

Abstract
Hepatitis B virus “e-antigen” is thought to be a soluble dimeric protein that is associated with chronic infection. It shares 149 residues with the viral capsid protein “core-antigen”, but has an additional ten-residue, hydrophobic, cysteine-containing amino-terminal propeptide whose presence correlates with physical, serological, and immunological differences between the two proteins. In core-antigen dimers, the subunits pair by forming a four-helix bundle stabilized by an inter-molecular disulfide bond. The structure of e-antigen is probably similar, but instead has two intra-molecular disulfide bonds involving the propeptide. To compare the proteins directly, and thereby clarify the role of the propeptide, mutations and solution conditions were identified that render both proteins as either soluble dimers or assembled capsids. Thermally induced unfolding monitored by circular dichroism, and electrophoresis of oxidized and reduced dimers, showed that the propeptide has a destabilizing effect, and that the intra-molecular disulfide bond forms preferentially and blocks the formation of the inter-molecular disulfide bond that otherwise stabilizes the dimer. The e-antigen capsids are less regular than core-antigen capsids; nevertheless, cryo-EM reconstructions confirm that they are constructed of dimers resembling those of core-antigen capsids. In them, a portion of the propeptide is visible near the dimer interface, suggesting that it intercalates there, consistent with the known formation of a disulfide bond between C(−7) in the propeptide and C61 in the dimer interface. However, this intercalation distorts the dimer into an assembly-reluctant conformation.
Keywords: HBeAg, HBcAg, thermal stability, circular dichroism, cryo-EM
INTRODUCTION
Hepatitis B virus (HBV) causes 350 – 400 million chronic infections worldwide, and approximately 1 million deaths annually.1 Infected hepatocytes express two closely related viral antigens; the capsid or core-antigen (HBcAg), and a secreted protein known as e-antigen (HBeAg). Unlike HBcAg, HBeAg is not essential for infection, replication, or assembly,2; 3; 4 yet it is conserved in all members of the Hepadnaviridae. It is thought to modulate both the innate 5; 6; 7 and adaptive 8; 9; 10 immune responses so as to favor persistent infection.11; 12; 13
HBcAg and HBeAg are both derived from the pre-C/C gene but they are initiated from different in-frame start codons. The first residue of HBcAg is designated as (+1) and the wild-type protein terminates at residue 183. HBeAg on the other hand is initiated at an upstream start site and its first residue is by convention designated as (−29). This precursor is processed amino-terminally with the removal of a 19-residue leader peptide, and carboxy-terminally at position 154.14 Although the amino-terminus of serum-derived HBeAg has not yet been determined directly, it is assumed to be (−10) and HBeAg is therefore believed to have the form (−10)154, which is however susceptible to adventitious processing to the form (−10)149.14
HBcAg and HBeAg therefore share a common sequence, except for 10 and 29 (or 34) residues at their amino- and carboxy-termini, respectively, and both proteins are considered to be dimeric.15; 16 In the case of HBcAg the two polypeptide chains pair through the formation of a four-helix bundle and the resulting dimers polymerize into capsids17 whereas in HBeAg, despite a very similar primary structure, the dimers remain soluble. HBcAg and HBeAg also have several cysteine residues (reviewed in16). Crucial among these are C61, which can oxidize to form an inter-molecular disulfide bond across the dimer interface, and C(−7) in HBeAg, which can form an alternative intra-molecular disulfide bond with C61.16 These cysteines play a central role in the structure and secretion of HBeAg.18; 19; 20 The structural and physiological differences between HBcAg and HBeAg are therefore attributable to the propeptide and its cysteine residue.
The goal of this study was to prepare soluble (dimeric) and particulate (capsid) forms of both HBcAg and HBeAg, thereby allowing direct comparison of the two proteins and insight as to the role of the propeptide. To this end, we expressed recombinant forms of HBcAg and HBeAg in Escherichia coli. For HBcAg we employed a protein consisting of only the first 149 residues of the 183-residue wild-type protein, as these are known to suffice for capsid assembly.16 This protein forms capsids with two morphologies; one composed of 90 dimers (with T = 3 icosahedral symmetry, and a diameter of 32 nm) and one composed of 120 dimers (with T = 4 symmetry, and a diameter of 35 nm).21 These capsids are devoid of nucleic acid but otherwise appear to be indistinguishable – at least on their outer surface - from wild-type (Cp183) capsids, and are therefore well suited for structural studies.16 For HBeAg we employed a construct with the same 149-residue sequence as above but extended at its amino-terminus with the 10-residue propeptide, in other words, identical to the (−10)149 form of HBeAg (Fig. 1).
Fig. 1. The wild-type sequence of the Cp(−10)149 variant used in this study.

All cysteine residues are present. The Cp149 sequence begins at position (+1). Mutations and truncations of this basic construct are given in Table 1. The foreign octapeptide employed previously in lieu of the normal (−10) propeptide30 is shown below. In that construct the first ten residues were derived from E. coli beta-galactosidase.
The cryo-EM structures of the T = 4 capsid22; 23; 24 have shown that dimers cluster around the 2- and 5-fold axes of symmetry and the crystal structure17 reveals that interactions between neighboring dimers are mediated by residues in helix 5 (residues 112–127), the proline rich loop (128–136), and the carboxy-terminal arm (137–142). Accordingly, we mutated residues in these regions, aiming to produce protein that forms capsids less readily, i.e. more soluble dimers.15; 25 With both proteins available as soluble dimers, the cysteine residues were then systematically mutated to investigate their contributions, and that of the propeptide, to protein stability. Stability was assessed in terms of thermal denaturation temperature and monitored by circular dichroism spectroscopy. As noted above, the native HBeAg is probably inherently dimeric; nevertheless, we determined conditions in which dimers can be induced to assemble into capsids and used these to localize the residual propeptide (residues −10 to −1) by cryo-electron microscopy.
The mutant proteins used in this study are named in accordance with previous nomenclature, slightly extended.15 Cp183 refers to the wild-type polypeptide chain of HBcAg. Cp149 refers to a polypeptide consisting of the first 149 residues of Cp183, and corresponds to the assembly domain of that protein. Cp(−10)149 refers to HBeAg. Dimer and capsid forms of both proteins are indicated by the subscripts “d” and “c”, respectively. The names of mutations are appended to that of the basic construct, for instance, Cp(−10)149d.G123A. Disulfide bonds are specified by superscripts. For example, the above protein, with an intra-molecular disulfide bond between C(−7) in the propeptide and C61 in the assembly domain, is specified as Cp(−10)149d.G123AC(−7)–C61. In the constructs used, the cysteines at positions 48 and 107, which are not involved in disulfide bonds,17 have been mutated to alanine, although this is not indicated explicitly in the name.
