

Abstract
Receptor-interacting protein 140 (RIP140) is abundantly expressed in mature adipocyte and modulates gene expression involved in lipid and glucose metabolism. Protein kinase C epsilon and protein arginine methyltransferase 1 can sequentially stimulate RIP140 phosphorylation and then methylation, thereby promoting its export to the cytoplasm. Here we report a lipid signal triggering cytoplasmic accumulation of RIP140, and a new functional role for cytoplasmic RIP140 in adipocyte to regulate lipolysis. Increased lipid content, particularly an elevation in diacylglycerol levels, promotes RIP140 cytoplasmic accumulation and increased association with lipid droplets (LDs) by its direct interaction with perilipin. By interacting with RIP140, perilipin more efficiently recruits hormone-sensitive lipase (HSL) to LDs and enhances adipose triglyceride lipase (ATGL) forming complex with CGI-58, an activator of ATGL. Consequentially, HSL can more readily access its substrates, and ATGL is activated, ultimately enhancing lipolysis. In adipocytes, blocking cytoplasmic RIP140 accumulation reduces basal and isoproterenol-stimulated lipolysis and the pro-inflammatory potential of their conditioned media (i.e. activating NF-κB and inflammatory genes in macrophages). These results show that in adipocytes with high lipid contents, RIP140 increasingly accumulates in the cytoplasm and enhances triglyceride catabolism by directly interacting with perilipin. The study suggests that reducing nuclear export of RIP140 might be a useful means of controlling adipocyte lipolysis.
Keywords: adipocyte, post-translational modification, diacylglyceride, lipid droplet, lipase, lipid, RIP140
1. Introduction
Adipocytes store energy as triglyceride (TG) in lipid droplets (LDs) under a nutrient-excess condition, and triglyceride can be used as the energy source through lipolysis in a nutrient-deprived state [1]. In healthy individuals, adipocytes alter their lipid storage, lipolysis, and glucose uptake according to the nutritional status and hormonal fluctuations. Defects in homeostasis of the adipose tissue (e.g., glucose uptake, proper lipid storage, adiponectin secretion) can contribute to the initiation and progression of metabolic disorders, including type 2 diabetes mellitus (T2DM) [2–4]. Increased lipolysis following a high fat diet (HFD) can also contribute to T2DM as a result of lipotoxicity, which can manifest in the liver, muscle and heart and cause insulin resistance and cardiomyopathy [2, 5–8]. Free fatty acids (FFA) released by increased lipolysis can also enhance low-grade chronic inflammation by activating adipocyte tissue macrophages (ATMs), which further suppresses insulin sensitivity and adipocyte function [6, 9]. However, the exact mechanism contributing to the dysregulation of lipolysis remains elusive [2, 10].
During fasting, or in time of energy demand, TG is hydrolyzed into fatty acids and glycerol to provide a source of energy [2]. Lipolysis is a tightly regulated process, modulated by catecholamine, insulin and natriuretic peptides [1, 11]. Perilipin, a structural protein associated with LDs and involved in their formation, is the major regulator of lipolysis and can modulate the basal and stimulated lipolytic rates. In resting adipocytes, perilipin reduces lipolysis and increases lipid storage, partly by sequestering comparative gene identification-58 (CGI-58), an activator of adipose triglyceride lipase (ATGL). Normally, the cytoplasmic hormone-sensitive lipase (HSL) cannot access its substrates within the LDs, but is catecholamine-stimulated adipocytes, activated protein kinase A (PKA) phosphorylates both perilipin and HSL. Phosphorylation of perilipin frees CGI-58 to stimulate ATGL activity, whereas phosphorylation of HSL increases its association with the phosphorylated perilipin on LDs, thereby enhancing its access to substrate [12, 13]. Both mechanisms are promoted by perilipin phosphorylation, but it is less clear if the actions of perilipin can be modulated by other mechanisms.
Receptor-interacting protein 140 (RIP140) is well known as a co-regulator for numerous transcription factors and nuclear receptors. It is abundantly expressed in various tissues including ovary, uterus, and testis, as well as in metabolic tissues/organs such as adipose tissue, liver and muscle [14, 15]. Studies of RIP140 knockout mice demonstrated that RIP140 plays roles in numerous biological processes such as ovulation and metabolism [14, 16, 17]. In the nucleus, it recruits additional cofactors such as histone deacetylases (HDACs) and C-terminal binding protein (CtBP) for transcriptional regulation [15, 18]. Depleting RIP140 from adipocytes leads to decreased TG accumulation and a higher rate of fatty acid oxidation [19, 20]. Conversely, RIP140 expression is elevated during adipogenesis. Following a series of post-translational modifications (PTMs), RIP140 is increasingly exported to the cytoplasm [15, 21, 22]. In animals, HFD can induce cytoplasmic accumulation of RIP140 in adipocytes, but the signal triggering cytoplasmic accumulation of RIP140 is unclear [23]. Mechanistically, the export of RIP140 to the cytoplasm is stimulated by nuclear protein kinase C epsilon (PKCε)-elicited serine phosphorylation, followed by protein arginine methyltransferase 1 (PRMT1)-stimulated arginine methylation of RIP140 [22]. Predictably, cytoplasmic RIP140 performs functions different from those involved in gene regulation [19, 20, 22]. For example, cytoplasmic RIP140 can negatively regulate glucose transporter type 4 (GLUT4) trafficking by interacting with the 160-KDa Akt substrate (AS160), thereby reducing glucose uptake [23]. Questions remain to be answered concern the possible roles of cytoplasmic RIP140 in lipid-loaded adipocytes, especially with regards to their lipid metabolism, and the identity of specific mediators that might transmit lipid signals to promote aberrant accumulation of RIP140 in the cytoplasm of fully differentiated, lipid-loaded adipocytes after a HFD feeding.
