

Abstract
Hypothalamic proopiomelanocortin (POMC) neurons play a key role in regulating energy balance and neuroendocrine function. Much attention has been focused on regulation of POMC gene expression with less emphasis on regulated peptide processing. This is particularly important given the complexity of posttranslational POMC processing which is essential for the generation of biologically active MSH peptides. Mutations that impair POMC sorting and processing are associated with obesity in humans and in animals. Specifically, mutations in the POMC processing enzymes prohormone convertase 1/3 (PCI/3) and in carboxypeptidase E (CPE) and in the α-MSH degrading enzyme, PRCP, are associated with changes in energy balance. There is increasing evidence that POMC processing is regulated with respect to energy balance. Studies have implicated both the leptin and insulin signaling pathways in the regulation of POMC at various steps in the processing pathway. This article will review the role of hypothalamic POMC in regulating energy balance with a focus on POMC processing.
Index Words: Proopiomelanocortin, POMC, α-MSH, prohormone convertases, carboxypeptidase E, obesity
1. Introduction
Proopiomelanocortin (POMC) is a 31-kDa prohormone precursor protein that is synthesized in the pituitary, in the arcuate nucleus of the hypothalamus, in the medulla and in several peripheral tissues. POMC is then processed in a tissue specific manner to yield biologically active peptides (Figure 1). In the brain POMC neurons play a critical role in regulating energy balance via interactions of the POMC-derived MSH peptides with brain melanocortin receptors. For review see (Lee and Wardlaw, 2007). The POMC-derived endogenous opioid peptide, β-endorphin (β-EP), can also affect energy balance (Bodnar, 2004). The MC4 receptor and to a lesser extent the MC3 receptor are responsible for mediating the central effects of the MSH peptides on energy balance. The activity of the brain melanocortin receptors can be further modulated by the MSH antagonist, agouti-related protein (AgRP). α-MSH inhibits feeding and stimulates energy expenditure while AgRP is orexigenic and decreases energy expenditure. α-MSH and AgRP are synthesized in distinct neuronal populations in the arcuate nucleus of the hypothalamus that project to other hypothalamic regions, including the paraventricular nucleus and lateral hypothalamus, and to the brainstem, areas that are particularly important in regulating energy balance (Cowley et al., 1999; Elmquist et al., 1999). Some POMC is also synthesized in the nucleus of the solitary tract in the medulla. POMC and AgRP neurons can act as sensors of peripheral energy stores and respond to a variety of nutrient, neuronal and hormonal signals, including leptin and insulin. In rodents, genetic or pharmacological inactivation of POMC or the MC4 receptor results in hyperphagia and obesity as does overexpression of AgRP (Challis et al., 2004; Graham et al., 1997; Huszar et al., 1997; Ollmann et al., 1997; Yaswen et al., 1999). Targeted deletion of the MC3 receptor also causes an obesity phenotype (Butler et al., 2000; Chen et al., 2000). This system is highly relevant to human energy balance as defects in POMC synthesis and processing and haploinsufficiency of the MC4 receptor have all been reported in human obesity syndromes (Coll et al., 2004). This article will review the role of POMC in regulating energy balance with a focus on the role of POMC processing.
Figure 1. POMC processing.

Schematic diagram of the POMC precursor molecule and the major peptide products that are derived from this precursor by endoproteolytic cleavage. (JP = Joining peptide; LPH= Lipotropin; EP = Endorphin; CLIP= Corticotropin-like-intermediate lobe peptide; da-α–MSH = desacetyl α–MSH; PC1/3 and PC2= Prohormone convertases 1/3 and 2; CPE= Carboxypeptidase E; N-AT= N-acetyltransferase; PAM= Peptidyl α-amidating monooxygenase; PRCP = Prolylcarboxypeptidase).
2. POMC Mutations and Obesity
Mice with Pomc gene deletion or with POMC neuron ablation are obese. Two POMC-null mutant mouse models have been created and both have an obese phenotype despite profound adrenal insufficiency (Challis et al., 2004; Yaswen et al., 1999). In the first model, the entire third exon of Pomc was deleted, thus removing the coding region for the relevant POMC-derived peptides but the first 18 amino acids of POMC still remained (Yaswen et al., 1999). In the second model the entire POMC sequence was deleted (Challis et al., 2004). In both models, homozygous Pomc null mice have abnormal adrenal development, altered pigmentation and develop hyperphagia and obesity. Serum levels of corticosterone were undetectable in the mutant mice. Yaswen et al. noted increased weight gain in Pomc −/− mice in the second postnatal month and by the third postnatal month, weights were about twice those of the wild-type mice. Body length also increased significantly, as is the case for Mc4 receptor knockout mice. Serum leptin levels were markedly increased in homozygous Pomc mutant mice. Leptin levels were also increased in heterozygous mutant mice although body weight was normal. Treatment of Pomc null mice with a synthetic α-MSH agonist caused a decrease in food intake and substantial weight loss. After two weeks of treatment, the mutant mice lost 46% of their excess body weight and when the α-MSH agonist injections were stopped, the mice returned to their pretreatment weight. Challis et al. noted an increase in body weight in the Pomc −/− mice after 8 weeks of age. Both fat and lean body mass were increased relative to wild-type mice and basal metabolic rate was decreased by 23%. Although Pomc +/− mice were not obese on a standard chow diet, they did become obese on a 45% high fat diet. Thus haploinsufficiency of this gene can cause obesity but only when exposed to a high fat diet, in contrast to haploinsufficiency of the MC4 receptor. The obesity of Pomc −/− mice and its associated metabolic complications were markedly exacerbated by either replacement with glucocorticoids or by selective transgenic restoration of pituitary Pomc (Coll et al., 2005; Smart et al., 2006). In another model, POMC neurons were progressively ablated by deleting the mitochondrial transcription factor A (Tfam) gene using a Cre-lox approach. These mice developed an obesity syndrome similar to that described for Pomc null mice (Xu et al., 2005a).
In contrast, genetic models of POMC overexpression have been shown to protect from obesity. Transgenic neuronal overexpression of Pomc has been shown to attenuate obesity in ob/ob mice (Mizuno et al., 2003). Central Pomc gene delivery via recombinant adeno-associated virus has also been shown to reduce food intake and adiposity in obese Zucker rats (Li et al., 2003). Overexpression of an N-terminal POMC transgene, that includes both α-MSH and γ3-MSH, reduced weight gain and adiposity in male mice (C57BL/6 background) on a normal diet and attenuated obesity in male and female db3J/db3J mice (Savontaus et al., 2004). This transgene also protected male and female mice from weight gain and increased adiposity when exposed to a high fat diet (Lee et al., 2007).
