

Summary
Cell cycle checkpoints are surveillance mechanisms that safeguard genome integrity. While the extrinsic pathways that halt the cell cycle in response to DNA damages have been well documented, the intrinsic pathways that ensure orderly progression of cell cycle events are not well understood. We demonstrate that Drosophila MEK and ERK constitute an essential intrinsic checkpoint pathway that restrains cell cycle progression in the absence of DNA damage and also responds to ionizing radiation to arrest the cell cycle. Embryos lacking MEK exhibit faster and extra division cycles and fail to undergo timely midblastula transition (MBT) or arrest following ionizing radiation. Conversely, constitutively activated MEK causes cell cycle arrest. Further, MEK activation in the early embryo is cell cycle-dependent and Raf independent and increases in response to ionizing radiation or in the absence of Chk1. Thus, MEK/ERK activation is required for multiple checkpoints and is essential for orderly cell cycle progression.
Introduction
Cell cycle checkpoints control critical cell cycle events such as DNA replication and chromosome segregation to ensure genome integrity. Cell cycle checkpoints can be divided into intrinsic regulatory pathways that ensure the orderly progression of cell cycle events and extrinsic pathways that are activated only when DNA damages are detected (Elledge, 1996; Hartwell and Weinert, 1989). It has been well documented that the ATM and ATR checkpoint kinases mediate the extrinsic pathways that halt the cell cycle when DNA damages are detected (Kastan and Bartek, 2004). In contrast, the intrinsic pathways that monitor the orderly and timely progression of cell cycle events are not well understood.
The mitogen-activated protein kinase kinase (MEK) and its substrate ERK are essential components of the evolutionarily conserved Ras/Raf/MEK/ERK mitogenic signaling cascade (Wellbrock et al., 2004). The primary function of this signaling pathway in stimulating cell proliferation is evidenced by the fact that gain-of-function mutations in Ras and Raf have been identified in a variety of human cancers with high frequency (Brose et al., 2002; Davies et al., 2002). In Drosophila, the Ras/Raf/MEK/ERK cascade mediates RTK signaling for cell fate specification during normal development, and causes overproliferation when overactivated (Li, 2005; Rubin et al., 1997). However, it has also been reported that the Ras/Raf/MEK/ERK pathway can exert antiproliferative effects under high-intensity Raf signaling (Roovers and Assoian, 2000) and that activation of MEK and ERK facilitates DNA damage-induced checkpoint responses (Wu et al., 2005). Intriguingly, activated MEK and ERK have been detected in the nucleus associated with mitotic apparatus during mitosis, raising the possibility that they may directly participate in cell cycle progression (Shapiro et al., 1998). Indeed, MEK/ERK activation has been implicated in the spindle checkpoint controlling mitotic progression (Guadagno and Ferrell, 1998). Mechanistic understanding of the function of MEK/ERK in directly regulating cell cycle progression is confounded by the multitude of transcription targets of this signaling pathway that may be differentially expressed in different cellular contexts, indirectly influencing the cell cycle.
The early Drosophila embryo is an ideal model system to investigate cell cycle regulation by the in the Ras/Raf/MEK/ERK pathway in the absence of transcription (Foe et al., 1993; Li, 2005; Orr-Weaver, 1994). Drosophila utilizes a fast and transcription-independent cell cycle program for early divisions, with each cycle consisting of mainly S and M phases and lasting 10 min on average (Foe et al., 1993; Orr-Weaver, 1994). The first 13 synchronous divisions are controlled solely by maternally deposited components. Following the 13th mitosis, a significant lengthening of the 14th interphase (to >60 min), accompanied by cellularization and the initiation of zygotic transcription, marks the midblastula transition (MBT) that switches embryogenesis to zygotic control (Foe et al., 1993). MBT is enforced by the Grapes (Grp)/Chk1 DNA replication checkpoint system, as grp mutant embryos continue the maternally controlled cell cycle program and fail to lengthen the interphase to allow completion of DNA replication (Sibon et al., 1997). However, grp mutants exhibit normal early cycles prior to mitosis 12 (Ji et al., 2004), and the premature entry into the 13th mitosis leads to anaphase chromosome segregation failure (Sibon et al., 2000; Takada et al., 2003). Thus, Grp/Chk1-independent checkpoint pathways should exist that control the orderly progression of early cycles and block chromosome segregation in late cycles of grp mutants.
In this study, we demonstrate that the MEK/ERK pathway plays an essential role in an intrinsic checkpoint pathway that restrains cell cycle progression even in the absence of DNA damage to ensure that initiation of cell cycle events depends on the completion of an earlier event. In addition, we show that this pathway is also required for the DNA replication or damage checkpoint responses, in conjunction with or in parallel to the Grp/Chk1 and Mnk/Chk2 pathways.
Results
Defective Early Nuclear/Cell Divisions and MBT in MEK Embryos
To investigate the direct role of the Ras/Raf/MEK/ERK signaling cascade in cell cycle regulation, we examined embryos lacking the maternal contribution of Drosophila Raf (Draf; encoded by pole hole [phl]) and MEK (encoded by Dsor1), referred to as Draf and MEK embryos, respectively (see Experimental Procedures). We found that MEK, but not Draf, embryos exhibit striking defects in the pattern and density of cortical nuclei in the early embryo. Following 13 cycles of synchronous divisions, wild-type embryos exhibit a cortical nuclear monolayer with a regular distribution pattern (Figure 1A; also see Foe et al., 1993; Orr-Weaver, 1994). MEK embryos, in contrast, exhibited a higher nuclear density (or DNA content) and an irregular cortical nuclear distribution, with massive nuclei falling into the interior yoke region (Figures 1A and 1C), suggesting a “mitotic catastrophe,” by which irreparably damaged nuclei are eliminated from the cortex (Takada et al., 2003). Draf embryos, however, exhibited normal nuclear patterns except in the posterior region, where falling nuclei were detected due to defective Torso signaling (Li, 2005) (data not shown; see Figure 1C; also see Ambrosio et al., 1989). Multiple defects were also seen in MEK embryos throughout early division cycles before cycle 13. These include fragmentation of nuclei, multispindle mitotic figures, and chromosomal bridges (Figure 1B), suggesting that the mitotic catastrophe (falling nuclei) observed in MEK embryos might be a consequence of accumulated mitotic errors in earlier stages.