RESULTS
Mutations that affect protein solubility
The expression in E. coli of Cp183 or Cp149 results in the formation of capsids in the cytoplasm.16 This self-assembly process affords a simple means for screening mutants that affect capsid formation. When only assembly is blocked, dimers accumulate, whereas, when the mutation perturbs protein folding, insoluble aggregates form. These expression products may be distinguished using a simple differential centrifugation scheme (see Materials and Methods). Briefly, cell homogenates are subjected to low speed centrifugation which pellets highly aggregated protein and any unbroken cells. Centrifugation of the supernatant at higher speed pellets capsids while soluble protein remains in the supernatant. The results from a series of Cp149 mutants and carboxy-terminal deletions are summarized in Table 1.
Table 1.
Mutations affecting solubility and capsid assembly
| Mutant proteins | Expression in E. coli | Structural Locationb | ||
|---|---|---|---|---|
| Insoluble | Capsid | Solublea | ||
| Cp149 | ++++ | |||
| Cp143 | +++ | + | ||
| Cp141 | ++ | ++ | ||
| Cp140 | ++++ | |||
| Cp139 | ++++ | |||
| Cp149.T142A | ++++ | Ext | ||
| Cp149.S141A | +++ | + | Ext | |
| Cp149.L140A | ++++ | Ext | ||
| Cp149.N136A | ++++ | PR | ||
| Cp149.Y132A | ++++ | PR | ||
| Cp149.R127A | ++++ | 5α | ||
| Cp149.G123A | ++++ | 5α | ||
| Cp149.Y132A. | ++++ | 5α | ||
| G123A.R127A | ||||
| Cp149.R127A.G123A | ++++ | 5α | ||
| Cp149.TI14A | + | +++ | 5α | |
| Cp149.G123A.T114A | ++++ | 5α | ||
| Cp149.C48A.C107A | ++++ | |||
| Cp149.C48A.C61A.C107A | ++++ | |||
| Cp(−10)149 | ++++ | |||
| Cp(−10)149.G123A | ++ | ++ | ||
The soluble protein is always dimeric.
Structural locations based on the atomic structure17 where Ext refers to the extended region (residues 137–142); PR, the proline rich region (residues 128–136); and 5α, helical region 5 (residues112–127).
As shown previously, truncation of the capsid protein from 183 to 149 residues still allows capsid formation.22 Further carboxy-terminal truncation from 149 down to 140 residues yields progressively higher proportions of T = 3 capsids22and capsids (of both sizes) with lower stability. Truncation beyond 140 residues causes misfolding of the monomer and aggregation of the protein. Accordingly, all mutations were assessed in the context of the 149-residue assembly domain (Table 1).
These observations demonstrate that mutations in the carboxy-terminal region, especially in helix 5 (residues 112–127) and the proline-rich loop (residues 128–136), have a profound affect on capsid assembly while having no apparent affect, as judged by circular dichroism, on the overall fold of the polypeptide chain. It is worth noting that the mutations G123A and R127A which block capsid assembly by Cp149 do not affect Cp183 in the same way. Like wild-type Cp183, these mutant capsids cannot be dissociated under non-denaturing conditions, however, they exhibit an increased tendency to aggregate (data not shown).
The disulfide bonding patterns of the proteins will be discussed in detail below but we mention here that all three cysteines in Cp149 (C48, C61, and C107) can be substituted with alanine both individually and jointly without affecting the expression and assembly of capsids, although it increases the proportion of T = 3 capsids.22; 26 Furthermore, these mutations can be introduced into the protein together with mutations such as G123A without affecting expression or solubility of the protein.
Characterization of Cp149
We have previously shown that Cp183c are extremely stable, requiring protein denaturation for dissociation.27 Cp149c, on the other hand, are less stable and can be dissociated under mildly-denaturing conditions such as elevated pH and low concentrations of urea. Further, it was established by analytical ultracentrifugation that the resulting dissociated capsid protein is dimeric.27 These dimers are quite stable and further dissociation to monomers requires denaturation with SDS and reduction with DTT. Analysis by sedimentation equilibrium ultracentrifugation of the proteins in Table 1 showed that they were all dimeric. For example, sedimentation equilibrium ultracentrifugation (at pH 7.5) of Cp149.G123A indicated a mass of 32,744 Da (see Fig. S2 in15), consistent with a dimer, and with no indication of further association.15 Single symmetrical peaks eluting in gel filtration, with the protein loaded at high concentrations (>10 mg/ml), also indicated that the protein is dimeric. The overall secondary structure of the mutant dimers was assessed by both near- and far-UV circular dichroism. As was the case with Cp149d generated by dissociation of Cp149c,27 the helical content (far-UV CD) and tertiary conformation (near-UV CD) of the mutants was similar to that of wild-type Cp149d (Fig. S1). This indicates that the disruption of assembly was the result of small localized perturbations rather than large conformational changes typical of protein denaturation. This is also consistent with the expression and solubility behavior of the mutant proteins.
Expression and purification of Cp(−10)149
When Cp(−10)149 is expressed in E. coli it is in an insoluble form. This insolubility, however, does not appear to be due to protein misfolding, which usually leads to inclusion body formation, but instead seems to be due to the low solubility of the construct and/or its association with bacterial cytoplasmic components. This was shown by the extraction of the protein under mildly-denaturing conditions, for example, 3 M urea at pH 9.5 (i.e. the same conditions that are used to dissociate Cp149c). Once extracted, the protein can be purified by conventional chromatographic procedures as described in Materials and Methods. During purification the protein was well behaved and showed no tendency to either aggregate or associate. The deletion of the first three residues (SKL) of the amino-terminal (−10) sequence slightly improved the solubility of the expressed protein, but substituting individual residues within the (−10) sequence with alanine had either little or no effect on the solubility of the expressed protein (data not shown). The solubility was, however, improved by incorporating the same mutations which affected the assembly of the Cp149d. Thus, Cp(−10)149 with the G123A and/or R127A mutation(s), exhibited mixed expression, with both soluble and poorly soluble protein accumulating rather than only insoluble material. Further, the insoluble protein present was far more readily solubilized than the wild-type protein, and the resulting fully purified protein was stably soluble to at least 10 mg/ml at neutral pH.
Characterization of Cp(−10)149
The molecular weight of purified Cp(−10)149 was measured by sedimentation equilibrium ultracentrifugation. Over the pH range of 7.0–9.5 the protein behaved as an ideal dimer with no evidence of a monomer-dimer transition (Fig. 2). The helical content and conformation are similar to that of Cp149, as assessed by near- and far-UV circular dichroism (Fig. S1). The assembly of Cp149d is ionic strength and pH dependent, and at pH 7 and 150 mM NaCl assembly to Cp149c is both rapid and complete.27; 28 Under these same conditions, Cp(−10)149d remains soluble and >350 mM NaCl is required to induce formation of Cp(−10)149c. Electron microscopy of these capsids shows them to be essentially identical to Cp149c (discussed further below). Most (~90%) of the Cp(−10)149c have T = 3 morphology, unlike Cp149c where >90% are of the T = 4 form.22 These results show that the 10-residue amino-terminal extension of Cp(−10)149 favors the accumulation of Cp(−10)149d under near-physiological conditions, but does not completely block assembly into Cp(−10)149c. The introduction of mutations in the helix-5 domain, especially G123A and R127A, improved the solubility of Cp(−10)149, and protein with these and other mutations was used for the biophysical experiments described below.