POMC mutations have also been reported in obese humans. In 1998, Krude et al. reported two patients from Germany with genetic POMC deficiency characterized by adrenal insufficiency, red hair pigmentation and early-onset obesity (Krude et al., 1998). The first patient was found to be a compound heterozygote for two mutations in exon 3 that resulted in ACTH and α-MSH deficiency. She had a normal birth weight and was diagnosed with adrenal insufficiency at 3 weeks of age when she developed cholestasis. Increased appetite and obesity was first noted at 4 months of age. The second patient was homozygous for a mutation in exon 2 which abolishes POMC translation. His birth weight was normal and obesity was first noted at 5 months of age. Adrenal insufficiency was diagnosed at 12 months of age when he developed hypoglycemia and hyponatremia. Subsequent development in both children was normal except for the abnormal eating behavior and obesity. Both children had pale skin and red hair color. The heterozygous parents in both families had normal adrenal function and did not have obesity or red hair. Remarkably, the children were obese despite adrenal insufficiency which in other circumstances would be expected to cause anorexia and weight loss. The contrast between these patients with generalized POMC deficiency and patients with pituitary disease who have POMC deficiency limited to the pituitary, underscores the critical role that hypothalamic POMC plays in regulating energy balance. Three other patients with a similar POMC deficiency syndrome were subsequently described by the same group (Krude et al., 2003). A novel homozygous frameshift mutation in POMC, leading to the loss of all POMC-derived peptides, has also been found in a child of Turkish origin with adrenal insufficiency and severe obesity (Farooqi et al., 2006). In this family, of the 12 relatives that were heterozygous for the POMC mutation, 11 were obese. In contrast, of the 7 relatives that were wild-type only one was obese. Thus, in humans, as in mice, the loss of one copy of the POMC gene may predispose to obesity. Several other groups have reported additional POMC mutations associated with obesity (Clement et al., 2008; Creemers et al., 2008; Dubern et al., 2008). Two groups have reported POMC variants that implicate β-MSH in the control of human body weight regulation (Biebermann et al., 2006; Lee et al., 2006). β-MSH is a normal POMC processing product in the human but not in the rodent. In one study, 538 patients with severe, early-onset obesity were screened for POMC mutations and 5 unrelated probands, who were heterozygous for a rare missense variant in the region coding for β-MSH, were identified (Lee et al., 2006). In the other study, a similar mutation was found during a screen of 15 severely obese children. Compared to wild-type β-MSH, the ability of the variant peptide to bind to and activate the MC4 receptor was impaired. A missense mutation that disrupts the dibasic amino acid cleavage site between β-MSH and β-EP has also been reported to occur more frequently in obese children (Challis et al., 2002). Heterozygous β-MSH mutations are also associated with obesity (Biebermann et al., 2006; Challis et al., 2002; Lee et al., 2006).
Evidence that variant alleles of POMC may modulate weight level in humans is provided by a study in a population of Mexican Americans showing a linkage of serum leptin levels and fat mass to an interval on chromosome 2 which includes the POMC locus (Comuzzie et al., 1997). Subsequent studies in a French and in an African-American population have reported similar associations (Hager et al., 1998; Rotimi et al., 1999).
3. POMC Regulation and Energy Balance
POMC is regulated in the arcuate nucleus by a number of hormones, nutrients, neuropeptides and neurotransmitters, many of which are known to affect energy balance. These include leptin, insulin, and dietary nutrients. POMC expression in arcuate neurons is suppressed during fasting and stimulated when energy stores are increased. Levels of peripheral energy stores are sensed by leptin receptors on POMC neurons. There is extensive evidence documenting the activation of POMC neurons by leptin as shown by the induction of Fos, SOCS-3, STAT3 phosphorylation, and increase in POMC heteronuclear RNA and mRNA levels (Elias et al., 1999; Korner et al., 1999; Munzberg et al., 2003; Schwartz et al., 1997). Electrophysiology studies have shown that leptin stimulates POMC neuron firing (Cowley et al., 2001) and regulates activity of potassium channels in POMC neurons thus modulating neuronal excitability (Jobst et al., 2004). In addition, mice with selective deletion of leptin receptors on POMC neurons are obese (Balthasar et al., 2004). Leptin can also affect the development of POMC neuronal projections and can modulate the number of excitatory and inhibitory synapses on POMC neurons (Bouret et al., 2004; Pinto et al., 2004). Stimulatory effects of insulin on POMC gene expression have also been demonstrated (Benoit et al., 2002). Insulin binds to its receptors on POMC neurons and stimulates phosphatidylinositol-3 kinase (PI3K). This leads to phosphorylation of AKT and subsequent phosphorylation and exclusion of FoxO1 from the nucleus of POMC neurons (Fukuda et al., 2008). Although leptin and insulin can act by distinct signaling pathways, there is evidence for shared intracellular signaling pathways with respect to PI3K stimulation in POMC neurons (Xu et al., 2005b). Electrophysiological studies also demonstrate opposing actions of leptin and insulin in distinct populations of arcuate POMC neurons (Williams et al., 2010). Mice lacking both insulin and leptin receptors on POMC neurons have significant systemic insulin resistance that is distinct from the phenotype seen with deletion of either receptor alone (Hill et al., 2010). POMC neurons can also respond to dietary nutrients; they are glucose responsive (Ibrahim et al., 2003) and can respond to leucine via stimulation of the mTOR pathway (Blouet et al., 2009; Cota et al., 2006). As described above, much attention has been focused on POMC neuronal activation and on the regulation of POMC gene expression. However as outlined in the following sections, the posttranslational processing of POMC is critical for the generation of biologically active peptides and there is evidence that this may be regulated with respect to energy balance.
