

Abstract
Filamentous phages are now the most widely used vehicles for phage display, and provide an efficient means for epitope identification. However, the peptides they display are not very immunogenic because they normally fail to present foreign epitopes at the very high densities required for efficient B-cell activation. Meanwhile, systems based on virus-like particles (VLPs) permit the engineered high-density display of specific epitopes, but are incapable of peptide library display and affinity selection. We developed a new peptide display platform based on VLPs of the RNA bacteriophage MS2. It combines the high immunogenicity of MS2 VLPs with the affinity selection capabilities of other phage display systems. Here we describe plasmid vectors that facilitate the construction of high complexity random sequence peptide libraries on MS2 VLPs and that allow control of the stringency of affinity selection through the manipulation of display valency. We used the system to identify epitopes for several previously characterized monoclonal antibody targets, and showed that the VLPs thus obtained elicit antibodies in mice whose activities mimic those of the selecting antibodies.
Keywords: virus-like particle, phage display, epitope vaccine
Introduction
Phage display is a well-established technology for the identification of peptide epitopes. Filamentous phages are the most widely used platforms for phage display and affinity selection using monoclonal antibodies has enabled the isolation from random sequence peptide libraries of numerous epitopes and epitope mimics (i.e. mimotopes). For example, filamentous phage display has identified the epitopes of numerous monoclonal antibodies1, including rituximab (CD20) 2; 3; 4, Herceptin (Her-2/neu) 5, and cetuximab (EGF-R) 6; 7.Mimics of discontinuous conformational epitopes, and even of non-peptide epitopes (e.g. carbohydrates) have been found, and in some cases have been used as immunogens to elicit antibodies that recognize the native epitope on the original antigen. These studies raise the possibility that epitopes identified by phage display might also be used as the basis for vaccines.However, as a tool for vaccine development filamentous phage display has deficiencies. For example, filamentous phages do not readily permit the display of foreign sequences at the high densities necessary for potent immunogenicityand generally are unable to elicit high titer antibodies to the foreign antigens they display.Therefore, peptides identified by phage display must usually be produced synthetically and then linked to a carrier protein that necessarily displays the epitope in a structural context unrelated to the one in which it was selected. It is frequently observed that the affinities of peptides for their targets are highly dependent on their structural contexts, a fact reflected in the observation that isolated synthetic peptides seldom maintain the affinity they showed when present on the phage particle, and often lose the ability to induce antibodies whose characteristics mimic those of the selecting antibody.
In contrast, peptides displayed on RNA bacteriophage virus-like particles (VLPs)are presented in a densely repetitive structure highly stimulatory to the immune system, especially to B cells 8, and are thereforehighly immunogenic. The enormous enhancement of immunogenicity when epitopes are presented polyvalently on VLPs has been well documented (see, for example, refs. 9; 10).We have created a system based on bacteriophage MS2 that combines the affinity-selection capabilities of conventional phage display with the potent immunogenicity of a VLP.In previous reports, we showed that a single-chain dimer version of coat protein shows high tolerance to foreign peptide insertions in one of its surface loops, making possible the display of such sequences at high density on the surface of MS2 VLPs8; 11; 12. Moreover, these VLPs encapsidate their own mRNA, making possible the amplification by reverse transcription and PCR of sequences affinity-selected from peptide libraries. Here we describe the creation of plasmid vectors that facilitate the construction of high complexity random sequence peptide libraries, and which permit the control of the stringency of affinity selection by controlling peptide display valency. Further, wedemonstrate the utility of MS2 VLPs foraffinity-selection using several previously characterizedmonoclonal antibodies.
Results
Testing alternative peptide insertion sites in MS2 coat protein
The MS2 coat protein AB-loop consists of gly13, gly14 and thr15, and is our preferred site of peptide display. We previously made insertions at a KpnI site introduced by two silent mutations in codons 14 and 15. Insertions result in duplication of amino acids 14 and 15 at the insertion boundaries. Since the insertions are flanked on the N-terminal side by residue 15 and by amino acid 14 on the C-terminal side, we call this the 15/14 insertion mode. Insertions of 6, 8 or 10 NNY triplets in this mode yielded libraries in which about 90-95% of recombinant proteins were able to repress translation and to form a VLP8. From the beginning, however, we recognized that other AB-loop insertion modes were possible, and that they might be more desirable than 15/14, which necessarily flanks the insertion with several small amino acids that might confer an undesired flexibility to the polypeptide chain at these points. Therefore, before constructing random sequence libraries for affinity-selection, we wanted to test some alternative insertion modes.
The plasmid p2MS3 contains a single-chain dimer with a unique SalI site near the AB-loop of its downstream half. It is highly similar to the previously described p2MCTk3 8. We constructedlibraries with insertions of eight copies of the NNY triplet between residues 13 and 14 (the 13/14 mode), between 13 and 16 (the 3/16 mode), and between 12 and 17 (the 12/17 mode), and tested them for the frequency of clones producing correctly folded coat protein by the translational repression and VLP assembly assays. Note that NNY (where N = A, C, G, T and Y = C, T) has the possibility of encoding 15 amino acids, but no stop codons. On a second plasmid (pRZ5) a DNA sequence encoding the translational operator of the MS2 replicase cistron is fused to a version of the E. colilacZ gene lacking its own translation initiation codon13. Since the two plasmids are members of different incompatibility groups, and because they express resistance to different antibiotics, they are readily maintained together in the same bacterial strain, where the coat protein expressed from p2MS3 translationally represses the synthesis of ß-galactosidase expressed from pRZ5. As we showed previously 8, merely counting the proportion of white versus blue colonies in a library plated on medium containing x-gal provides a simple means of determining the frequency with which random sequence AB-loop insertions disrupt coat protein folding. The absence of stop codons in an NNY library eliminates a trivial cause of loss of coat protein function. The ability of coat protein from white colonies to assemble into VLPs is readily confirmed by agarose gel electrophoresis of crude cell extracts. The results (Figure 1) demonstrate that these alternative display modes show insertion tolerances at least as high as those of the 15/14 configuration8. Judging from the percentage of clones competent for translational repression, the 13/14 mode was tolerant to about 98-99% of 8mer insertions, while 13/16 and 12/17 tolerated 98% and 92%, respectively. When lysates from cells grown from two-dozen repressor-competent (i.e. white) colonies of each insertion mode were subjected to agarose gel electrophoresis, virtuallyall yielded detectable VLPs.
Figure 1.