Fig. 2. Sedimentation equilibrium centrifugation of Cp(−10)149.
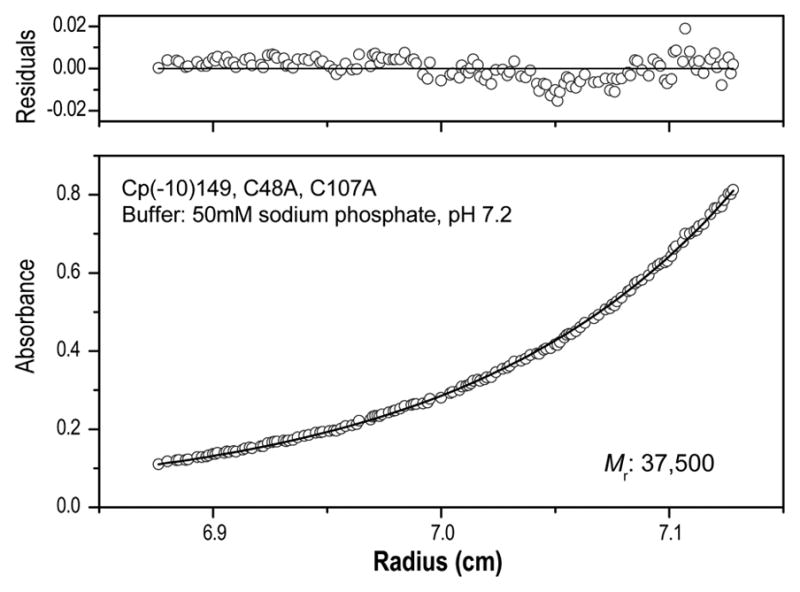
The molecular weight of Cp(−10)149d was determined under nondenaturing conditions (50 mM NaHPO4 buffer, pH 7.2). The open circles represent experimental data and the solid line represents a curve fitted using a single-species model. The upper panel shows the residuals (difference between the fitted and experimental values) as a function of radial position. The molecular weight determined (37,500) is close to that predicted (36,000) for a stable dimer. In the example shown, the Cp(−10)149.C48A.C107A mutant was used. Similar results were obtained for the other Cp(−10)149d constructs described including one, for example, with solubility enhancing mutations G123A.R127A.15
Disulfide bonding and thermal stability of Cp149 and Cp(−10)149
Previous studies have shown that the introduction of cysteine mutations into the Cp183 and Cp149 constructs has no affect on either protein expression or the assembly of capsids (see16 and references therein). We have probed the stability of Cp149d and Cp(−10)149d using far-UV circular dichroism at 222 nm to monitor the helix-coil transitions upon thermal denaturation. A series of cysteine mutants which restrict disulfide bond formation to defined pairings were used in these measurements (Table 2). In Cp149d, if cysteines at positions 48 and 107 are mutated to alanine, disulfide bond formation is limited to C61 which readily forms an intermolecular link with C61 on the adjacent monomer (this disulfide bond is also formed in Cp183c and presumably exists in virions). The oxidized protein runs as a dimer on non-reducing SDS-PAGE (Fig. 3) and melts at 65 °C (Table 3). In Cp149d containing alanine substitutions at all three cysteine positions, the melting temperature is reduced by ~2 °C (fully reduced wild type protein also melts at this temperature27). In Cp(−10)149d there is normally an intramolecular disulfide formed between the cysteines at positions (−7) and 61.18; 19 In constructs with only these cysteines present, oxidized protein is monomeric under nonreducing SDS-PAGE, with the band mobility slightly greater than control reduced protein, as is typical for proteins with intramolecular disulfides29 (Fig. 3). In the absence of denaturant the protein is dimeric, i.e. oxidation or reduction of the C(−7)–C61 disulfide bond has no effect on its dimeric status. Oxidized Cp(−10)149d has a melting temperature of ~51 °C and is significantly less stable than either the reduced or oxidized Cp149d, which melt 12 and 14 °C higher, respectively (Table 3). When formation of the intramolecular C(−7)–C61 disulfide bond is prevented, either by substituting all cysteines with alanine or by reduction, the stability is further reduced by ~2 °C. Taken together, these results show that in Cp(−10)149d the formation of the intramolecular disulfide bond between C(−7) and C61 occurs in preference to the intermolecular C61-C61 bond which normally occurs in Cp149. However, if C(−7) is mutated to alanine while C61 is retained (in a C48A, C107A background) then dimeric protein is observed on nonreducing SDS-PAGE, indicating intermolecular disulfide bond formation between C61 on adjacent monomers. The melting temperature of the oxidized C(−7)A protein was ~63 °C, similar that of the Cp149d (Table 3). Upon reduction, the stability reverted to that of all other reduced Cp(−10)149 variants.
Table 2.
Proteins with mutated cysteine residues
| Mutant protein | Cysteine Residues | 5α mutationsa | ||||
|---|---|---|---|---|---|---|
| −7 | 48 | 61 | 107 | 123 | 127 | |
| Cp149 | - | C | C | C | G | R |
| Cp149.4A | - | A | C | A | A | A |
| Cp149.5A | - | A | A | A | A | A |
|
| ||||||
| Cp(−10)149 | C | C | C | C | G | R |
| Cp(−10)149.4A | C | A | C | A | A | A |
| Cp(−10)149.5A | A | A | C | A | A | A |
| Cp(−10)149.6A | A | A | A | A | A | A |
Refers to helical region 5 (residues 112–127) of HBcAg.17
Fig. 3. SDS-PAGE of reduced and oxidized Cp149d and Cp(−10)149d.
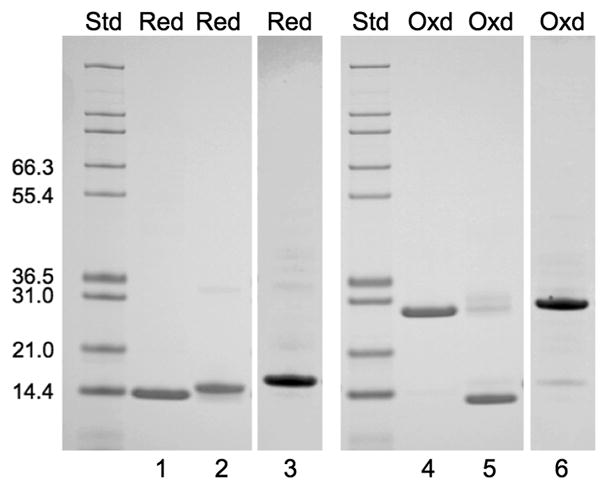
Lanes 1, 2 and 3 refer to Cp149d.C61.C48A.C107A; Cp(−10)149d.C(−7).C61.C48A.C107A; and Cp(−10)149d.C(−7)A.C61.C48A.C107A, respectively. The previously oxidized proteins were treated with DTT (Red) prior to electrophoresis. Lanes 4, 5, and 6 are the same proteins, but not reduced (Oxd). All three proteins migrate as monomers when reduced but only Cp(−10)149d.C(−7).C61.C48A.C107A migrates as a monomer when oxidized. Note that the oxidized from migrates with slightly higher mobility than the reduced form (compare lanes 2 and 5) due to the intra-molecular (C(−7)-C(61) disulfide bond, and that no dimers or tetramers are formed, indicating that there is no C(−7)-C(−7) bond formation, either within or between dimers. All proteins, whether oxidized or reduced, are dimeric under native conditions as shown, for example, in Fig. 2.
