

Abstract
The potential for relapse following cessation of drug use can last for years, implying the induction of stable changes in neural circuitry. In hippocampal slices from rats treated with nicotine for 1 week, withdrawal from nicotine in vivo produces an increase in CA1 pyramidal cell excitability that persists up to 9 months. Immediately upon drug cessation, the enhanced excitability depends on input from regions upstream of CA1, while the long-term excitability change (> 4 weeks) is expressed as an increase in the intrinsic excitability of CA1 neurons. Re-exposure to nicotine in vitro restores hippocampal function to control levels via activation of high-affinity nicotinic acetylcholine receptors after 1 d of withdrawal, but not at times >4 weeks. Thus, nicotine in vivo first induces homeostatic adaptations followed by other more robust neural changes. These mechanisms may contribute to hippocampal localized cue-motivated reinstatement of drug-seeking and/or cognitive deficits observed during withdrawal.
Introduction
One of the most challenging aspects of drug addiction treatment is the long-term susceptibility to relapse (Hyman et al., 2006). For tobacco use, the intensity of craving predicts relapse in ex-smokers (Killen and Fortmann, 1997; Etter and Hughes, 2006), and, since it persists longer than other known nicotine withdrawal symptoms, craving likely results in relapse to smoking following prolonged periods of abstinence (Durcan et al., 2002). The frequent pairing of smoking with everyday activities leads to the presence of unavoidable smoking-related cues, and cue-induced craving is correlated with increased activation of brain regions involved in associative learning including the hippocampus (Smolka et al., 2006; Franklin et al., 2007). Furthermore, the hippocampus is activated in smokers' brains when subjects are exposed to smoking-related images (Due et al., 2002), and direct stimulation of the hippocampus can reinstate drug-seeking behavior in animals previously trained to lever-press for addictive substances (Vorel et al., 2001). These observations imply that chronic exposure to drugs of abuse, such as nicotine, may induce lasting changes in hippocampal function.
Nicotine addiction, viewed as a form of associative learning (Kelley, 2004), may share many of the cellular and molecular mechanisms that underlie associations formed during more routine learning tasks (Kauer and Malenka, 2007). In the hippocampus, both acute in vitro and chronic in vivo nicotine treatments alter the induction thresholds for various forms of synaptic plasticity (Fujii et al., 1999; Ji et al., 2001; Welsby et al., 2006; Yamazaki et al., 2006a,b). The lowered threshold for long-term potentiation observed in the dentate gyrus after chronic in vivo nicotine exposure can be attributed specifically to elevated expression of α4β2* nicotinic acetylcholine receptors (nAChRs) on perforant path inputs (Nashmi et al., 2007), thereby linking the well established nicotine-induced nAChR upregulation (Marks et al., 1983; Schwartz and Kellar, 1983) with more robust downstream effects. While these nicotine-induced cellular alterations could contribute to the learning deficits observed on immediate withdrawal from nicotine (Davis et al., 2005), it is unlikely that they can be reconciled with more persistent behavioral effects, especially given that the changes in nAChR number and synaptic plasticity return to baseline shortly after drug cessation (Marks et al., 1985; Collins et al., 1988; Yamazaki et al., 2006b; Gao et al., 2010). To explain the lifelong aspects of addiction, it has been proposed that drugs of abuse, including nicotine, do not simply induce homeostatic adaptations that would return to baseline soon after removal of drug, but change the homeostatic reward set point, which then provides a continuous addictive drive (Koob and Bloom, 1988; Koob and Le Moal, 2001, 2005; DiFranza and Wellman, 2005; Hyman et al., 2006). The physiological substrate of these drug-induced allostatic states is currently not defined.
Here we show that withdrawal of nicotine results in increased excitability of principal neurons in the hippocampus. As predicted by behavioral studies, the changes are initially homeostatic and are reversed by re-exposure to nicotine in vitro, but, rather than reverting to control levels after an extended period of withdrawal, they become transformed into a more persistent and nonhomeostatic form.
Materials and Methods
Animals and treatment
Animals were individually housed on a standard 12 h light/dark cycle and given food and water ad libitum. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Animals and were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham. Six-week-old male Sprague Dawley rats (Harlan and Charles River Laboratories) weighing 150–225 g were implanted subcutaneously under isoflurane anesthesia with an osmotic mini pump (2ML1, Alzet) that delivered 9 mg/kg/d nicotine hydrogen tartrate (3.15 mg/kg/d nicotine base) or saline for 7 d, after which the pump was removed. This concentration of nicotine has been previously shown to result in plasma levels of nicotine in the rats similar to those found in human smokers (Murrin et al., 1987; Benowitz, 1988; Epping-Jordan et al., 1998). Experiments were performed at 1 d, 4 weeks (4–6 weeks), 3 months (3–4 months), 6 months (6–7 months), or 9 months (9–10 months) following osmotic pump removal.
Slice preparation
Acute transverse hippocampal slices (400 μm) were prepared from nicotine- or saline-treated rats at various time points after cessation of treatment (see Animals and treatment). Rats were deeply anesthetized with isoflurane and decapitated. The brain was rapidly removed from the skull and submerged in icy cold sucrose solution consisting of the following (in mm): 85 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 d-glucose, 75 sucrose, 4 MgSO4, and 0.5 CaCl2. The solution was bubbled with 95% O2/5% CO2 before and during use. The hippocampi were dissected out, and slices were made with a McIlwain tissue chopper. Slices were incubated at 33°C in an interface holding chamber containing sucrose solution for 30 min and then transferred to an interface holding chamber containing artificial CSF (ACSF) at room temperature for at least an additional 30 min. The ACSF consisted of the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 d-glucose, 1.3 MgSO4, and 2.5 CaCl2 and was continuously bubbled with 95% O2/5% CO2.
Electrophysiology
Slices were transferred to an interface recording chamber (Fine Science Tools) perfused at 1.5 ml/min with oxygenated ACSF at 31−33°C 15 min before recording. In some experiments, a scalpel blade was used to sever connections between CA3 and dentate gyrus or remove the CA3 region immediately before placing the slices in the recording chamber.
Field potential recordings.
Field potential recordings were made with glass pipettes filled with ACSF in stratum radiatum and stratum pyramidale of CA1 using an Axoclamp 2B amplifier and pClamp 9.2 software (Molecular Devices). Data were sampled at 4 kHz and filtered at 1 kHz. The fiber volley and EPSP (fEPSP) were recorded from stratum radiatum, and the population spike (pSpike) was recorded from stratum pyramidale following 0.05 Hz stimulation of the Schaffer collateral/commissural pathway with a bipolar nickel-chromate wire electrode. In a subset of experiments, stimulation of the alvear pathway was alternated with Schaffer collateral/commissural stimulation and the antidromic pSpike (aSpike) was recorded from stratum pyramidale. After a 20 min stable baseline was established using a subthreshold stimulus intensity, a pseudorandom series of 15–30 different intensity stimuli was used to collect input–output (I-O) data. For drug applications, the response was allowed to restabilize for 10–20 min following the initial I-O data collection, and drugs were bath applied in ACSF for 60 min. Following this, another set of I-O data was collected.
The amplitude of the fiber volley was measured as the difference between the early positivity and the negative peak of the initial small deflection. The fEPSP slope was measured as the fit of a straight line to the trace in the 0.5 ms interval immediately following the fiber volley. The absolute value of the fEPSP slope was reported. The pSpike amplitude was measured as the difference between the first positive peak and the negative peak of the pSpike. The aSpike amplitude was measured as the difference between the most positive point immediately after the stimulus artifact and the negative peak of the aSpike.
Individual I-O and EPSP–spike (E-S) curves were fit with a Boltzmann equation using Kaleidagraph (Synergy Software). Any slices with <2 mV initial maximal pSpike amplitude in ACSF were excluded from analysis.
Intracellular recordings.
Current-clamp recordings were made from CA1 pyramidal cells using microelectrodes filled with 2 m KMeSO4 or 3 m K-acetate (resistance 70–130 MΩ, pH 7.2–7.4) using an Axoclamp 2B amplifier in bridge mode and pClamp 9.2 software (Molecular Devices). Recordings used for assessing action potential characteristics were sampled at 25 kHz and filtered at 10 kHz; recordings of spontaneous activity were sampled at 10 kHz and filtered at 3 kHz; all other data were sampled at 4 kHz and filtered at 1 kHz. The bridge balance was carefully monitored and adjusted before each measurement. Only cells with a stable resting membrane potential (RMP) more negative than −55 mV, input resistance (RN) >20 MΩ, and evoked spikes that overshot across a 0 mV level were used. Once the neuron had stabilized for 10 min after the initial impalement and met the cell health criteria, the membrane properties were measured. Resting membrane potential values were measured in the absence of any injected current and were corrected according to the tip potential recorded extracellularly immediately after termination of the intracellular recording. The cell input resistance was measured by injecting hyperpolarizing current pulses (−0.5 nA, 400 ms) and calculated by dividing the steady-state voltage response by the current pulse amplitude (RN = V/I). The sag ratio was the ratio of the steady-state voltage deflection to the peak voltage deflection during hyperpolarizing current pulses (−0.5 nA, 400 ms).
The current–voltage (I–V) relationship was studied following injection of hyperpolarizing and subthreshold depolarizing current pulses (500 ms duration). The steady-state voltage deflection with respect to the resting membrane potential (ΔV) was plotted against the amount of current injected. Current–spike response curves were constructed from data obtained using either 100 or 500 ms depolarizing current steps (0 to 1.0 nA in 0.1 nA increments). The postburst medium afterhyperpolarization (mAHP) was studied using a 100 ms depolarizing current step that reliably elicited a burst of four action potentials. The peak mAHP amplitude was calculated as the maximum negative voltage deflection from the baseline potential during the first 250 ms after the current offset.
