc Chem Res. 2001;34:595. doi: 10.1021/ar000064e.Also see: Davies HML. In: Advances in Cycloaddition. Harmata M, editor. Vol. 5. JAI Press; 1998. p. 119.West FG. In: Advances in Cycloaddition. Lautens M, editor. Vol. 4. JAI Press; Greenwich: 1997. p. 1.Rigby JH, Pigge FC. Org React. 1997;51:351.Harmata M. Tetrahedron. 1997;53:6235.
- 3.For leading examples of oxygen-substituted oxyallyl cations, see: Sáez JA, Arnó M, Domingo LR. Tetrahedron. 2005;61:7538.Harmata M, Kahraman M, Adenu G, Barnes CL. Heterocycles. 2004;62:583.Sáez JA, Arnó M, Domingo LR. Org Lett. 2003;5:4117. doi: 10.1021/ol035652h.Funk RL, Aungst RA. Org Lett. 2001;3:3553. doi: 10.1021/ol016668f.Harmata M, Sharma U. Org Lett. 2000;2:2703. doi: 10.1021/ol006281x.Lee K, Cha JK. Org Lett. 1999;1:523. doi: 10.1021/ol990709e.Masuya K, Domon K, Tanino K, Kuwajima I. J Am Chem Soc. 1998;120:1724.Harmata M, Elomari S, Barnes CJ. J Am Chem Soc. 1996;118:2860.and references cited within.
- 4.For examples of sulfur-substituted oxyallyl cations, see: Hardinger SA, Bayne C, Kantorowski E, McClellan LL, Nuesse MA. J Org Chem. 1995;60:1104.Harmata M, Gamlath CB. J Org Chem. 1988;53:6156.
- 5.For examples of halogen-substituted oxyallyl cations, see: Harmata M, Wacharasindhu S. Org Lett. 2005;7:2563. doi: 10.1021/ol050598l.Lee K, Cha JK. Org Lett. 1999;1:523. doi: 10.1021/ol990709e.
- 6.For oxidopyridinium ions: Peece KM, Gin DY. Org Lett. 2005;7:3323. doi: 10.1021/ol051184v.Sung MJ, Lee HI, Chong Y, Cha JK. Org Lett. 1999;1:2017. doi: 10.1021/ol9911932.Dennis N, Ibrahim B, Katritzky AR. J Chem Soc, Perkin Trans. 1976;1:2307.For phthalamide-substituted systems: Walters MA, Arcand HR. J Org Chem. 1996;61:1478.and references therein.
- 7.For leading examples of nitrogen-stabilized oxyallyl cations in [4 + 3] cycloadditions, see: Walters MA, Arcand HR, Lawrie DJ. Tetrahedron Lett. 1995;36:23.Walters MA, Arcand HR. J Org Chem. 1996;61:1478.Arcand HR. PhD Dissertation Thesis. Dartmouth College; Hanover, NH: 1996. Investigation of the reactivity of nitrogen-substituted oxyallyl cations (asymmetry, stereoselective)MaGee DI, Godineau E, Thornton PD, Walters MA, Sponholtz DJ. Eur J Org Chem. 2006:3667.Myers AG, Barbay JK. Org Lett. 2001;3:425. doi: 10.1021/ol006931x.
- 8.Xiong H, Hsung RP, Berry CR, Rameshkumar C. J Am Chem Soc. 2001;123:7174. doi: 10.1021/ja0108638. [DOI] [PubMed] [Google Scholar]
- 9.For leading reviews on allenamides, see: Hsung RP, Wei LL, Xiong H. Acc Chem Res. 2003;36:773. doi: 10.1021/ar030029i.Standen PE, Kimber MC. Curr Opin Drug Discov & Devel. 2010;13:645.Also see: Tracey MR, Hsung RP, Antoline J, Kurtz KCM, Shen L, Slafer BW, Zhang Y. In: Science of Synthesis, Houben-Weyl Methods of Molecular Transformations. Weinreb Steve M., editor. Vol. 21.4. Georg Thieme Verlag KG; 2005. Deagostino A, Prandi C, Tabasso S, Venturello P. Molecules. 2010;15:2667. doi: 10.3390/molecules15042667.
- 10.(a) Lohse AG, Krenske E, Houk KN, Hsung RP. Org Lett. 2010;12:5506. doi: 10.1021/ol1023745. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Antoline JE, Hsung RP. Synlett. 2008:739. [Google Scholar]
- 11.For our work on nitrogen-stabilized oxyallyl cations in intermolecular [4 + 3] cycloadditions with furans and pyrroles, see: Krenske EK, Houk KN, Lohse AG, Antoline JE, Hsung RP. Chem Science. 2010;1:387. doi: 10.1039/C0SC00280A.Antoline JE, Hsung RP, Huang J, Song Z, Li G. Org Lett. 2007;9:1275. doi: 10.1021/ol070103n.Rameshkumar C, Xiong H, Tracey MR, Berry CR, Yao LJ, Hsung RP. J Org Chem. 2002;67:1339. doi: 10.1021/jo011048d.For our work in intramolecular [4 + 3] cycloadditions, see: Rameshkumar C, Hsung RP. Angew Chem Int Ed. 2004;43:615. doi: 10.1002/anie.200352632.Xiong H, Huang J, Ghosh S, Hsung RP. J Am Chem Soc. 2003;125:12694. doi: 10.1021/ja030416n.For a recent account on intramolecular [4 + 3] cycloadditions of allenyl dienes employing PtCl2 as a catalyst, see: Trillo B, López F, Gulías M, Castedo L, Mascareñas JL. Angew Chem Int Ed. 2007;47:951. doi: 10.1002/anie.200704566.