Figure 1. Mitotic Division Defects in MEK Embryos.
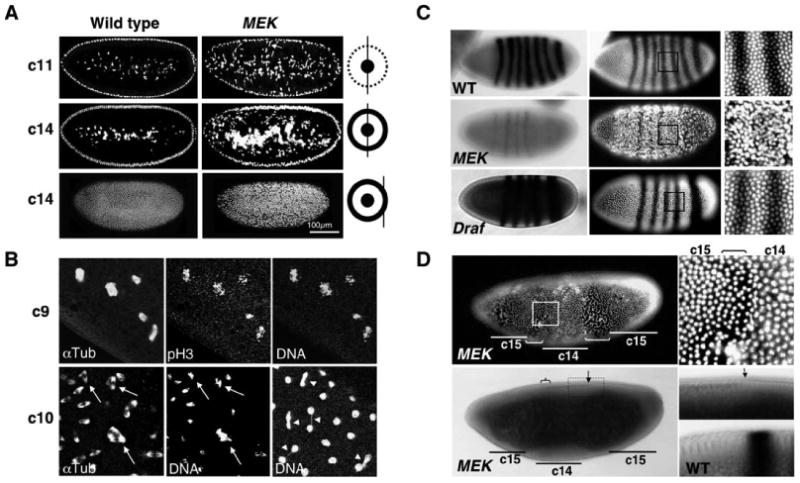
(A) Wild-type and MEK (Dsor1LH110) embryos were stained with propidium iodide to detect DNA. The diagram illustrates the position of confocal optical section (black straight line) relative to the cortical layer (outer circle) and the yolk region (inner circle) of the embryo. c11 and c14 indicate the nuclear division cycles 11 and 14. Note that MEK embryos exhibit an abnormally large number of cortical nuclei falling into the yolk (arrow).
(B) A cycle-9 MEK embryo contains catastrophic nuclei seen as fragmented pH3-positive chromosomes tangled with α-tubulin. A cycle-10 embryo (bottom left and center) contains multiple spindle mitotic figures (arrow). Another cycle-10 embryo (bottom right) contains nuclei connected by chromosomal bridges (unresolved chromosomes; arrowhead).
(C) Cycle-14 wild-type, MEK (Dsor1LH110), and Draf (Draf11-29) embryos showing ftz mRNA (stripes in left panels) and DNA (white in right panels). Note the very diminished levels of ftz expression, a lack of detectable cellularization, and irregular nuclear spacing in MEK, but not Draf, embryos.
(D) Top: The 14th mitosis in a MEK embryo is seen as mitotic waves (brackets), which appear as gaps due to cytoplasmic contractions during mitosis, advancing from poles toward the center, leaving behind a cycle-15 (c15) cortical nuclear layer. Bottom: The same embryo in a light micrograph showing that the central region (c14) has initiated cellularization (furrows and thickening of light subcortical layer in bottom panels) and ftz expression (brace), which is disrupted by the advancing 14th mitotic waves. The arrow marks the front of the advancing posterior mitotic wave. A higher magnification is shown to the right. The bottom right panel shows the same region from a cellularized wild-type embryo with a stripe of ftz expression.
During the long pause period following the 13th mitosis, wild-type embryos undergo midblastular transition (MBT), initiating zygotic gene transcription and morphological changes, such as cellularization and gastrulation (Figure 1C; also see Foe et al., 1993; Sibon et al., 1997). However, MEK, but not Draf, embryos fail to undergo timely MBT, lacking significant zygotic transcription and cellularization (Figure 1C). Interestingly, cellularization and low levels of zygotic transcripts were seen in areas of MEK embryos with cycle 14 nuclear density but not in areas with cycle 15 nuclear density (Figure 1D). We deduce that in MEK embryos cellularization and zygotic gene transcription could initiate, but were later disrupted by an additional round of nuclear division. Chromosomal bridges and abnormal chromosomal segregation could result from premature entry and exit from mitosis, and a lack of MBT and the presence of a large number of catastrophic nuclei suggest a failure of cell cycle checkpoints (Takada et al., 2003).
An Intrinsic Cell Cycle Checkpoint Pathway Mediated by MEK and ERK
To investigate the cause of the mitotic defects seen in MEK embryos, we directly observed and measured the durations of the nuclear/cell division cycles in MEK embryos by time-lapse confocal microscopy following injection of a fluorescent DNA dye (see Experimental Procedures). Consistent with previous reports (Foe et al., 1993; Sibon et al., 1997), wild-type embryos exhibit short early division cycles with increasing length and a long interphase 14 (>60 min) (Figure 2A). MEK embryos, however, exhibited significantly shorter durations for each phase of the division cycle, lacked the long interphase-14 pause, and underwent an extra round of synchronized division (Figure 2A). These observations suggest that MEK embryos lack the intrinsic cell cycle checkpoints responsible for delaying each phase of the cell cycle until crucial events such as DNA replication are completed, and the fast division cycles result in premature entry and exit from mitosis, leading to the observed mitotic defects in later cycles of MEK embryos.