This study shows that the adipocyte fat content can trigger cytoplasmic accumulation of RIP140, and demonstrates a new functional role for cytoplasmic RIP140 in adipocyte; specifically, it positively modulates lipolysis through its direct interaction with perilipin. The physiological relevance of this pathway is validated by examining the pro-inflammatory potential of conditioned media collected from adipocyte cultures with altered cytoplasmic RIP140 accumulation. Our findings provide new insights into the roles of cytoplasmic RIP140 in HFD-induced adipocyte dysfunction and support the notion that targeting cytoplasmic RIP140 could be a therapeutic strategy in managing T2DM or other metabolic syndromes.
2. Materials and methods
2.1. Cell Culture and Treatment
3T3-L1 cells were maintained and differentiated as described [24]. For DAGK inhibitor, mature 3T3-L1 adipocytes were treated with R59022 for 24 h and cell lysates were collected. For lipolysis, mature 3T3-L1 adipocytes were starved in serum-free medium for 3 h and then incubated with serum-free medium with or without 10 μM isoproterenol for another 2 h.
2.2. Reagents and Transfection
Antibodies for actin, lamin, CGI-58 and PKCε were from Santa Cruz Biotechnology. Anti-flag, anti-alpha tubulin, anti-calnexin and anti-perilipin A were from Sigma Aldrich. Anti-Oxophos complex IV antibody was from Upstate. Anti-HSL, anti-ATGL and anti-Phospho-PKA substrate antibodies were from Cell signaling. Anti-perilipin A, anti-giantin and anti-calreticulin antibodies were from Abcam. Anti-RIP140 (ab42126) was from Abcam and its specificities in immunofluoresence and immunoblotting were determined in previous report [23]. Anti-cyclophilin A antibody was from calbiochem. siRNAs were from Qiagen. Insulin was from Sigma Aldrich. Isoproterenol was from Cayman. BODIPY 493/503 was from Molecular Probes (Invitrogen). siRNA transfect ion was conducted by DeliverX Plus siRNA transfection kit (Panomics) as manufacturer’s instruction. Plasmid of full length and different fragments of Flag-RIP140 constructs were as described [23]. Plasmid of full length and different fragment of perilipin were cloned from cDNA of 3T3-L1 adipocyte and then cloned into pCMV-PL vector-containing 3xFlag tags.
2.3. Western Blotting, Immunoprecipitation and Immunofluoresence
Western blotting was conducted as described previously [24]. For immunoprecipitation, 500 μg whole cell lysates were incubated with 5 μg indicated antibodies for 2~3 h in 500 μl Co-IP buffer (50mM Tris-HCl pH 8.0, 10% glycerol, 100mM NaCl, 1mM EDTA and 0.1% NP-40) and then incubated with protein G beads (Upstate) overnight. After centrifugation, beads were washed using a Co-IP buffer three times, and the precipitates were subjected into SDS-PAGE for western blotting. Immunofluorescence assay was conducted as a previous report [23]. For co-staining with lipid droplet, BODIPY 493/503 and fluorescence-conjugated secondary antibodies were co-incubated with cells for 1~3 h. Images were acquired by Olympus FluoView1000 IX2 inverted confocal microscope. Colocalization analysis was performed by Manders Coefficients in Image J as previous described [25].
2.4. Cell fractionation
Nuclear and cytoplasmic fractionation, cells were collected and fractions were collected as in a previous report [23]. Forty 10-cm plates with mature 3T3-L1 adipocytes were collected for isolating organelles as reported (31). 80 μg proteins from indicated organelles were subjected into western blotting. Organelles isolation was performed as previous report [26].
2.5. TG Content Measurement and Lipolysis Assay
TG content was assayed as described previously [24]. For lipolysis assay, cells were starved in serum-free medium and then stimulated with or without isoproterenol for 2 h. Media were collected and glycerol levels within the media were determined by adipolysis assay kit (Cayman) as manufacture’s instruction. The glycerol levels were normalized to the protein amounts of the cell lysates.
2.6. In Vitro GST Pull-down Assay
GST-RIP140 and GST-perilipin were produced by BL-21 strain. Expression was induced by IPTG at 20°C overnight. Bacteria pellets were lysed in PBS. For GST-perilipin, lysate was collected and incubated with GST beads to purify GST-perilipin. For GST-RIP140, after centrifugation, insoluble pellet was lysed by Inclusion Body Solubilization Reagent (Thermo Scientific) and lysate was dialyzed as instruction. Dialyzed lysate was incubated with GST beads to purify refolded GST-RIP140. For pull-down assay, it was conducted as a previous report with modifications [23]. In GST-RIP140 set, the washing condition was 50mM Tris-HCl pH 8.0, 10% glycerol, 100mM NaCl, 1mM EDTA and 2% NP-40. Peptide fragments were synthesized in vitro by TNT assay kit as previous report [23].
2.7. Mice
Male mice (C56BL/6J) (Jackson Laboratory) were housed in a temperature-controlled environment with 12-hr light/dark photocycle and fed with a normal diet (5% fat) (#2018, Harlan Teklad) or a high-fat diet (HFD) (60% fat) (#F3282, Bio-Serv). Animal experiments were conducted in procedures approved by University of Minnesota Institutional Animal Care and Use Committee.
2.8. Tissue Collection and Immunohistochemical Staining
Epididymal adipose tissues were fixed, embedded and sectioned by the Histology & Microscopy Core Facility (University of Minnesota). Immunohistochemical staining of RIP140 and perilipin was performed as previously described [22]. The nuclei of sections were stained with DAPI then mounted (Vector Laboratories) for microscopic analysis (Olympus FluoView1000 IX2 inverted confocal microscope).