- 12.For recent diastereoselective [4 + 3] cycloadditions, see: Chung WK, Lam SK, Lo B, Liu LL, Wong WT, Chiu P. J Am Chem Soc. 2009;131:4556. doi: 10.1021/ja807566t.Craft DT, Gung BW. Tetrahedron Lett. 2008;49:5931. doi: 10.1016/j.tetlet.2008.07.155.Davies HML, Dai X. J Am Chem Soc. 2004;126:2693.Prié G, Prévost N, Twin H, Fernandes SA, Hayes JF, Shipman M. Angew Chem Int Ed. 2004;43:6517. doi: 10.1002/anie.200461084.Grainger RS, Owoare RB, Tisselli P, Steed JW. J Org Chem. 2003;68:7899. doi: 10.1021/jo034356f.Montanã AM, Grima PM. Tetrahedron. 2002;58:4769.Beck H, Stark CBW, Hoffman HMR. Org Lett. 2000;2:883. doi: 10.1021/ol991386p.and reference 11 cited within Harmata M, Rashatasakhon P. Synlett. 2000:1419.Cho SY, Lee JC, Cha JKJ. Org Chem. 1999;64:3394. doi: 10.1021/jo990262n.Harmata M, Jones DE, Kahraman M, Sharma U, Barnes CL. Tetrahedron Lett. 1999;40:1831.Kende AS, Huang H. Tetrahedron Lett. 1997;38:3353.Harmata M, Jones DE. J Org Chem. 1997;62:4885.
- 13.Huang J, Hsung RP. J Am Chem Soc. 2005;127:50. doi: 10.1021/ja044760b. [DOI] [PubMed] [Google Scholar]
- 14.For an account on asymmetric [4 + 3] cycloadditions that predated ours, see: Harmata M, Ghosh SK, Hong X, Wacharasindu S, Kirchhoefer P. J Am Chem Soc. 2003;125:2058. doi: 10.1021/ja029058z.For an enantioselective formal [4 + 3] cycloaddition, see: Dai X, Davies HML. Adv Synth Catal. 2006;348:2449.
- 15.For some examples, see: Grainger RS, Owoare RB. Org Lett. 2004;6:2961. doi: 10.1021/ol048911r.Vidal PM, Proemmel S, Beil W, Wartchow R, Hoffmann HMR. Org Lett. 2004;6:4155. doi: 10.1021/ol048603t.Gillingham DG, Kataoka O, Garber SB, Hoveyda AH. J Am Chem Soc. 2004;126:12288. doi: 10.1021/ja0458672.Hoegermeier J, Reissig HU, Bruedgam I, Hartl H. Adv Synth Catal. 2004;346:1868.Lautens M, Fagnou K, Yang D. J Am Chem Soc. 2003;125:14884. doi: 10.1021/ja034845x.
- 16.Due to rotamers, many of the signals were not well resolved and/or line-broadened and we were unable to obtain 13C NMR data.
- 17.Lohse AG, Hsung RP. Org Lett. 2009;11:3430. doi: 10.1021/ol901283m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The base-promoted isomerization of allenamides with α- or γ-substituents require higher temperatures and longer reactions times.
- 19.For a review on Hoffmann’s notation, see: Hoffmann HMR. Angew Chem. 1973;85:877.Angew Chem Int Ed Engl. 1973;12:819.see also: Hoffmann HMR, Joy DR. J Chem Soc B. 1968:1182.
- 20.DFT calculations were performed at the B3LYP/6-31G(d) level using Spartan ‘10 Release 1.0.1v4, Wavefunction Inc., Irvine, CA, 92612.
- 21.Webster R, Lautens M. Org Lett. 2009;11:4688. doi: 10.1021/ol9019869. [DOI] [PubMed] [Google Scholar]
- 22.(a) Sobhana Babu B, Balasubramanian KK. J Org Chem. 2000;65:4198. doi: 10.1021/jo000074t. [DOI] [PubMed] [Google Scholar]; (b) Porzelle A, Gordon VA, Williams CM. Synlett. 2007:1619. [Google Scholar]
- 23.Martinelli MJ, Nayyar NK, Moher ED, Dhokte UP, Pawlak JM, Vaidyanathan R. Org Lett. 1999;1:447. [Google Scholar]
- 24.Due to the instability of the tosylate, it was carried on without further purification.
- 25.Hashmi ASK, Ata F, Haufe P, Rominger F. Tetrahedron. 2009;65:1919. [Google Scholar]
- 26.For reviews on enzyme catalysis with furfural, see: Gregory RJH. Chem Rev. 1999;99:3649. doi: 10.1021/cr9902906.Wajant H, Effenberger F. Biol Chem. 1996;377:611.
- 27.For leading references, see: Bushey ML, Haukaas MH, O’Doherty GA. J Org Chem. 1999;64:2984. doi: 10.1021/jo990095r.Harris JM, Keranen MD, O’Doherty GA. J Org Chem. 1999;64:2982. doi: 10.1021/jo990410+.Also see: Taniguchi T, Nakamura K, Ogasawara K. Synlett. 1996;971Taniguchi T, Ohnishi H, Ogasawara K. Chem Commun. 1996:1477.
- 28.Shen L, Hsung RP, Zhang Y, Antoline JE, Zhang X. Org Lett. 2005;7:3081. doi: 10.1021/ol051094q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.