Figure 2. MEK and ERK Are Essential for the Intrinsic Cell Cycle Checkpoints.
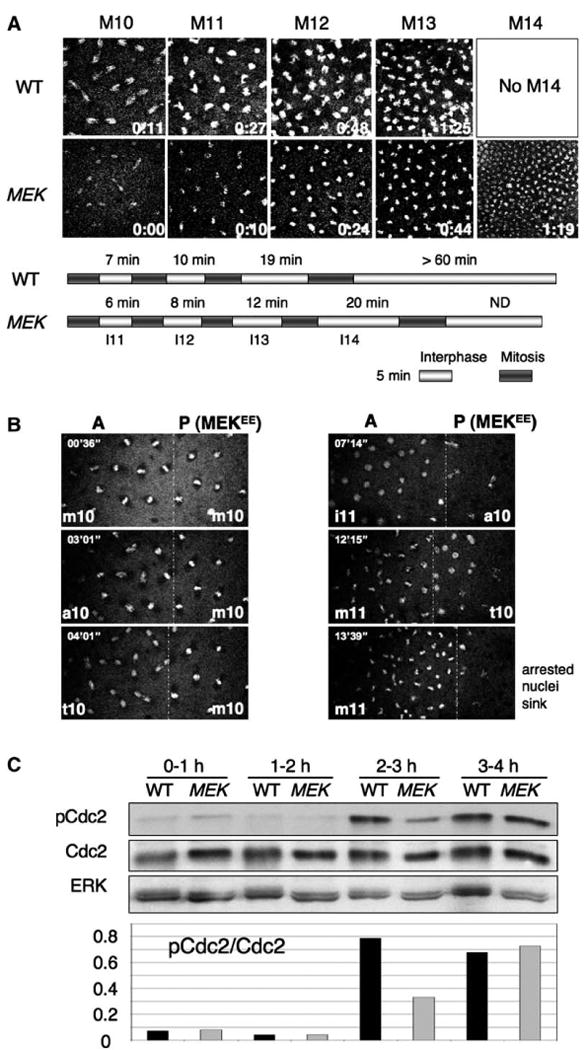
(A) Wild-type and MEK (Dsor1LH110) embryos were injected with the DNA dye oli-green and observed by time-lapse confocal microscopy. Frames of metaphase (or anaphase) nuclei of indicated division cycles are shown with a timestamp. Bars represent the durations of each indicated interphase and mitosis. Note the shortened division cycle phases and the presence of an extra mitosis (M14) in MEK embryos.
(B) mRNA for constitutively active MEK (MEKEE) together with oli-green was locally injected into wild-type embryos at the posterior (P). Due to retarded diffusion, locally injected mRNA was confined and compartmentalized at the posterior. The anterior region (A) was not affected and served as an internal control. Note the nuclei in the posterior region (site of injection) arrest at metaphase 10 (m10) and eventually sink into the yolk, while those in the anterior region continue to divide.
(C) Total protein extracts from wild-type and MEK embryos at indicated time intervals following egg deposition were subjected to SDS-PAGE and blotted with anti-pY15-Cdc2. The membrane was stripped and re-blotted sequentially with anti-Cdc2 and anti-ERK. Note the presence of a lower amount of pCdc2 at the onset of MBT (2–3 hr AED) in MEK embryos (lane 6) compared with wild-type control embryos (lane 5), while the amounts of total Cdc2 are similar.
To confirm that the defects are indeed due to a lack of MEK activity and to investigate the downstream targets of MEK in checkpoint function, we examined the consequences of perturbing the MEK/ERK pathway by injecting early embryos with the following reagents. To inhibit the MEK/ERK pathway, we injected the MEK inhibitor, U0126, and mRNA encoding Mkp3, which is an ERK-specific phosphatase that inhibits ERK activity (Kim et al., 2004). To activate the pathway, we injected mRNA encoding a constitutively activated MEK, MEKEE (see Experimental Procedures). Time-lapse confocal microscopy demonstrated that constitutive activation of MEK delays and arrests the cell cycle (Figure 2B), and inhibition of MEK or ERK results in faster and shorter division cycles (Figure S1; see the Supplemental Data available with this article online), similar to those observed in MEK embryos. Since these defects were not found in Draf embryos, it thus appears that the function of MEK and ERK in cell cycle checkpoint control is independent of Draf (see Discussion).
Normal MBT and the lengthening of the cell cycle result from activation of a DNA replication checkpoint and are associated with an increase in the inhibitory tyrosine phosphorylation of the cyclin-dependent kinase Cdc2 (pCdc2) (Sibon et al., 1997). Indeed, 2–3 hr after egg deposition (AED; corresponding to MBT initiation), wild-type embryos exhibit a significant increase in the levels of pCdc2 (Figure 2C). Similar to grp mutant embryos that lack of normal MBT, MEK embryos do not exhibit significant increase in the level of pCdc2 at 2–3 hr AED (Figure 2C; cf. lanes 5 and 6). Thus, MEK is required for normal MBT and the DNA replication checkpoint.
Taken all together, these results suggest that the Draf-independent MEK/ERK pathway plays an essential role in an intrinsic cell cycle checkpoint control mechanism that operates during early embryogenesis in the absence of any induced DNA damage.