4. POMC Processing
The POMC precursor protein is synthesized in the endoplasmic reticulum and moves to the Golgi complex where it is sorted for delivery to secretory granules. The POMC precursor contains an N-terminal sequence that acts as a sorting signal to secretory granules in the regulated secretory pathway. Membrane carboxypeptidase E (CPE) has been shown to bind this N-terminal POMC sequence and to serve as a sorting receptor (Cool et al., 1997). During this trafficking process POMC is proteolytically cleaved into a number of biologically active peptides (Figure 1). The differential expression of prohormone convertases (PCs) in various tissues leads to tissue specific posttranslational processing of POMC (Castro and Morrison, 1997; Smith and Funder, 1988). Functionally active peptides are produced by endoproteolytic cleavage at adjacent pairs of basic amino acids by the prohormone convertases, PC1/3 and PC2 (Benjannet et al., 1991). In the anterior pituitary, POMC is processed predominantly to ACTH, β-lipotropin (LPH) and a 16 kDa N-terminal fragment. ACTH is critical for the maintenance of adrenocortical function. In the hypothalamus and in the intermediate lobe of the pituitary (which is prominent in the rodent), POMC is more extensively processed: ACTH is further processed to produce α-MSH and corticotropin-like-intermediate lobe peptide (CLIP); β-LPH is processed to γ-LPH and β-EP; N-terminal POMC is processed to γ3-MSH (Emeson and Eipper, 1986; Pritchard and White, 2007). In the human, γ-LPH can be further processed to β-MSH (Biebermann et al., 2006). A scheme of POMC processing in the hypothalamus is depicted in Figure 1. POMC is initially cleaved by PC1/3 to yield pro-ACTH and β-LPH. Pro-ACTH is then cleaved by PC1/3 to ACTH and N-terminal POMC. Further processing by PC2 yields ACTH 1–17 and CLIP as well as γ-LPH and β-EP 1–31. Another processing enzyme, carboxypeptidase E (CPE), removes the C-terminal basic amino acid residues from ACTH 1–17 to form ACTH 1–13 which is then amidated by the enzyme peptidyl α-amidating monooxygenase (PAM) to generate desacetyl α-MSH. Desacetyl α-MSH can then be acetylated by an N-acetyltransferase to form α-MSH. Desacetyl α-MSH is, however, the predominant form of α-MSH in rodent hypothalamic extracts, in contrast to the intermediate lobe of the pituitary where N-acetyl α-MSH is the predominant form (Wilkinson, 2006). Recently a new processing enzyme, prolylcarboxypeptidase (PRCP) has been identified that is responsible for inactivation of α-MSH by removal of the C-terminal valine (Wallingford et al., 2009). Processing of β-LPH by PC2 yields γ-LPH and β-EP 1–31, one of the endogenous opioid peptides. β-EP can also be acetylated resulting in loss of opioid activity. As with α-MSH, acetylation of β-EP occurs to a large extent in the intermediate lobe of the pituitary but not in the hypothalamus. Although there is little acetylation of β-EP in the hypothalamus, β-EP 1–31 can be cleaved by PC2 and CPE to β-EP 1–27 and 1–26 which have markedly reduced opioid activity (Nicolas and Li, 1985).
POMC is processed to a number of peptides that can affect feeding behavior and energy balance. Intracerebroventricular (icv) injection of α-MSH and other synthetic MSH agonists have been shown to suppress food intake in the rodent and in the monkey (Koegler et al., 2001; McMinn et al., 2000). Furthermore, injection of synthetic α-MSH antagonists, increases food intake, indicating a role for endogenous α-MSH in appetite control. In addition to suppressing food intake, α-MSH can affect energy expenditure, oxygen consumption and fuel oxidation, all of which contribute to overall changes in energy balance. Thus α-MSH has a clear role in regulating energy balance but there is also evidence that other POMC-derived MSH peptides, including β-MSH and perhaps γ–MSH, may play a role in this process. In addition, the POMC-derived opioid peptide, β-EP, can affect energy balance. Regulation of POMC processing is particularly important because a number of peptides are produced with very different (and even opposing) biological activities. For example, α-MSH can attenuate the effects of β-EP on gonadotropin and prolactin release (Shalts et al., 1992; Wardlaw and Ferin, 1990; Wardlaw et al., 1986) and can also attenuate β-EP and morphine-induced analgesia (Contreras and Takemori, 1984; Sandman and Kastin, 1981). With respect to feeding, α-MSH is inhibitory while β-EP and other opioids have well documented stimulatory effects (Bodnar, 2004). The role of opioids with respect to energy balance is, however, complex as demonstrated by recent studies in β-EP null mice (Appleyard et al., 2003; Low et al., 2003). Opioid receptors, like melanocortin receptors, are G-protein coupled but they are coupled to inhibitory rather than stimulatory G proteins. Several studies have reported interactions between the melanocortin and opioid pathways with respect to feeding and weight gain (Grossman et al., 2003; Olszewski et al., 2001).
5. AgRP Processing
AgRP also undergoes posttranslational processing but in contrast to POMC this is not essential to generate biologically activity peptides as the precursor itself possesses considerable biological activity. AgRP is processed to a C-terminal biologically active fragment, AgRP83–132 (Breen et al., 2005; Li et al., 2000; Rossi et al., 1998; Xiao et al., 2010). The majority of the AgRP immunoactivity detected in the hypothalamus appears to be AgRP83–132 with only a small portion of full length AgRP. Creemers et al have shown PC1/3 is expressed in AgRP neurons and is responsible for the generation of AgRP83–132 (Creemers et al., 2006). Both RNA interference and overexpression studies demonstrate that PC1/3 is primarily responsible for cleavage of AgRP in vitro. In addition, hypothalamic extracts from PC1/3 null mice contained 3-fold more unprocessed full-length AgRP compared to wild-type mice, demonstrating that PC1/3 plays a role in AgRP cleavage in vivo (Creemers et al., 2006). AgRP83–132 has been shown to be a more potent MC-4R antagonist than full-length AgRP (Creemers et al., 2006). Thus the C-terminal processing of full–length AgRP may serve to increase the biological activity of AgRP. However in contrast to POMC, unprocessed AgRP is biologically active and functions as a potent MC4 receptor antagonist.
6. POMC processing and energy balance
6.1 Overview POMC processing and obesity
Abnormal POMC processing is associated with obesity in human and in animal models. Mutations in a highly conserved N-terminal region of POMC that impair POMC sorting to the regulated secretory pathway have been reported in 2 subjects with severe, early-onset obesity (Creemers et al., 2008). Heterozygous mutations that disrupt the dibasic cleavage site between β-MSH and β-EP are also associated with obesity (Challis et al., 2002). In addition, two patients with PC1/3 deficiency and obesity have been identified (Jackson et al., 2003; Jackson et al., 1997). Obesity has also been reported in Pcsk1 mutant mice (Lloyd et al., 2006) and in mice with deletion of the transcription factor, Nhlh2, that regulates PC1/3 and PC2 expression (Jing et al., 2004). CPE deficient mice are also obese (Naggert et al., 1995). Furthermore, deletion of an enzyme, prolylcarboxypeptidase (PRCP), that degrades α-MSH, promotes a lean phenotype (Wallingford et al., 2009). There is also evidence to suggest that POMC processing is regulated with respect to energy balance (Perello et al., 2007; Pritchard et al., 2003; Sanchez et al., 2004). The ratio of the POMC precursor to the processed peptides was increased during fasting in the rat consistent with less POMC processing (Pritchard et al., 2003). Parallel changes in PC1/3 and PC2 have also been reported during fasting, some of which are reversed by leptin (Perello et al., 2007; Sanchez et al., 2004). Short-term treatment with leptin was reported to increase the proportion of acetylated α-MSH in the hypothalamus of ob/ob mice but studies from different laboratories did not find significant acetylation of α-MSH in the hypothalamus or the regulation of α-MSH acetylation by leptin (Guo et al., 2004; Perello et al., 2007) (Wilkinson, 2006). A more detailed discussion of PC1/3, CPE, PRCP and POMC processing as related to changes in energy balance and to leptin and insulin action is provided below and is depicted schematically in Figure 2.
Figure 2. Parallel regulation of POMC gene transcription and peptide processing.