Single action potential waveform characteristics and firing probabilities were obtained from action potentials elicited by 10 trials of 10 ms depolarizing current injections (0 to 1.9 nA in 0.1 nA increments). The I0.5 indicates the current required to evoke a spike with a probability of 0.5 and was calculated by fitting individual firing probability curves with a Boltzmann equation using Kaleidagraph (Synergy Software). Action potential threshold was determined as the point where the first derivative of the action potential was equal to 20 mV/ms. Spike height was defined as the peak relative to the most negative voltage reached during the fast AHP (fAHP) immediately after the spike. Spike half-width was measured as the width at half-maximal spike amplitude. The fAHP was measured as the potential attained at the end of the spike downstroke. In all cases, the spikes were evoked from resting membrane potential.
Intracellular E-S data were collected by stimulating the Schaffer collateral/commissural pathway at 0.05 Hz with a bipolar nickel-chromate wire electrode. A pseudorandom series of 10 trials of 10 different intensity stimuli was used to collect data both subthreshold and suprathreshold for firing. The EPSP slope was measured as the fit of a straight line to the trace in the 1.0 ms interval where the trace starts sloping upward after the stimulus artifact. The EPSP amplitude was measured as the difference between the resting membrane potential measured immediately before the stimulus artifact and the potential measured at peak of the EPSP. The presence or absence of an action potential was visually determined.
The frequency of spontaneous events was determined using the event detection analysis in Clampfit 9.2 (Molecular Devices). For individual cells, representative spontaneous events were averaged and used as a template for searching the traces.
Extracellular spontaneous multiunit recordings.
Field potential recordings were made with glass pipettes filled with ACSF in stratum pyramidale of CA3 and stratum granulosum of dentate gyrus using an Axoclamp 2B amplifier and pClamp 9.2 software (Molecular Devices). Data were sampled at 10 kHz and bandpass filtered (0.5–3 kHz). Before recording activity, a “noise” recording was made with the electrode placed in the bath to determine the noise level. Spikes were detected by threshold set above the recorded noise level and individually confirmed by inspection.
Statistics
Statistical significance was determined for the spontaneous EPSP (sEPSP) and spike frequencies, membrane properties, and action potential characteristics using a two-sample Student's unpaired t test. Mean extracellular I-O and E-S curves data were constructed by averaging data at each stimulus intensity. Mean intracellular E-S curves were constructed by binning EPSP slope data in 0.5 mV/ms bins and determining the firing probability within each bin. Mean current–spike response curves and I–V curves were constructed by averaging data at each current injection. Statistical significance was determined using regression analysis of the curve fits in SPSS (SPSS). All data are expressed as mean ± SEM, with n indicating the number of slices or cells. p < 0.05 is considered significant. Example traces of fEPSPs are averages of two to three sweeps.
Drug and solutions
All drugs were bath applied in ACSF at the concentrations indicated. d-amino-5-phosphonovaleric acid (d-APV), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) disodium salt, dihydro-β-erythroidine (DHβE), and tetrodotoxin (TTX) were obtained from Tocris Bioscience. All other drugs and chemicals were obtained from Sigma-Aldrich.
Results
Chronic nicotine treatment persistently increases the excitability of CA1 pyramidal cells
To study neuroadaptations following chronic in vivo nicotine treatment, young adult rats were treated with 9 mg/kg/d nicotine hydrogen tartrate (equivalent to 3.15 mg/kg/d nicotine base) or saline for 1 week, and acute hippocampal slices were prepared 1 d to 9 months after drug cessation (see Materials and Methods). This dose of nicotine has been previously shown to result in plasma levels of nicotine in the rats similar to those found in human smokers (Murrin et al., 1987; Benowitz, 1988). Importantly, both somatic and affective withdrawal symptoms can be induced in rats following cessation of similar nicotine treatments (Epping-Jordan et al., 1998) (for review, see Markou, 2008). It is noted that in the current study a single continuous exposure to nicotine was used, and a more intermittent nicotine administration protocol may result in somewhat different levels of nicotine and therefore different effects (Matta et al., 2007).
Although direct modification of synaptic efficacy is the most commonly studied form of associative plasticity, it is not the only type of long-lasting change observed in the hippocampus. The propensity of a given synaptic potential to trigger action potential firing—so-called E-S coupling—can also be altered in an activity-dependent manner (Bliss and Lomo, 1973). As activity within the brain is dependent on both the synaptic input and the spiking output of neurons, changes in the excitability of neurons may also be an important adaptation involved in addiction. E-S coupling was assessed at the single-cell level using intracellular current-clamp recordings of CA1 pyramidal cells following stimulation of the Schaffer collateral/commissural pathway.
For a given EPSP slope measured intracellularly, there was an increase in firing probability in cells from nicotine-treated animals (nic-cells) compared with cells from saline-treated animals (sal-cells) following both 1 d (Fig. 1B) (p < 0.05) and long-term (Fig. 1A,C) (p < 0.05) withdrawal. We also assessed excitability in the absence of experimenter-evoked synaptic input using current injections to determine firing probability. The I0.5 (injected current required for a firing probability of 0.5) was reduced in nic-cells compared with sal-cells at both 1 d (nic-cells: 0.68 ± 0.10 nA, n = 13; sal-cells: 1.21 ± 0.11 nA, n = 15; p < 0.05) and long-term (nic-cells: 0.48 ± 0.06 nA, n = 14; sal-cells: 0.92 ± 0.07 nA, n = 13; p < 0.05) withdrawal.
Figure 1.
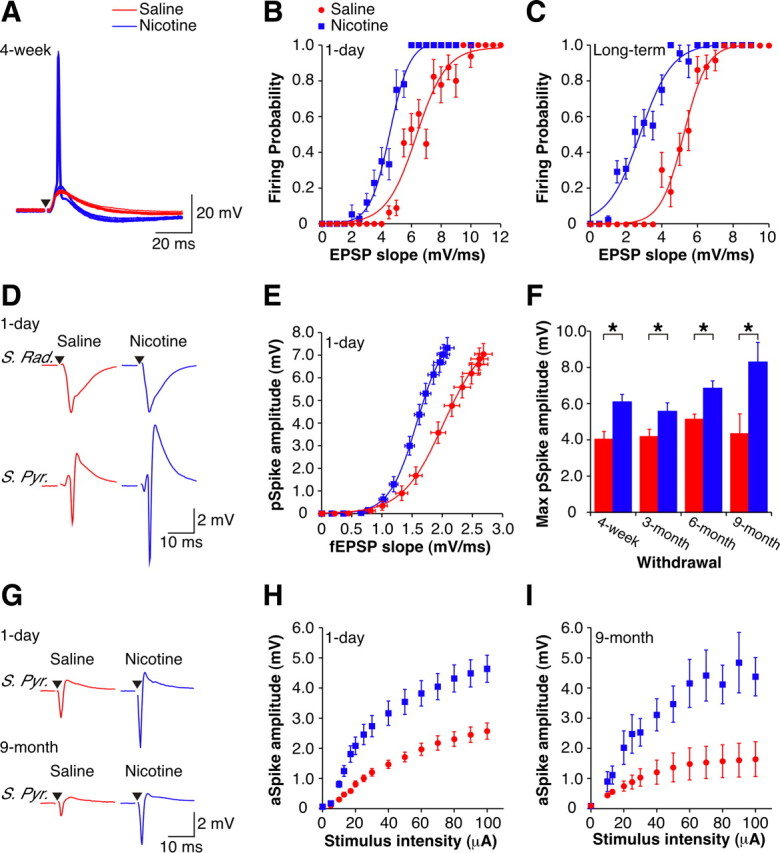
Chronic nicotine induces persistent changes in EPSP–Spike coupling. A, Examples of orthodromically evoked EPSPs and action potentials recorded intracellularly from CA1 pyramidal cells in current-clamp with matching EPSP slopes recorded from slices from 4 week withdrawn animals. The nic-cell (blue) has a firing probability of 0.5, while the sal-cell (red) is subthreshold for action potential firing. Stimulus artifact has been removed for clarity, and a black triangle represents time of stimulus. B, C, Mean intracellular E-S curves from 1 d withdrawn (B) (sal-cells, n = 7; nic-cells, n = 5) and long-term (4-week + 3-month) withdrawn (C) (sal-cells, n = 4; nic-cells, n = 7) animals. D, Examples of orthodromically evoked suprathreshold fEPSPs recorded in stratum radiatum (top traces) and pSpikes recorded in stratum pyramidale (bottom traces) from 1 d withdrawn animals (sal-slice: red; nic-slice: blue). For a given fEPSP slope, nic-slices have an increased pSpike amplitude. Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. E, Mean extracellular E-S curves from 1 d withdrawn animals (sal-slices, n = 37; nic-slices, n = 36). F, Histogram showing the maximal pSpike amplitude for different periods of withdrawal: 4 week (sal-slices, n = 12; nic-slices, n = 20); 3 month (sal-slices, n = 17; nic-slices, n = 22); 6 month (sal-slices, n = 6; nic-slices, n = 10); and 9 month (sal-slices, n = 4; nic-slices, n = 5). G, Examples of aSpikes recorded in stratum pyramidale from 1 d (top) and 9 month (bottom) withdrawn animals (sal-slices: red; nic-slices: blue). Stimulus artifacts have been removed for clarity and black triangles represent time of stimulus. H, I, Mean aSpike I-O curves from 1 d withdrawn (H) (sal-slices, n = 20; nic-slices, n = 18) and 9 month withdrawn (I) (sal-slices, n = 4; nic-slices, n = 5) animals. Data are presented as the mean ± SEM. S. Rad., Stratum radiatum; S. Pyr., stratum pyramidale. *p < 0.05.
