

Abstract
Objective
Knockout of the neural and cardiac expressed transcription factor HF-1b causes electrophysiological abnormalities including fatal ventricular arrhythmias that occur with increasing frequency around the 4th week of postnatal life. This study addresses factors that may contribute to conduction disturbance in the ventricle of the HF-1b knockout mouse. Disruptions to gap junctional connexin40 (Cx40) have been reported in distal (i.e., apically located), but not proximal His–Purkinje conduction tissues of the HF-1b knockout mouse. This abnormality in myocardial Cx40 led us to address whether 4-week-old HF-1b knockout postnates display other disruptions to ventricular structure and function.
Methods
Western blotting and immunoconfocal quantification of Cx43 and coronary arteriole density and function were undertaken in the ventricle. Electrical activation was described by optical mapping.
Results
Western blotting and immunoconfocal microscopy indicated that overall levels of Cx43 (p <0.001) and percent of Cx43 localized in intercalated disks (p <0.001) were significantly decreased in the ventricular myocardium of knockouts relative to wildtype littermate controls. Analysis of the reduction in Cx43 level by basal and apical territories revealed that the decrease was most pronounced in the lower, apical half of the ventricle of knockouts relative to controls (p <0.001). Myocyte size also showed a significant decrease in the knockout, that was more marked within the apical half of the ventricle (p <0.05). Optical recordings of ventricular activation indicated apically localized sectors of slowed conduction in knockout ventricles not occurring in controls that could be correlated directly to tissues showing reduced Cx43. These discrete sectors of abnormal conduction in the knockout heart were resolved following point stimulation of the ventricular epicardium and thus were not explained by dysfunction of the His–Purkinje system. To further probe base-to-apex abnormalities in the HF-1b knockout ventricle, we analyzed coronary arterial structure and function. These analyses indicated that relative to controls, the apical ventricular territory of the HF-1b knockout had reductions in the density of small resistance vessels (p <0.01) and deficits in arterial function as assayed by bead perfusion (p <0.01).
Conclusion
The HF-1b knockout ventricle displays abnormalities in Cx43 level, myocyte size, activation spread and coronary arterial structure and function. These abnormalities tend to be more pronounced in the apical territory of the ventricle and seem likely to be factors contributing to the pathological disturbance of cardiac conduction that characterizes the heart of the HF-1b knockout mouse.
Keywords: Transgenic animal models, Arrhythmia mechanisms, Arteries, Sudden death, Gap junctions
1. Introduction
Sudden cardiac death is a leading health problem in Western nations, taking the lives of hundreds of thousands of people each year [1,2]. The etiology of sudden cardiac death is multifactorial, though most commonly it is associated with cardiovascular disease, and in particular, coronary artery disease. While mutations in ion channels have been implicated in a number of cases, the degree to which other genetic and epigenetic factors predispose the remainder of this population to develop a deadly arrhythmia remain to be fully characterized. A transgenic mouse model of sudden cardiac death, the HF-1b homozygous knockout mouse (HF1-b−/−) [3], has been reported. HF-1b is a zinc finger transcription factor (Sp-1 related) that is preferentially expressed in neural [4] and myocardial [3] lineages. HF-1b−/− mice survive to birth and display relatively normal cardiac morphology. During postnatal life, HF-1b−/− animals of either sex develop complete penetrance of a severe disturbance to cardiac conduction, symptoms of which include secondary and tertiary atrioventricular blocks, spontaneous ventricular tachycardia, asystole and sudden cardiac death [3]. The mechanism leading to these defects of activation and the extent to which the cardiac pathogenesis of the HF-1b−/− knockout mouse can aid our understanding of arrhythmogenesis in the human heart remain to be determined.
In addition to a tendency to develop lethal arrhythmias, other abnormalities have been identified in the ventricle of the HF-1b knockout mouse [3]. These included increased action potential duration heterogeneity among isolated myocytes, up-regulation of minK mRNA and abnormalities in the expression of the gap junction protein connexin40 (Cx40) in conduction tissues. Interestingly, disruption of Cx40 in the HF-1b knockout was largely confined to Purkinje fiber cells in the most distal parts of the conduction system. In mouse, distal conduction cells are localized almost exclusively in the apical half of the ventricle [5]. The apical localization of Cx40 abnormality led us to address whether the ventricular myocardium of the HF-1b knockout displayed other spatial and/or territory specific variations in ventricular structure and function. We thus undertook analyses of Cx43 level and distribution, myocyte size variation, optically mapped activation spread and coronary arterial structure and function in the knockout heart, focusing in particular on base-to-apex variations in the parameters studied. These analyses revealed multiple pathological changes in the structure and activation of the HF-1b knockout heart that were consistently more marked in the apical territory of the ventricle than in the base.