Table 3.
Effects of cysteine mutations on thermal stability of the dimeric proteins
| Mutant protein | Disulfide Linkage
|
SDS-PAGEa | Melting Temp (°C) | |
|---|---|---|---|---|
| Residues | Type | |||
| Cp149.4A | C61-C61 | inter | dimer | 65 |
| Cp149.5A | none | monomer | 63 | |
| Cp(−10)149.4A | C(−7)–C61 | intra | monomer | 51 |
| Cp(−10)149.5A | C61-C61 | inter | dimer | 60–64 |
| Cp(−10)149.6A | none | monomer | 49 | |
SDS-PAGE was performed under non-reducing conditions. The monomer or dimer form is inferred from the apparent molecular weights, ~17 kDa and ~35 kDa respectively, of the proteins. Under reducing conditions all proteins are monomeric.
Localization of the Cp(−10)149 propeptide by cryo-EM and image reconstruction
Cp(−10)149d, although indisposed to capsid formation, can nevertheless be induced to assemble by increasing the ionic strength. Such capsids are primarily of the T = 3 form and although they appear by EM to be similar to Cp149c, they are less regular in shape (Fig. 4) and more susceptible to aggregation. We have determined the structure of Cp(−10)149c capsids from cryo-EM data to ~12 Å resolution. Comparison with Cp149c at the same resolution reveals extra density in the form of small spur-like projections at the base of the spikes (Fig. 5). Such spurs are also observed in the Cp(−10)149c assembled from oxidized dimers (Fig. S4). The spurs are located similarly to those seen in Cp149c with other amino-terminal extensions,30; 31; 32 and we assign the spur density to a portion of the propeptide. By modeling a dimer from the HBV capsid atomic structure (PDB: 1QGT17) into the T = 3 density map33 we observe that the amino-terminus of each subunit is located within 10 Å of a spur, suggesting that Cp(−10)149 protein is folded slightly differently in this region due to the presence of the attached propeptide. The proximity of the spurs to the four-helix bundle of the spike indicates that the propeptide is favorability positioned to allow formation of a disulfide bond between its C(−7) and the C61 residue of either Cp subunit within the four-helix bundle. The tip of the spur is ~20 Å distant from the C61 on the same subunit, corresponding to a minimum of five residues in an extended conformation, and slightly closer to the C61 on the second subunit of the dimer. SDS PAGE of oxidized Cp(−10)149 indicates that it is the intra-molecular disulfide bond that is formed.
Fig. 4. Cryo-electron micrograph of Cp(−10)149c.
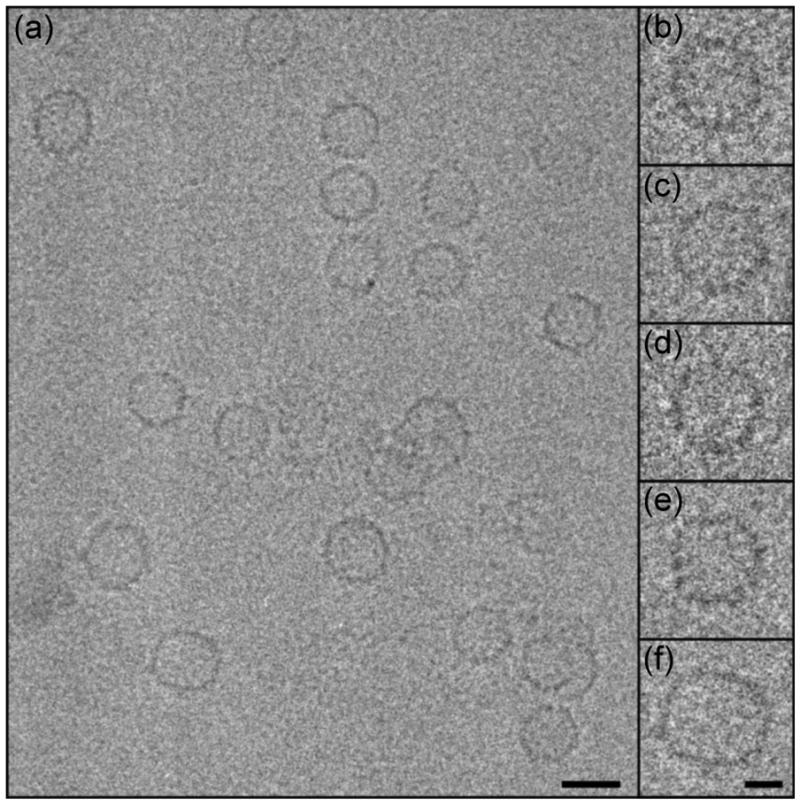
Shown is a representative field of capsids (a), including a number that are somewhat irregular. On the right are enlarged views of uniform T = 3 particles (b-d), and non-uniform T = 3 (e) and T = 4 (f) particles. Bar = 250 Å, 100 Å in the inset.
Fig. 5. Representations of Cp(−10)149c T = 3.

(a) Surface views of the Cp(−10)149c (upper-left) and control Cp149c (lower-right). Additional spurs of density on the Cp(−10)149c are colored according to proximity to fitted Cp subunit amino-termini, and following a previously established coloring scheme: AB-dimer subunits are green and yellow, respectively; the CC-dimer subunits for T = 3 capsids are red.33 Bar = 50 Å. (b) A close-up view of the CC-dimer with the surface replaced by a mesh shows the fitted Cp subunits and the locations of their amino-termini (as indicated) in relation to the density spurs. Residue C61 colored light blue. Bar = 10 Å. The relationship of the Cp amino-termini and spurs to residue C61 (colored purple) is shown in a side-view (c) and a top-view (d) of the AB-dimer. Scale same as (b). As indicated in Materials and Methods, capsids were assembled from both reduced and oxidized dimers, and T = 3 and T = 4 particles were reconstructed from each condition, for a total of four reconstructions. However, as the locations of the spurs were very similar in all four cases only one example of each of the two capsid morphologies is shown, i.e. a T = 3 capsid assembled from reduced dimers is shown here, and a T = 4 capsid assembled from oxidized dimers is shown in Fig. S4.