The increased excitability of individual cells was reflected in the CA1 population, assessed by comparing the fEPSP recorded in the CA1 dendritic layer (stratum radiatum) to the pSpike—a measure of the number of synchronously firing cells (Andersen et al., 1971)—recorded in the somatic layer (stratum pyramidale). We found that in slices from nicotine-treated animals (nic-slices), the same size fEPSP slope yielded a larger pSpike compared with that in slices from saline-treated animals (sal-slices) (Fig. 1D). Thus, following 1 d withdrawal from nicotine there was a leftward shift in the E-S curve (Fig. 1E) (p < 0.05). This enhanced excitability persisted for up to at least 9 months after drug cessation, shown here as a larger maximal pSpike in nic-slices (Fig. 1F) with no difference in maximal fEPSP slope (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). To assess excitability independent of experimenter-evoked synaptic input at the population level, extracellularly measured aSpikes were evoked by CA1 pyramidal cell axon stimulation in the alveus (Fig. 1G). Following 1 d of withdrawal, aSpikes were enhanced in nic-slices compared with sal-slices (Fig. 1G, top, H) (p < 0.05). Similar to E-S potentiation, nicotine-induced augmentation of the aSpike persisted in nic-slices from animals withdrawn for up to 9 months (Fig. 1G, bottom, I) (p < 0.05). Together, these data establish that there is a long-lasting increase in the excitability of CA1 pyramidal cells in animals previously treated with nicotine.
Increased spontaneous excitatory synaptic transmission drives the enhanced excitability of CA1 pyramidal cells immediately following drug cessation
Following 1 d of withdrawal, nic-cells have a significantly depolarized resting membrane potential compared with sal-cells (Fig. 2A). There was a trend toward an increase in the input resistance of the nic-cells compared with sal-cells (supplemental Table S1, available at www.jneurosci.org as supplemental material). However, this is not surprising given that in CA1 pyramidal cells the input resistance is not a purely passive property and varies with the resting membrane potential (Spruston and Johnston, 1992). Indeed, in a subset of nic-cells artificially hyperpolarized by ∼5 mV to a level similar to that of sal-cells (RMP: −64.8 ± 3.3 mV; hyperpolarized cells: −69.3 ± 3.5 mV; n = 4; p < 0.05) the RN was significantly reduced (RMP RN = 35.3 ± 4.2 MΩ; hyperpolarized potentials RN = 32.0 ± 4.6 MΩ; n = 4; p < 0.05). As no other measured properties of these cells differ (supplemental Table S1, available at www.jneurosci.org as supplemental material), the depolarized resting membrane potential results in nic-cells being closer to action potential threshold; therefore, less current is required to induce action potential firing (Fig. 2B,C) (p < 0.05). Following long-term potentiation induction, changes in E-S coupling have been found to be partly due to changes in the balance of excitation and inhibition (Staff and Spruston, 2003; Marder and Buonomano, 2004). Since the increased excitability is expressed in the absence of experimenter evoked synaptic input (Fig. 1G), if altered synaptic transmission underlies the withdrawal-induced depolarized membrane potential, it must be spontaneous in nature. Intracellular recordings of spontaneous activity indicate that the frequency, but not the amplitude, of sEPSPs is increased in nic-cells compared with sal-cells (Fig. 2D–F), while the frequency (nic-cells: 0.15 ± 0.02 Hz; sal-cells: 0.11 ± 0.01 Hz; p > 0.05) and amplitude (nic-cells: −1.16 ± 0.10 pA; sal-cells: −1.08 ± 0.08 pA; p > 0.05) of spontaneous IPSPs remain unchanged. It is possible that the increase in sEPSP frequency may not fully account for the entire depolarization and that additional mechanisms, such as depolarized-activated voltage-dependent channels (e.g., sodium or calcium channels) or the activation of extrasynaptic receptors, may contribute to the translation of increased CA3 activity into a depolarization of CA1 pyramidal cells.
Figure 2.

Enhanced excitability at 1 d withdrawal is dependent on an increase in glutamatergic synaptic transmission. A, Mean and individual resting membrane potentials of CA1 pyramidal cells from 1 d withdrawn animals (sal-cells, n = 22; nic-cells, n = 25). B, Examples of subthreshold depolarizations and action potentials evoked by depolarizing current pulses (0.1 nA steps, 100 ms) up to threshold from CA1 pyramidal cells from a 1 d withdrawn saline-treated animal (red, 0.4 nA at threshold) and a 1 d withdrawn nicotine-treated animal (blue, 0.2 nA at threshold). Capacitance artifacts have been removed for clarity. C, Mean current–spike response curves for 100 ms depolarizing current pulses (sal-cells, n = 21; nic-cells, n = 24). D, Top, Examples of spontaneous EPSPs recorded in current-clamp from CA1 pyramidal cells from 1 d withdrawn animals. Bottom, Example sEPSPs (average of 5 events) presented on an expanded scale (sal-cell, red; nic-cell, blue). E, F, Mean spontaneous EPSP frequency (E) and amplitude (F) in CA1 pyramidal cells from 1 d withdrawn animals (sal-cells, n = 14; nic-cells, n = 14). G, Time course of the resting membrane potential during CNQX + d-APV wash-in (sal-cells, n = 5; nic-cells, n = 5). Error bars are only shown once per minute for clarity. H, Time course of the normalized fEPSP slope during CNQX + d-APV wash-in (sal-slices, n = 5; nic-slices, n = 4). Inset, Example fEPSPs in the presence of ACSF and after 30 min wash-in of CNQX + d-APV. Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. I, Mean current required for a firing probability of 0.5 (I0.5) for CA1 pyramidal cells in ACSF (sal-cells, n = 15; nic-cells, n = 13) and CNQX + d-APV (sal-cells, n = 5; nic-cells, n = 4). Data are presented as mean ± SEM. *p < 0.05.
If the depolarized resting membrane potential and enhanced excitability in nic-cells are due to the increase in sEPSPs, then we would expect the effects to be reversed by blockers of excitatory synaptic transmission. Accordingly, antagonism of glutamate receptors with CNQX (10 μm) and d-APV (50 μm) resulted in hyperpolarization of nic-cells (ACSF: −62.6 ± 1.7 mV; CNQX + d-APV: −67.8 ± 2.9 mV; n = 5; p < 0.05) with no effect on the resting membrane potential of sal-cells (ACSF: −71.2 ± 1.4 mV; CNQX + d-APV: −71.0 ± 1.1 mV, n = 5; p > 0.05) (Fig. 2G). Application of CNQX and d-APV also resulted in a decrease in the input resistance of the nic-cells (ACSF: RN = 39.2 ± 4.4 MΩ; CNQX + d-APV: RN = 33.8 ± 4.2 MΩ; n = 5; p < 0.05) with no effect on the input resistance of the sal-cells (ACSF: RN = 31.0 ± 2.2 MΩ; CNQX + d-APV: RN = 31.4 ± 2.4 MΩ; n = 5; p > 0.05), consistent with the current injected hyperpolarization data reported above. While it might be expected that blockade of sEPSPs would increase the input resistance of the nic-cells alone, given the hyperpolarization observed following perfusion with CNQX and d-APV and the dependence of the input resistance on the resting membrane potential in these cells (Spruston and Johnston, 1992), a decrease in input resistance is not unexpected. The differential effects of the drugs on nic-cells and sal-cells were not due to a difference in the efficacy of receptor blockade in cells from the different treatment groups, as inhibition of the extracellular fEPSPs recorded during drug application was similar between the two groups (Fig. 2H). As expected, in the presence of CNQX and d-APV, the increased firing probability observed in nic-cells was also restored to control levels (Fig. 2I), indicating that the enhanced excitability of the nic-cells was likely due to an increased background release of glutamate. We next measured spontaneous activity in the presence of TTX (1 μm) to determine whether the elevated glutamatergic transmission was action potential dependent. The frequency of synaptic events remaining in TTX was not different between nic-cells and sal-cells (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material), and application of TTX also hyperpolarized nic-cells (ACSF: −65.3 ± 1.0 mV; TTX: −68.8 ± 1.4 mV; n = 4; p < 0.05) (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material) and decreased the input resistance of nic-cells (ACSF: RN = 33.8 ± 3.2 MΩ; TTX: RN = 30.3 ± 3.3 MΩ; n = 4; p < 0.05). Importantly, the I–V curves of 1 d withdrawn cells were not significantly different in the presence of CNQX + d-APV or TTX (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material), providing additional evidence that the enhanced excitability of the CA1 pyramidal cells immediately after nicotine treatment is due to a change in synaptic inputs and not intrinsic excitability. These results imply that the depolarized resting membrane potential and the resulting enhanced cellular excitability rely on action potential-dependent spontaneous activity that may arise as a result of changes upstream of CA1.
An increase in the firing rate of CA3 pyramidal cells has been shown to boost subthreshold evoked synaptic inputs to CA1 pyramidal cells (Stacey and Durand, 2001), possibly via an augmented transfer of current from synapse to soma (Hô and Destexhe, 2000) and/or dendritic spiking (Kamondi et al., 1998), which culminates in an increase in the probability of action potential firing (Destexhe et al., 2001). If an increase in the firing of CA3 pyramidal neurons is causing the enhanced CA1 excitability, then nic-slices lacking input from these neurons should behave like sal-slices. In confirmation of this suggestion, E-S curves constructed at 1 d of withdrawal in slices with the CA3 region completely removed were no longer significantly different between nicotine- and saline-exposed animals (Fig. 3A) (p > 0.05).