2. Methods
2.1. Animals, tissue collection and PCR genotyping
HF-1b+/+ (wildtype), HF-1b+/− (heterozygous) and HF-1b−/− (knockout) mice were obtained from a breeding colony using animals produced by homologous recombination as previously described by Nguyen-Tran et al. [3]. Genotype for each mouse was determined using PCR analysis of genomic DNA from tail biopsies. All animals were treated and cared for in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals” (NIH Publication No. 85-23, revised 1996, National Research Council, Washington, DC).
2.2. Western blotting
Western blot analyses of Cx43 levels were undertaken on alkaline membrane extracts from 4-week-old mouse ventricles and HeLa cells using approaches previously described [6,7]. Alkaline treatment provided for enrichment of the pool of membrane-localized Cx43 gap junctions similar to that quantified in the immunoconfocal Cx43 content analyses. Non-transfected (wildtype) human cervix carcinoma HeLa cells and transfected cells expressing mouse Cx43 were kindly provided by Professor Klaus Willecke. Western blotting of Cx43 was repeated on independent blots from at least 3 animals per genotype. The polyclonal antibody (Rb3) against Cx43 used in this study was raised to a peptide corresponding to amino acid residues 252–270 of rat Cx43 using a customized service (Research Genetics, Inc.). The serum was then affinity purified against the peptide coupled to an activated chromatography matrix also using a customized service (Research Genetics, Inc.). This peptide is a commonly used Cx43 immunogen in production of monoclonal and polyclonal antibodies against Cx43 [8]. The specificity of the antibody for Cx43 in mouse and HeLa cells expressing Cx43 was confirmed in Western blotting and immunohistochemical tests (Fig. 1).
Fig. 1.
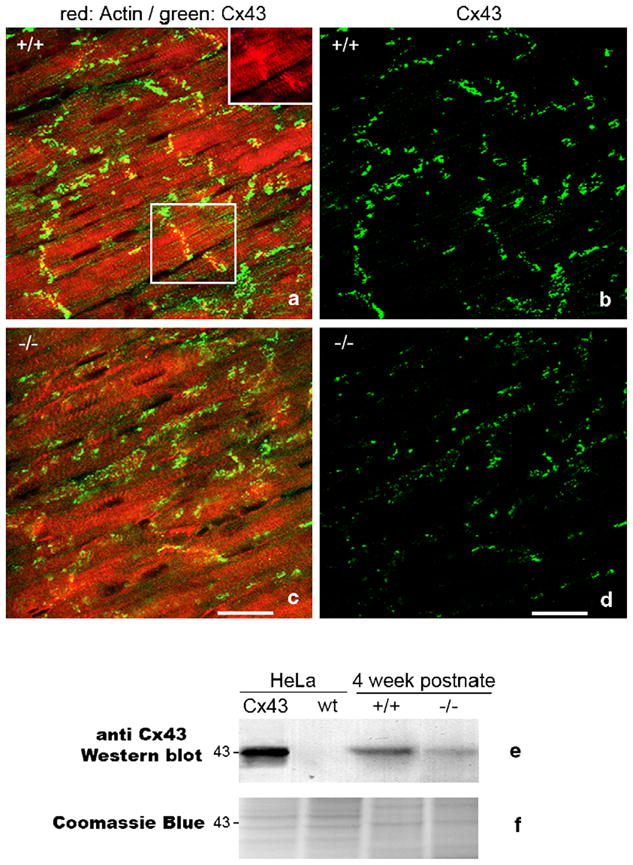
Spatial heterogeneities in ventricular Cx43 in HF-1b knockout myocardium. (a–d) Double labeling for Cx43 (green) and rhodamine–phalloidin (red) in posterior apical myocardium of the ventricle of the HF-1b wildtype (a) and knockout (c). The inset in the upper right corner of (a) shows rhodamine–phalloidin delineation of intercalated disks shown in the boxed region within (a). Rhodamine–phalloidin labeling was used in the identification and quantification of percent Cx43 area localized at intercalated disks (see Table 1). (b, d) Cx43 signal from images in (a) and (c) are shown without rhodamine–phallodin staining to improve illustration of the differences in Cx43 level and distribution between wildtype and knockout tissues. Scale bars=25 μm. (e) Cx43 Western blots of wildtype (non-Cx43 expressing) HeLa cells and HeLa cells stably transfected with Cx43, ventricular myocardium (VM) of 4-week postnate HF-1b knockout and wildtype littermates. (f) Coomassie blue-stained gel equivalent to those transferred to membrane for probing with Cx43 antibody shows equal loading of lanes.