DISCUSSION
It is almost 40 years since the “e-antigen” of HBV was first described,34 and almost 30 years since it was isolated from serum and found to be closely related to core-antigen.35; 36 In the interim, the termini of e-antigen have been identified,14; 37 and it has been established that the protein is probably a dimer and not a monomer.15; 16 However, it has not become clear how the ten residues of the e-antigen propeptide can so profoundly affect the physical and functional properties of this protein.15 In this study, we have compared the two proteins (with and without propeptide) in the same physical state, i.e. either as soluble dimers or as assembled capsids. Knowing the structure of the propeptide-free protein at high resolution,17; 38 we can now begin to understand how the propeptide exerts its effects.
Preparation and characterization of the Cp149 and Cp(−10)149 proteins
When Cp183 is expressed in E. coli it forms RNA-containing capsids that cannot be dissociated without resorting to strongly denaturing conditions. The positively charged carboxy-terminal domains (residues 149–183) are sequestered inside Cp183c where they interact with the oppositely charged RNA, thereby stabilizing the capsid.39; 40 However, most structural studies have instead employed assembly-competent constructs such as Cp149 that lack the carboxy-terminal domain. The resulting capsids can be dissociated under mild conditions that preserve the fold of the protein.27 These Cp149d afford a useful starting point for investigating e-antigen (Cp(−10)149).
Referring to the structure of Cp149c,17 we designed mutations aimed at blocking assembly and confirmed that single mutations in the carboxy-terminal region (residues 123–132), especially G123A, had this effect (Table 1). Cp149 constructs with these mutations were dimeric, and had native-like conformations. As anticipated, the introduction of the assembly-blocking mutations into Cp(−10)149 enhanced the solubility of this protein, as manifested in its ease of extraction from cell lysates and purification. We have previously shown that Cp(−10)149 is dimeric under native conditions and monomeric in both reducing and non-reducing SDS-PAGE.15 Nevertheless, wild-type Cp(−10)149d can be induced to assemble into capsids. These particles, though non-physiological, have been useful for assessing the effects of assembly on the antigenic properties of the protein,15 and as discussed below, for structural studies.
Structural roles of cysteine residues in Cp149 and Cp(−10)149
Previously we reviewed the structural roles of cysteine residues in HBV capsid protein.16 Cp183 has cysteines at positions 48, 61, 107 and 183. In brief, C61 forms an intra-dimer [C61-C61] disulfide bond, whereas C183 forms an inter-dimer [C183-C183] disulfide bond. As the two remaining cysteines, C48 and C107, do not engage in disulfide bonds in the native conformations17 we mutated them to alanine in most constructs used (Table 2). In Cp149d, the C61-C61 bond is also formed. However, Cp(−10)149 has a cysteine at position (−7), which forms an intra-monomer disulfide with C61, and the C61-C61 linkage observed in Cp149d is thereby blocked. However, if C(−7) is mutated to alanine then the C61-C61 bond does form (Table 3). Thus, while the conformation of Cp(−10)149d places C(−7) in a position favorable for oxidation with C61, there is enough flexibility at the dimer interface to allow the C61-C61 bond to form if the two cysteines are available.
The thermal stabilities of the dimers with various oxidation patterns may be ranked as follows:
It follows that: first, Cp149d is more stable than Cp(−10)149d regardless of oxidation state, and the propeptide has an overall destabilizing effect. Second, the conformation of Cp(−10)149d positions C(−7) close to C61 on the same polypeptide chain, and that [C(−7)–C61] bonds form faster than [C61-C61] bonds (the latter rate is almost zero, based on timed SDS-PAGE analysis, data not shown). However, if C(−7) is unavailable, then formation of the C61-C61 bond proceeds. Given that the overall shape of the dimer is preserved in Cp(−10)149c and, by extension, in Cp(−10)149d, it follows that there must be sufficient conformational flexibility at the dimer (helix 3α) interface for the propeptide to insert so that C(−7) can engage C61. Third, when the formation of the C61-C61 bond in Cp(−10)149d does occur, it overrides the destabilizing effect of the propeptide, and the protein is then as stable as Cp149d.
Location of the Cp(−10)149 propeptide
Previously we determined a cryo-EM structure at 11 Å resolution from capsids assembled from Cp149 with a foreign octapeptide appended to its amino-terminus.30 The amino-terminal extension was visualized as a curved strand of density emanating from a site near the bottom of the spike, where residue 1 is assigned in the crystal structure. Our reconstructions of Cp(−10)149c at a nominal resolution of ~12 Å (Fig. 5 and Fig. S4), reveal a small projection (the “spur”) with the same attachment point as the appended octapeptide. The spur is a very small feature to be detected at this relatively modest resolution but as it appears consistently at quasi-equivalent (and independently calculated) sites on both capsids, there is little doubt that it is real. The most straightforward interpretation is that it represents part of the propeptide with the remainder (not visualized due to flexibility) feeding into the dimer interface to consummate the C(−7)–C61 bond. Consistent with this inference, the propeptide is significantly more hydrophobic (+0.660 average hydropathicity) than the foreign octapeptide (+0.125 average hydropathicity) used in the earlier analysis, which remained external to the spike. We further infer that the location of the propeptide in the dimer interface has a perturbing effect on the capsid structure, accounting for the generally less regular nature of Cp(−10)149c as compared to Cp149c.
Thus, Cp(−10)149 is destabilized by the presence of the propeptide, whether reduced or oxidized, but reconstructions of capsids formed from these two forms of the dimer reveal no great differences (compare Fig. 5 and Fig. S4). This suggests that the propeptide binds to the dimer structure in both cases, and that the disulfide bond can form under oxidizing conditions without any additional large-scale structural changes.
On the basis of cryo-EM reconstructions, the HBV capsid has been described as a series of α-helical rods and flexible links which can transmit conformational information from the carboxy-terminal regions (residues 132–142) to the spikes on the outside of the shell.40; 41 A similar linkage has also been proposed from a comparison of the atomic structures of the capsid and unassembled dimer.38 By analogy, distortion at the dimer interface, as induced by the presence of the hydrophobic propeptide, could effect the stability and assembly of Cp(−10)149d.
MATERIALS AND METHODS
Screening of mutations that affect protein solubility
The proteins listed in Table 1 were prepared as described previously15 and analyzed by differential centrifugation. Briefly, E. coli were grown in 25 ml of Luria broth supplemented with ampicillin and induced with IPTG, yielding approximately 100 mg of cells. The cells were suspended in 0.7 ml of 50 mM Tris-HCl (pH 7.4), 0.1 mM EDTA, broken by sonication, and centrifuged for 45 minutes in a microcentrifuge to remove cell debris and aggregated material. The supernatants were then centrifuged in a Beckman TL-100 centrifuge with a TL 100.3 rotor for 1 hr at 135,000xg to pellet capsids and aggregates following which the pellets were suspended in 0.5 ml of the above buffer. Pellets and supernatants were analyzed by SDS-PAGE.