MEK Embryos Are Sensitive to Irradiation and Lack DNA Damage Checkpoints
To investigate whether the MEK/ERK pathway is also required for responses to DNA damage in an extrinsic checkpoint pathway, we exposed early embryos to low-dose X-ray radiation (150 rad). Upon examination 35 min after irradiation, we found that the majority of wild-type embryos had halted nuclear/cell divisions and arrested at mitosis, as the nuclei failed to increase in number and remained positive for the phospho-histone H3 (pH3) antigen (Figure 3A). In contrast, a significant number of MEK embryos failed to arrest the division cycle, exhibiting a higher density of pH3-negative nuclei in the whole embryo (data not shown) or in small patches on the cortical layer (Figure 3A). A partial failure of MEK embryos to arrest division following irradiation is also evident by the higher ratio of embryos in interphase (pH3-negative) versus in mitosis (pH3-positive) than that of wild-type after X-ray treatment (Figure 3B). Consistent with an essential role of MEK in the activation of a DNA damage-induced checkpoint pathway that halts the cell cycle, immediately following X-ray irradiation (within 10 min), a marked increase in pCdc2 levels was detected in wild-type, but not MEK, embryos (Figure 3C).
Figure 3. MEK Is Essential for DNA Damage-Induced Checkpoint Response.
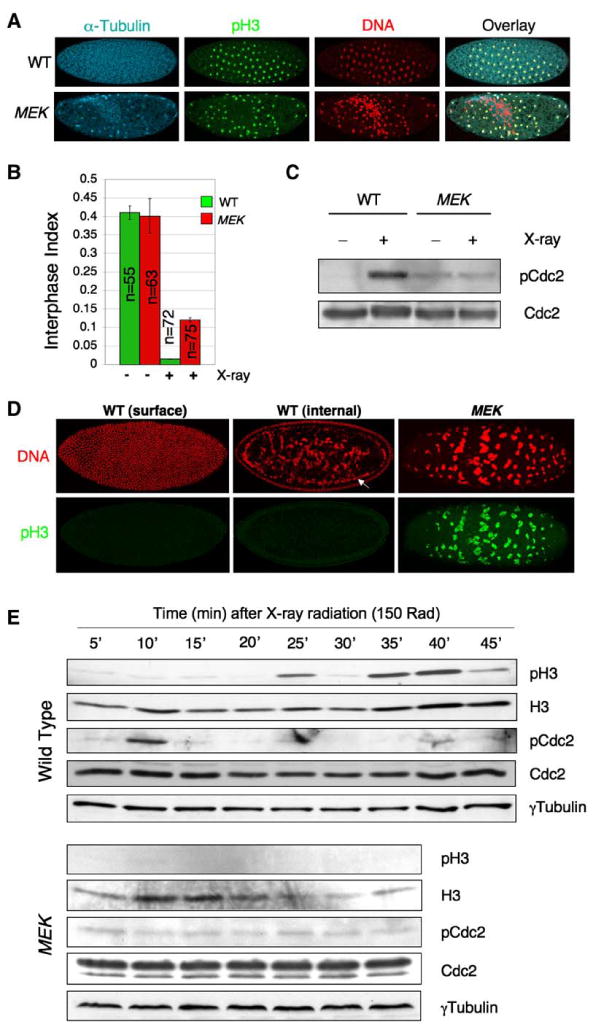
(A) Embryos (0–1 hr AED) were treated with low-dose X-ray radiation (150 rad), allowed to develop for 35 min at 25°C, and then fixed and stained with propidium iodide (red) and antibodies for α-Tubulin (cyan) and phospho-Histone H3 (pH3; green).
(B) Interphase index of embryos 35 min following the treatment in (A). The interphase index is defined as the percentage of pH3-negative embryos of the total. “n” denotes the number of embryos counted. Error bars represent standard deviation.
(C) Immediately following X-ray irradiation (within 10 min) as in (A), embryo lysates were subjected to SDS-PAGE and blotted sequentially with indicated antibodies.
(D) 60 min following irradiation as in (A), embryos were fixed and stained with propidium iodide (red) and anti-pH3 (green). The arrow points to a second nuclear layer formed by falling nuclei (middle). Note the unusually large and fragmented DNA (pH3-positive) in the MEK embryo.
(E) Freshly laid eggs (0–1 hr AED) of indicated genotypes were treated with 150 rad X-ray, and then ten embryos were homogenized at each indicated time point. Embryo lysates were subjected to SDS-PAGE and blotted sequentially with indicated antibodies. The membrane was stripped of antibodies between blotting.
Halting the cell cycle following DNA damage allows for proper repair of damaged DNA (Kastan and Bartek, 2004); a failure to arrest the cell cycle would prevent such DNA repair, leading to accumulation of damage and “mitotic catastrophe” (Takada et al., 2003). Indeed, wild-type, but not MEK, embryos appeared to be able to recover from low-dose X-ray radiation, presumably following proper repair of DNA damage. One hour following irradiation, wild-type embryos appeared to have resumed normal nuclear/cell divisions, as evidenced by the appearance of regularly patterned cortical nuclear monolayer consisting of high density pH3-negative nuclei (Figure 3D, left; cf. Figure 3A), although these embryos apparently had eliminated many nuclei that presumably suffered irreparable DNA damage, which were seen as having fallen into the interior (Figure 3D, middle; cf. Figure 1A). MEK embryos, in contrast, appeared highly sensitive to the same low-dose X-ray treatment, and the majority of these embryos exhibited hallmarks of “mitotic catastrophe,” such as aggregated and fragmented DNA that remained pH3 positive (Figure 3D, right) or complete loss of the cortical nuclear layer (data not shown). Thus, it appeared that in response to low-dose irradiation, wild-type syncytial embryos immediately respond by inhibiting Cdc2 activity (G2 arrest) but temporarily arrest at metaphase and resume division later on. In contrast, MEK embryos fail in each of these steps.