Figure 3.
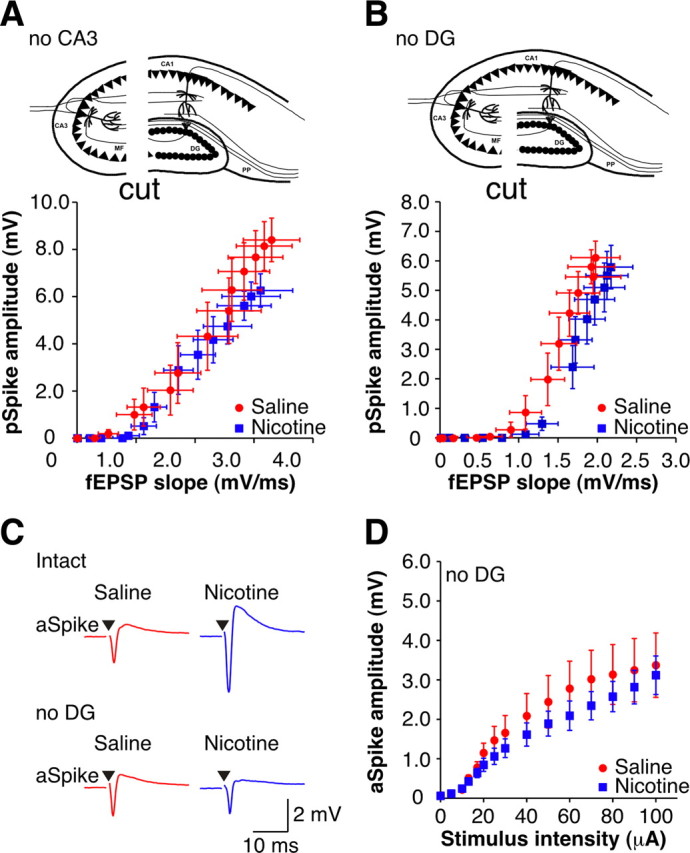
Enhanced excitability in CA1 requires upstream connections with dentate gyrus at early withdrawal. A, Top, Diagram of no CA3 slice. The scalpel cut is indicated. Bottom, Mean extracellular E-S curves from 1 d withdrawn no CA3 slices (sal-slices, n = 8; nic-slices, n = 7). B, Top, Diagram of no DG slice. The scalpel cut is indicated. Bottom, Mean extracellular E-S curves from 1 d withdrawn no DG slices (sal-slices, n = 7; nic-slices, n = 8). C, Examples of aSpikes from intact (top) and no DG (bottom) slices from 1 d withdrawn animals (sal-slice, red; nic-slice, blue). Stimulus intensity: 40 μA. Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. D, Mean I-O curves for aSpikes from no DG slices (sal-slices, n = 6; nic-slices, n = 6). Data are presented as mean ± SEM. No CA3, Slices with the CA3 region removed; no DG, slices with connections severed between CA3 and dentate gyrus.
While the dentate gyrus does not directly innervate the CA1 region, it has been shown that a trisynaptically activated response can be measured in CA1 following stimulation of the perforant path fibers in an acute hippocampal slice (Sirvio et al., 1996). Activation requires sequential excitatory connections from dentate granule cells to CA3 pyramidal neurons and then from CA3 to CA1 pyramidal cells, indicating that there is sufficient functional connectivity present in an acute transverse hippocampal slice for activity within dentate gyrus to influence responses recorded in CA1. After 1 d of withdrawal, in slices with inputs from dentate gyrus severed, similar to CA3 removal, the E-S curves in nic-slices and sal-slices were no longer significantly different (Fig. 3B) (p > 0.05). In addition, removing input from dentate gyrus eliminated the aSpike potentiation measured in nic-slices (Fig. 3C,D) (p > 0.05), implying that all the changes in CA1 are driven by changes in spontaneous slice activity that originates in the dentate gyrus.
To directly measure the spontaneous firing activity within CA3, we recorded multiunit activity extracellularly in CA3 stratum pyramidale. As predicted by the above data, the frequency of spikes recorded in CA3 was significantly enhanced in nic-slices compared with sal-slices (Fig. 4A, top, B). Moreover, the increased CA3 activity was not present in slices with inputs from dentate gyrus severed (Fig. 4A, bottom, B). It is possible that the reliability of extracellular stimulation to initiate action potentials in the Schaffer collateral fibers is reduced during such elevated periods of CA3 activity (Soleng et al., 2003). In this regard, we observed a decrease in the amplitude of the fiber volley, a measure of the number of activated axons (Andersen et al., 1978), in nic-slices (Fig. 4C,D) (p < 0.05) and a consequent decrease in the initial slope of the stratum radiatum fEPSP (Fig. 4C,E) (p < 0.05). Consistent with these latter changes also being caused by the increase in spontaneous firing of the CA3 pyramidal neurons, they were eliminated in slices with no input from regions upstream of CA1 (Fig. 4F–H) (p > 0.05). Thus, while there is a decrease in the evoked population synaptic response, this is not a true change in synaptic efficacy (i.e., long-term depression). In sum, we show that the enhanced excitability of CA1 neurons immediately after withdrawal from nicotine is due to altered signaling originating in the dentate gyrus that permeates as increased excitation throughout the entire hippocampal circuit. Additional support for this idea comes from data indicating that spontaneous firing activity recorded from stratum granulosum in the dentate gyrus exhibit a trend toward an increased frequency of spikes recorded from nic-slices compared with sal-slices in both intact slices and slices with dentate gyrus isolated from the rest of the trisynaptic pathway (supplemental Fig. S4, available at www.jneurosci.org as supplemental material).
Figure 4.
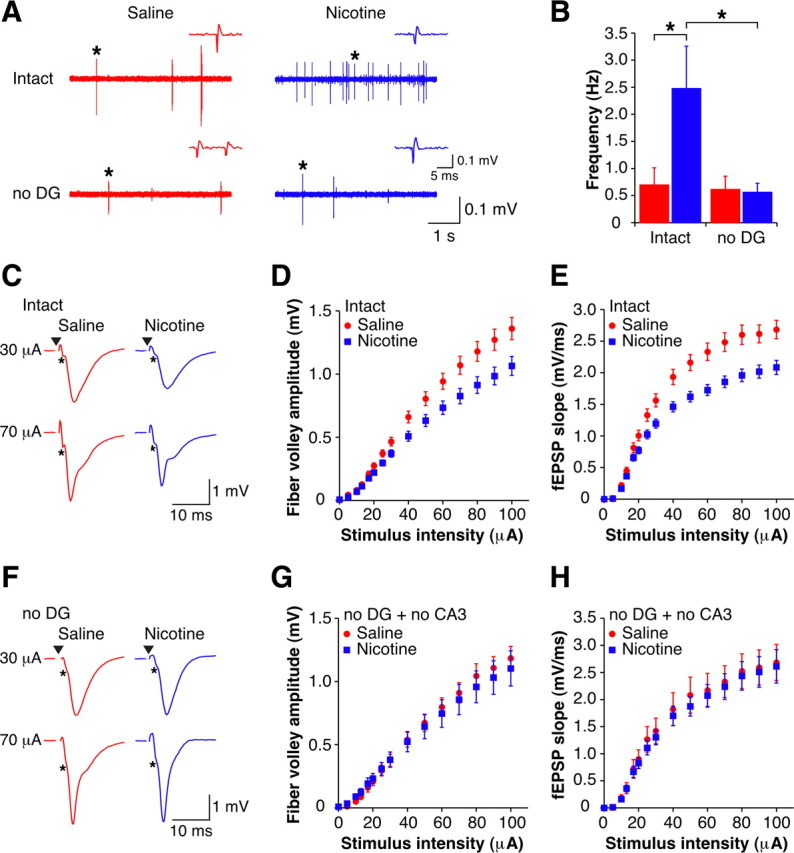
CA3 pyramidal cells have increased spontaneous firing dependent on inputs from dentate gyrus at early withdrawal. A, Examples of spontaneous extracellular spikes recorded in CA3 stratum pyramidale in intact (top) and no DG (bottom) slices from 1 d withdrawn animals (sal-slice, red; nic-slice, blue). Insets are examples of spikes, indicated by asterisks presented on an expanded scale. B, Mean frequencies of spontaneously occurring spikes recorded from CA3 stratum pyramidale in both intact (sal-slices, n = 11; nic-slices, n = 16) and no DG (sal-slices, n = 11; nic-slices, n = 11) slices from 1 d withdrawn animals. C, Examples of orthodromically evoked fEPSPs recorded in CA1 stratum radiatum from intact slices from 1 d withdrawn animals (sal-slices: red; nic-slices: blue). Fiber volleys are indicated by asterisks. Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. D, E, Mean fiber volley amplitude (D) and fEPSP slope (E) I-O curves for intact slices from 1 d withdrawn animals (sal-slices, n = 37; nic-slices, n = 36). F, Examples of orthodromically evoked fEPSPs recorded in CA1 stratum radiatum from no DG slices from 1 d withdrawn animals (sal-slices: red; nic-slices: blue). Fiber volleys are indicated by asterisks. Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. G, H, Mean fiber volley amplitude (G) and fEPSP slope (H) I-O curves for no DG + no CA3 slices from 1 d withdrawn animals (sal-slices, n = 15; nic-slices, n = 15). Data are presented as mean ± SEM. *p < 0.05. No DG, Slices with connections severed between CA3 and dentate gyrus; no CA3, slices with the CA3 region removed.