Expression and purification of Cp149 and Cp(−10)149 and their mutant derivatives
The proteins listed in Table 2 were expressed in E. coli as previously described.15 Cell paste (~50 g) was suspended in 120 ml of 50 mM Tris-HCl (pH 7.5), 2 mM EDTA, 2 mM DTT (Buffer A). Three protease inhibitor cocktail tablets (Complete EDTA-free: Roche Diagnostics) were added and the cell suspension was lysed by two passes through a French Press at 12,000 lb/in2 followed by 2 minutes of sonication. The lysed cell suspension was diluted to 250 ml with Buffer A. The suspension was then centrifuged at 25,000 ×g for one hour at 4 °C. At this point the Cp149-related proteins were in the supernatant and Cp(−10)149-related proteins were in the pellet.
The Cp149-related proteins were concentrated by precipitation with 40% (NH4)2SO4. The pellet was dissolved in 100 ml of 100 mM NaHCO3 (pH 9.6), 2 mM DTT (Buffer B) containing 3 M urea, then centrifuged at 25,000 ×g for 2 hours at 4 °C. The supernatant was applied in 50 ml aliquots to a Superdex 200 column (95 cm × 5 cm diameter) equilibrated with Buffer B. The pooled fractions from the Superdex 200 column were loaded onto a Q-Sepharose column (10 cm × 3.5 cm diameter) equilibrated with 50 mM Tris-HCl (pH 8.0), 2 mM DTT (Buffer C). After a two column-volume wash with Buffer C, a 1600-ml linear gradient from 0 to 1 M NaCl in Buffer C was applied to elute the protein. Selected fractions were pooled and concentrated to 40 ml in an Amicon stirred cell with a YM10 membrane (Millipore). The pH was titrated to 9.6 with NaOH and urea was added to a final concentration of 3 M. The material was then re-chromatographed on a Superdex 200 column equilibrated in Buffer B as described above. The protein was stored at −80 °C.
The Cp(−10)149-related protein pellets were homogenized in Buffer B containing 3 M urea and processed as described above for Cp149. All column fractions were monitored by SDS-PAGE. For all proteins approximately 1.5 mg of purified protein was recovered per gram wet weight of E. coli cells, a recovery of about 30% based on ~5% expression levels.
Oxidation of Cp(−10)149
Reductant was removed by gel filtration on Sephadex G25 and aqueous CuCl2 was added to a final concentration of 1 μM, the sample was incubated for 30 minutes at 4 °C and then 5 mM EDTA was added.42 The protein solution was concentrated with an Amicon stirred cell with a YM10 membrane and then chromatographed on a Superdex S200 column equilibrated in 50 mM NaHPO4 (pH 7.5).
Analytical ultracentrifugation
Analytical ultracentrifugation was carried out using a Beckman Optima XL-I analytical ultracentrifuge; absorption optics, an An-60 Ti rotor, and standard double-sector centerpiece cells were used. Equilibrium measurements were made after 16–20 hours at 21,500 rpm and 20 °C. Baselines were established by over-speeding at 45,000 rpm for 4 hours. Data were analyzed using the standard Optima XL-I data analysis software. The protein partial specific volume was calculated from the amino acid composition.43 Solvent density was estimated as described previously.44
Thermal denaturation
Measurements were made at 222 nm using a Jasco-720 spectropolarimeter equipped with a PTC-343 Peltier-type thermostatic cell holder and a 115B-QS 1-cm path length cell with a Teflon stopper, volume = 400 μl and outside dimensions H × W × D = 40 × 12.5 × 12.5 mm (Hellma Cells, Inc). Cooling circulating water (20 °C) was supplied using a Mulitemp III thermostatic circular (Amersham Biotech). Protein at ~1.0 mg/ml in 50 mM NaHPO4 (pH 7.5) was heated: the temperature was raised 1 °C per minute with a temperature slope of 20–100 °C. The step resolution was 1 °C, the response time 1 sec, the bandwidth 1 nm, and the sensitivity 200 mdeg.
Circular Dichroism
Spectra were recorded using a Jasco-720 spectropolarimeter. Measurements in the near-ultraviolet region (240–340 nm) were made using protein solutions in 50 mM NaHPO4 (pH 7.0) or in 100 mM NaHCO3 (pH 9.6), and a 1-cm path length cell. Measurements in the far-ultraviolet region (180–260 nm) were made in the same buffers using a 0.01-cm path length cell. For both near- and far-UV CD, protein solutions having 1.0–1.8 OD at 280 nm were used with a bandwidth of 1 nm. Solutions were filtered with a Millex-GV 0.22 μm filter (Millipore) and degassed prior to use. Secondary structures were estimated using standard methods.45; 46; 47
Assembly of Cp(−10)149c
Cp(−10)149c.C48A.C107A were assembled in two different ways; either from reduced dimers or from oxidized dimers. For assembly from reduced dimers, the protein prepared in Buffer B was used directly. Capsid assembly was initiated by dilution into assembly buffer, giving 0.2 mg/ml protein in 100 mM HEPES (pH 8), 350 mM NaCl, and incubation for 30 minutes at 37 ° before placing on ice. For assembly from oxidized dimers, the protein was oxidized as described above except that it was treated with 5 μM CuCl2. Oxidation and formation of the intramolecular disulfide bond was assessed by non-reducing SDS-PAGE. Assembly was then initiated as described for reduced dimers but without warming to 37 °. Capsid assembly and aggregation was assessed in both cases by negative stain electron microscopy.
Cryo-electron microscopy and image reconstruction
Samples were frozen in thin films for cryo-EM as described previously.30 Images of T = 4 capsids were analyzed from micrographs collected on an FEI CM200 FEG microscope operating at 120 kV at a nominal magnification of 38,000x. Data from an FEI Polara FEG microscope operating at 300 kV and 59,000x magnification were used for the T = 3 capsid analysis. Each field was imaged twice at different defocus settings, ranging from 1–2 μm from 7 pairs of micrographs taken on the TF20, and 1–6 μm from 10 pairs taken on the Polara. Micrographs were digitized on a Zeiss SCAI scanner with a fixed 7 um step, giving nominal sampling rates of 1.84 Å/pixel (T = 4) and 1.19 Å/pixel (T = 3). Capsid images were selected from the scanned micrographs with the x3dpreprocess software48 and image alignment and reconstructions were performed with the AUTO3DEM suite.49 Contrast transfer function parameters were estimated with BSOFT50 to enable compensation for phase reversals and astigmatism throughout the analysis. Defocus pairs of capsid images were computationally combined after initial orientations were determined, as previously detailed.48 The T = 4 dataset included 2691 pairs of images, of which 1267 were selected for the final reconstruction with an estimated resolution (FSC-0.5) of 11.9 Å. The T = 3 dataset included 871 pairs of images, of which 429 were selected for the final reconstruction with an estimated resolution (FSC-0.5) of 13.7 Å. Image processing was carried out on 32-core and 16-core Opteron-based computers running the Linux operating system and equipped with 128 GBytes and 40 GBytes of internal memory, respectively.