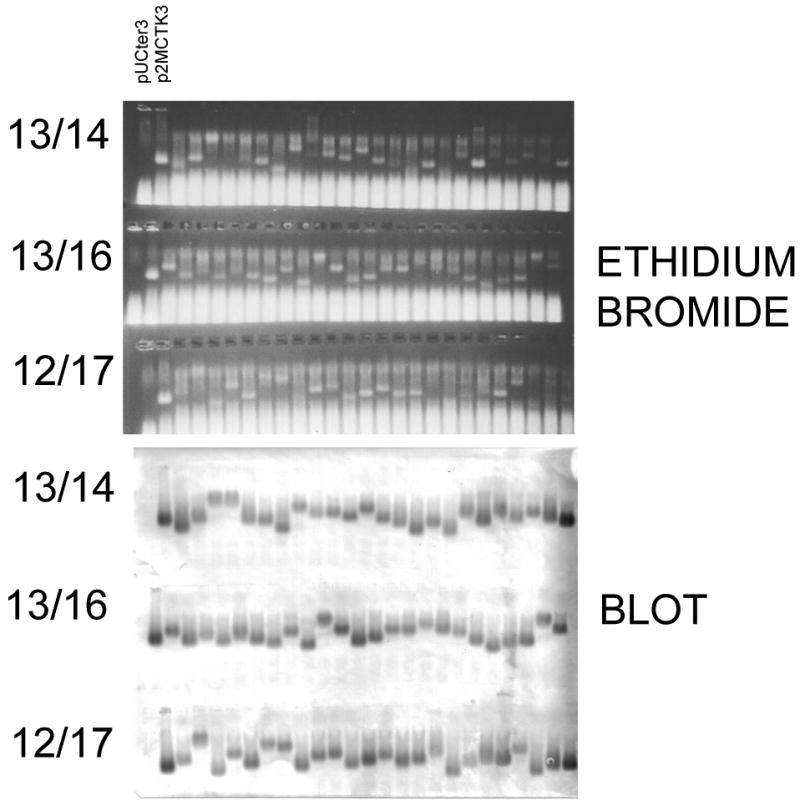
The MS2 single chain coat protein is broadly tolerant of random sequence peptide insertions in the 13/14, 13/16 and 12/17 as indicated by the presence of VLPs in crude cell lysates of a number of individual clones picked randomly from the three different peptide 8-mer libraries. The top panel shows gels stained with ethidium bromide. In the bottom panel, the gels were blotted to nitrocellulose and probed with anti-MS2 serum. Lanes marked pUCter3 and p2MCTK3 denote control lysates that respectively lack and contain MS2 VLPs lacking an inserted peptide.
We emphasize that the background of clones without an inserted peptide is extremely low in our libraries. Control ligations lacking the inserted fragments typically yielded at least 1000-fold fewer colonies upon transformation of bacteria. Moreover, we isolated plasmids from each of the several dozen white colonies we picked for agarose gel electorphoresis (Figure 1) andfound that all contained an insertion.
The pDSP1 and pDSP1(am) plasmid vectors
To facilitate the construction of random sequence peptide libraries in a system that expresses coat protein at high levels, we created the plasmid vectors illustrated in Figure 2. Each contains a single-chain dimer sequence expressed under control of the T7 transcription initiation and termination signals. The plasmids also contain ColE1-type replication origins and each expresses resistance to kanamycin. The pDSP1 vector was produced to facilitate the cloning of random peptide sequence insertions in the AB-loop sequence of the downstream half of the coat protein single-chain dimer by simply replacing the Sal I – Bam HI fragment (Figure 2) with a PCR fragment generated using a 5′ primer that attaches a SalI site and a random 30-nucleotide sequence to amino acid 16 of the coat sequence. The 3′ primer anneals downstream of the Bam HI site. Other designs that insert a different number of amino acids in slightly different locations of the AB-loop have also been employed using the scheme shown here. Amplification produces a fragment that can be simply ligated to pDSP1 cleaved with SalI and BamHI.
Figure 2.

The basic features of the plasmids, pDSP1 and pDSP62, used in this study for library construction and synthesis of VLPs for affinity selection.The plasmid pDSP1 is suitable for library construction by simple insertion of a Sal I – Bam HI fragment to which random sequences have been attached by PCR. On the other hand, pDSP62 allows extension of a mutagenic primer on a single-stranded circular template to introduce insertions by the site-directed mutagenesis method of Kunkel et al. 15 as implemented for library construction in filamentous phage by Sidhu et al. 14. The upstream half of the single-chain dimer contains a sufficient number of silent mutations (codon juggled) that a mutagenic primer can be directed to anneal specifically to the downstream half.
VLPs synthesized from pDSP1 display foreign peptides at 90 per particle, but it is well-known from filamentous phage display that high-valency display complicates the selection of high affinity ligands, because even peptides with low intrinsic affinity bind tightly when presented multiply(i.e. avidity vs. affinity). To control the average valencyof VLP peptide display we constructed pDSP1(am), a variant of pDSP1 that contains an amber codon at the junction between the two halves of the single-chain dimer sequence (Figure 3A). The plasmid we call pNMsupA produces an alanine-inserting amber suppressor tRNA from the E. colilac promoter on a chloramphenicol resistant plasmid with a p15A origin of replication. In the absence of the suppressor, ribosomes terminate translation at the amber codon and produce a VLP containing only wild-type coat protein. In suppressor presence a few percent of ribosomes read through the terminator to produce the single-chain dimer with its peptide passenger. This relatively small quantity of single-chain dimer co-assembles with the excess wild-type protein to produce a VLP that displays an estimated average of about 3 peptides per particle (Figure 3A). We describe below the construction of libraries designed to randomize sequences surrounding a tripeptide sequence (DKW) representing the core of the epitope recognized by the anti-HIV monoclonal antibody, 2F5. Since all the peptides in the population contain the DKW core, most (if not all) are expected to exhibit some affinity for the antibody. To test the effects on relative antibody binding we measured the ability of a synthetic peptide containing the wild-type 2F5 epitope to compete with the VLP library displaying the DKW-containing peptide at both high and low valency. Comparing the ability of the peptide to inhibit binding of the low- and high-density peptide populations should serve as a measure of their relative abilities to bind the antibody. Figure3B reveals about a 1,000-fold difference in the peptide concentration inhibiting half of the maximal binding.
Figure 3.