Increased intrinsic excitability of CA1 pyramidal cells following long-term withdrawal
Similar to changes after 1 d of withdrawal, the changes in CA1 following long-term withdrawal (4 weeks to 9 months) are manifest as an increase in both E-S coupling and firing probability, again consistent with an increase in pyramidal cell excitability (Fig. 1). Unlike the results at 1 d of withdrawal, following long-term withdrawal nic-cells are not depolarized (nic-cells: −67.3 ± 0.9 mV, n = 17; sal-cells: −66.1 ± 1.2 mV, n = 14; p > 0.05), indicating that while the overall effect may be the same, the precise mechanism underlying the enhanced excitability of CA1 pyramidal neurons is likely different. Both theoretical models and experimental plasticity data show that changes in voltage-dependent ion channels may account for E-S potentiation (Taube and Schwartzkroin, 1988; Wathey et al., 1992). Moreover, lasting alterations in the function of ion channels that control membrane excitability occur in prefrontal cortex after repeated cocaine administration (Nasif et al., 2005b), and cortical voltage-dependent Ca2+ channel function is enhanced after nicotine exposure (Katsura et al., 2002). Such changes in voltage-dependent ion channel function can amplify subthreshold EPSPs, as assessed by comparing the EPSP slope to the EPSP amplitude (Campanac and Debanne, 2008). Consistent with this idea, we find an increased amplitude/slope ratio of EPSPs recorded from nic-cells compared with sal-cells (nic-cells: 4.24 ± 0.21, n = 7; sal-cells: 3.28 ± 0.35, n = 4; p < 0.05), indicating possible voltage-dependent boosting. In support of a change in the intrinsic excitability of CA1 pyramidal cells at these later time points, we find that surgical removal of CA3 does not abolish the difference in excitability between the two treatment groups as illustrated by the continued presence of E-S potentiation (supplemental Fig. S5A, top, B, available at www.jneurosci.org as supplemental material) (p < 0.05) and aSpike potentiation (supplemental Fig. S5A, bottom, C, available at www.jneurosci.org as supplemental material) (p < 0.05) in nic-slices. Furthermore, during perfusion with blockers of synaptic transmission (10 μm CNQX; 50 μm d-APV; 10 μm bicuculline), nic-cells fired more spikes for a given current injection than sal-cells (Fig. 5A), and this resulted in a current–spike response curve that was shifted upward (Fig. 5C, solid symbols) (p < 0.05). Moreover, there was a significant increase in the input resistance of nic-cells compared with sal-cells (supplemental Table S2, available at www.jneurosci.org as supplemental material), and the I–V curves were significantly different between the two groups (Fig. 5D,E) (p < 0.05).
Figure 5.
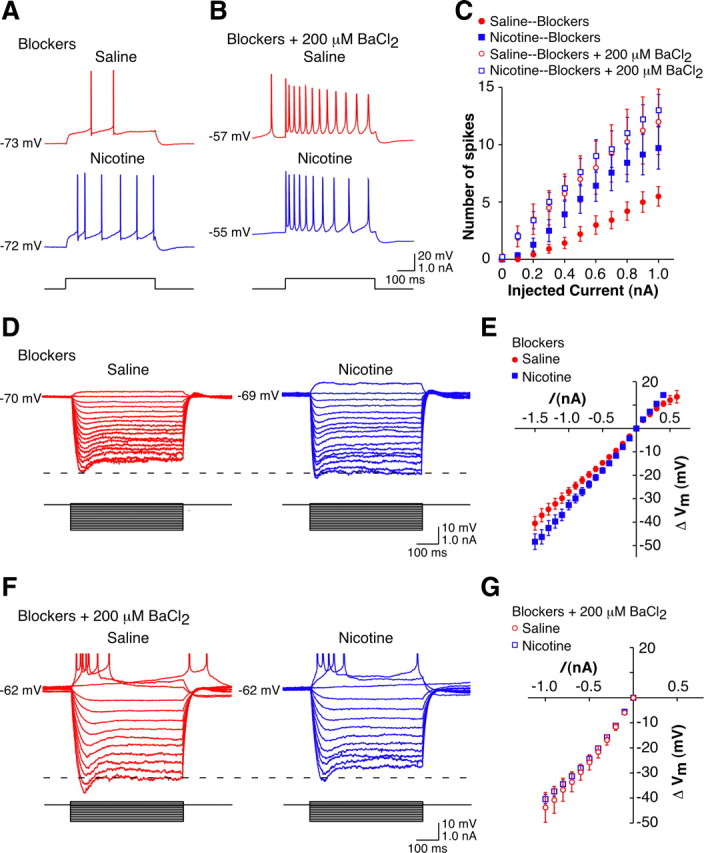
Enhanced excitability is due to a change in the intrinsic excitability of the CA1 pyramidal cells at late withdrawal. A, Example action potentials evoked by depolarizing current pulses (0.6 nA, 500 ms) in CA1 pyramidal cells from a long-term withdrawn saline-treated animal (red) and a long-term withdrawn nicotine-treated animal (blue). Experiments were conducted in the presence of blockers. Capacitance artifacts have been removed for clarity. B, Example action potentials evoked by depolarizing current pulses (0.6 nA, 500 ms) in CA1 pyramidal cells from a long-term withdrawn saline-treated animal (red) and a long-term withdrawn nicotine-treated animal (blue). Experiments were conducted in the presence of blockers + 200 μm BaCl2. Capacitance artifacts have been removed for clarity. C, Mean current–spike response curves for 500 ms depolarizing current pulses in CA1 pyramidal cells from long-term withdrawn animals in the presence of blockers (closed symbols; sal-cells, n = 14; nic-cells, n = 14) and blockers + 200 μm BaCl2 (open symbols; sal-cells, n = 4; nic-cells, n = 5). D, Example traces of the membrane potential response of CA1 pyramidal cells from a long-term withdrawn saline-treated animal (red) and a long-term withdrawn nicotine-treated animal (blue) to current steps (−1.5 to 0.1 nA, 500 ms). Experiments were conducted in the presence of blockers. Capacitance artifacts have been removed for clarity. E, Mean I–V curves for 500 ms hyperpolarizing and subthreshold depolarizing pulses in CA1 pyramidal cells from long-term withdrawn animals in the presence of blockers (sal-cells, n = 14; nic-cells, n = 14). F, Example traces of the membrane potential response of CA1 pyramidal cells from a long-term withdrawn saline-treated animal (red) and a long-term withdrawn nicotine-treated animal (blue) to current steps (−1.0 to 0.2 nA, 500 ms). Experiments were conducted in the presence of blockers + 200 μm BaCl2. Capacitance artifacts have been removed, and action potentials have been truncated for clarity. G, Mean I–V curves for 500 ms hyperpolarizing pulses in CA1 pyramidal cells from long-term withdrawn animals in the presence of blockers + 200 μm BaCl2 (sal-cells, n = 4; nic-cells, n = 5). Data are presented as the mean ± SEM. Blockers, 10 μm CNQX + 50 μm d-APV + 10 μm bicuculline.
Prior research has shown an increase in the input resistance of prefrontal cortical neurons following cocaine administration that was due to a decrease in Ba2+-sensitive potassium currents (Nasif et al., 2005a). The disparate I–V curves are consistent with the suggestion that a decrease in potassium currents may underlie the increased excitability in the nic-cells. Under the same experimental conditions as cited above (i.e., 10 μm CNQX; 50 μm d-APV; 10 μm bicuculline) to examine intrinsic excitability, application of BaCl2 (200 μm), a relatively nonspecific potassium channel blocker, normalized the number of spikes in response to a given input between nic-cells and sal-cells (Fig. 5B,C, open symbols) (p > 0.05). In addition, while the resting membrane potentials of nic-cells (−58.0 ± 1.9 mV, n = 5) and sal-cells (−60.0 ± 2.8 mV, n = 4) were depolarized to a similar extent in the presence of BaCl2, there was a greater effect on sal-cells with respect to input resistance such that in the presence of BaCl2 both groups had similar voltage response families (Fig. 5F) and I–V curves (Fig. 5G). These results imply that, following long-term withdrawal from nicotine, the enhanced excitability of CA1 pyramidal neurons may be mediated by a reduction in a Ba2+-sensitive potassium current.
Reversal of enhanced excitability by nicotine in vitro
The initial changes following drugs of abuse are often the result of homeostatic adaptations to the presence of the drug (Koob and Bloom, 1988). If the neuroadaptations revealed after withdrawal from nicotine are the result of a homeostatic mechanism, then re-exposure of the slices to nicotine should return function to control levels (Pu et al., 2002). To test this, 500 nm nicotine, a concentration that given our treatment paradigm should approximate that found in vivo (Henningfield et al., 1993), was bath applied for 1 h. Based on the changes in CA1 physiology after 1 d of withdrawal, treatment with nicotine in vitro should have the following two prominent effects: (1) increase the fEPSP slope measured in stratum radiatum; and (2) shift the E-S curve to the right. These effects were fully realized. First, in nic-slices, but not sal-slices, the fEPSP slope was enhanced during application of nicotine (Fig. 6A), making it no longer depressed compared with sal-slices (Fig. 6B) (p > 0.05). Second, E-S curves from the two treatment groups were no longer significantly different in the presence of 500 nm nicotine (Fig. 6C,D) (p > 0.05). These findings imply that the changes in hippocampal function upon immediate withdrawal from nicotine arise as a direct homeostatic adaptation to the presence of nicotine in vivo.
Figure 6.