3. Results
3.1. Fat content modulates sub-cellular distribution of RIP140 in adipocytes
Previously, we reported that, in adipocyte, RIP140 could be exported to the cytoplasm by PKCε- and PRMT1-mediated post-translational modifications [15, 21, 22]. In mature adipocyte, nuclear export of RIP140 is profound, and a HFD feeding dramatically increases cytoplasmic RIP140 levels in the animals’ adipose tissues [22, 23]. These observations suggest that fat content within adipocytes may be involved in cytoplasmic accumulation of RIP140. To investigate the relationship between fat content and RIP140 sub-cellular localization, we monitored RIP140 localization and the size of LDs in 3T3-L1 adipocytes. Those differentiated 3T3-L1 adipocytes containing larger LDs exhibited stronger cytoplasmic RIP140 staining (Fig. 1A). Approximately 80% of adipocytes containing large LDs displayed cytoplasmic RIP140, whereas only ~15 % of the adipocytes containing small LDs had RIP140 in the cytoplasm, suggesting that an increase in fat content stimulates cytoplasmic accumulation of RIP140. It has been shown that the DAG level is elevated when adipocytes extensively accumulate lipid [10, 27]; further, DAG is an important activator for PKCε activation which is involved in nuclear export of RIP140 [22]. We then hypothesized that, in adipocyte, an increase in DAG levels may promote cytoplasmic accumulation of RIP140. To investigate this possibility, we treated fully differentiated 3T3-L1 adipocytes with a non-selective DAG kinase inhibitor (R59022) to block DAG metabolism (thus increasing DAG levels). This treatment reduced nuclear RIP140 levels and increased cytoplasmic RIP140 levels (Fig. 1B). DAG kinase alpha (DGKα) is the most common form in the DAG kinase family and is mainly expressed in the cytoplasm. In contrast, DAG zeta (DGKζ) is a nuclear form of the DAG kinase family and is involved in ET-1-induced cardiomyote hypertrophy [28, 29]. To confirm the functional role for DAG kinases in the accumulation of cytoplasmic RIP140, we used siRNAs to knock down these two DAG kinases (Fig. 1C). The data show that knocking down either DGKα or DGKζ substantially increased the accumulation cytoplasmic RIP140 but reduced nuclear RIP140 level. Together, these results suggest that changes in DAG levels can modulate RIP140’s sub-cellular localization. Therefore, in adipocytes, an increase in fat content, or more specifically an increase in intracellular DAG levels, can promote cytoplasmic accumulation of RIP140.
Fig. 1.
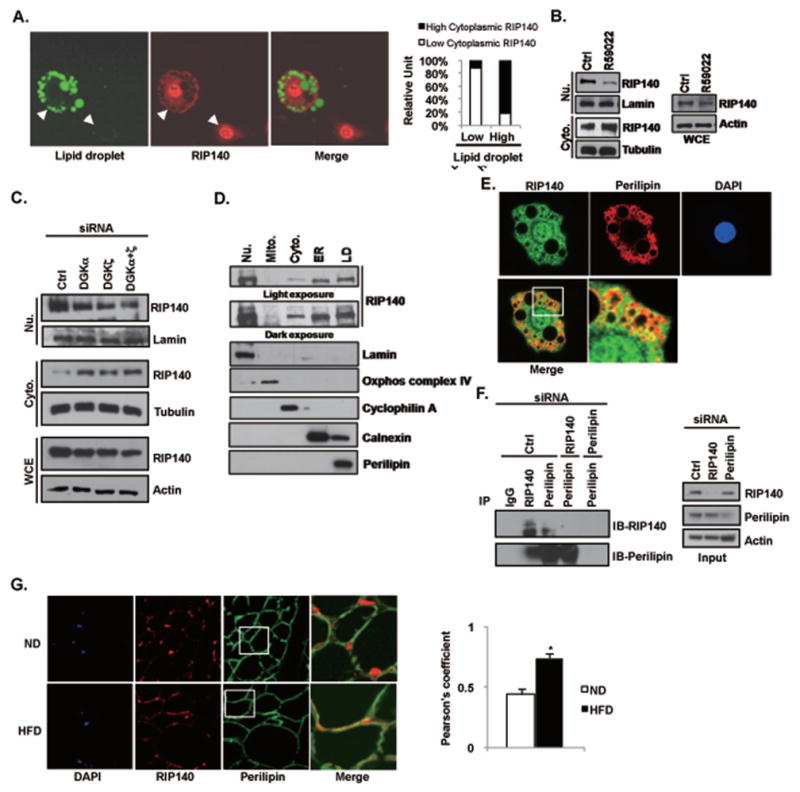
Fat content in adipocytes affects RIP140 localization. (A) Immunofluorescence analysis of RIP140 distribution in mature 3T3-L1 adipocytes. Left: Green: lipid droplet staining; red: RIP140 staining. Right: Quantitative analysis of RIP140 distribution and lipid droplet size. Data are the percentage of cell in each category. (B) The DAG kinase inhibitor R59022 enhances cytoplasmic RIP140 distribution but reduces nuclear RIP140 in mature 3T3-L1 adipocytes (left), whereas total RIP140 levels remain unchanged (right). (C) Knocking down DGKa and DGKz increases the cytoplasmic RIP140 level, but decreases the nuclear RIP140 level in mature 3T3-L1 adipocytes. (D) Immunoblot analysis of the sub-cellular distribution of RIP140. Fractions were identified by probing with the indicated organelle-specific markers. Nu: nuclei; Mito: mitochondria; Cyto: cytosol; ER: endoplasmic reticulum; LD: lipid droplets. (E) RIP140 colocalizes with perilipin in adipocytes. The yellow indicates complete colocalization of RIP140 with perilipin. (F) RIP140 and perilipin form immunocomplexes in vivo. Knocking down either RIP140 or perilipin reduces immunocomplex formation. (G) Short-term HFD feeding promotes the colocalization of RIP140 with perilipin in mouse epididymal adipose tissue. ND: normal diet; HFD: high-fat diet. Right: the quantification of co-localization. Data show the averages of Pearson’s coefficient for the indicated group. *: p value < 0.05 compared to the ND group.