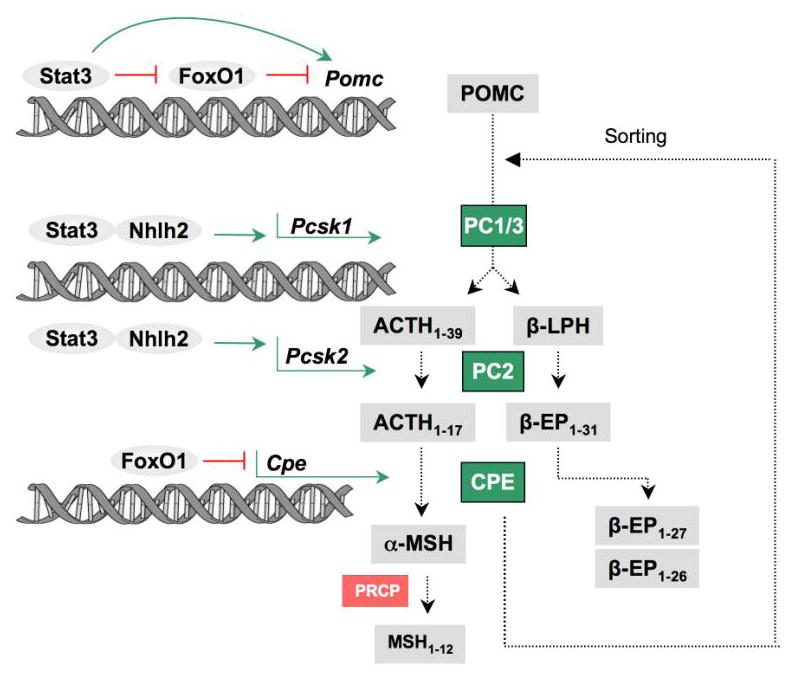
Diagram showing the interaction of the leptin and insulin signaling pathways in both the transcriptional regulation of Pomc and in the regulation of POMC procesing and the enzymes responsible for POMC procesing. Leptin (via STAT3) stimulates Pomc gene transcription but also stimulates the genes Psck1 and Psck2 for the procesing enzymes, PC1/3 and PC2. The transcription factor, Nhlh2, also stimulates Psck1 and Psck2. FoxO1, part of the insulin signaling pathway, inhibits Pomc gene transcription and also inhibits the processing enzyme, CPE which is responsible for the generation of α-MSH from ACTH 1–17 and for the C-terminal cleavage of β-EP 1–31 to β-EP 1–27/1–26. CPE also serves as a sorting receptor that sorts POMC to the regulated secretory pathway. PRCP is an enzymes responsible for the degradation of α-MSH. (EP = Endorphin; PC1/3 and PC2= Prohormone convertases 1/3 and 2; CPE= Carboxypeptidase E; PRCP = Prolylcarboxypeptidase; nhlh2= nescient helix-loop-helix2).
6.2 PC1/3 and energy balance
PC1/3 deficiency is associated with obesity in humans and in rodents. Two patients with PC1/3 deficiency have been described (Jackson et al., 2003; Jackson et al., 1997). Both were compound heterozygotes for PC1/3 loss of function mutations and had severe early-onset obesity. The first patient also had hypogonadotropic hypogonadism, postprandial hypoglycemia, and low cortisol and was shown to have impaired POMC and proinsulin processing. The second patient also had small intestinal absorption dysfunction and abnormal gut hormone processing. It is likely that the obesity in these subjects is related to the impairment of POMC processing however, the processing of a number of other hormones is also abnormal and could potentially contribute to the obesity phenotype. Similarly, mice with a novel Pcsk1 mutation (N222D) are obese and hyperphagic (Lloyd et al., 2006). However Pcsk1 knockout mice do not develop obesity (Zhu et al., 2002). The null mice are runted with associated defective pro-GHRH processing. In contrast mice with the N222D mutation have reduced but not absent PC1/3 activity and grow normally. These mice have normal pro-GHRH processing but defective POMC and proinsulin processing, similar to the human phenotype. Defective POMC processing leads to reduced levels of α-MSH in the hypothalamus of the Pcsk1(N222D) mutant mice. PC2 null mice are not obese and no humans have yet been identified with PC2 mutations. PC2 null mice do have low levels of α-MSH in the hypothalamus but they also have reduced growth rate and hypoglycemia (Furuta et al., 1997).
The neuronal transcription factor, nescient helix-loop-helix2 (Nhlh2), regulates the transcription of PC1/3 and PC2 and has also been implicated in obesity (Fox et al., 2007). Nhlh2 is expressed throughout the hypothalamus and specifically in POMC neurons. Deletion of Nhlh2 reduces expression of PC1 and PC2 mRNA in the hypothalamus and results in adult onset obesity (Jing et al., 2004). The null mice have normal POMC mRNA levels in the arcuate but have reduced levels of α-MSH with relatively more ACTH and pro-ACTH. Nhlh2 expression in the hypothalamus of wild-type mice is reduced during fasting and increased in response to leptin and food intake. Notably, the reduction in Nhlh2 expression during fasting coincides with the reduction in POMC-derived peptides levels in the arcuate nucleus (Jing et al., 2004). Fasting and leptin have also been shown to affect PC1/3 and PC2 expression. In the rat, fasting causes a decrease in PC1/3 and PC2 mRNA and protein expression in the PVN; leptin administration partially reversed the decrease in PC1/3 (Sanchez et al., 2004). In another study, POMC mRNA, POMC-derived peptide levels and PC1/3 all decreased in the arcuate nucleus after fasting and administration of leptin reversed these effects (Perello et al., 2007). Leptin has also been shown to increase PC1/3 and PC2 mRNA and protein expression in hypothalamic neuronal cells in culture (Sanchez et al., 2004). Furthermore, leptin increased PC1/3 and PC2 promoter activities (via STAT3) in transfected 293T cells (Sanchez et al., 2004).
6.3 Carboxypeptidase E (CPE) and energy balance
CPE is a metallocarboxypeptidase with neuroendocrine distribution. It cleaves C-terminal amino acid residues (usually Lys, Arg) after initial cleavage by PC1/3 and PC2. It is responsible for the C-terminal trimming of these basic residues on many peptides including the POMC-derived peptides. CPE also functions as a regulated secretory pathway sorting receptor which binds secretory proteins, including POMC, for sorting to the regulated secretory pathway. (Cool et al., 1997). Mice with mutant Cpe, Cpefat are obese, diabetic (with hyperproinsulinemia) and infertile. A heterozygous CPE variant with altered enzyme activity has also been reported in the human and is associated with early-onset type 2 diabetes (Chen et al., 2001). Cpefat mice have low levels of α-MSH in the hypothalamus and also have decreased C-terminal processing of β-EP 1–31 to β-EP 1–27 and β-EP 1–26 (Berman et al., 2001). There is also evidence that POMC is missorted to the constitutive secretory pathway in Cpefat mice (Cool et al., 1997). Cpefat mice also have decreased levels of PC1/3 protein levels due to accumulation of immature PC1/3 rather than reduction of PC1/3 mRNA (Berman et al., 2001).