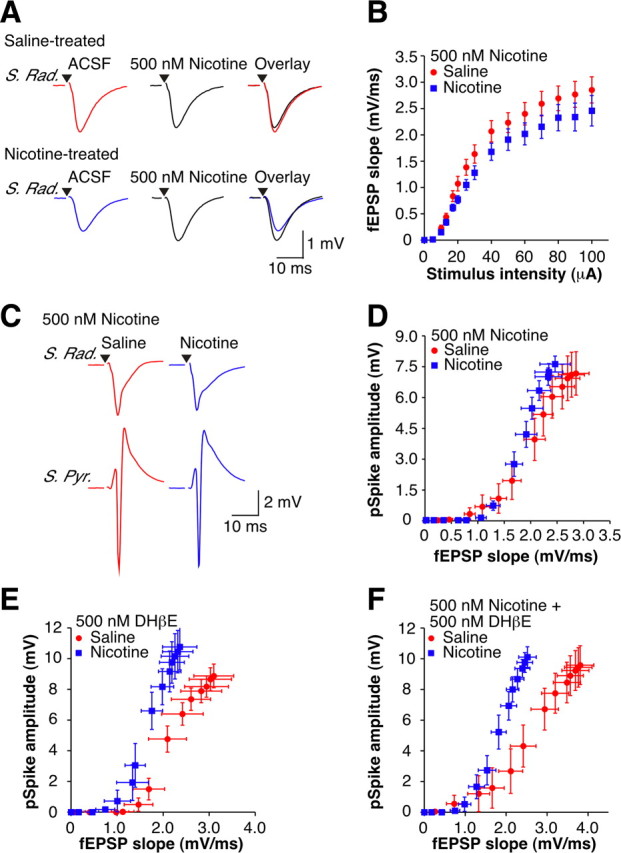
In vitro nicotine restores normal function at early withdrawal. A, Top, Examples of orthodromically evoked subthreshold fEPSPs recorded in stratum radiatum from 1 d withdrawn saline-treated animals in ACSF (red) and 500 nm nicotine (black). Bottom, Examples of orthodromically evoked subthreshold fEPSPs recorded in stratum radiatum from 1 d withdrawn nicotine-treated animals in ACSF (blue) and 500 nm nicotine (black). Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. B, Mean fEPSP slope I-O curves in the presence of 500 nm nicotine (sal-slices, n = 11; nic-slices, n = 11). C, Examples of orthodromically evoked suprathreshold fEPSPs recorded in stratum radiatum (top traces) and pSpikes recorded in stratum pyramidale (bottom traces) from 1 d withdrawn animals in the presence of 500 nm nicotine (sal-slices: red; nic-slices: blue). Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. D–F, Mean extracellular E-S curves in the presence of 500 nm nicotine (D) (sal-slices, n = 11; nic-slices, n = 11), 500 nm DHβE (E) (sal-slices, n = 5; nic-slices, n = 5), and 500 nm nicotine + 500 nm DHβE (F) (sal-slices, n = 9; nic-slices, n = 10). Data are presented as the mean ± SEM.
Nicotine-seeking behavior in rodents is dependent on nicotine-induced activation of high-affinity α4β2* nAChRs (Picciotto et al., 1998; Tapper et al., 2004; Pons et al., 2008). Based on our observations that acute nicotine has little or no effect on excitatory synaptic transmission in hippocampal slices from saline-treated animals, it is plausible that the in vitro homeostatic effects are the result of nicotine interacting with nAChRs upregulated by the in vivo nicotine exposure (Marks et al., 1983; Schwartz and Kellar, 1983). Although receptor desensitization will predominate at the low concentration of nicotine (500 nm) used here, some activation of these receptors would also be expected (Fenster et al., 1997). If the restoration of normal function seen following re-exposure to nicotine is due to nicotine re-desensitizing these receptors, then we might expect an antagonist to have the same restorative effect as nicotine. However, following application of 500 nm DHβE, a concentration that is relatively selective for the α4β2* nAChR (Alkondon and Albuquerque, 1993), there was still a significant difference between the E-S curves from sal-slices and nic-slices (Fig. 6E) (p < 0.05). Furthermore, when nicotine was introduced in the presence of DHβE, the previously observed homeostatic action of nicotine was no longer present (Fig. 6F) (p < 0.05). These data imply a critical role of α4β2* nAChR activation (rather than desensitization) for the restorative effects of nicotine.
Chronic nicotine treatment has been shown to upregulate α4β2* nAChRs specifically in the dentate gyrus of adult animals (Nguyen et al., 2003; Nashmi et al., 2007). Consistent with the enhanced excitability being a homeostatic response to nicotine interacting with nAChRs in this area in vivo, we found that the observed changes in CA1 physiology immediately following chronic nicotine treatment are dependent on the dentate gyrus (Figs. 3, 4). As would then be expected, administration of 500 nm nicotine in vitro no longer had any effects on the activity within the CA1 region in nic-slices deprived of upstream input (supplemental Fig. S6, available at www.jneurosci.org as supplemental material) (p > 0.05), consistent with the suggestion that the initial homeostatic changes arise as a direct result of changes in the function of nAChRs specifically in the dentate gyrus of nicotine-treated animals.
After longer drug-free periods (>4 weeks) and in contrast to the results observed at 1 d of withdrawal, re-exposure to nicotine was unable to reduce the E-S potentiation observed in nic-slices back to control levels (Fig. 7) (p < 0.05). The inability of in vitro nicotine to restore normal levels of excitability at longer times of drug withdrawal is consistent with both the idea that these changes are no longer homeostatic and the previously reported observation that the once upregulated high-affinity nAChRs have returned to baseline levels during the first 2 weeks of drug cessation (Marks et al., 1985; Collins et al., 1988).
Figure 7.

In vitro nicotine no longer restores normal function after long-term withdrawal. A, Examples of orthodromically evoked suprathreshold fEPSPs recorded in stratum radiatum (top traces) and pSpikes recorded in stratum pyramidale (bottom traces) from 3 month withdrawn animals in the presence of 500 nm nicotine (sal-slices: red; nic-slices: blue). Stimulus artifacts have been removed for clarity, and black triangles represent time of stimulus. B, Mean extracellular E-S curves from long-term withdrawn animals in the presence of 500 nm nicotine (sal-slices, n = 16; nic-slices, n = 16). Data are presented as the mean ± SEM.
Discussion
Following withdrawal from chronic nicotine in vivo, there is a persistent change in the excitability of CA1 pyramidal neurons, such that for a given synaptic input there is an increased action potential output. Although this form of neuronal plasticity is observed at all times (up to 9 months) after removal of drug, it has two distinct mechanistic phases. An initial homeostatic adaptation to the presence of nicotine, involving nAChRs located upstream of CA1, is followed by a second and more permanent phase that results in a relocalization of the plasticity to the CA1 region, and is associated with an altered pyramidal cell K+ conductance. These changes represent novel physiological adaptations to the presence of chronic nicotine and its withdrawal, and therefore may provide neural circuit explanations for certain aspects of nicotine dependence and withdrawal-induced behavioral deficits.
Most molecular and cellular theories of nicotine addiction begin with the well established upregulation of high-affinity α4β2* nAChRs (Marks et al., 1983; Schwartz and Kellar, 1983). More recent neurophysiological assessments of how nicotine interferes with the synaptic control of midbrain dopaminergic cells have extended these theories beyond the level of nAChRs. In particular, the concomitant downregulation of GABAergic synapses and strengthening of glutamatergic synapses onto ventral tegmental area (VTA) neurons by the prolonged application of nicotine in vitro provides a cellular model for nicotine-induced sensitization of VTA output (Mansvelder and McGehee, 2002; Vezina et al., 2007). By incorporating the reported upregulation of α4β2* nAChRs on VTA GABA interneurons into this scheme, these findings provide a model explaining how withdrawal from nicotine could lead to a “craving state” by increasing local inhibition and depressing dopamine output (Nashmi et al., 2007). While such models start to integrate the cellular and behavioral aspects of nicotine addiction, additional mechanisms will be required to account for the more persistent nature of relapse to this drug, especially given the transient increases in both nAChR number (Marks et al., 1985; Collins et al., 1988) and synaptic strength (Yamazaki et al., 2006b; Gao et al., 2010). Our current data show that continuous exposure to nicotine in vivo can produce long-lasting changes in neuronal excitability in an area of brain known to be important for drug context—the hippocampus (Kelley, 2004).
Immediately after withdrawal from nicotine, we describe a novel increase in CA1 pyramidal cell excitability that involves α4β2* nAChRs within the dentate gyrus. We conclude that these effects must depend on a gain of function within the dentate gyrus, as severing connections between dentate gyrus and CA3 abolishes, rather than mimics, the increased excitability of both the CA3 and CA1 regions in nicotine-treated slices. Dentate granule cells provide the only known output to CA3 from the dentate gyrus and form more synapses onto inhibitory interneurons within CA3 than onto pyramidal cells (Acsády et al., 1998). Due to a difference in the short-term plasticity of mossy fiber synapses onto CA3 pyramidal cells and interneurons (Jonas et al., 1993; Salin et al., 1996; Toth et al., 2000; Lawrence et al., 2004), the output of the dentate gyrus depends on the firing frequency of granule cells, switching from inhibitory to excitatory with increasing frequency (Henze et al., 2002). It follows that an increase in the burst firing behavior of granule cells could account for the increased excitability of CA3 pyramidal cells after chronic nicotine, and, given the interconnectivity of CA3 pyramidal cells (Chamberlin et al., 1990), a small change in the synaptic excitation of these cells may be sufficient to result in a large change in their excitability.