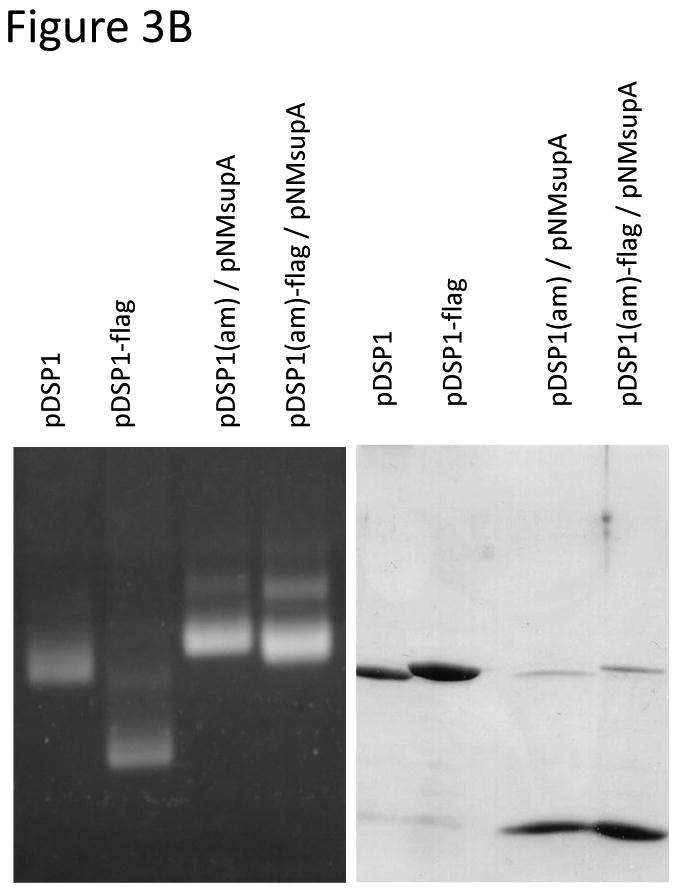
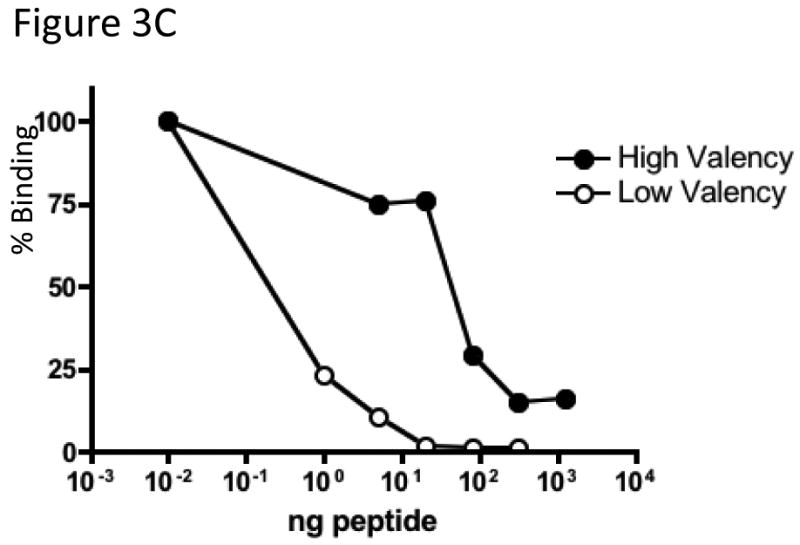
A. To reduce peptide display valency an amber codon was introduced at the junction of the two halves of the single-chain dimer in pDSP1(am) or pDSP62(am). These plasmids produce a large amount of wild-type coat protein, but occasional readthrough of the stop codon, mediated by a suppressor tRNA expressed from pNMsupA, produces small amounts of the single-chain with its guest peptide. The two proteins coassemble into a mosaic VLP whose peptide display valency depends on the efficiency of suppression. B. In the left panel an ethidium bromide-stained agarose gel of purified VLPs shows that the Flag-displaying VLP migrates faster due to the presence of aspartic acid residues in the peptide. A slight shift in the mobility of pDSP1(am)-flag VLPs is consistent with the presentation of some small number of peptides. The same particles were subjected to SDS gel electrophoresis and stained with coomassie brilliant blue (right panel). The relative amounts of wild-type and single-chain dimer species indicate the level of nonsense suppression and allow us to estimate an average valency of about three peptides per VLP. C.Low-valency particles displaying the DKW tripeptide bind less tightly than high-valency VLPs to a monoclonal antibody. Libraries displaying DKW-containing peptides (see text for details) at high- and low-valency were compared by competition ELISA for their abilities to compete with an epitope-containing peptide for binding to mAb 2F5.
The pDSP62 and pDSP62(am) plasmids
The scheme described above for pDSP1 and pDSP1(am) works well for the production of libraries with moderate complexities (e.g. 107 -108 members). However, this approach depends on gel purification of DNA fragments and their subsequent ligation before introduction into E. coli. These procedures are inconvenient to scale up, and ligation reactions can yield significant amounts of unproductive side products. A more efficient approach has been described by Sidhu et al. 14, who took advantage of the site-directed mutagenesis method of Kunkel et al. 15 to produce more complex libraries in filamentous phage. The idea is to prepare single-stranded, dUTP-substituted phagemid templates from a dut-, ung- host after super-infection with a M13 helper phage (e.g. M13K07). Random sequence insertions are produced by annealing a mutagenic primer to the single-stranded circular template, which is then converted to a covalently closed double-stranded circle by the action in vitro of DNA polymerase and DNA ligase. The reaction can easily be scaled up to produce tens of micrograms of cccDNA product, which is then introduced by electroporation into ung+E. coli, where the dUTP-containing parental strand is preferentially destroyed. In this way high rates of insertion (as much as 90%) are obtained in libraries containing as many as 1011 individual members. To adapt pDSP1 for this approach: (i) We introduced a M13 origin of replication into pDSP1 for production of single-strand phagemid DNAs, and (ii) replaced the upstream half of the single-chain dimer with a synthetic “codon-juggled” sequence containing the maximum possible number of silent mutations. This ensures that annealing of the mutagenic primer is directed to only one half of the single-chain dimer. These changes resulted in the production of derivatives of pDSP1 and pDSP1(am) we call pDSP62 (see Figure 2) and pDSP62(am). (iii) Since these plasmids already confer resistance to kanamycin, we were unable to use the usualkanamycin resistant M13KO7 as helper phage. This necessitated the construction of M13CM1, which replaces kanamycin with the chloramphenicol resistance marker of pACYC184. Using these methods random sequence peptide libraries with complexities in excess of 2-3×1010 have been easily constructed. With a little effort libraries withsignificantly higher complexities are attainable.
Flag epitope selections
To establish the efficacy of affinity selection on the MS2 VLP platform we used the well-characterized M2 anti-Flag monoclonal antibody as a target, and a library constructed in pDSP1 that contains ten NNS triplets inserted in the 13/16 mode. [It should be noted that libraries based on NNS triplets can encode all 20 amino acids, unlike the NNY libraries used above in experiments testing the peptide insertion tolerance of the coat proetin AB-loop.] This particular library contained about 108 independent clones and displayed foreign peptides at high valency. Since then, more complex libraries have been constructed using pDSP62 (see above), but the pDSP1 library was deemed sufficiently complex to give a reasonable probability of encountering some version of the Flag epitope. The first selection round was conducted against 250ng of the antibody immobilized by adsorption to plastic wells, with an estimated ten-fold mass excess of VLPs over antibody molecules. After extensive washing, bound VLPs were eluted and then subjected to reverse transcription and PCR using the primers described in Materials and Methods. The PCR products were digested with SalI and BamHI and cloned in pDSP62 for production of VLPs for use in round 2. In this, and in all subsequent rounds, cloning of the selectants yielded at least 5 × 106 independent clones. The second selection was conducted under the same conditions as round 1. Products of the second and third rounds were cloned in pDSP62(am) and the VLPs were produced in the presence of the amber suppressor (pNMsupA) described above. Thus, peptides were displayed at high-valency in rounds 1 and 2 and at low-valency in rounds 3 and 4. In the fourth round the amount of antibody was reduced to 50ng, so that VLPs were present at about 50-fold excess compared to antibody. Fourth round selectants were cloned in pDSP62 for high valency display in anticipation of overproduction and purification of VLPs for immunization experiments. Sequences of a few selectants from each round are shown in Figure 4. Those obtained in early rounds show only limited similarity to the known Flag epitope, DYKDDDDKL, but certain key elements arealready evident, including especially the YK dipeptide. By round three, all the sequences show the DYK element together with at least one downstream D. By round four only one sequence was obtained among the 7 clones we sequenced, and of all the clones we characterized, it had the greatest sequence similarity to the Flag epitope.