A recent study has shown that CPE may link FoxO1 (a transcription factor that mediates effects of insulin) in POMC neurons with regulation of food intake (Plum et al., 2009). Mice with FoxO1 ablation in POMC neurons have a lean phenotype with decreased food intake and increased CPE expression in the arcuate. Analysis of POMC peptides in these mice revealed an anorexigenic profile with a selective increase of α-MSH and HPLC analysis demonstrated relatively more β-EP 1–27 and β-EP1–26 compared to β-EP 1–31; this is opposite to the pattern seen in CPE-deficient mice. Since β-EP 1–31 (like other opioids) has stimulatory effects on food intake, C-terminal cleavage to peptides with markedly reduced opioid activity, could serve to enhance α-MSH activity. Regulated CPE-dependent cleavage of α-MSH and β-EP could thus contribute to the phenotype of Pomc-FoxO1−/− mice. However CPE-dependent sorting of POMC to the regulated secretory pathway may also play a role in this process. FoxO1 has been shown to directly interact with and regulate Cpe (Plum et al., 2009). Chromatin immunoprecipitation assay of the Cpe promoter in Neuro2A cells demonstrated occupancy of the Cpe promoter by FoxO1. Moreover constitutively nuclear FoxO1 suppressed reporter gene activity driven by a Cpe promoter. This study clearly links the insulin signaling pathway to CPE dependent POMC processing. This is analogous to the link between leptin, STAT3 and PC1/3 dependent POMC processing. Thus in addition to regulating POMC gene expression both leptin and insulin signaling pathways can affect POMC peptide processing.
6.4 Prolylcarboxypeptidase (PRCP) and α-MSH degradation
A number of studies have focused on the enzymes involved with the production of α-MSH but little is known about how α-MSH is degraded. Recently Wallingford and colleagues have identified an enzyme, prolylcarboxypeptidase (PRCP), that inactivates α-MSH by removing the C-terminal valine residue (Wallingford et al., 2009). The resulting truncated peptide, α-MSH 1–12, was shown to be ineffective in reducing food intake when injected icv compared to α-MSH 1–13. Prcp null mice had a lean phenotype and had elevated levels of α-MSH in the hypothalamus. Small molecule inhibitors of PRCP activity also decreased food intake. PRCP is widely distributed both within the brain and in many peripheral tissues. In the hypothalamus PRCP is highly expressed in the lateral hypothalamus and dorsomedial nucleus and to lesser extent in the arcuate. Thus the release of PRCP from other neuronal populations that send efferents to areas where α-MSH is released from axon terminals could serve to modulate the strength of the α-MSH signal at the MC4 receptor. PRCP is thus a potential new drug target that could be used to enhance α-MSH signaling in the hypothalamus. However given its widespread distribution, there are likely many other peptides substrates that would also be affected. At present it is unknown if human PRCP mutations are associated with body weight or adiposity.
7. Conclusion
POMC plays a critical role in regulating energy balance but must be properly sorted to the secretory pathway and processed to its biologically active peptide products in order to exert its effects on food intake, body weight and adiposity. There is considerable evidence that abnormalities in the POMC processing pathway can lead to obesity. Mutations that impair POMC sorting or the processing of MSH peptides are associated with obesity as are mutations in POMC processing enzymes themselves. Obesity is seen with PC1/3 mutations, with deletion of the transcription factor, Nhlh2, that regulates PC1/3 and with CPE deficiency. There is also evidence that POMC processing is regulated with respect to energy balance. Changes in the ratio of the POMC precursor to the processed peptides and in POMC processing enzyme levels have been reported in the hypothalamus during fasting, re-feeding and with leptin treatment. Leptin has been shown to increase PC1/3 and PC2 expression in hypothalamic neuronal cells and to increase Pcsk1 and Pcsk2 promoter activities. A recent study has shown that CPE may link the insulin signaling pathway in POMC neurons with regulation of food intake. FoxO1, a transcription factor that mediates effects of insulin, has been shown to directly interact with and regulate Cpe and when deleted from POMC neurons appears to alter CPE dependent POMC processing. Thus in addition to regulating POMC gene expression, both leptin and insulin signaling pathways can affect POMC processing. Until now, little has been known about the process of MSH peptide degradation. However, a recent discovery has shown that the enzyme, PRCP, is responsible for α-MSH degradation and that deletion of PRCP promotes a lean phenotype. A summary showing the parallel regulation of POMC gene expression and peptide processing is shown in Figure 2. There is thus considerable evidence that abnormalities in the POMC processing pathway can lead to obesity and that the POMC processing is regulated with respect to energy balance at multiple steps. A more detailed understanding of the control of this pathway will hopefully lead to effective new therapies for human obesity.