3.2. RIP140 localizes to lipid droplet surfaces and interacts directly with perilipin
Although it was clear that RIP140 could be exported into the cytoplasm in adipocytes, the specific cytoplasmic localization of RIP140 was unknown. We then performed sub-cellular fractionation, by ultracentrifugation, of mature 3T3-L1 adipocytes. RIP140 was then detected by immunoblotting of these sub-cellular fractions. The result shows that RIP140 is detected in cytosol, endoplasmic reticulum, and LD (Fig. 1D), but not in mitochondria, or Golgi (Fig. S1). Interestingly, the LD fraction had particularly abundant RIP140. We then examined if RIP140 could associate with perilipin, a LD-associated protein and an important regulator of the formation and maintenance of LDs. Intriguingly, RIP140 can be colocalized with perilipin around LDs and formed immuno-complexes with perilipin in vivo, and knocking down either component reduced the formation of these complexes (Fig. 1E and F). The quality and specificity of RIP140 antibody in these experiments have been described previously [23]. Note that in the cytoplasm, RIP140 is wide distributed; therefore it is only partially co-localized with perilipin that is much more enriched in LDs. We have shown that RIP140 accumulated in the cytoplasm of adipose tissue in animals after a HFD feeding [23], we then evaluated if RIP140 could colocalize with perilipin in the primary adipocytes of animals fed a HFD. Indeed, RIP140 also partially colocalized with perilipin in epididymal adipose tissue from mice fed with a HFD (Fig. 1G, the lower boxed area showing prominent yellow signal indicative of co-localization), whereas no such colocalization was seen in adipocytes from mice fed a normal diet (Fig. 1G, the upper boxed area showing little yellow signal). The degree of colocalization in ND versus HFD animals was examined and indicated with Pearson’s coefficient (Fig. 1G, right panel). All together, the results show that cytoplasmic RIP140 can associate with perilipin around LDs in adipocyte, which can be enhanced by a HFD feeding in animals.
In vitro reciprocal protein interaction assays further confirmed that RIP140 interacted directly with perilipin (Fig. 2A). We then examined the perilipin-interacting domain of RIP140 by GST pull-down assay, and found that the perilipin-interacting domain of RIP140 was located in the amino terminus (amino acids 1–350) of RIP140, which contains the repressive domain 1 (RD1) (Fig. 2B). As shown in the reciprocal GST pull-down assay, the amino terminus (1–160) of perilipin could not, but all the remaining fragments of perilipin could, interact with GST-RIP140 (Fig. 2C). Interestingly, the amino terminal portion (amino acids 1–405) that extends to the hydrophobic regions of perilipin interacted weakly with RIP140 as compared to fragments of 1–250 residues and 251-the end. It is possible that these hydrophobic regions could form structural barriers, reducing perilipin’s interaction with RIP140 in the in vitro assay [30]. This result suggests that the central portion (amino acids 161–300) of perilipin is the major RIP140-interacting domain and the hydrophobic region of perilipin may be involved in the regulation of this interaction. Importantly, although the amino terminus of perilipin (amino acids 17–121) is the most highly conserved domain in this protein family [31], this region failed to interact with RIP140, suggesting that interaction between perilipin and RIP140 is highly specific, but not a general phenomenon common to the perilipin protein family.
Fig. 2.
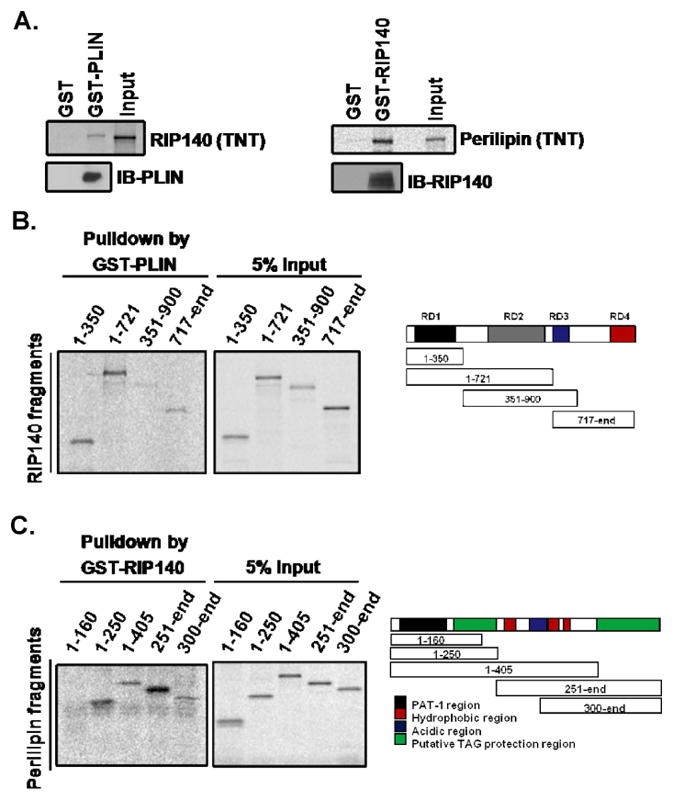
Interacting domains of RIP140 and perilipin, as determined by glutathione S-transferase (GST) pull-down assays. (A) RIP140 directly interacts with GST-perilipin (left) and perilipin (PLIN) directly interacts with GST-RIP140 (right). (B) Interacting domains of RIP140. Right: Schematic of the in vitro-produced RIP140 peptide fragments. RD: repressive domain. (C) Interacting domains of perilipin. Right: Schematic of the in vitro-produced perilipin peptide fragments.