Figure 4.

Sequences of some peptides obtained after selection with the Flag-specific M2 mAb. Six peptides from rounds one and two already show some features of the wild-type epitope. All fiveof the round three sequences we determined contain the DYK tripeptide and at least one downstream D residue. All seven peptides sequenced from round four show a single sequence, which, of all the sequences encountered, is the best match to the Flag epitope.
Affinity selectionusing a mAb against anthrax protective antigen
Protective antigen protein is the principle component of current anthrax vaccines. A number of neutralizing epitopes have been identified, including one with the sequence ASFFD, which maps to the antigen's 2ß2-2ß3 loop 16; 17. Although the F20G77 epitope has been mapped using overlapping peptides that scan through the sequence of protective antigen, it should be emphasized that the essential elements of the ASFFD sequence have not been previously determined either by mutation or phage display, so we expected to obtain selectants with sequence similarity to the epitope, but were unsure which elements would be conserved. Selections were conducted in the same manner as the Flag selections described above, using the same random 10mer library. A number of clones from each selection were subjected to DNA sequence analysis and the peptides they encode are shown in Figure 5. The first and second round selectants represent a complex population containing many members with somesimilarity to the ASFFD sequence. In particular, the A, S and D elements were already present. By rounds three and four,more obvious similarities to the mapped epitope have appeared, with one of the sequences (ASYFD) yielding a near perfect fit (only one conservative substitution), and the vast majority containing the apparently crucial FD dipeptide recognition element. Combining the round 3 and 4 sequences suggests the consensus for the F20G77 epitope: (A/S)(S/T)(F/L/A)FD. Note also the persistent presence of P two residues before the start of this pentamer consensus.
Figure 5.

Peptides obtained after selection with a monoclonal antibody directed to the ASFFD epitope of anthrax protective antigen. The round three and four sequences were combined to derive the consensus sequence shown.
Construction of DKW libraries and affinity selection using the 2F5 mAb
Broadly neutralizing antibodies for HIV occur only rarely. One approach to control of HIV would be to find a vaccine capable of eliciting antibodies that mimic their activities. The monoclonal known as 2F5 is one such antibody. It recognizes a previously mapped epitope contained within the sequence ELDKWAL, the essential feature of which is the DKW sequence. The role of surrounding amino acids seems to be to favor a conformation that presents the DKW sequence in a particular ß-turn-like conformation recognized by the antibody. We constructed libraries of 8-amino acid insertions, comprised of the DKW sequence surrounded by random amino acids. Four separate libraries were constructed following the designs described in the Materials and Methods, which vary the position of the DKW sequence within the insertion. Specifically, the libraries were of the following four types:Library I - XDKWXXXX; library II - XXDKWXXX; library III - XXXDKWXX; and library IV – XXXXDKWX.Since the DKW tripeptide is present in our library from the beginning, most members of the initial population are likely to interact with 2F5 at some level. Therefore, all four rounds of selection were conducted at low valency. To enhance the stringency of selection the reactions contained varying amounts of a competing linear synthetic peptide, increasing to a maximum of 5ug peptide in the fourth round. Otherwise, the conditions of selection were the same as those described above. The progress of selection was confirmed by competition ELISA in which we measured the ability of each pool of VLPs to bind to 2F5 in the presence of different concentrations of the 2F5-binding competitor peptide (Figure 6). Each round of selection resulted in the generation of a population of VLPs with higher avidity for 2F5.
Figure 6.
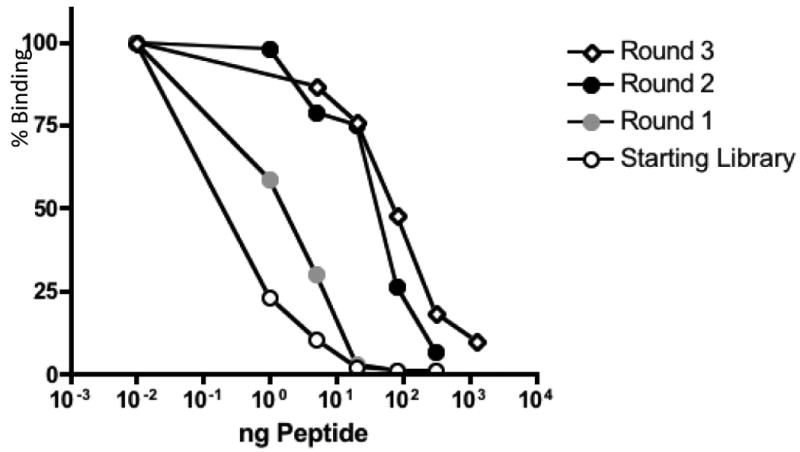
The progress of affinity-selection is demonstrated by the increasing ability of VLPs from rounds one through four to compete with an epitope binding peptide for binding to the 2F5 mAb. Pools of selected VLPs (all at low valency) were bound to 2F5 in ELISA in the presence of increasing concentrations of the competitor peptide LLELDKWASLWNWFD.
Sequence analysis revealed the recovery of two different peptide sequence families in rounds 3 and 4 (Figure 7). Both show certain obvious similarities to the epitope in HIV gp41. Family 1 emphasizes the importance of the A and L residues, which appear at positions one and three amino acids after the DKW core. This family also frequently preserves an acidic residue in the position just preceding DKW, but in our case it is always D rather than the E encountered in the wild-type sequence. It nearly also has a G in the first position of the peptide. Family 2 presents a slightly different picture. Like Family 1 it universally conserves the A residue immediately following the DKW, but it has other characteristics it doesn't share either with Family 1, or with the wild-type epitope itself, even though these features appear repeatedly in a large number of individual Family 2 members. For example, Y always appears in the first position immediately preceding the DKW core. P nearly always appears three amino acids after DKW. Several other residues appear at much higher than expected frequencies, allowing the identification of the consensus sequence shown in the figure. Notably, although the DKW sequence appears in a variety of positions in round 1 and 2 selectants, its location is always restricted to residues 3-5 or to 2-4 in Families 1 and 2 respectively. These differences may reflect peculiar requirements for 2F5 recognition of the epitope in the specific environment of the AB-loop.