Molecular Modeling
Density maps and atomic models were visualized in UCSF Chimera.51 The T = 4 capsid reconstruction was scaled to match the HBcAg (1–149) atomic model17 (PDB: 1QGT) and the T = 3 map scaled to a previously assembled T = 3 pseudo-atomic model based on the 1QGT coordinates.33; 52 Molecular graphics was performed on Apple Macintosh computers running OSX 10.5 and 10.6 operating systems.
Supplementary Material
Acknowledgments
We wish to thank Dr. Adam Zlotnick for helpful discussions, Joshua Kaufman and Ira Palmer for technical assistance, and Philip Greer and Douglas Bevan for aid with computer resources. This research was supported by the NIAMS Intramural Research Program of the National Institutes of Health, and by the Commonwealth of Pennsylvania, SAP 4100031302 (JFC).
Abbreviations
- HBV
Hepatitis B virus
- HBcAg
core-antigen
- HBeAg
e-antigen
- Cp
capsid protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lesmana LA, Leung NW, Mahachai V, Phiet PH, Suh DJ, Yao G, Zhuang H. Hepatitis B: overview of the burden of disease in the Asia-Pacific region. Liver Int. 2006;26(Suppl 2):3–10. [Google Scholar]
- 2.Chang C, Enders G, Sprengel R, Peters N, Varmus HE, Ganem D. Expression of the precore region of an avian hepatitis B virus is not required for viral replication. Journal of Virology. 1987;61:3322–5. doi: 10.1128/jvi.61.10.3322-3325.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlicht HJ, Salfeld J, Schaller H. The duck hepatitis B virus pre-C region encodes a signal sequence which is essential for synthesis and secretion of processed core proteins but not for virus formation. J Virol. 1987;61:3701–3709. doi: 10.1128/jvi.61.12.3701-3709.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen HS, Kew MC, Hornbuckle WE, Tennant BC, Cote PJ, Gerin JL, Purcell RH, Miller RH. The precore gene of the woodchuck hepatitis virus genome is not essential for viral replication in the natural host. Journal of Virology. 1992;66:5682–4. doi: 10.1128/jvi.66.9.5682-5684.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riordan SM, Skinner N, Nagree A, McCallum H, McIver CJ, Kurtovic J, Hamilton JA, Bengmark S, Williams R, Visvanathan K. Peripheral blood mononuclear cell expression of toll-like receptors and relation to cytokine levels in cirrhosis. Hepatology. 2003;37:1154–64. doi: 10.1053/jhep.2003.50180. [DOI] [PubMed] [Google Scholar]
- 6.Visvanathan K, Skinner NA, Thompson AJ, Riordan SM, Sozzi V, Edwards R, Rodgers S, Kurtovic J, Chang J, Lewin S, Desmond P, Locarnini S. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology. 2007;45:102–10. doi: 10.1002/hep.21482. [DOI] [PubMed] [Google Scholar]
- 7.Durantel D, Zoulim F. Innate response to hepatitis B virus infection: observations challenging the concept of a stealth virus. Hepatology. 2009;50:1692–5. doi: 10.1002/hep.23361. [DOI] [PubMed] [Google Scholar]
- 8.Chen MT, Billaud JN, Sallberg M, Guidotti LG, Chisari FV, Jones J, Hughes J, Milich DR. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc Natl Acad Sci U S A. 2004;101:14913–8. doi: 10.1073/pnas.0406282101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen M, Sallberg M, Hughes J, Jones J, Guidotti LG, Chisari FV, Billaud JN, Milich DR. Immune tolerance split between hepatitis B virus precore and core proteins. Journal of Virology. 2005;79:3016–27. doi: 10.1128/JVI.79.5.3016-3027.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang CF, Lin SS, Ho YC, Chen FL, Yang CC. The immune response induced by hepatitis B virus principal antigens. Cell Mol Immunol. 2006;3:97–106. [PubMed] [Google Scholar]
- 11.Ou JH. Molecular biology of hepatitis B virus e antigen. J Gastroenterol Hepatol. 1997;12:S178–87. doi: 10.1111/j.1440-1746.1997.tb00499.x. [DOI] [PubMed] [Google Scholar]
- 12.Milich D, Liang TJ. Exploring the biological basis of hepatitis B e antigen in hepatitis B virus infection. Hepatology. 2003;38:1075–86. doi: 10.1053/jhep.2003.50453. [DOI] [PubMed] [Google Scholar]
- 13.Yang CY, Kuo TH, Ting LP. Human hepatitis B viral e antigen interacts with cellular interleukin-1 receptor accessory protein and triggers interleukin-1 response. J Biol Chem. 2006;281:34525–36. doi: 10.1074/jbc.M510981200. [DOI] [PubMed] [Google Scholar]
- 14.Messageot F, Salhi S, Eon P, Rossignol JM. Proteolytic processing of the hepatitis B virus e antigen precursor. Cleavage at two furin consensus sequences. J Biol Chem. 2003;278:891–5. doi: 10.1074/jbc.M207634200. [DOI] [PubMed] [Google Scholar]
- 15.Watts NR, Vethanayagam JG, Ferns RB, Tedder RS, Harris A, Stahl SJ, Steven AC, Wingfield PT. Molecular basis for the high degree of antigenic cross-reactivity between hepatitis B virus capsids (HBcAg) and dimeric capsid-related protein (HBeAg): insights into the enigmatic nature of the e-antigen. Journal of Molecular Biology. 2010;398:530–41. doi: 10.1016/j.jmb.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steven AC, Conway JF, Cheng N, Watts NR, Belnap DM, Harris A, Stahl SJ, Wingfield PT. Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv Virus Res. 2005;64:127–165. doi: 10.1016/S0065-3527(05)64005-5. [DOI] [PubMed] [Google Scholar]
- 17.Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3:771–80. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 18.Wasenauer G, Kock J, Schlicht HJ. A cysteine and a hydrophobic sequence in the noncleaved portion of the pre-C leader peptide determine the biophysical properties of the secretory core protein (HBe protein) of human hepatitis B virus. J Virol. 1993;66:5338–5346. doi: 10.1128/jvi.66.9.5338-5346.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nassal M, Rieger A. An intramolecular disulfide bridge between Cys-7 and Cys61 determines the structure of the secretory core gene product (e antigen) of hepatitis B virus. J Virol. 1993;67:4307–4315. doi: 10.1128/jvi.67.7.4307-4315.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schödel F, Peterson D, Zheng J, Jones JE, Hughes JL, Milich DR. Structure of hepatitis B virus core and e-antigen. A single precore amino acid prevents nucleocapsid assembly. J Biol Chem. 1993;268:1332–7. [PubMed] [Google Scholar]