The observation that activation of α4β2* nAChRs is sufficient for nicotine-seeking behavior in rodents (Tapper et al., 2004) implies that these receptors may be persistently stimulated by nicotine in vivo, despite high receptor occupation and the predomination of desensitization (Quick and Lester, 2002; Brody et al., 2006). Consistent with these behavioral data, we find that reactivation of α4β2* nAChRs in the dentate gyrus in vitro is capable of restoring normal excitability to the CA1 region. Since in vitro nicotine has no overall effect on excitability in slices from saline-treated animals, the homeostatic effect of nicotine presumably arises from its interaction with functionally altered nAChRs. Importantly, α4 subunit-containing nAChRs, localized to where the perforant path terminates in the dentate gyrus, are particularly sensitive to upregulation by nicotine in vivo (Nashmi et al., 2007), thereby providing the molecular and cellular basis for our finding that the initial neuroadaptation induced by nicotine is dependent specifically on this region. Overall, we propose that chronic nicotine acting in vivo at upregulated α4β2* nAChRs on perforant path axons (Nashmi et al., 2007) and/or directly on GABAergic interneurons (Alkondon and Albuquerque, 2005) alters the excitatory/inhibitory balance in the dentate gyrus to favor inhibition, resulting in a decreased excitatory output. Adaptations to the constant presence of nicotine in vivo such as an increase in the intrinsic and/or synaptic excitability of the granule cells would constitute an appropriate homeostatic restoration of function, thereby keeping neuronal communication within a stable operational range (Turrigiano and Nelson, 2000). However, during the early phase of withdrawal, the inhibitory drive provided by nicotine is removed, revealing an enhanced dentate granule cell excitability, which then propagates to the CA1 region.
During the initial 2–3 weeks of withdrawal in vivo, nicotine will not be present to maintain α4β2* nAChRs in their upregulated state, and normal function should be restored within the dentate gyrus as these receptors return to their original condition (Marks et al., 1985; Collins et al., 1988). According to the simplest homeostatic models of addiction (Koob and Bloom, 1988), it follows that the downstream changes in CA1 pyramidal cell excitability should also reverse shortly after cessation of drug. However, this is not what we observe: the increased neuronal excitability is maintained, albeit through a different mechanism. Secondary adaptations that occur during abstinence from drugs of abuse may somewhat counteract the withdrawal symptoms but do not return the brain to its predrug state (Koob and Bloom, 1988; Koob and Le Moal, 2001, 2005; DiFranza and Wellman, 2005; Hyman et al., 2006). Thus, withdrawal from drug and associated global stress responses may be responsible for the generation of a chronically altered brain through the induction of an allostatic state (Koob and Le Moal, 2001). Together, these arguments imply that evaluation of the period of abstinence after withdrawal of nicotine may be just as relevant as the immediate effects of drug use, as demonstrated by the finding that pharmacological intervention during withdrawal may prevent some of the long-lasting neuronal rewiring (Francesconi et al., 2009).
We speculate that these mechanisms likely induce further plasticity that accounts for the additional persistent effects in the CA1 region not readily explained by homeostatic adaptations. Our data show that a decrease in Ba2+-sensitive K+ currents may underlie the enhanced excitability of CA1 pyramidal cells following long-term withdrawal from nicotine, in agreement with prior research showing that similar conductances are altered in prefrontal cortical neurons following cocaine administration (Nasif et al., 2005a). Such ionic changes can also explain our extracellular data, as E-S potentiation may be produced by a change in voltage-dependent ion channels (Taube and Schwartzkroin, 1988; Wathey et al., 1992). The observation that multiple phases of plasticity are apparent on withdrawal from nicotine is consistent with proposed theories of drug dependence and addiction that suggest the initial induction mechanisms are drug specific and the subsequent enduring changes are independent of the primary drug targets (Koob and Bloom, 1988; Hyman et al., 2006).
Although other studies of nicotine addiction have highlighted the importance of drug-induced modulation of nAChRs and synaptic transmission in the central reward pathways (Mansvelder and McGehee, 2002), it is not known whether these effects are long lasting or whether they would be sufficient to sustain drug seeking (Hyman et al., 2006). It is thought that hypersensitivity of certain cortical structures to drug-associated cues results in altered glutamatergic control of the nucleus accumbens during addiction (Kalivas and Volkow, 2005). Indeed, glutamate receptor antagonism in the nucleus accumbens can prevent reinstatement of drug-seeking behavior while agonist application in this structure reinstates drug-seeking behavior (Cornish and Kalivas, 2000). The nucleus accumbens receives highly convergent glutamatergic input from prefrontal cortex and hippocampus (O'Donnell and Grace, 1995), and hippocampal input is required for switching nucleus accumbens neurons to the depolarized up-state, which allows prefrontal cortical inputs to trigger action potential firing in these cells (O'Donnell and Grace, 1995). Action potential firing of nucleus accumbens neurons is associated with drug-seeking behavior (Peoples et al., 2004), and, following 1 month withdrawal, activation of nucleus accumbens neurons by drug-associated stimuli is enhanced (Hollander and Carelli, 2007). It is, therefore, tempting to suggest that the increased excitability of the hippocampus observed here could contribute to the increased spiking of nucleus accumbens neurons by increasing the ability of these neurons to respond to prefrontal cortical input. Moreover, the persistent nicotine-induced alteration of the input–output properties of CA1 pyramidal neurons may help to explain why a few cigarettes can produce lifelong behavioral changes (Russell, 1990) such as the long-term susceptibility to relapse following cessation of nicotine.
Footnotes
This research was supported by Public Health Service National Institute on Drug Abuse Grant DA11940. We thank Dr. H. A. Lester for helpful comments and suggestions on the manuscript. We also acknowledge Dr. T. J. Gould for helpful comments on a previous version of the manuscript.
References
- Acsády L, Kamondi A, Sík A, Freund T, Buzsáki G. GABAergic cells are the major postsynaptic targets of mossy fibers in the rat hippocampus. J Neurosci. 1998;18:3386–3403. doi: 10.1523/JNEUROSCI.18-09-03386.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Nicotinic receptor subtypes in rat hippocampal slices are differentially sensitive to desensitization and early in vivo functional up-regulation by nicotine and to block by bupropion. J Pharmacol Exp Ther. 2005;313:740–750. doi: 10.1124/jpet.104.081232. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- Andersen P, Bliss TV, Skrede KK. Unit analysis of hippocampal population spikes. Exp Brain Res. 1971;13:208–221. doi: 10.1007/BF00234086. [DOI] [PubMed] [Google Scholar]
- Andersen P, Silfvenius H, Sundberg SH, Sveen O, Wigström H. Functional characteristics of unmyelinated fibres in the hippocampal cortex. Brain Res. 1978;144:11–18. doi: 10.1016/0006-8993(78)90431-6. [DOI] [PubMed] [Google Scholar]
- Benowitz NL. Drug therapy. Pharmacologic aspects of cigarette smoking and nicotine addition. N Engl J Med. 1988;319:1318–1330. doi: 10.1056/NEJM198811173192005. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63:907–915. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Debanne D. Spike timing-dependent plasticity: a learning rule for dendritic integration in rat CA1 pyramidal neurons. J Physiol. 2008;586:779–793. doi: 10.1113/jphysiol.2007.147017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Traub RD, Dingledine R. Role of EPSPs in initiation of spontaneous synchronized burst firing in rat hippocampal neurons bathed in high potassium. J Neurophysiol. 1990;64:1000–1008. doi: 10.1152/jn.1990.64.3.1000. [DOI] [PubMed] [Google Scholar]
- Collins AC, Romm E, Wehner JM. Nicotine tolerance: an analysis of the time course of its development and loss in the rat. Psychopharmacology. 1988;96:7–14. doi: 10.1007/BF02431526. [DOI] [PubMed] [Google Scholar]
- Cornish JL, Kalivas PW. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JA, James JR, Siegel SJ, Gould TJ. Withdrawal from chronic nicotine administration impairs contextual fear conditioning in C57BL/6 mice. J Neurosci. 2005;25:8708–8713. doi: 10.1523/JNEUROSCI.2853-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Rudolph M, Fellous JM, Sejnowski TJ. Fluctuating synaptic conductances recreate in vivo-like activity in neocortical neurons. Neuroscience. 2001;107:13–24. doi: 10.1016/s0306-4522(01)00344-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFranza JR, Wellman RJ. A sensitization-homeostasis model of nicotine craving, withdrawal, and tolerance: integrating the clinical and basic science literature. Nicotine Tob Res. 2005;7:9–26. doi: 10.1080/14622200412331328538. [DOI] [PubMed] [Google Scholar]
- Due DL, Huettel SA, Hall WG, Rubin DC. Activation in mesolimbic and visuospatial neural circuits elicited by smoking cues: evidence from functional magnetic resonance imaging. Am J Psychiatry. 2002;159:954–960. doi: 10.1176/appi.ajp.159.6.954. [DOI] [PubMed] [Google Scholar]
- Durcan MJ, Deener G, White J, Johnston JA, Gonzales D, Niaura R, Rigotti N, Sachs DP. The effect of bupropion sustained-release on cigarette craving after smoking cessation. Clin Ther. 2002;24:540–551. doi: 10.1016/s0149-2918(02)85130-x. [DOI] [PubMed] [Google Scholar]
- Epping-Jordan MP, Watkins SS, Koob GF, Markou A. Dramatic decreases in brain reward function during nicotine withdrawal. Nature. 1998;393:76–79. doi: 10.1038/30001. [DOI] [PubMed] [Google Scholar]