Figure 7.

Peptides obtained after affinity selection with the broadly neutralizing anti-HIV monoclonal antibody, 2F5.Consensus sequences for the two families were determined from round four peptides.
Immunogenicity of VLPs selected using monoclonal antibodies against Flag and Protective Antigen
To test the immunogenicity of selected VLPs, groups of mice were immunized with a Round 4 Flag selectant (Rd4 Flag: GDYTDYKSDD), a Round 4 F20G77 selectant (Rd4 PA: GSAVASFYDR), or with a negative control (wild-type MS2 VLPs). As shown in Figure 8, both selected VLPs elicited appropriate antibody responses. We had previously cloned the wild-type Flag epitope into the coat protein of bacteriophage PP7. Because anti-MS2 antibodies do not cross-react with PP7, we used Flag-PP7 VLPs to assess whether the Flag selectants could elicit IgG that specifically binds to the Flag epitope. As shown in Figure 8A, sera from mice immunized with the Round 4 Flag selectant, but not sera from mice immunized with wild-type MS2 VLPs, bound to Flag-PP7 VLPs. As an additional control, we determined that Flag selectant sera do not bind to wild-type PP7 VLPs (data not shown). Similarly, mice immunized with the F20G77-selected VLPs elicited antibodies that could bind to recombinant anthrax protective antigen (Figure 8B). These assays gave end-point dilution titers of 103-104. Using standard curves generated with the monoclonal antibodies, we estimate that these titers correspond to serum antibody concentrations of about 2-3 μg/mL. These data demonstrate that recombinant VLPs selected using monoclonal antibodies can induce antibodies that can cross-react with the cognate antigen.
Figure 8.
Serum antibody responses in mice immunized with VLPs taken from round four of selections against the Flag M2 and anthrax protective antigen F20G77 mAbs. A. shows the response to VLPs displaying the sequence GDYTDYKSDD, and B shows the response to VLPs with GSAVASFYDR.
Immunization of mice with representative VLPs from either family of the 2F5 selectants yielded high titers of antibodies that in ELISA recognized a synthetic peptide containing the epitope, but failed to detectably interact with native gp41 (results not shown).
Discussion
The purpose of this work was to establish an expression system that facilitates the production of peptide libraries on MS2 VLPs, to develop methods for recovery of affinity selected sequences from those libraries, and to demonstrate whether these methods faithfully recover known epitopes. The plasmid pDSP1 has features that make it simple to construct libraries through procedures like that in Figure 2, in which a fragment containing synthetic sequences is generated by PCR and then the complete single-chain dimer gene is reconstructed by conventional cloning between the SalI and BamHI sites of the plasmid. These methods are adequate for libraries of moderate complexity, and the libraries utilized for the present work were constructed in this manner. The plasmid called pDSP62, however, makes possible the utilization of the methods described by Sidhu et al. to allow production of libraries with complexities that may exceed 1011 individual members14. It differs from pDSP1 by the presence of an origin of replication from phage M13, which allows the production of single-stranded phagemid DNA by superinfection with a chloramphenicol resistant helper phage we call M13CM1. In this scheme, library construction relies on the site-directed mutagenesis method of Kunkel, et al. 15, in which mutagenic primers are annealed to dUTP-substituted template DNA and extended with DNA polymerase in the presence of T4 DNA ligase to produce closed circular DNA. These methods are easily scaled up to the level of 10-20 ug, making it straightforward to obtainlarge numbers of independent transformants by electroporation. We showed previously that coat protein folding best tolerates foreign peptides when they are inserted into only the downstream half of the single-chain dimer. The presence in pDSP62 of a “codon juggled” version of coat protein in the upstream half allows the design of mutagenic primers that anneal specifically to the downstream sequence.
It is well known that peptide display valency is an important consideration in any effort to identify high-affinity ligands by phage display, because when displayispolyvalent it is difficult to discriminate intrinsically high-affinity interactions from weaker ones that benefit from simultaneous interaction at many sites (i.e. affinity vs. avidity).A generally accepted approach in filamentous phage display is to conduct early rounds of selection using polyvalent display, thus enriching for peptides with some minimal affinity for a target, while maintaining substantial diversity in the selectant population18; 19. Switching to low valency in subsequent rounds allows the identification of the strongest binders. In filamentous phages this is typically achieved by fusing peptides to pVIII (high copy) or pIII (low copy). Our system offers a similarly simple means of controlling display valency, but with the added benefit that we can modulate valency without altering the structural context of the displayed peptide. We constructed variants of the pDSP1 and pDSP62 vectors with UAG nonsense codons at the junctions of the two halves of the single-chain dimer. In particles expressed from pDSP1(am) and pDSP62(am), the average display valency is controlled by the efficiency of nonsense suppression mediated by an alanine-inserting suppressor tRNA 20; 21, which we expressed from a plasmid we call pNMsupA. This plasmid uses the origin of replication and chloramphenicol resistance determinant of pACYC18422, making it easy to stably maintain the plasmid in strains that also contain pDSP1(am) or pDSP62(am). We estimate that under the conditions we use, pNMsupA supports the production of particles with an average of about three peptides per VLP. Although we have not yet determined whether this is an optimal copy number,the results of affinity selection with the anti-Flag monoclonal antibody show that the method readily identifies epitopes that are very close matches to the native sequence. Moreover, the binding to 2F5 of low valency DKW VLP libraries is about 1,000-fold more sensitive to competition from a synthetic epitope-containing peptide than is a high-valency DKW library (Figure 5B).
The M2 anti-Flag monoclonal antibody offered an opportunity to compare the results of MS2 VLP selections from those obtained by filamentous phage display. Sequences obtained from each selection round show a clear progression toward a better sequence match with the known epitope. Rounds one and two yielded selectants possessing some of the key features of the Flag epitope, and by round 3, the selection seems nearly complete. At that point a number of sequences are still present, but all show high similarity to the native epitope. After round four, all seven sequenced clones contained the same peptide, which was also the closest match to the wild-type epitope of all the sequences we found. It was present as a minority species after round three, but became the predominant, if not the only peptide in round four. New England Biolabs published the sequences of a series of third round selectants obtained from a 7mer library displayed on pIII(five copies per virion), and the same M2 monoclonal antibody (see NEB Transcript, Summer 1996).They arrived at the consensus sequence DYKXXD. Combining the results of our rounds 3 and 4 (Figure 6) suggests a preferred sequence that could be written DYKSDD. Although there are a number of experimental differences that make it difficult to make direct comparisons, it seems clear that the results of MS2 VLP display are essentially comparableto those obtained with filamentous phage.In fact, the sequences of our selectants more closely match that of the wild-type epitope than those reported previously.