- 21.Uetrecht C, Versluis C, Watts NR, Roos WH, Wuite GJ, Wingfield PT, Steven AC, Heck AJ. High-resolution mass spectrometry of viral assemblies: molecular composition and stability of dimorphic hepatitis B virus capsids. Proc Natl Acad Sci U S A. 2008;105:9216–20. doi: 10.1073/pnas.0800406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry. 1996;35:7412–7421. doi: 10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- 23.Bottcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 24.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy [see comments] Nature. 1997;386:91–4. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 25.Bourne CR, Katen SP, Fulz MR, Packianathan C, Zlotnick A. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry. 2009;48:1736–42. doi: 10.1021/bi801814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng J, Schödel F, Peterson DL. The structure of hepadnaviral core antigens: identification of free thiols and determination of the disulfide bonding pattern. J Biol Chem. 1992;267:9422–9429. [PubMed] [Google Scholar]
- 27.Wingfield PT, Stahl SJ, Williams RW, Steven AC. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry. 1995;34:4919–4932. doi: 10.1021/bi00015a003. [DOI] [PubMed] [Google Scholar]
- 28.Ceres P, Zlotnick A. Weak protein-protein interactions are sufficient to drive assembly of hepatitis B virus capsids. Biochemistry. 2002;41:11525–31. doi: 10.1021/bi0261645. [DOI] [PubMed] [Google Scholar]
- 29.Allore RJ, Barber BH. A recommendation for visualizing disulfide bonding by one-dimensional sodium dodecyl sulfate--polyacrylamide gel electrophoresis. Anal Biochem. 1984;137:523–7. doi: 10.1016/0003-2697(84)90121-0. [DOI] [PubMed] [Google Scholar]
- 30.Conway JF, Cheng N, Zlotnick A, Stahl SJ, Wingfield PT, Steven AC. Localization of the N terminus of hepatitis B virus capsid protein by peptide-based difference mapping from cryoelectron microscopy. Proc Nat’l Acad Sci USA. 1998;95:14622–7. doi: 10.1073/pnas.95.25.14622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tan SW, McNae IW, Hob KI, Walkinshaw MD. Crystallization and X-ray analysis of the T = 4 particle of Hepatitis B capsid protein with an N-terminal extension. Acta Crystallogr F. 2007;63:642–7. doi: 10.1107/S1744309107033726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cotelesage JJ, Osiowy C, Lawrence C, Devarennes SL, Teow S, Beniac DR, Booth TF. Hepatitis B Virus Genotype G forms core-like particles with unique structural properties. J Viral Hepat. 2010 doi: 10.1111/j.1365-2893.2010.01330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Conway JF, Watts NR, Belnap DM, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Characterization of a conformational epitope on hepatitis B virus core antigen and quasi-equivalent variations in antibody binding. J Virol. 2003;77:6466–6473. doi: 10.1128/JVI.77.11.6466-6473.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magnius LO, Espmark JA. New specificities in Australia antigen positive sera distinct from the Le Bouvier determinants. J Immunol. 1972;109:1017–21. [PubMed] [Google Scholar]
- 35.Ferns RB, Tedder RS. Monoclonal antibodies to hepatitis Be antigen (HBeAg) derived from hepatitis B core antigen (HBcAg): their use in characterization and detection of HBeAg. J Gen Virol. 1984;65:899–908. doi: 10.1099/0022-1317-65-5-899. [DOI] [PubMed] [Google Scholar]
- 36.MacKay P, Lees J, Murray K. The conversion of hepatitis B core antigen synthesized in E coli into e antigen. J Med Virol. 1981;8:237–43. doi: 10.1002/jmv.1890080404. [DOI] [PubMed] [Google Scholar]
- 37.Nassal M. Conserved cysteines of the hepatitis B virus core protein are not required for assembly of replication-competent core particles nor for their envelopment. Virology. 1992;190:499–505. doi: 10.1016/0042-6822(92)91242-m. [DOI] [PubMed] [Google Scholar]
- 38.Packianathan C, Katen SP, Dann CE, 3rd, Zlotnick A. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. Journal of Virology. 2010;84:1607–15. doi: 10.1128/JVI.02033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watts NR, Conway JF, Cheng N, Stahl SJ, Belnap DM, Steven AC, Wingfield PT. The morphogenic linker peptide of HBV capsid protein forms a mobile array on the interior surface. EMBO J. 2002;21:876–884. doi: 10.1093/emboj/21.5.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roseman AM, Berriman JA, Wynne SA, Butler PJ, Crowther RA. A structural model for maturation of the hepatitis B virus core. Proc Natl Acad Sci U S A. 2005;102:15821–6. doi: 10.1073/pnas.0504874102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bottcher B, Vogel M, Ploss M, Nassal M. High plasticity of the hepatitis B virus capsid revealed by conformational stress. Journal of Molecular Biology. 2006;356:812–22. doi: 10.1016/j.jmb.2005.11.053. [DOI] [PubMed] [Google Scholar]
- 42.Nassal M, Rieger A, Steinau O. Topological analysis of the hepatitis B virus core particle by cysteine-cysteine cross-linking. J Mol Biol. 1992;225:1013–1025. doi: 10.1016/0022-2836(92)90101-o. [DOI] [PubMed] [Google Scholar]
- 43.Cohn EJ, Edsall JT. Proteins, amino acids and peptides. Van Nostrand-Reinhold; Princeton, N.J: 1943. [Google Scholar]
- 44.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical Centrifugation in Biochemistry and Polymer Science. Royal Society for Chemistry; Cambridge, U.K: 1992. pp. 90–125. [Google Scholar]
- 45.Perczel A, Park K, Fasman GD. Analysis of the circular-dichroism spectrum of proteins using the convex constraint algorithm - a practical guide. Analytical Biochemistry. 1992;203:83–93. doi: 10.1016/0003-2697(92)90046-a. [DOI] [PubMed] [Google Scholar]
- 46.Sreerama N, Woody RW. A self-consistent method for the analysis of protein secondary structure from circular dichroism. Anal Biochem. 1993;209:32–44. doi: 10.1006/abio.1993.1079. [DOI] [PubMed] [Google Scholar]
- 47.Chang CT, Wu CS, Yang JT. Circular dichroic analysis of protein conformation: inclusion of the beta-turns. Anal Biochem. 1978;91:13–31. doi: 10.1016/0003-2697(78)90812-6. [DOI] [PubMed] [Google Scholar]
- 48.Conway JF, Steven AC. Methods for reconstructing density maps of “single particles” from cryoelectron micrographs to subnanometer resolution. J Struct Biol. 1999;128:106–118. doi: 10.1006/jsbi.1999.4168. [DOI] [PubMed] [Google Scholar]
- 49.Yan X, Sinkovits RS, Baker TS. AUTO3DEM--an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol. 2007;157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heymann JB, Belnap DM. Bsoft: Image processing and molecular modeling for electron microscopy. J Struct Biol. 2007;157:3–18. doi: 10.1016/j.jsb.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 51.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 52.Belnap DM, Watts NR, Conway JF, Cheng N, Stahl SJ, Wingfield PT, Steven AC. Diversity of core antigen epitopes of hepatitis B virus. Proc Nat’l Acad Sci USA. 2003;100:10884–9. doi: 10.1073/pnas.1834404100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.