- Etter JF, Hughes JR. A comparison of the psychometric properties of three cigarette withdrawal scales. Addiction. 2006;101:362–372. doi: 10.1111/j.1360-0443.2005.01289.x. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA. Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci. 1997;17:5747–5759. doi: 10.1523/JNEUROSCI.17-15-05747.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francesconi W, Berton F, Repunte-Canonigo V, Hagihara K, Thurbon D, Lekic D, Specio SE, Greenwell TN, Chen SA, Rice KC, Richardson HN, O'Dell LE, Zorrilla EP, Morales M, Koob GF, Sanna PP. Protracted withdrawal from alcohol and drugs of abuse impairs long-term potentiation of intrinsic excitability in the juxtacapsular bed nucleus of the stria terminalis. J Neurosci. 2009;29:5389–5401. doi: 10.1523/JNEUROSCI.5129-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin TR, Wang Z, Wang J, Sciortino N, Harper D, Li Y, Ehrman R, Kampman K, O'Brien CP, Detre JA, Childress AR. Limbic activation to cigarette smoking cues independent of nicotine withdrawal: a perfusion fMRI study. Neuropsychopharmacology. 2007;32:2301–2309. doi: 10.1038/sj.npp.1301371. [DOI] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Gao M, Jin Y, Yang K, Zhang D, Lukas RJ, Wu J. Mechanisms involved in systemic nicotine-induced glutamatergic synaptic plasticity on dopamine neurons in the ventral tegmental area. J Neurosci. 2010;30:13814–13825. doi: 10.1523/JNEUROSCI.1943-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- Henze DA, Wittner L, Buzsáki G. Single granule cells reliably discharge targets in the hippocampal CA3 network in vivo. Nat Neurosci. 2002;5:790–795. doi: 10.1038/nn887. [DOI] [PubMed] [Google Scholar]
- Hô N, Destexhe A. Synaptic background activity enhances the responsiveness of neocortical pyramidal neurons. J Neurophysiol. 2000;84:1488–1496. doi: 10.1152/jn.2000.84.3.1488. [DOI] [PubMed] [Google Scholar]
- Hollander JA, Carelli RM. Cocaine-associated stimuli increase cocaine seeking and activate accumbens core neurons after abstinence. J Neurosci. 2007;27:3535–3539. doi: 10.1523/JNEUROSCI.3667-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW, Volkow ND. The neural basis of addiction: a pathology of motivation and choice. Am J Psychiatry. 2005;162:1403–1413. doi: 10.1176/appi.ajp.162.8.1403. [DOI] [PubMed] [Google Scholar]
- Kamondi A, Acsády L, Buzsáki G. Dendritic spikes are enhanced by cooperative network activity in the intact hippocampus. J Neurosci. 1998;18:3919–3928. doi: 10.1523/JNEUROSCI.18-10-03919.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsura M, Mohri Y, Shuto K, Hai-Du Y, Amano T, Tsujimura A, Sasa M, Ohkuma S. Up-regulation of L-type voltage-dependent calcium channels after long term exposure to nicotine in cerebral cortical neurons. J Biol Chem. 2002;277:7979–7988. doi: 10.1074/jbc.M109466200. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci. 2007;8:844–858. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Killen JD, Fortmann SP. Craving is associated with smoking relapse: findings from three prospective studies. Exp Clin Psychopharmacol. 1997;5:137–142. doi: 10.1037//1064-1297.5.2.137. [DOI] [PubMed] [Google Scholar]
- Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Lawrence JJ, Grinspan ZM, McBain CJ. Quantal transmission at mossy fibre targets in the CA3 region of the rat hippocampus. J Physiol. 2004;554:175–193. doi: 10.1113/jphysiol.2003.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Cellular and synaptic mechanisms of nicotine addiction. J Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- Marder CP, Buonomano DV. Timing and balance of inhibition enhance the effect of long-term potentiation on cell firing. J Neurosci. 2004;24:8873–8884. doi: 10.1523/JNEUROSCI.2661-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markou A. Review. Neurobiology of nicotine dependence. Philos Trans R Soc Lond B Biol Sci. 2008;363:3159–3168. doi: 10.1098/rstb.2008.0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MJ, Burch JB, Collins AC. Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther. 1983;226:817–825. [PubMed] [Google Scholar]
- Marks MJ, Stitzel JA, Collins AC. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 1985;235:619–628. [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology. 2007;190:269–319. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- Murrin LC, Ferrer JR, Zeng WY, Haley NJ. Nicotine administration to rats: methodological considerations. Life Sci. 1987;40:1699–1708. doi: 10.1016/0024-3205(87)90020-8. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, Collins AC, Marks MJ, Lester HA. Chronic nicotine cell specifically upregulates functional α4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasif FJ, Sidiropoulou K, Hu XT, White FJ. Repeated cocaine administration increases membrane excitability of pyramidal neurons in the rat medial prefrontal cortex. J Pharmacol Exp Ther. 2005a;312:1305–1313. doi: 10.1124/jpet.104.075184. [DOI] [PubMed] [Google Scholar]
- Nasif FJ, Hu XT, White FJ. Repeated cocaine administration increases voltage-sensitive calcium currents in response to membrane depolarization in medial prefrontal cortex pyramidal neurons. J Neurosci. 2005b;25:3674–3679. doi: 10.1523/JNEUROSCI.0010-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther. 2003;307:1090–1097. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Synaptic interactions among excitatory afferents to nucleus accumbens neurons: hippocampal gating of prefrontal cortical input. J Neurosci. 1995;15:3622–3639. doi: 10.1523/JNEUROSCI.15-05-03622.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peoples LL, Lynch KG, Lesnock J, Gangadhar N. Accumbal neural responses during the initiation and maintenance of intravenous cocaine self-administration. J Neurophysiol. 2004;91:314–323. doi: 10.1152/jn.00638.2003. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391:173–177. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, Changeux JP, Maskos U, Fratta W. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G. Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci. 2002;22:1914–1921. doi: 10.1523/JNEUROSCI.22-05-01914.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick MW, Lester RA. Desensitization of neuronal nicotinic receptors. J Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- Russell MA. The nicotine addiction trap: a 40-year sentence for four cigarettes. Br J Addict. 1990;85:293–300. doi: 10.1111/j.1360-0443.1990.tb03085.x. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci U S A. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz RD, Kellar KJ. Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science. 1983;220:214–216. doi: 10.1126/science.6828889. [DOI] [PubMed] [Google Scholar]
- Sirvio J, Larson J, Quach CN, Rogers GA, Lynch G. Effects of pharmacologically facilitating glutamatergic transmission in the trisynaptic intrahippocampal circuit. Neuroscience. 1996;74:1025–1035. doi: 10.1016/0306-4522(96)00170-4. [DOI] [PubMed] [Google Scholar]
- Smolka MN, Bühler M, Klein S, Zimmermann U, Mann K, Heinz A, Braus DF. Severity of nicotine dependence modulates cue-induced brain activity in regions involved in motor preparation and imagery. Psychopharmacology. 2006;184:577–588. doi: 10.1007/s00213-005-0080-x. [DOI] [PubMed] [Google Scholar]
- Soleng AF, Chiu K, Raastad M. Unmyelinated axons in the rat hippocampus hyperpolarize and activate an H current when spike frequency exceeds 1 Hz. J Physiol. 2003;552:459–470. doi: 10.1113/jphysiol.2003.048058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol. 1992;67:508–529. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- Stacey WC, Durand DM. Synaptic noise improves detection of subthreshold signals in hippocampal CA1 neurons. J Neurophysiol. 2001;86:1104–1112. doi: 10.1152/jn.2001.86.3.1104. [DOI] [PubMed] [Google Scholar]
- Staff NP, Spruston N. Intracellular correlate of EPSP-spike potentiation in CA1 pyramidal neurons is controlled by GABAergic modulation. Hippocampus. 2003;13:801–805. doi: 10.1002/hipo.10129. [DOI] [PubMed] [Google Scholar]
- Tapper AR, McKinney SL, Nashmi R, Schwarz J, Deshpande P, Labarca C, Whiteaker P, Marks MJ, Collins AC, Lester HA. Nicotine activation of α4* receptors: sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- Taube JS, Schwartzkroin PA. Mechanisms of long-term potentiation: EPSP/spike dissociation, intradendritic recordings, and glutamate sensitivity. J Neurosci. 1988;8:1632–1644. doi: 10.1523/JNEUROSCI.08-05-01632.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth K, Suares G, Lawrence JJ, Philips-Tansey E, McBain CJ. Differential mechanisms of transmission at three types of mossy fiber synapse. J Neurosci. 2000;20:8279–8289. doi: 10.1523/JNEUROSCI.20-22-08279.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Vezina P, McGehee DS, Green WN. Exposure to nicotine and sensitization of nicotine-induced behaviors. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1625–1638. doi: 10.1016/j.pnpbp.2007.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL. Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science. 2001;292:1175–1178. doi: 10.1126/science.1058043. [DOI] [PubMed] [Google Scholar]
- Wathey JC, Lytton WW, Jester JM, Sejnowski TJ. Computer simulations of EPSP-spike (E-S) potentiation in hippocampal CA1 pyramidal cells. J Neurosci. 1992;12:607–618. doi: 10.1523/JNEUROSCI.12-02-00607.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby P, Rowan M, Anwyl R. Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. Eur J Neurosci. 2006;24:3109–3118. doi: 10.1111/j.1460-9568.2006.05187.x. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Jia Y, Niu R, Sumikawa K. Nicotine exposure in vivo induces long-lasting enhancement of NMDA receptor-mediated currents in the hippocampus. Eur J Neurosci. 2006a;23:1819–1828. doi: 10.1111/j.1460-9568.2006.04714.x. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Fujii S, Jia Y, Sumikawa K. Nicotine withdrawal suppresses nicotinic modulation of long-term potentiation induction in the hippocampal CA1 region. Eur J Neurosci. 2006b;24:2903–2916. doi: 10.1111/j.1460-9568.2006.05160.x. [DOI] [PubMed] [Google Scholar]