Acknowledgments
The support of NIH DK08003 and the Atkins Foundation is gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Appleyard SM, Hayward M, Young JI, Butler AA, Cone RD, Rubinstein M, Low MJ. A role for the endogenous opioid beta-endorphin in energy homeostasis. Endocrinology. 2003;144:1753–1760. doi: 10.1210/en.2002-221096. [DOI] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SCJ, Elmquist JK, Lowell BB. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Benjannet S, Rondeau N, Day R, Chretien M, Seidah NG. PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:3564–3568. doi: 10.1073/pnas.88.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit SC, Air EL, Coolen LM, Strauss R, Jackman A, Clegg DJ, Seeley RJ, Woods SC. The catabolic action of insulin in the brain is mediated by melanocortins. J Neurosci. 2002;22:9048–9052. doi: 10.1523/JNEUROSCI.22-20-09048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman Y, Mzhavia N, Polonskaia A, Devi LA. Impaired prohormone convertases in Cpe(fat)/Cpe(fat) mice. The Journal of Biological Chemistry. 2001;276:1466–1473. doi: 10.1074/jbc.M008499200. [DOI] [PubMed] [Google Scholar]
- Biebermann H, Castaneda TR, van Landeghem F, von Deimling A, Escher F, Brabant G, Hebebrand J, Hinney A, Tschop MH, Gruters A, Krude H. A role for beta-melanocyte-stimulating hormone in human body-weight regulation. Cell Metabolism. 2006;3:141–146. doi: 10.1016/j.cmet.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Blouet C, Jo YH, Li X, Schwartz GJ. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J Neurosci. 2009;29:8302–8311. doi: 10.1523/JNEUROSCI.1668-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar RJ. Endogenous opioids and feeding behavior: a 30-year historical perspective. Peptides. 2004;25:697–725. doi: 10.1016/j.peptides.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Bouret SG, Draper SJ, Simerly RB. Trophic action of leptin on hypothalamic neurons that regulate feeding. Science. 2004;304:108–110. doi: 10.1126/science.1095004. [DOI] [PubMed] [Google Scholar]
- Breen TL, Conwell IM, Wardlaw SL. Effects of fasting, leptin, and insulin on AGRP and POMC peptide release in the hypothalamus. Brain Research. 2005;1032:141–148. doi: 10.1016/j.brainres.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- Castro MG, Morrison E. Post-translational processing of proopiomelanocortin in the pituitary and in the brain. Crit Rev Neurobiol. 1997;11:35–57. doi: 10.1615/critrevneurobiol.v11.i1.30. [DOI] [PubMed] [Google Scholar]
- Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O’Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36) Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4695–4700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis BG, Pritchard LE, Creemers JW, Delplanque J, Keogh JM, Luan J, Wareham NJ, Yeo GS, Bhattacharyya S, Froguel P, White A, Farooqi IS, O’Rahilly S. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Human Molecular Genetics. 2002;11:1997–2004. doi: 10.1093/hmg/11.17.1997. [DOI] [PubMed] [Google Scholar]
- Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao L, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van Der Ploeg LH. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nature Genetics. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- Chen H, Jawahar S, Qian Y, Duong Q, Chan G, Parker A, Meyer JM, Moore KJ, Chayen S, Gross DJ, Glaser B, Permutt MA, Fricker LD. Missense polymorphism in the human carboxypeptidase E gene alters enzymatic activity. Human Mutation. 2001;18:120–131. doi: 10.1002/humu.1161. [DOI] [PubMed] [Google Scholar]
- Clement K, Dubern B, Mencarelli M, Czernichow P, Ito S, Wakamatsu K, Barsh GS, Vaisse C, Leger J. Unexpected endocrine features and normal pigmentation in a young adult patient carrying a novel homozygous mutation in the POMC gene. The Journal of Clinical Endocrinology and Metabolism. 2008;93:4955–4962. doi: 10.1210/jc.2008-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coll AP, Challis BG, Lopez M, Piper S, Yeo GS, O’Rahilly S. Proopiomelanocortin-deficient mice are hypersensitive to the adverse metabolic effects of glucocorticoids. Diabetes. 2005;54:2269–2276. doi: 10.2337/diabetes.54.8.2269. [DOI] [PubMed] [Google Scholar]
- Coll AP, Farooqi IS, Challis BG, Yeo GS, O’Rahilly S. Proopiomelanocortin and energy balance: insights from human and murine genetics. The Journal of Clinical Endocrinology and Metabolism. 2004;89:2557–2562. doi: 10.1210/jc.2004-0428. [DOI] [PubMed] [Google Scholar]
- Comuzzie AG, Hixson JE, Almasy L, Mitchell BD, Mahaney MC, Dyer TD, Stern MP, MacCluer JW, Blangero J. A major quantitative trait locus determining serum leptin levels and fat mass is located on human chromosome 2. Nature Genetics. 1997;15:273–276. doi: 10.1038/ng0397-273. [DOI] [PubMed] [Google Scholar]
- Contreras PC, Takemori AE. Antagonism of morphine-induced analgesia, tolerance and dependence by alpha-melanocyte-stimulating hormone. J Pharmacol Exp Ther. 1984;229:21–26. [PubMed] [Google Scholar]
- Cool DR, Normant E, Shen F, Chen HC, Pannell L, Zhang Y, Loh YP. Carboxypeptidase E is a regulated secretory pathway sorting receptor: genetic obliteration leads to endocrine disorders in Cpe(fat) mice. Cell. 1997;88:73–83. doi: 10.1016/s0092-8674(00)81860-7. [DOI] [PubMed] [Google Scholar]
- Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–930. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Pronchuk N, Fan W, Dinulescu DM, Colmers WF, Cone RD. Integration of NPY, AGRP, and melanocortin signals in the hypothalamic paraventricular nucleus: evidence of a cellular basis for the adipostat. Neuron. 1999;24:155–163. doi: 10.1016/s0896-6273(00)80829-6. [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- Creemers JW, Lee YS, Oliver RL, Bahceci M, Tuzcu A, Gokalp D, Keogh J, Herber S, White A, O’Rahilly S, Farooqi IS. Mutations in the amino-terminal region of proopiomelanocortin (POMC) in patients with early-onset obesity impair POMC sorting to the regulated secretory pathway. The Journal of Clinical Endocrinology and Metabolism. 2008;93:4494–4499. doi: 10.1210/jc.2008-0954. [DOI] [PubMed] [Google Scholar]
- Creemers JW, Pritchard LE, Gyte A, Le Rouzic P, Meulemans S, Wardlaw SL, Zhu X, Steiner DF, Davies N, Armstrong D, Lawrence CB, Luckman SM, Schmitz CA, Davies RA, Brennand JC, White A. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–1631. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- Dubern B, Lubrano-Berthelier C, Mencarelli M, Ersoy B, Frelut ML, Bougle D, Costes B, Simon C, Tounian P, Vaisse C, Clement K. Mutational analysis of the pro-opiomelanocortin gene in French obese children led to the identification of a novel deleterious heterozygous mutation located in the alpha-melanocyte stimulating hormone domain. Pediatric Research. 2008;63:211–216. doi: 10.1203/PDR.0b013e31815ed62b. [DOI] [PubMed] [Google Scholar]