3.3. Silencing RIP140 expression or inhibiting its export reduces lipolysis
Perilipin contributes to LD formation and controls lipolytic rates [1, 13, 31]. Nuclear RIP140 can promote lipid accumulation in adipocytes through its gene regulatory activities, thus nuclear export of RIP140 would reduce the lipid content in adipocytes [16, 21, 22]. However, it was elusive whether other mechanisms could also be contributed by cytoplasmic RIP140 to regulate lipid metabolism in adipocyte. The association of RIP140 with perilipin around LDs suggests that RIP140 may modulate lipolysis by regulating the activity of perilipin, or its associated lipases. To test this hypothesis, we manipulated adipocyte’s RIP140 level or its nuclear export (stimulated by PKCε) and monitored lipolysis, indicated by the reduction in TG content under isoproterenol treatment. This assay monitors changes in the percentage of TG content under a 2h isoproterenol treatment, reflecting, primarily, the effects via lipolysis. We found that reducing cytoplasmic RIP140 levels, by direct silencing or blocking its nuclear export (through silencing PKCε), significantly reduced isoproterenol-stimulated lipolysis (Fig. 3A). But the basal TG content (reflecting, primarily, lipid synthesis) was reduced only in RIP140-silenced, but not in PKCε-silenced (only blocking RIP140 export) adipocytes (Fig. S2). The nuclear RIP140 is known to regulate genes that modulate lipogenic capacity in adipocytes [20, 22, 32], which is blocked only when its expression, rather than its export, is reduced. Furthermore, direct assessment of lipolysis by measuring the amount of glycerol released to the culture medium showed that silencing RIP140 or PKCε significantly reduced both the basal and stimulated glycerol release relative to the control silencing (Fig. 3B). Interestingly, silencing RIP140 or PKCε did not affect the expression of key lipolytic enzymes/regulators (HSL, ATGL, cofactor CGI-58, and perilipin) (Fig. 3C). Of note, isoproterenol effectively induced glycerol release in either RIP140- or PKCε-silencing adipocytes, although PKCε-silencing seemed to cause a more profound effect. However, basal glycerol release was significantly reduced by silencing either RIP140 or PKCε. These results suggest that cytoplasmic RIP140 might regulate the basal lipolytic machinery but not isoproterenol-stimulated signaling pathways. Altogether, these results indicate that cytoplasmic RIP140 positively modulates the lipolytic machinery, but does not seem to affect the expression of lipolytic proteins.
Fig. 3.
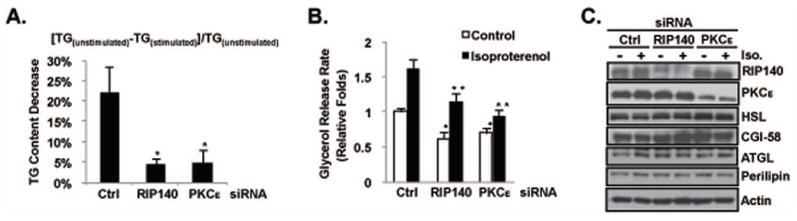
Cytoplasmic RIP140 facilitates lipolysis. (A) Silencing RIP140 or PKCε reduces the isoproterenol-triggered decrease of triglyceride (TG) content in adipocytes. Bars indicate the means ± SD of the ratios of reduced TG : total TG content from untreated cells ( as calculated by the equation above the figure). *: p value < 0.05 compared to the control (Ctrl) group. (B) Silencing RIP140 or PKCε decreases basal and isoproterenol-stimulated glycerol release from cultured adipocyte. Bars indicate the means ± SD of the relative amounts of released glycerol. *: p value < 0.05 compared to the control siRNA in the control group. **: p value < 0.05 compared to the control siRNA in the isoproterenol group. (C) Targeting RIP140 or PKCε did not change the expression profile of lipolysis-related proteins. Expression levels of indicated proteins in whole cell lystaes were determined by immunoblotting.
3.4. Effects of cytoplasmic RIP140 on the formation of lipolytic enzyme complexes
Lipolysis is controlled, primarily, by the formation of perilipin-HSL and CGI-58-ATGL enzyme complexes [1, 31]. In resting adipocytes, inactive HSL is dispersed throughout the cytoplasm whereas ATGL is localized on LDs and remains inactive because it is segregated from its perilipin-associated activator CGI-58. In catecholamine-stimulated adipocytes, perilipin on LDs recruits HSL to access its substrates, and CGI-58 dissociates from perilipin to activate ATGL [1, 12, 13]. We first asked if cytoplasmic RIP140 can affect the interaction of perilipin with HSL. It appears that silencing RIP140 or PKCε markedly reduced the formation of HSL-perilipin complexes in co-immunoprecipitation assay (Fig. 4A). Importantly, silencing RIP140, or PKCε, had no effect on perilipin phosphorylation (Fig. 4B) or HSL phosphorylation (Fig. 4C). These results further support that cytoplasmic RIP140 does not alter PKA signaling which is in agreement with the observation shown in Fig. 3B that cytoplasmic RIP140 does not affect isoproterenol-stimulated signaling pathways. Instead, cytoplasmic RIP140 appears to promote lipolysis by facilitating the formation of perilipin-HSL complexes. Finally, silencing RIP140 or PKCε enhanced complex formation between CGI-58 and perilipin (Fig. 4D), thus reducing ATGL activation. Taken together, the data show that cytoplasmic RIP140 elevates lipolysis by promoting the formation of HSL/perilipin and ATGL-CGI-58 complexes, which increases the accessibility of HSL with its substrate, and enhance ATGL activity, respectively.
Fig. 4.