To confirm that low-dose X-ray treatment causes wild-type, but not MEK, syncytial embryos to undergo sequential arrests at G2/M and metaphase, respectively, we irradiated newly deposited eggs (0–1 hr AED) and examined cell cycle markers at 5 min intervals. Consistent with our postulation, Cdc2 inhibition (pCdc2) was seen at 10 min, and phospho-H3 levels peaked at 35–40 min following X-ray in treated wild-type embryos, but these markers remained unchanged in MEK embryos (Figure 3E). Taken together, these results suggest that the MEK/ERK pathway is required for both extrinsic and intrinsic cell cycle checkpoint mechanisms in Drosophila embryos.
MEK Activation Is Cell Cycle-Dependent and Increases in Response to Ionizing Radiation or in grp Mutants
To investigate whether MEK activation (phosphorylation) correlates with its requirement in checkpoint control, we examined embryos with antibodies that recognize phospho-MEK (pMEK). Indeed, we detected increased pMEK signals during late interphase and late mitosis (Figures 4A and 4B). The late interphase pMEK signals were found more concentrated on the nuclear envelope immediately before it breaks down, corresponding to the G2-M transition (Figure 4A). The mitotic pMEK signals were found to colocalize with anaphase/telophase chromosomes (Figure 4B). Increased pMEK signals during mitosis were also found in Drosophila S2 cells (Figure 4C) in patterns similar to those found in mammalian cells (Shapiro et al., 1998). Although cell cycle-dependent MEK/ERK activation has previously been reported (Guadagno and Ferrell, 1998; Hayne et al., 2004; Shapiro et al., 1998), it is also reported that pMEK antibodies cross-react with Nucleophosmin/B23 (Hayne et al., 2004). However, Drosophila Nucleophosmin/B23 lacks the N-terminal epitope recognized by pMEK antibodies (Figure S2) and thus is unlikely to account for the pMEK signals in immunostaining. Moreover, the peptide sequence used for pMEK antibody production is not homologous to any sequences other than MEK itself in the Drosophila genome by Blast search (data not shown). The cell cycle-specific MEK activation and its subnuclear localization are consistent with a role of the MEK/ERK pathway in an intrinsic cell cycle checkpoint regulatory mechanism.
Figure 4. MEK Activation during Cell Cycle Progression.
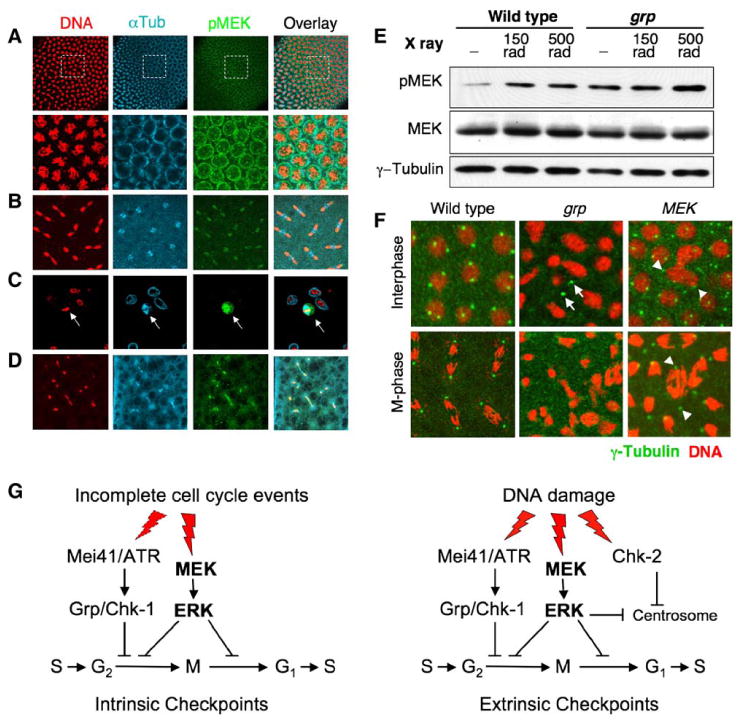
(A) A division cycle-12 wild-type embryo was fast-fixed and stained with indicated antibodies. Note that a band of higher pMEK signals (green) was seen coinciding with prophase or late interphase nuclei. Lower panels: higher magnification of boxed areas of top panels.
(B) A cycle-10 wild-type embryo during anaphase/telophase.
(C) An S2 cell during mitosis (arrow).
(D) A cycle-9 grp embryo during anaphase/telophase.
(E) Wild-type and grp mutant embryos were irradiated with 0, 150, or 500 rad of X-ray, immediately subjected to SDS-PAGE, and blotted sequentially with indicated antibodies. Note the increased pMEK levels in irradiated embryos, and in grp mutant embryos even without irradiation (cf. lanes 1 and 4).
(F) Chk2-dependent centrosome inactivation fails in MEK embryos. Cycle-13 embryos of indicated genotypes were stained for DNA (red) and γ-tubulin (green). Note the centrosome foci (bright green spots) present in wild-type and MEK embryos but absent in grp embryos. Arrows point to free centrosome foci not associated with the nucleus in the grp embryo. Arrowheads point to centrosome foci associated with morphologically abnormal nuclei or chromosomes in the MEK embryo.
(G) A model for the MEK/ERK pathway in cell cycle checkpoints. Cell cycle checkpoints are regulatory mechanisms that can be classified as intrinsic (left) and extrinsic (right) pathways. The intrinsic pathways ensure the orderly progression of cell cycle events and are activated at different transition points to prevent premature entry into the next cell cycle event. Activation of the MEK/ERK pathway at entry into and exit from mitosis represents such an intrinsic checkpoint regulatory mechanism. The ATR/Chk1 pathway may act in conjunction with MEK. The extrinsic pathways are activated only when DNA damage is detected. The MEK/ERK pathway acts in conjunction with or in parallel to the Chk1 and Chk2 pathways to halt the cell cycle progression in response to DNA damage.