The anthrax F20G77 selectionsgave similar results (Figure 5). In this case no prior phage display result or mutational analysis is available for comparison, but the peptides we found have obvious similarities to the native ASFFD sequence previously mapped using overlapping peptides derived from protective antigen. Changes that accompany the selection process give us some clues as to the essential features of the epitope. In early rounds the A, S and D components are already present in most isolates, suggesting that they (and especially the D) are important elements for establishing basic recognition by the antibody. In later rounds the A is still present in some isolates, but S increasingly predominates there. The Fresidue that immediately precedes the Donly appears in rounds three and four, suggesting that it plays an important role in higher affinity binding. Thewild-type epitope's other F, on the other hand, appears in only two of the peptides, and only in round four. In the absence of affinity measurements we cannot say what its contribution to binding is, or whether its appearance in round four indicates a trend, but it is notable that throughout the selection, hydrophobic residues predominate at this position, and that by the last stage leucine has emerged as the clear favorite. Interestingly, our selectants also show a strong preference for a proline residue two amino acids upstream of the ASFFD homology. This is not a feature of the natural protective antigen sequence and may reflect structural requirements for presentation of the peptide in an optimal conformation for antibody binding in the context of the coat protein AB-loop.
The 2F5 selectants were obtained from a biased library where the core epitope sequence (DKW) was present(albeit in four distinct locations)in all clones of the starting population. Our object was to identify peptides able to present the DKW sequence in a structural context that favored strong antibody binding. Remember that in this case all four rounds were conducted at low valency. As with the other selections, the populations became less complex as the selection proceeded, until two different sequence families were obtained. The fact that each family contains a number of members suggests that they were not obtained by simply passing through a selection bottleneck that arbitrarily restricted the population to these two general types. Family 2 showed more extensive similarity with the native epitope sequence than did Family 1. This was mainly evident from the common occurrence in Family 2 of a leucine at the third residue downstream of the DKW. Family 1, on the other hand, shareswith the native epitope the presence of an alanine just after the DKW core,butyields a very clear consensus sequence that deviates from it significantly in allthe other positions. Especially notable is the universal presence of tyrosine preceding DKW and a very strong preference for proline at the third position C-terminal to the tripeptide core.
Immunization of mice with representative VLPs from either of the two families of 2F5 selectants yielded high titers of antibodies that in ELISA recognized a synthetic peptide containing the epitope, but they failed to detectably interact with native gp41 (results not shown). Two possible explanations come to mind: (i) The environment of the coat protein AB-loop may be inappropriate for mimicking the relevant native conformation of the epitope. In its complex with antibody, the epitope peptide adopts a predominantly extended conformation, with the DKW sequence forming a kink in the form of a type I ß-turn in the middle of the peptide 23. This extended conformation would seem to be inconsistent with the constraints imposed by the AB-loop. (ii) The human-derived 2F5 antibody itself has a peculiar structure characterized by an extended CDR3 region 24. Antibodies of this type may be extremely rare or even non-existent in the mouse repertoire.
It is frequently observed that peptides optimized by affinity selection in one structural context may lose much of their binding affinity when moved to new contexts (see 25, for examples). We hope that the ability to directly select peptides specific for monoclonal antibodies on a platform with high intrinsic immunogenicity will provide a better means for identification of mimotope-based vaccines by obviating the necessity to conjugate synthetic versions of affinity-selected peptides to more immunogenic carriers. As shown here, selected VLPs are potent immunogens, capable of inducing an antibody response against the target of the selecting monoclonal antibody, at least when that antibody recognizes a linear epitope. Future work will determine whether the MS2 VLP platform represents an improved platform for the identification effective mimicsof more complex epitopes.
Materials and Methods
Assessing coat protein function by translational repression and VLP assembly
We have described previously the use of in vivo translational repression assays and of gel electrophoresis of VLPs to confirm the correct folding and assembly of coat protein recombinants 8; 13. Briefly, coat protein normally represses viral replicase synthesis in infected cells by inhibiting ribosome binding the replicase cistron. Functional coat proteins expressed from the lac promoter on plasmids derived from pUC119 can repress translation of a replicase-ß-galactosidase fusion protein expressed from a second, compatible plasmid, yielding white colonies on medium containing x-gal (4-chloro-3-indolyl-ß-D-galctoside). Blue colonies indicate a failure of the protein to properly fold. The presence of VLPs in crude cell lysates can be determined by electrophoresis on agarose gels, which are stained with ethidium bromide to reveal the RNA-containing particle and then transferred to nitrocellulose for probing with anti-MS2 serum 8; 26.
Construction of plasmids, phages and random sequence libraries
The plasmids and phages described here were constructed using standard molecular biology methods and have the characteristics described in the text and illustrated in Figure 2. Briefly, pDSP1, pDSP62 and their derivatives contain the phage T7 promoter and terminator regions of pET3d, and the kanamycin resistance gene and replication origin of pET9a (from Novagen). In a precursor common both to pDSP1 and pDSP62, an unwanted Sal I site and other nearby extraneous plasmid sequences were removed by Bal 31 deletion. Compared to pDSP1, pDSP62 contains two additional features. The first is the M13 origin of replication taken from pUC119, and the second is the replacement of the upstream half of the single-chain dimer sequences with a synthetic “codon-juggled” version of coat protein. This sequence was designed using the web-based program GeneDesign available at http://genedesign.thruhere.net/gdo/index.html, and synthesized by assembly PCR from synthetic oligonucleotides 27. The detailed structures of the plasmids are available from the authors upon request.
The plasmids known as pDSP1(am) and pDSP62(am) were constructed by site-directed mutagenesis of pDSP1 and pDSP62 to introduce an amber codon at the junction between the two halves of the single-chain dimer. To allow for low level suppression of the stop codon, we constructed pNMsupA, which uses the replication origin and chloramphenicol resistance of pACYC18422, and the lac promoter of pUC19 to express an alanine-inserting amber suppressing tRNA 20; 21.
The helper phage called M13CM1 was constructed from M13K07 by replacement of the kanamycin resistance gene with the chloramphenicol resistance determinant of pACYC184, taking advantage of conveniently situated Xho I and Sac I sites in the M13K07 sequence.
The plasmid vectors described here may be requested from the authors.
Libraries and production of VLPs
To test the tolerance of coat protein to peptide insertions between residues 13 and 14, between13 and 16, or between 12 and 17 (i.e. the 13/14, 13/16 and 12/17 insertion modes) we created upstream PCR primers that introduced eight repeats of the NNY triplet at the desired sites. Each preserves the coat gene's native Sal I site.
13/14: CCCGTCGACAATGGC(NNY)8GGAACTGGCGACGTGACTGTC
13/16: CCCGTCGACAATGGC(NNY)8GGCGACGTGACTGTCGCCCCA
12/17: CCCGTCGACAAT(NNY)8GACGTGACTGTCGCCCCAAGC
A downstream primer was produced that annealed to plasmid sequences (p2MS3 is a pUC119 derivative) downstream of the Bam HI site. The products of PCR were digested with Sal I and Bam HI and inserted between these sites in p2MS3, whose properties are similar to the previously described p2MCTK3 8 and are further described in the text. The resulting libraries were introduced by transformation into strain CSH41F-(pRZ5) 13 and the percentage of blue vs. white colonies was determined by counting a few hundred colonies. At least 1,000-fold fewer colonies were obtained from ligation reactions lacking the PCR-generated fragment and restriction digests of plasmid minipreps from several dozen individual members of each of these libraries confirmed that virtually all colonies contained a foreign peptide sequence.