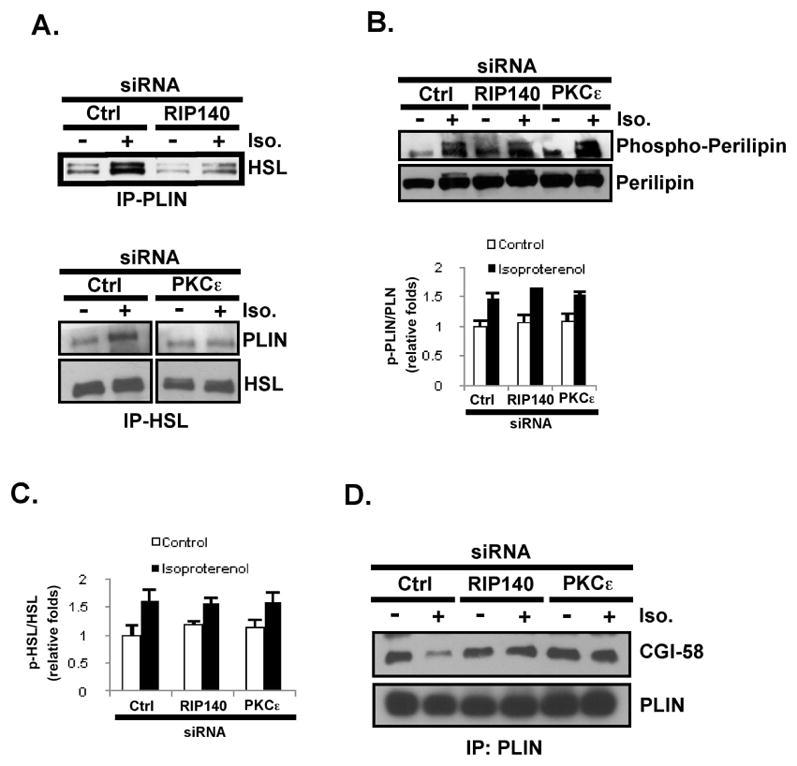
Silencing cytoplasmic RIP140 reduces isoproterenol-stimulated HSL-perilipin colocalization and complex formation, as well as CGI-58 association with ATGL. (A) Isoproterenol-stimulated HSL-perilipin complex formation, in vivo, is reduced by silencing RIP140 (left) or PKCε (right). (B) Upper: Basal and stimulated phosphorylation status of perilipin under RIP140 or PKCε silencing. Lower: Quantified result is presented as mean ± SD of phosphor-perilipin/perilipin ratios. (C) Neither RIP140- or PKCε-silencing can alter the basal or stimulated phosphorylation status of HSL. Quantified result is presented as mean ± SD of phosphor-HSL/HSL ratios. (D) silencing RIP140 or PKCε blocks the isoproterenol-stimulated dissociation of CGI-58 from perilipin.
3.5. Conditioned medium from cytoplasmic RIP140-deficient adipocyte cultures is less effective in eliciting a pro-inflammatory response in macrophages
Free fatty acids released by adipocytes can be proinflammatory for macrophages, i.e., they trigger M1 activation of macrophages via a TLR4- NF-κB-dependent mechanism, thus contributing to diabetic progression [9]. Based on the role of cytoplasmic RIP140 in regulating lipolysis, we then examined the effect of cytoplasmic RIP140 in modulating the proinflammatory potential of adipocyte-conditioned media, assessed by M1 activation of macrophages. As predicted, the conditioned media from isoproterenol-treated control cultures (increased lipolysis renders free fatty acid release) more effectively induced NF-κB activity in macrophage cultures than did the medium from the unstimulated controls (Fig. 5A). In contrast, the conditioned media from RIP140- or PKCε-silenced adipocyte cultures (blocking cytoplasmic RIP140 to reduce lipolysis, resulting in less free fatty acid release) exerted much weaker effects on NF-κB activation in macrophages. Furthermore, the conditioned media from RIP140- or PKCε-silenced adipocyte cultures were less effective than the controls in elevating the expression of endogenous proinflammatory genes IL-1β and IL-6 in macrophages (Fig. 5B). Together, these data show that the conditioned media of adipocyte cultures with reduced cytoplasmic RIP140 accumulation is less proinflammatory.
Fig. 5.

Reducing cytoplasmic RIP140 in adipocytes suppresses the inflammatory properties of their conditioned media. (A) Silencing RIP140 or PKCε in adipocytes reduces the ability of the conditioned media to activate NF-κB activity in cultured Raw264.7 macrophages. *: p value < 0.05 compared to the control siRNA in the control group. **: p value < 0.05 compared to the control siRNA in the isoproterenol group. (B) Silencing RIP140 or PKCε in 3T3-L1 adipocytes reduces the ability of their conditioned media to activate expression of endogenous proinflammatory genes in Raw264.7. *: p value < 0.05 compared to the control group of IL-1 beta. **: p value < 0.05 compared to the control group of IL-6. (C) Schematic presentation of the role of cytoplasmic RIP140 in modulating lipolysis in adipocytes. After HFD feeding, the DAG-PKCε signaling cascade promotes nuclear export of RIP140. Cytoplasmic RIP140 then interacts with perilipin to enhance lipolysis by promoting the formation of perilipin-HSL and ATGL/CGI-58 complexes.
4. Discussion
HFD feeding promotes nuclear export of RIP140 in adipocytes [23]. Although RIP140, as a nuclear coregulator, has been known for its role in lipid accumulation in the adipose tissue, its functional role in the cytoplasm of adipocyte, with regards to lipid metabolism, was unclear. This study aimed to determine the signals promoting cytoplasmic RIP140 accumulation, and to determine the role of cytoplasmic RIP140 in lipid metabolism of adipocytes. Here, we demonstrate that fat content, especially an elevation in DAG levels (such as those triggered by a high fat content), facilitated RIP140 nuclear export and its localization on LDs. Cytoplasmic RIP140 facilitated lipolysis neither by affecting the expression nor the phosphorylation of key lipolytic enzymes, but rather via its direct interaction with perilipin that ultimately activates HSL and ATGL, two key lipolytic enzymes (Fig. 5C). Thus, RIP140’s cytoplasmic functions appear to rely primarily on its ability to interact with various proteins: In glucose uptake, RIP140 interferes with GLUT4 trafficking via its direct interaction with AS160 [23], whereas in lipolysis it interacts with perilipin associated with LDs.