In addition, increased pMEK signals were also detected in grp mutant embryos (Figures 4D and 4E) and in embryos exposed to X-ray (Figure 4E). The higher levels of MEK activation in grp mutants suggest that MEK activation may compensate for a lack of the Grp/Chk1 pathway. Furthermore, it has been shown that DNA damage or a failure in the Grp/Chk1 pathway leads to Chk2-dependent centrosome inactivation and chromosomal segregation defects (Sibon et al., 2000; Takada et al., 2003). We examined the Chk2-dependent centrosome inactivation by γ-tubulin staining and found that, in contrast to grp mutants, centrosome inactivation fails in cycle 13 MEK mutant embryos, despite the presence of damaged chromosomes/DNA (Figure 4F), suggesting that lack of MEK also leads to a failure of Chk2-dependent centrosome inactivation. Thus, MEK appears to also act in an extrinsic pathway, in conjunction with or in parallel to the Chk1 and Chk2 pathways.
Discussion
We have investigated the direct role of the MEK/ERK pathway in regulating cell cycle progression. We found evidence of an acute requirement for this pathway to, unexpectedly, slow down cell cycle progression, suggesting that the MEK/ERK pathway is required for an intrinsic checkpoint pathway. The faster and extra rounds of division cycles exhibited by MEK mutant embryos suggest a failure at checkpoints that control entry into and exit from mitosis, i.e., the DNA replication, spindle assembly, and centrosome-inactivation checkpoints. The lack of these checkpoints in MEK embryos results in severe chromosomal and mitotic defects. In contrast to the established notion that there are no gap phases in the early Drosophila division cycles (Foe et al., 1993), our study suggests that very short gap phases (especially G2) do exist and are eliminated in the absence of MEK. This short G2 appears to be an essential “wait period” enforced by the intrinsic MEK/ERK checkpoint pathway, allowing for monitoring the completion of DNA synthesis and repairing any damages (Figure 4G).
We found evidence that MEK/ERK acts in conjunction with or in parallel to the Chk1 and Chk2 pathways. The severe chromosomal defects associated with the accelerated division cycles of MEK embryos and the detection of increased MEK activation in grp mutants (see Figure 4E) suggest that the MEK/ERK pathway is more essential and may compensate for a defect in the Grp/Chk1 pathway. Indeed, grp mutant embryos exhibit only moderate cell cycle acceleration phenotypes in the early division cycles (prior to cycle 11) (Fogarty et al., 1994; Ji et al., 2004; this study). In addition, grp mutant embryos appear to arrest at the 13th metaphase (Fogarty et al., 1994; Ji et al., 2004) or their chromosome segregation is blocked (Sibon et al., 2000; Takada et al., 2003), whereas MEK embryos undergo an extra round of synchronized division (see Figures 1D and 2A). It has been shown that the chromosomal segregation failure following the 13th mitosis in grp mutant embryos is due to accumulation of DNA damage and consequent activation of the Chk2 pathway, resulting in centrosome inactivation (Takada et al., 2003). The ability of MEK embryos to continue nuclear division in spite of accumulated DNA damage indicates that the Chk2 pathway fails to activate in these embryos. Indeed, the centrosome complex (γ-tubulin focus), which is absent in grp mutant embryos, is present in MEK embryos (see Figure 4F). Thus, both the Chk1 and Chk2 pathways fail to execute checkpoint functions in the absence of the MEK/ERK pathway. We propose that MEK and ERK activation acts in conjunction with both the Chk1 and Chk2 pathways to exert intrinsic checkpoint control over normal cell cycles and also extrinsic checkpoint function in response to DNA damage (Figure 4G). We are currently investigating the relationship between the MEK/ERK pathway and the Chk1 and Chk2 pathways.
Cell cycle-dependent activation of the MEK/ERK pathway has previously been observed in cultured mammalian cells (Shapiro et al., 1998), and a role for MEK/ERK activation in the spindle assembly checkpoint during normal mitotic progression and in Cdc20 phosphorylation, which is required for the execution of the spindle checkpoint, has been established in Xenopus egg extracts (Chung and Chen, 2003; Guadagno and Ferrell, 1998). It has recently been shown in Xenopus that the protein kinase Mos, but not Raf, mediates cell cycle-dependent MEK/ERK activation (Yue and Ferrell, 2004). Mos is a protooncoprotein and MAP Kinase Kinase Kinase, as is Raf. We have found that Drosophila Raf is not involved in the intrinsic cell cycle checkpoint pathway mediated by MEK and ERK. Although it yet to be shown whether Drosophila Mos is responsible for MEK activation during cell cycle progression, there appears to be intriguing parallels between Drosophila and vertebrates.
In summary, our results demonstrate the presence of an intrinsic cell cycle checkpoint mechanism mediated by the MEK/ERK pathway in the Drosophila early embryo that act at multiple checkpoints to ensure orderly progression of the cell cycle, avoiding potential DNA damage. The MEK/ERK checkpoint pathway is also essential to mediate an extrinsic pathway, halting cell cycle progression in response to ionizing radiation.