For affinity-selection experiments, two different libraries were constructed in pDSP1 using a PCR/cloning scheme similar to the one described above. First, a random sequence 10-mer library with 5 × 108 individual recombinants was constructed by inserting ten copies of the triplet NNS (N=A, C, G, T and S=G, C) in the 13/16 mode.Note that these NNS libraries are distinct from the NNY libraries described above, which can encode only 15 of the 20 amino acids. These NNS libraries have the ability to encode all 20 amino acids. Plasmid libraries were produced and amplified by electroporation of strain 10G (from Lucigen), and then VLP libraries were produced by introduction of the amplified libraries into C41(DE3) (also from Lucigen). Second, libraries of DKW-containing 8-mer peptides were introduced by a similar method into pDSP1, also in the 13/16 mode. We actually created four different DKW libraries, each of which places the tripeptide core sequence in a different position within an otherwise randomized 8mer peptide. They were constructed to display the following sequences: Library I - XDKWXXXX; library II - XXDKWXXX; library III - XXXDKWXX; and library IV - XXXXDKWX.Transformation of bacterial strain 10G (Lucigen, Inc.) with each of the four libraries yielded in excess of 108 independent recombinants. The libraries were separately amplified in liquid culture, plasmids were extracted, and introduced by electroporation into strain C41(DE3) for production of VLPs. Care was taken to ensure no loss of complexity in the second transformation. The resulting population of VLPs was purified by Sepharose CL4B chromatography13. Each DKW library contained greater than 108 individual members and VLPs were mixed in equal quantities prior to affinity-selection.
Affinity Selections
We conducted selection using three different monoclonal antibodies. The M2 anti-Flag monoclonal was purchased from Sigma-Aldrich. The broadly neutralizing anti-HIV antibody known as 2F5 was obtained from the NIH HIV AIDS Research and Reference Reagents Program. The F20G77 antibody was provided by Jody D. Berry of the National Microbiology Laboratory of the Public Health Agency of Canada and recognizes an epitope of anthrax protective antigen 17.
Selections were conducted against monoclonal antibodies adsorbed to the surface of plastic wells (96-well Immulon 2, Thermo Scientific). In the first three rounds of selection,250 ng of an antibody in PBS were adsorbed to a well overnight at 4°C. The wells were subsequently blocked by incubation for two hours at room temperature with 0.5% non-fat dry milk in PBS, and a VLP library prepared as described above was added and incubated at room temperature for two hours. The binding reactions were conducted in 50ul, with an estimated 2-5ug of VLP. Wells were washed ten times with PBS, and bound particles were eluted for 5 minutes in 50ul of 0.1M glycine, pH 2.7. The eluted VLPs were neutralized by addition of 5ul 1M Tris, pH 9.0, and 10ul of eluate were subjected to reverse transcription for one hour in a 20ul reaction with MMLV reverse transcriptase (Promega) and 2 pmol of a primer (E2: 5′- TCAGCGGTGGCAGCAGCCAA-3′) that anneals 3′ of the coat protein coding sequence. The product of reverse transcription was amplified by PCR using Taq DNA polymerase and primer E3 (5′-CGGGCTTTGTTAGCAGCCGG-3′), which anneals just upstream of E2, and J2 (5′-ACTCCGGCCTCTACGGCAAC-3′), which anneals specifically to the junction sequence between the two halves of the single-chain dimer. The resulting PCR product was digested with SalI and BamHI and cloned in pDSP62 for production of VLPs for use in a second round of selection, conducted identically to the first. Note that amplification of sequences produced from pDSP62 and pDSP62(am) permits the replacement of the J2 primer with one that anneals upstream of the single-chain dimer junction in the codon-juggled half (62up: CTATGCAGGGGTTGTTGAAG). Round 2 and 3 selectants were cloned in pDSP62(am) for production of VLPs displaying their peptides at low valency. Apart from this reduction in the level of display valency, the conditions of selection in rounds 3 and 4 were similar to those of the previous rounds, except that in round 4 the amount of antibody was reduced to 50ng. In the 2F5 selections, to facilitate the selection of tight interactions, a competing peptide of sequence LLELDKWASLWNWFD (Anaspec) was included at increasing amounts (100ng round 1, 500ng round 2, 1ug round 3, and 5ug round 4).
Relative Affinity Measurements
The relative affinities of VLPs for the monoclonal antibody 2F5 were determined by competition ELISA. Briefly, 250 ng of antibody was absorbed to the wells of an ELISA plate. Following blocking (with 0.5% milk in PBS), ∼2 μg VLPs were added to wells in the presence of varying amounts of linear peptide (LLELDKWASLWNWFD, mentioned above) that can bind to 2F5. Following this incubation, bound VLPs were detected by incubating wells with rabbit polyclonal anti-MS2 antiserum (1 hour, 1:1000 dilution), followed by a peroxidase-labeled goat ant-rabbit IgG antibody (Jackson Immunoresearch; 1 hour, 1:2000 dilution). Binding was detected using the substrate ABTS, followed by detection at OD405.
Immunogenicity of selected recombinant VLPs
Groups of Balb/c mice were given three intramuscular doses at two-week intervals of 10 ug VLPs formulated 1:1 in incomplete Freund's adjuvant (Sigma-Aldrich). Two weeks following the final vaccination, sera was taken and IgG antibody levels in dilutions of sera were measured by ELISA as previously described 8. Anti-Flag antibody levels were measured by testing the reactivity of sera to bacteriophage PP7 VLPs engineered to display the Flag epitope 12. As a control for this ELISA, we also tested the reactivity of sera to wild-type non-recombinant PP7 VLPs. Anti-anthrax protective antigen (PA) antibody levels were measured by testing the reactivity of sera to recombinant PA (List Biological Laboratories).
Acknowledgments
The authors wish to thank Jody Berry for providing mAb F20G77. This study was supported by the NIH (R01 GM042901 to D.S.P. and R01 AI08335 to B.C.) and by the Bill and Melinda Gates Foundation (Explorations Grant #5311 to B.C. and D.S.P.).