- Elias CF, Aschkenasi C, Lee C, Kelly J, Ahima RS, Bjorbaek C, Flier JS, Saper CB, Elmquist JK. Leptin differentially regulates NPY and POMC neurons projecting to the lateral hypothalamic area. Neuron. 1999;23:775–786. doi: 10.1016/s0896-6273(01)80035-0. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Emeson RB, Eipper BA. Characterization of pro-ACTH/endorphin-derived peptides in rat hypothalamus. J Neurosci. 1986;6:837–849. doi: 10.1523/JNEUROSCI.06-03-00837.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi IS, Drop S, Clements A, Keogh JM, Biernacka J, Lowenbein S, Challis BG, O’Rahilly S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006;55:2549–2553. doi: 10.2337/db06-0214. [DOI] [PubMed] [Google Scholar]
- Fox DL, Vella KR, Good DJ. Energy balance pathways converging on the Nhlh2 transcription factor. Front Biosci. 2007;12:3983–3993. doi: 10.2741/2365. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Jones JE, Olson D, Hill J, Lee CE, Gautron L, Choi M, Zigman JM, Lowell BB, Elmquist JK. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640–13648. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta M, Yano H, Zhou A, Rouille Y, Holst JJ, Carroll R, Ravazzola M, Orci L, Furuta H, Steiner DF. Defective prohormone processing and altered pancreatic islet morphology in mice lacking active SPC2. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6646–6651. doi: 10.1073/pnas.94.13.6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of Agrt leads to obesity in transgenic mice. Nature Genetics. 1997;17:273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- Grossman HC, Hadjimarkou MM, Silva RM, Giraudo SQ, Bodnar RJ. Interrelationships between mu opioid and melanocortin receptors in mediating food intake in rats. Brain Research. 2003;991:240–244. doi: 10.1016/s0006-8993(03)03442-5. [DOI] [PubMed] [Google Scholar]
- Guo L, Munzberg H, Stuart RC, Nillni EA, Bjorbaek C. N-acetylation of hypothalamic alpha-melanocyte-stimulating hormone and regulation by leptin. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11797–11802. doi: 10.1073/pnas.0403165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager J, Dina C, Francke S, Dubois S, Houari M, Vatin V, Vaillant E, Lorentz N, Basdevant A, Clement K, Guy-Grand B, Froguel P. A genome-wide scan for human obesity genes reveals a major susceptibility locus on chromosome 10. Nature Genetics. 1998;20:304–308. doi: 10.1038/3123. [DOI] [PubMed] [Google Scholar]
- Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, Lauzon D, Lee CE, Coppari R, Richardson JA, Zigman JM, Chua S, Scherer PE, Lowell BB, Bruning JC, Elmquist JK. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metabolism. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- Ibrahim N, Bosch MA, Smart JL, Qiu J, Rubinstein M, Ronnekleiv OK, Low MJ, Kelly MJ. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- Jackson RS, Creemers JW, Farooqi IS, Raffin-Sanson ML, Varro A, Dockray GJ, Holst JJ, Brubaker PL, Corvol P, Polonsky KS, Ostrega D, Becker KL, Bertagna X, Hutton JC, White A, Dattani MT, Hussain K, Middleton SJ, Nicole TM, Milla PJ, Lindley KJ, O’Rahilly S. Small-intestinal dysfunction accompanies the complex endocrinopathy of human proprotein convertase 1 deficiency. The Journal of Clinical Investigation. 2003;112:1550–1560. doi: 10.1172/JCI18784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, Hutton JC, O’Rahilly S. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nature Genetics. 1997;16:303–306. doi: 10.1038/ng0797-303. [DOI] [PubMed] [Google Scholar]
- Jing E, Nillni EA, Sanchez VC, Stuart RC, Good DJ. Deletion of the Nhlh2 transcription factor decreases the levels of the anorexigenic peptides alpha melanocyte-stimulating hormone and thyrotropin-releasing hormone and implicates prohormone convertases I and II in obesity. Endocrinology. 2004;145:1503–1513. doi: 10.1210/en.2003-0834. [DOI] [PubMed] [Google Scholar]
- Jobst EE, Enriori PJ, Cowley MA. The electrophysiology of feeding circuits. Trends in Endocrinology and Metabolism: TEM. 2004;15:488–499. doi: 10.1016/j.tem.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Koegler FH, Grove KL, Schiffmacher A, Smith MS, Cameron JL. Central melanocortin receptors mediate changes in food intake in the rhesus macaque. Endocrinology. 2001;142:2586–2592. doi: 10.1210/endo.142.6.8198. [DOI] [PubMed] [Google Scholar]
- Korner J, Chua SC, Jr, Williams JA, Leibel RL, Wardlaw SL. Regulation of hypothalamic proopiomelanocortin by leptin in lean and obese rats. Neuroendocrinology. 1999;70:377–383. doi: 10.1159/000054499. [DOI] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nature Genetics. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- Krude H, Biebermann H, Schnabel D, Tansek MZ, Theunissen P, Mullis PE, Gruters A. Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4-10. The Journal of Clinical Endocrinology and Metabolism. 2003;88:4633–4640. doi: 10.1210/jc.2003-030502. [DOI] [PubMed] [Google Scholar]
- Lee M, Kim A, Chua SC, Jr, Obici S, Wardlaw SL. Transgenic MSH overexpression attenuates the metabolic effects of a high-fat diet. American Journal of Physiology. 2007;293:E121–131. doi: 10.1152/ajpendo.00555.2006. [DOI] [PubMed] [Google Scholar]
- Lee M, Wardlaw SL. The central melanocortin system and the regulation of energy balance. Front Biosci. 2007;12:3994–4010. doi: 10.2741/2366. [DOI] [PubMed] [Google Scholar]
- Lee YS, Challis BG, Thompson DA, Yeo GS, Keogh JM, Madonna ME, Wraight V, Sims M, Vatin V, Meyre D, Shield J, Burren C, Ibrahim Z, Cheetham T, Swift P, Blackwood A, Hung CC, Wareham NJ, Froguel P, Millhauser GL, O’Rahilly S, Farooqi IS. A POMC variant implicates beta-melanocyte-stimulating hormone in the control of human energy balance. Cell Metabolism. 2006;3:135–140. doi: 10.1016/j.cmet.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Li G, Mobbs CV, Scarpace PJ. Central pro-opiomelanocortin gene delivery results in hypophagia, reduced visceral adiposity, and improved insulin sensitivity in genetically obese Zucker rats. Diabetes. 2003;52:1951–1957. doi: 10.2337/diabetes.52.8.1951. [DOI] [PubMed] [Google Scholar]
- Li JY, Finniss S, Yang YK, Zeng Q, Qu SY, Barsh G, Dickinson C, Gantz I. Agouti-related protein-like immunoreactivity: characterization of release from hypothalamic tissue and presence in serum. Endocrinology. 2000;141:1942–1950. doi: 10.1210/endo.141.6.7462. [DOI] [PubMed] [Google Scholar]
- Lloyd DJ, Bohan S, Gekakis N. Obesity, hyperphagia and increased metabolic efficiency in Pc1 mutant mice. Human Molecular Genetics. 2006;15:1884–1893. doi: 10.1093/hmg/ddl111. [DOI] [PubMed] [Google Scholar]
- Low MJ, Hayward MD, Appleyard SM, Rubinstein M. State-dependent modulation of feeding behavior by proopiomelanocortin-derived beta-endorphin. Ann N Y Acad Sci. 2003;994:192–201. doi: 10.1111/j.1749-6632.2003.tb03180.x. [DOI] [PubMed] [Google Scholar]
- McMinn JE, Wilkinson CW, Havel PJ, Woods SC, Schwartz MW. Effect of intracerebroventricular alpha-MSH on food intake, adiposity, c-Fos induction, and neuropeptide expression. Am J Physiol Regul Integr Comp Physiol. 2000;279:R695–703. doi: 10.1152/ajpregu.2000.279.2.R695. [DOI] [PubMed] [Google Scholar]