The underlying mechanisms by which RIP140 alters its molecular interactions with perilipin remain to be determined. The dissection of key interacting domains, with respect to each interacting partner, would be needed to address this question. To this end, it is known that perilipin interacts with CGI-58 through its C-terminal region (amino acids 382–429) and with HSL through both its N- (amino acids 141–200) and C-terminal (amino acids 406–480) regions [30, 31]. This study identified the putative TAG protection regions of perilipin – a region that partially overlaps with the HSL- and CGI-58-interacting domains- as its major RIP140-interaction domain. Upon recruitment of RIP140, conformational changes could occur on perilipin, promoting its interaction with HSL while reducing its ability to associate with CGI-58. Importantly, the regulation of perilipin interaction with HSL and CGI-58, by RIP140, does not involve perilipin phosporylation. Further studies are required to determine the molecular details of such conformational changes, and to evaluate whether other PTMs of RIP140 might regulate its interactions with these cytoplasmic factors.
Studies in transgenic mice identified DAG as an important regulator that promotes insulin resistance and T2DM. An over abundance of nutrition, especially by HFD feeding, dramatically increases DAG levels in adipose tissue, muscle cells and liver, which in turn activates several members of the PKC family to reduce insulin sensitivity [5, 27]. Among these, PKCε is a novel member that can be activated solely by DAG [27]. The exact mechanisms responsible for elevated DAG levels in fully differentiated adipocytes and in HFD-fed mice remain to be determined [10, 27]. Nevertheless, activation of PKCε by elevated DAG levels presents a likely mechanism by which HFD could stimulate the nuclear export of RIP140. Activated PKCε phosphorylates two specific serine residues on nuclear RIP140, which then stimulates arginine methylation of three arginine residues; this in turn increases RIP140’s interaction with exportin for nuclear export [15, 21, 22]. Conversely, blocking PKCε activation might retard RIP140 nuclear export and reduce its cytoplasmic activity. Consistent with this mechanism, promoting fatty acid oxidation by the mitochondrial uncoupler or inhibiting acetyl-CoA carboxylase-1 and -2 to reduce intracellular DAG levels prevents PKCε activation and enhances glucose uptake in adipose tissue [33, 34]. Thus, regulating RIP140 nuclear export could be an effective strategy to modulate glucose uptake and lipolysis in adipose tissue.
Other studies in transgenic mice suggest that the increased adipogenic capacity in adipose tissue is an anti-diabetes response [6–8]. Adipocytes normally expand in a healthy process, and reduced necrosis and lipolysis can be protective for liver, muscle and heart under over-nutrition [2, 6]. The increase in the lipolytic rate in both obese mice and patients contributes to the initiation and progression of several metabolic disorders. However, the mechanisms that regulate healthy adipocyte expansion and reduce unwanted lipolysis remain elusive. The adipose tissues of RIP140 knockout mice have reduced fat and increased β-oxidation. Some of these effects can be partially attributable to the transcriptional regulatory activity of nuclear RIP140 [20, 32]. In the nucleus, RIP140 regulates the expression of genes that are important for lipid accumulation and fatty acid oxidation [19, 32]. However, the discovery that RIP140 also modulates important biological processes in the cytoplasm justifies a re-evaluation of its possible contributions to the overall regulation of metabolic processes in whole animals and under diseased conditions. For example, increasing cytoplasm RIP140 could decrease glucose uptake (thus reducing the availability of acetyl-CoA) [23] and escalate lipolysis (enhancing TG degradation) (this study); this would presumably provide a counteracting mechanism to maintain the overall lipid homeostasis in adipocytes when they mature and begin to accumulate lipid. Interestingly, over-expression of RIP140 in mice results in impairment of cardiac functions and cardiac hypertrophy [35]. To this end, RIP140 might contribute to these cardiac defects by promoting lipotoxicity.
Overall, these studies support the hypothesis that RIP140 responds to changes in the nutritional status of an individual by altering its activity and sub-cellular distribution. The cytoplasmic RIP140 can retard glucose uptake by interacting with AS160 [23], which would reduce the availability of acetyl-CoA for lipid synthesis. This study further shows that the cytoplasmic RIP140 can promote lipolysis to reduce lipid load. Accordingly, it may be a vital strategy to target cytoplasmic RIP140, such as by preventing its nuclear export, to provide beneficial effects under certain pathological conditions, such as by enhancing glucose uptake and reducing lipolysis.
Supplementary Material
Acknowledgments
This study was supported in part by NIH grants DK54733, DK54733-09S1, DK60521, DK60521-07S1, DA11190, K02-DA13926, DA11806, and the Distinguished McKnight University Professorship to L.-N. Wei. American Heart Association Predoctoral fellowship to P.-C. Ho. We thank S.-C. Luo, A. Smith and S. C. Chan for technical help.