Abbreviations used
- VLPs
virus-like particles
- mAb
monoclonal antibody
- ELISA
enzyme-linked immunosorbent assay
- PA
protective antigen
- PCR
polymerase chain reaction
- x-gal
4-chloro-3-indolyl-ß-D-galactoside
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Riemer AB, Jensen-Jarolim E. Mimotope vaccines: epitope mimics induce anti-cancer antibodies. Immunol Lett. 2007;113:1–5. doi: 10.1016/j.imlet.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li M, Yan Z, Han W, Zhang Y. Mimotope vaccination for epitope-specific induction of anti-CD20 antibodies. Cell Immunol. 2006;239:136–43. doi: 10.1016/j.cellimm.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Perosa F, Favoino E, Caragnano MA, Dammacco F. CD20 mimicry by a mAb rituximab-specific linear peptide: a potential tool for active immunotherapy of autoimmune diseases. Ann N Y Acad Sci. 2005;1051:672–83. doi: 10.1196/annals.1361.112. [DOI] [PubMed] [Google Scholar]
- 4.Binder M, Otto F, Mertelsmann R, Veelken H, Trepel M. The epitope recognized by rituximab. Blood. 2006;108:1975–8. doi: 10.1182/blood-2006-04-014639. [DOI] [PubMed] [Google Scholar]
- 5.Riemer AB, Klinger M, Wagner S, Bernhaus A, Mazzucchelli L, Pehamberger H, Scheiner O, Zielinski CC, Jensen-Jarolim E. Generation of Peptide mimics of the epitope recognized by trastuzumab on the oncogenic protein Her-2/neu. J Immunol. 2004;173:394–401. doi: 10.4049/jimmunol.173.1.394. [DOI] [PubMed] [Google Scholar]
- 6.Riemer AB, Kurz H, Klinger M, Scheiner O, Zielinski CC, Jensen-Jarolim E. Vaccination with cetuximab mimotopes and biological properties of induced anti-epidermal growth factor receptor antibodies. J Natl Cancer Inst. 2005;97:1663–70. doi: 10.1093/jnci/dji373. [DOI] [PubMed] [Google Scholar]
- 7.Hartmann C, Muller N, Blaukat A, Koch J, Benhar I, Wels WS. Peptide mimotopes recognized by antibodies cetuximab and matuzumab induce a functionally equivalent anti-EGFR immune response. Oncogene. 2010;29:4517–27. doi: 10.1038/onc.2010.195. [DOI] [PubMed] [Google Scholar]
- 8.Peabody DS, Manifold-Wheeler B, Medford A, Jordan SK, do Carmo Caldeira J, Chackerian B. Immunogenic display of diverse peptides on virus-like particles of RNA phage MS2. J Mol Biol. 2008;380:252–63. doi: 10.1016/j.jmb.2008.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chackerian B, Lowy DR, Schiller JT. Conjugation of a self-antigen to papillomavirus-like particles allows for efficient induction of protective autoantibodies. J Clin Invest. 2001;108:415–23. doi: 10.1172/JCI11849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chackerian B. Virus-like particles: flexible platforms for vaccine development. Expert Rev Vaccines. 2007;6:381–90. doi: 10.1586/14760584.6.3.381. [DOI] [PubMed] [Google Scholar]
- 11.Peabody DS. Subunit fusion confers tolerance to peptide insertions in a virus coat protein. Arch Biochem Biophys. 1997;347:85–92. doi: 10.1006/abbi.1997.0312. [DOI] [PubMed] [Google Scholar]
- 12.Caldeira Jdo C, Medford A, Kines RC, Lino CA, Schiller JT, Chackerian B, Peabody DS. Immunogenic display of diverse peptides, including a broadly cross-type neutralizing human papillomavirus L2 epitope, on virus-like particles of the RNA bacteriophage PP7. Vaccine. 28:4384–93. doi: 10.1016/j.vaccine.2010.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peabody DS. Translational repression by bacteriophage MS2 coat protein expressed from a plasmid. A system for genetic analysis of a protein-RNA interaction. J Biol Chem. 1990;265:5684–9. [PubMed] [Google Scholar]
- 14.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Phage display for selection of novel binding peptides. Methods in Ezymology. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 15.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–92. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gubbins MJ, Berry JD, Corbett CR, Mogridge J, Yuan XY, Schmidt L, Nicolas B, Kabani A, Tsang RS. Production and characterization of neutralizing monoclonal antibodies that recognize an epitope in domain 2 of Bacillus anthracis protective antigen. FEMS Immunol Med Microbiol. 2006;47:436–43. doi: 10.1111/j.1574-695X.2006.00114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang J, Xu J, Li G, Dong D, Song X, Guo Q, Zhao J, Fu L, Chen W. The 2beta2-2beta3 loop of anthrax protective antigen contains a dominant neutralizing epitope. Biochem Biophys Res Commun. 2006;341:1164–71. doi: 10.1016/j.bbrc.2006.01.080. [DOI] [PubMed] [Google Scholar]
- 18.Wells JA. Hormone mimicry. Science. 1996;273:449–50. doi: 10.1126/science.273.5274.449. [DOI] [PubMed] [Google Scholar]
- 19.Lowman HB, Bass SH, Simpson N, Wells JA. Selecting high-affinity binding proteins by monovalent phage display. Biochemistry. 1991;30:10832–8. doi: 10.1021/bi00109a004. [DOI] [PubMed] [Google Scholar]
- 20.Kleina LG, Masson JM, Normanly J, Abelson J, Miller JH. Construction of Escherichia coli amber suppressor tRNA genes. II. Synthesis of additional tRNA genes and improvement of suppressor efficiency. J Mol Biol. 1990;213:705–17. doi: 10.1016/S0022-2836(05)80257-8. [DOI] [PubMed] [Google Scholar]
- 21.Normanly J, Kleina LG, Masson JM, Abelson J, Miller JH. Construction of Escherichia coli amber suppressor tRNA genes. III. Determination of tRNA specificity. J Mol Biol. 1990;213:719–26. doi: 10.1016/S0022-2836(05)80258-X. [DOI] [PubMed] [Google Scholar]
- 22.Chang AC, Cohen SN. Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol. 1978;134:1141–56. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ofek G, Tang M, Sambor A, Katinger H, Mascola JR, Wyatt R, Kwong PD. Structure and mechanistic analysis of the anti-human immunodeficiency virus type 1 antibody 2F5 in complex with its gp41 epitope. J Virol. 2004;78:10724–37. doi: 10.1128/JVI.78.19.10724-10737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zwick MB, Komori HK, Stanfield RL, Church S, Wang M, Parren PW, Kunert R, Katinger H, Wilson IA, Burton DR. The long third omplementarity-determining region of the heavy chain is important in the activity of the broadly neutralizing anti-human immunodeficiency virus type 1 antibody 2F5. J Virol. 2004;78:3155–61. doi: 10.1128/JVI.78.6.3155-3161.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sidhu SS. Phage display in biotechnology and drug discovery. CRC Press/Taylor & Francis; Boca Raton: 2005. (Drug discovery). [Google Scholar]
- 26.Peabody DS. The RNA binding site of bacteriophage MS2 coat protein. EMBO J. 1993;12:595–600. doi: 10.1002/j.1460-2075.1993.tb05691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assembly of a gene and entire plasmid from large numbers of oligodeoxyribonucleotides. Gene. 1995;164:49–53. doi: 10.1016/0378-1119(95)00511-4. [DOI] [PubMed] [Google Scholar]