- Mizuno TM, Kelley KA, Pasinetti GM, Roberts JL, Mobbs CV. Transgenic neuronal expression of proopiomelanocortin attenuates hyperphagic response to fasting and reverses metabolic impairments in leptin-deficient obese mice. Diabetes. 2003;52:2675–2683. doi: 10.2337/diabetes.52.11.2675. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C. Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology. 2003;144:2121–2131. doi: 10.1210/en.2002-221037. [DOI] [PubMed] [Google Scholar]
- Naggert JK, Fricker LD, Varlamov O, Nishina PM, Rouille Y, Steiner DF, Carroll RJ, Paigen BJ, Leiter EH. Hyperproinsulinaemia in obese fat/fat mice associated with a carboxypeptidase E mutation which reduces enzyme activity. Nature Genetics. 1995;10:135–142. doi: 10.1038/ng0695-135. [DOI] [PubMed] [Google Scholar]
- Nicolas P, Li CH. Beta-endorphin-(1-27) is a naturally occurring antagonist to etorphine-induced analgesia. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:3178–3181. doi: 10.1073/pnas.82.10.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen Y, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- Olszewski PK, Wirth MM, Grace MK, Levine AS, Giraudo SQ. Evidence of interactions between melanocortin and opioid systems in regulation of feeding. Neuroreport. 2001;12:1727–1730. doi: 10.1097/00001756-200106130-00042. [DOI] [PubMed] [Google Scholar]
- Perello M, Stuart RC, Nillni EA. Differential effects of fasting and leptin on pro-opiomelanocortin peptides in the arcuate nucleus and in the nucleus of the solitary tarct. American Journal of Physiology. 2007 doi: 10.1152/ajpendo.00466.2006. [DOI] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304:110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, Kim AJ, Cawley NX, Paik JH, Loh YP, Depinho RA, Wardlaw SL, Accili D. The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nature Medicine. 2009 doi: 10.1038/nm.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard LE, Oliver RL, McLoughlin JD, Birtles S, Lawrence CB, Turnbull AV, White A. Proopiomelanocortin-derived peptides in rat cerebrospinal fluid and hypothalamic extracts: evidence that secretion is regulated with respect to energy balance. Endocrinology. 2003;144:760–766. doi: 10.1210/en.2002-220866. [DOI] [PubMed] [Google Scholar]
- Pritchard LE, White A. Neuropeptide processing and its impact on melanocortin pathways. Endocrinology. 2007;148:4201–4207. doi: 10.1210/en.2006-1686. [DOI] [PubMed] [Google Scholar]
- Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, Bloom SR. A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology. 1998;139:4428–4431. doi: 10.1210/endo.139.10.6332. [DOI] [PubMed] [Google Scholar]
- Rotimi CN, Comuzzie AG, Lowe WL, Luke A, Blangero J, Cooper RS. The quantitative trait locus on chromosome 2 for serum leptin levels is confirmed in African-Americans. Diabetes. 1999;48:643–644. doi: 10.2337/diabetes.48.3.643. [DOI] [PubMed] [Google Scholar]
- Sanchez VC, Goldstein J, Stuart RC, Hovanesian V, Huo L, Munzberg H, Friedman TC, Bjorbaek C, Nillni EA. Regulation of hypothalamic prohormone convertases 1 and 2 and effects on processing of prothyrotropin-releasing hormone. The Journal of Clinical Investigation. 2004;114:357–369. doi: 10.1172/JCI21620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandman CA, Kastin AJ. Intraventricular administration of MSH induces hyperalgesia in rats. Peptides. 1981;2:231–233. doi: 10.1016/s0196-9781(81)80040-x. [DOI] [PubMed] [Google Scholar]
- Savontaus E, Breen TL, Kim A, Yang LM, Chua SCJ, Wardlaw SL. Metabolic effects of transgenic melanocyte-stimulating hormone overexpression in lean and obese mice. Endocrinology. 2004;145:3881–3891. doi: 10.1210/en.2004-0263. [DOI] [PubMed] [Google Scholar]
- Schwartz MW, Seeley RJ, Woods SC, Weigle DS, Campfield LA, Burn P, Baskin DG. Leptin increases hypothalamic pro-opiomelanocortin mRNA expression in the rostral arcuate nucleus. Diabetes. 1997;46:2119–2123. doi: 10.2337/diab.46.12.2119. [DOI] [PubMed] [Google Scholar]
- Shalts E, Feng YJ, Ferin M, Wardlaw SL. Alpha-melanocyte-stimulating hormone antagonizes the neuroendocrine effects of corticotropin-releasing factor and interleukin-1 alpha in the primate. Endocrinology. 1992;131:132–138. doi: 10.1210/endo.131.1.1319315. [DOI] [PubMed] [Google Scholar]
- Smart JL, Tolle V, Low MJ. Glucocorticoids exacerbate obesity and insulin resistance in neuron-specific proopiomelanocortin-deficient mice. The Journal of Clinical Investigation. 2006;116:495–505. doi: 10.1172/JCI25243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AI, Funder JW. Proopiomelanocortin processing in the pituitary, central nervous system, and peripheral tissues. Endocr Rev. 1988;9:159–179. doi: 10.1210/edrv-9-1-159. [DOI] [PubMed] [Google Scholar]
- Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu ZW, Gao XB, Diament A, Haus KA, Shariat-Madar Z, Mahdi F, Wardlaw SL, Schmaier AH, Warden CH, Diano S. Prolylcarboxypeptidase regulates food intake by inactivating alpha-MSH in rodents. The Journal of Clinical Investigation. 2009;119:2291–2303. doi: 10.1172/JCI37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardlaw SL, Ferin M. Interaction between beta-endorphin and alpha-melanocyte-stimulating hormone in the control of prolactin and luteinizing hormone secretion in the primate. Endocrinology. 1990;126:2035–2040. doi: 10.1210/endo-126-4-2035. [DOI] [PubMed] [Google Scholar]
- Wardlaw SL, Smeal MM, Markowitz CE. Antagonism of beta-endorphin-induced prolactin release by alpha-melanocyte-stimulating hormone and corticotropin-like intermediate lobe peptide. Endocrinology. 1986;119:112–118. doi: 10.1210/endo-119-1-112. [DOI] [PubMed] [Google Scholar]
- Wilkinson CW. Roles of acetylation and other post-translational modifications in melanocortin function and interactions with endorphins. Peptides. 2006;27:453–471. doi: 10.1016/j.peptides.2005.05.029. [DOI] [PubMed] [Google Scholar]
- Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao E, Kim AJ, Dutia R, Conwell I, Ferin M, Wardlaw SL. Effects of estradiol on cerebrospinal fluid levels of agouti-related protein in ovariectomized rhesus monkeys. Endocrinology. 2010;151:1002–1009. doi: 10.1210/en.2009-0853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS. Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol. 2005a;3:e415. doi: 10.1371/journal.pbio.0030415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. The Journal of Clinical Investigation. 2005b;115:951–958. doi: 10.1172/JCI24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nature Medicine. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- Zhu X, Zhou A, Dey A, Norrbom C, Carroll R, Zhang C, Laurent V, Lindberg I, Ugleholdt R, Holst JJ, Steiner DF. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10293–10298. doi: 10.1073/pnas.162352599. [DOI] [PMC free article] [PubMed] [Google Scholar]