Experimental Procedures
Fly Strains and Genetics
The following strong or null alleles were used in this study: Dsor1LH110, Dsor1r1, Draf11-29, and grp06034 (all from the Bloomington Drosophila Stock Center, Bloomington, IN). Oregon R flies were used as wild-type control. To produce embryos lacking maternal contributions of MEK or Draf, we used the dominant female sterile (DFS) technique (Chou and Perrimon, 1992) to generate germline clones (GLCs) homozygous for Dsor1LH110 or Draf11-29 as described (Li et al., 1998). For example, Draf11-29 FRT101/FM7 females were crossed to ovoD1 FRT101; hs-Flp38 males, and the resulting larvae were heat-shocked daily for 2 hr at 37°C during the 3rd instar and pupal stages. Adult Draf11-29 FRT101/ovoD1 FRT101 females with GLCs were mated to wild-type males, and the embryos were collected for experiments. Since both Dsor1 and Draf are on the X chromosome, 50% of the embryos will inherit a wild-type copy of the gene from the paternal X chromosome (paternally rescued), and the rest will inherit the Y chromosome (nonpaternally rescued). The latter class of embryos will be completely null for Dsor1 or Draf. Dsor1LH110 and Dsor1r1 exhibited identical early division phenotypes. Results from Dsor1LH110 are shown. Phenotypes shown in this study are from nonpaternally rescued Dsor1 or Draf embryos. grp mutant embryo were from grp06034 homozygotes.
In embryos derived from Dsor1 mutant germline cells, 50% exhibited cellularization and mitotic defects as shown in Figure 1. The other 50% showed normal cellularization and ftz expression. When allowed to develop to late stage, 50% embryos did not show any further differentiation and failed to develop cuticles, while the other half developed cuticle structures with typical torso class phenotypes (missing posterior terminal structures). Based on the presence of two distinct phenotypic classes, we deduce that the first 50% must be fertilized by the Y chromosome (male embryos) and thus represent maternal and zygotic null Dsor1 mutant embryos, while the other half must be paternally rescued by a wild-type X chromosome (female embryos).
Immunohistochemistry and Western Blots
Rabbit antibodies for nonphosphorylated MEK and phospho-MEK (pMEK) (Cell Signaling Technology; 1:1000), rabbit anti-Cdc2 (PSTAIRE; Santa Cruz Biotechnology; 1:500), rabbit antibodies against Tyr-15 phosphorylated Cdc2 (pCdc2; Cell Signaling Technology; 1:1000), mouse anti-α-tubulin (Sigma; 1:1000), rabbit anti-ERK (Sigma; 1:1000), and rabbit anti-phospho-histone H3 (Upstate Biotechnology; 1:1000) were used as primary antibodies for whole-mount immunostaining of embryos and Western blots. The following secondary antibodies were used for fluorescent immunostaining of embryos: goat anti-rabbit Alexa 488 (1:500), goat anti-mouse Alexa 594 (1:500), and goat anti-mouse Alexa 633 (1:500), all from Molecular Probes. Embryos were fixed in 37% formaldehyde for 5 min to preserve microtubule structures when necessary. Nucleus or DNA was stained with propidium iodide (Sigma) at 1 μg/ml after RNase treatment (at 400 μg/ml for 30 min at 37°C) or bizbenzimide (Hoechst 33258; Sigma; at 1 μg/ml). In situ hybridization was performed by standard methods using antisense RNA probes made by in vitro transcription from a ftz cDNA using Digoxingenin-UTP nucleotide mix (Roche). Stained embryos were analyzed and photographed with a Leica confocal microscope or an Axiophot compound microscope.
Time-Lapse Confocal Microscopy
Embryos (0–1 hr after egg deposition) produced by females of specified genotypes were manually dechorionated and injected with oli-green (Molecular Probes; 100 μg/ml in DMSO) or together with mRNA (0.5 mg/ml; mixed with oli-green immediately before injection) or drugs (U0126, 20 μM, Cell Signaling Technology). Injected embryos were observed with a Leica TCS-SP inverted laser-scanning confocal microscope and scanned at 30 s intervals. Images were assembled with QuickTime Pro. Quantification of cell cycle phases was as previously described (Sibon et al., 1997). Mitosis is defined to begin at nuclear envelope breakdown (influx of the fluorescent dye) and end when the chromosomes decondense and nuclei round up, forming new nuclear envelope. Interphase begins at this point till the start of the next mitosis.
Assay for DNA Damage-Induced Checkpoint Responses
Staged embryos were irradiated in an AXR Minishot X-ray machine (Associated X-Ray Corporation, East Haven, CT) at 150 rad (1 min at 150 kV, 3 mA) or otherwise specified. Irradiated embryos were either fixed for whole-mount immunostaining or homogenized for Western blots at specified time points.
Molecular Biology and mRNA Synthesis
Constitutively active Drosophila MEK (MEKEE) was created by in vitro mutagenesis replacing serine residues at positions 234 and 238 to negatively charged glutamate residues. These mutations mimic the effect of phosphorylation at positions equivalent to 218 and 222 of human MEK-1, resulting in constitutive activation (Huang and Erikson, 1994). MEKEE and Drosophila Mkp3 mRNAs were synthesized using the mMESSAGE mMACHINE T7 Kit (Ambion) according the manufacturer's manual.
Supplementary Material
Acknowledgments
We thank Dr. J. Chung (Mkp3) and the Bloomington Drosophila Stock Center for various reagents and Drosophila strains, and Dr. Jiyong Zhao for helpful comments on the manuscript. V.M. is a recipient of the Wilmot Cancer Postdoctoral Research Fellowship from the James P. Wilmot Foundation. This study was supported by research grants from the National Institutes of Health (R01GM65774; R01GM077046) and the American Cancer Society (RSG-06-196-01-TBE) to W.X.L.
Footnotes
Supplemental Data: Supplemental Data include two figures and can be found at http://www.developmentalcell.com/cgi/content/full/11/4/575/DC1/.