Abbreviations
- RIP140
receptor-interacting protein 140
- PKCε
protein kinase C epsilon
- LD
lipid droplet
- HSL
hormone-sensitive lipase
- ATGL
adipose triglyceride lipase
- CGI-58
comparative gene identification-58
- DAG
diacylglyceride
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Farese RV, Jr, Walther TC. Cell. 2009;139:855–860. doi: 10.1016/j.cell.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilherme A, Virbasius JV, Puri V, Czech MP. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lago F, Gomez R, Gomez-Reino JJ, Dieguez C, Gualillo O. Trends Biochem Sci. 2009;34:500–510. doi: 10.1016/j.tibs.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 4.Shibata R, Ouchi N, Murohara T. Circ J. 2009;73:608–614. doi: 10.1253/circj.cj-09-0057. [DOI] [PubMed] [Google Scholar]
- 5.Erion DM, Shulman GI. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Unger RH, Scherer PE. Trends Endocrinol Metab. 2010;21:345–352. doi: 10.1016/j.tem.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang MY, Grayburn P, Chen S, Ravazzola M, Orci L, Unger RH. Proc Natl Acad Sci U S A. 2008;105:6139–6144. doi: 10.1073/pnas.0801981105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. J Clin Invest. 2006;116:3015–3025. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaidhu MP, Anthony NM, Patel P, Hawke TJ, Ceddia RB. Am J Physiol Cell Physiol. 2010;298:C961–971. doi: 10.1152/ajpcell.00547.2009. [DOI] [PubMed] [Google Scholar]
- 11.Langin D. Cell Metab. 2010;11:242–243. doi: 10.1016/j.cmet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 12.Granneman JG, Moore HP. Trends Endocrinol Metab. 2008;19:3–9. doi: 10.1016/j.tem.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Guo Y, Cordes KR, Farese RV, Jr, Walther TC. J Cell Sci. 2009;122:749–752. doi: 10.1242/jcs.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christian M, White R, Parker MG. Trends Endocrinol Metab. 2006;17:243–250. doi: 10.1016/j.tem.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Mostaqul Huq MD, Gupta P, Wei LN. Curr Med Chem. 2008;15:386–392. doi: 10.2174/092986708783497382. [DOI] [PubMed] [Google Scholar]
- 16.Fritah A, Christian M, Parker MG. Am J Physiol Endocrinol Metab. 2010;299:E335–340. doi: 10.1152/ajpendo.00243.2010. [DOI] [PubMed] [Google Scholar]
- 17.White R, Leonardsson G, Rosewell I, Ann Jacobs M, Milligan S, Parker M. Nat Med. 2000;6:1368–1374. doi: 10.1038/82183. [DOI] [PubMed] [Google Scholar]
- 18.Lee CH, Wei LN. J Biol Chem. 1999;274:31320–31326. doi: 10.1074/jbc.274.44.31320. [DOI] [PubMed] [Google Scholar]
- 19.Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So PW, Medina-Gomez G, Vidal-Puig A, White R, Parker MG. Proc Natl Acad Sci U S A. 2004;101:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Powelka AM, Seth A, Virbasius JV, Kiskinis E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD, Parker MG, Czech MP. J Clin Invest. 2006;116:125–136. doi: 10.1172/JCI26040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mostaqul Huq MD, Gupta P, Tsai NP, White R, Parker MG, Wei LN. EMBO J. 2006;25:5094–5104. doi: 10.1038/sj.emboj.7601389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta P, Ho PC, Huq MD, Khan AA, Tsai NP, Wei LN. PLoS One. 2008;3:e2658. doi: 10.1371/journal.pone.0002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ho PC, Lin YW, Tsui YC, Gupta P, Wei LN. Cell Metab. 2009;10:516–523. doi: 10.1016/j.cmet.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ho PC, Gupta P, Tsui YC, Ha SG, Huq M, Wei LN. Cell Signal. 2008;20:1911–1919. doi: 10.1016/j.cellsig.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zinchuk V, Zinchuk O, Okada T. Acta Histochem Cytochem. 2007;40:101–111. doi: 10.1267/ahc.07002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho SY, Shin ES, Park PJ, Shin DW, Chang HK, Kim D, Lee HH, Lee JH, Kim SH, Song MJ, Chang IS, Lee OS, Lee TR. J Biol Chem. 2007;282:2456–2465. doi: 10.1074/jbc.M608042200. [DOI] [PubMed] [Google Scholar]
- 27.Schmitz-Peiffer C, Biden TJ. Diabetes. 2008;57:1774–1783. doi: 10.2337/db07-1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sakane F, Imai S, Kai M, Yasuda S, Kanoh H. Curr Drug Targets. 2008;9:626–640. doi: 10.2174/138945008785132394. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi H, Takeishi Y, Seidler T, Arimoto T, Akiyama H, Hozumi Y, Koyama Y, Shishido T, Tsunoda Y, Niizeki T, Nozaki N, Abe J, Hasenfuss G, Goto K, Kubota I. Circulation. 2005;111:1510–1516. doi: 10.1161/01.CIR.0000159339.00703.22. [DOI] [PubMed] [Google Scholar]
- 30.Shen WJ, Patel S, Miyoshi H, Greenberg AS, Kraemer FB. J Lipid Res. 2009;50:2306–2313. doi: 10.1194/jlr.M900176-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brasaemle DL. J Lipid Res. 2007;48:2547–2559. doi: 10.1194/jlr.R700014-JLR200. [DOI] [PubMed] [Google Scholar]
- 32.Christian M, Kiskinis E, Debevec D, Leonardsson G, White R, Parker MG. Mol Cell Biol. 2005;25:9383–9391. doi: 10.1128/MCB.25.21.9383-9391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Choi CS, Savage DB, Abu-Elheiga L, Liu ZX, Kim S, Kulkarni A, Distefano A, Hwang YJ, Reznick RM, Codella R, Zhang D, Cline GW, Wakil SJ, Shulman GI. Proc Natl Acad Sci U S A. 2007;104:16480–16485. doi: 10.1073/pnas.0706794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang D, Christianson J, Liu ZX, Tian L, Choi CS, Neschen S, Dong J, Wood PA, Shulman GI. Cell Metab. 2010;11:402–411. doi: 10.1016/j.cmet.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fritah A, Steel JH, Nichol D, Parker N, Williams S, Price A, Strauss L, Ryder TA, Mobberley MA, Poutanen M, Parker M, White R. Cardiovasc Res. 2010;86:443–451. doi: 10.1093/cvr/cvp418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.