References
- Ambrosio L, Mahowald AP, Perrimon N. l(1)pole hole is required maternally for pattern formation in the terminal regions of the embryo. Development. 1989;106:145–158. doi: 10.1242/dev.106.1.145. [DOI] [PubMed] [Google Scholar]
- Brose MS, Volpe P, Feldman M, Kumar M, Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. [PubMed] [Google Scholar]
- Chou TB, Perrimon N. Use of a yeast site-specific recombinase to produce female germline chimeras in Drosophila. Genetics. 1992;131:643–653. doi: 10.1093/genetics/131.3.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Chen RH. Phosphorylation of Cdc20 is required for its inhibition by the spindle checkpoint. Nat Cell Biol. 2003;5:748–753. doi: 10.1038/ncb1022. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664–1672. doi: 10.1126/science.274.5293.1664. [DOI] [PubMed] [Google Scholar]
- Foe VE, Odell GM, Edgar BA. Mitosis and morphogenesis in the Drosophila embryo: point and counterpoint. In: Bate M, Arias AM, editors. The Development of Drosophila melanogaster. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1993. pp. 149–300. [Google Scholar]
- Fogarty P, Kalpin RF, Sullivan W. The Drosophila maternal-effect mutation grapes causes a metaphase arrest at nuclear cycle 13. Development. 1994;120:2131–2142. doi: 10.1242/dev.120.8.2131. [DOI] [PubMed] [Google Scholar]
- Guadagno TM, Ferrell JE., Jr Requirement for MAPK activation for normal mitotic progression in Xenopus egg extracts. Science. 1998;282:1312–1315. doi: 10.1126/science.282.5392.1312. [DOI] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA. Checkpoints: controls that ensure the order of cell cycle events. Science. 1989;246:629–634. doi: 10.1126/science.2683079. [DOI] [PubMed] [Google Scholar]
- Hayne C, Xiang X, Luo Z. MEK inhibition and phosphorylation of serine 4 on B23 are two coincident events in mitosis. Biochem Biophys Res Commun. 2004;321:675–680. doi: 10.1016/j.bbrc.2004.07.024. [DOI] [PubMed] [Google Scholar]
- Huang W, Erikson RL. Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc Natl Acad Sci USA. 1994;91:8960–8963. doi: 10.1073/pnas.91.19.8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji JY, Squirrell JM, Schubiger G. Both cyclin B levels and DNA-replication checkpoint control the early embryonic mitoses in Drosophila. Development. 2004;131:401–411. doi: 10.1242/dev.00944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- Kim M, Cha GH, Kim S, Lee JH, Park J, Koh H, Choi KY, Chung J. MKP-3 has essential roles as a negative regulator of the Ras/mitogen-activated protein kinase pathway during Drosophila development. Mol Cell Biol. 2004;24:573–583. doi: 10.1128/MCB.24.2.573-583.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WX. Functions and mechanisms of receptor tyrosine kinase Torso signaling: lessons from Drosophila embryonic terminal development. Dev Dyn. 2005;232:656–672. doi: 10.1002/dvdy.20295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Melnick M, Perrimon N. Dual function of Ras in Raf activation. Development. 1998;125:4999–5008. doi: 10.1242/dev.125.24.4999. [DOI] [PubMed] [Google Scholar]
- Orr-Weaver TL. Developmental modification of the Drosophila cell cycle. Trends Genet. 1994;10:321–327. doi: 10.1016/0168-9525(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Roovers K, Assoian RK. Integrating the MAP kinase signal into the G1 phase cell cycle machinery. Bioessays. 2000;22:818–826. doi: 10.1002/1521-1878(200009)22:9<818::AID-BIES7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Chang HC, Karim F, Laverty T, Michaud NR, Morrison DK, Rebay I, Tang A, Therrien M, Wassarman DA. Signal transduction downstream from Ras in Drosophila. Cold Spring Harb Symp Quant Biol. 1997;62:347–352. [PubMed] [Google Scholar]
- Shapiro PS, Vaisberg E, Hunt AJ, Tolwinski NS, Whalen AM, McIntosh JR, Ahn NG. Activation of the MKK/ERK pathway during somatic cell mitosis: direct interactions of active ERK with kinetochores and regulation of the mitotic 3F3/2 phosphoantigen. J Cell Biol. 1998;142:1533–1545. doi: 10.1083/jcb.142.6.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibon OC, Stevenson VA, Theurkauf WE. DNA-replication checkpoint control at the Drosophila midblastula transition. Nature. 1997;388:93–97. doi: 10.1038/40439. [DOI] [PubMed] [Google Scholar]
- Sibon OC, Kelkar A, Lemstra W, Theurkauf WE. DNA-replication/DNA-damage-dependent centrosome inactivation in Drosophila embryos. Nat Cell Biol. 2000;2:90–95. doi: 10.1038/35000041. [DOI] [PubMed] [Google Scholar]
- Takada S, Kelkar A, Theurkauf WE. Drosophila checkpoint kinase 2 couples centrosome function and spindle assembly to genomic integrity. Cell. 2003;113:87–99. doi: 10.1016/s0092-8674(03)00202-2. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Wu D, Chen B, Parihar K, He L, Fan C, Zhang J, Liu L, Gillis A, Bruce A, Kapoor A, Tang D. ERK activity facilitates activation of the S-phase DNA damage checkpoint by modulating ATR function. Oncogene. 2005;25:1153–1164. doi: 10.1038/sj.onc.1209148. [DOI] [PubMed] [Google Scholar]
- Yue J, Ferrell JE., Jr Mos mediates the mitotic activation of p42 MAPK in Xenopus egg extracts. Curr Biol. 2004;14:1581–1586. doi: 10.1016/j.cub.2004.08.056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.