2.3. Quantificative analyses of Cx43, myocyte size and arterial density
Cx43 immunolabeling and quantification were done as follows: Frozen sections (10 μm) of 4-week-old wildtype, heterozygous and knockout hearts (3 per genotype) were labeled with the Rb3 anti-Cx43 antibody and rhodamine–phalloidin (Sigma) using standard protocols as previously described [6]. Rhodamine–phalloidin delineates myofibers and enabled precise discrimination of intercalated disk-localized Cx43 signal. Estimates of Cx43 content were derived from confocal optical sections by an investigator blind to genotype. The quantitation method has been validated by us [6,9,10] and others [11,12] in many studies, including by direct comparison to freeze-fracture morphometry of Cx43 GJs [13]. Cx43 immunolabeled sections used for the quantification were selected from posterior regions of the serially sectioned hearts. This region of the ventricle is flat and contains myofibers uniformly oriented across the left and right ventricles, providing for full-length frozen sections of the ventricle containing high densities of longitudinally arrayed cells (e.g., Fig. 1). Frozen sections of the ventricle were divided into basal and apical halves. Fifteen optical sections were taken at random within each of these apical and basal territories of the frozen section. This was repeated on frozen sections from each of the nine mouse hearts used (3 wildtype, 3 heterozygote, 3 knockout) to give a total of 270 optical sections sampled (i.e., 15 optical sections ×2 territories ×3 mice ×3 genotypes). Single optical sections were taken with a ×40 oil objective (Plan-Neofluor 40× 1.3 NA oil, Zeiss), using constant zoom, aperture and gain settings on a Bio-Rad MRC-1024 scanning laser confocal microscope (LSCM) within a mid-Z-plane of the frozen section. Measurement of Cx43 content within optical sections was performed using NIH Image. Following 3×3 median filtering, a pixel intensity threshold was set such that a binary image of the Cx43 signal was matched precisely to an underlying grayscale image of Cx43 immunolabeling. The area of Cx43 (i.e., content) within the optical section was derived from the binary image. Phalloidin-labeling was used to assist in discrimination of Cx43 signals immunolocalized at intercalated disks (i.e., percent disk-localized Cx43 area—see inset Fig. 1a) and also provide an estimate of myocardial area within the optical section to normalize Cx43 content measurements (i.e., Cx43 area per tissue area).
2.4. Myocyte size quantification
Frozen sections of 4-week-old wildtype and knockout hearts (3 per genotype) were labeled with FITC conjugated to wheat germ agglutinin (WGA–FITC, Molecular Probes) and Hoechst (Sigma) for delineation of myocyte borders and nuclei. For labeling, sections were incubated with a PBS solution containing WGA–FITC (1: 100) and Hoechst (1:20,000) for 10 min at 37 °C, washed in PBS and cover-slipped in anti-fade mounting medium (Molecular Probes). The sections used in this quantification were sister, or near sister sections, to those used for Cx43 quantification. Double-labeled fluorescent images for counting myocyte profiles were captured using a ×40 1.3 NA oil immersion objective on a Leica DML fluorescence microscope equipped with a Hammatsu C5180 digital camera attached to a video monitor (Sony Triniton). Images were projected on a video screen and cell diameter estimates were measured blind to genotype orthogonal to the long axis of the longitudinally arrayed cells delineated by WGA–FITC directly from the screen. Measurements were made only on those myocyte profiles containing Hoechst-labeled nuclei to ensure that the cell was measured at a mid-plane within its volume. The diameter measurement was taken at or near the mid-point of the myocyte nucleus. Diameter measurements were made at 5 random microscopic fields within each of the basal and apical halves of the ventricular sections. Myocytes (5–20) were measured within each of these fields. The design used could thus be summarized as 5–20 myocytes ×5 fields ×2 ventricular territories ×3 animals×2 genotypes.
2.5. Smooth muscle actin immunolabeling and arterial density quantification
Frozen sections of neonatal and 4-week-old wildtype and knockout hearts (on at least 3 per genotype) were immunolabeled with antibody against smooth muscle actin (1:1000, Sigma) using standard protocols [14]. The arterial density was measured, blind to genotype, on hearts from 4-week-old animals using stereological methods described previously [15]. In brief, the immunofluorescently labeled histological sections were assessed using a point-counting protocol [16]. The sections used in this quantification were sister, or near sister sections, to those used for the Cx43 and myocyte size assessments. Intersections of coronary arterial profiles and myocardial tissues (taken to normalize arterial area to myocardial tissue area) with a M168 Weibel grid were counted at 30 independent microscope fields (15 each at random locations in the apical and basal halves) within each histological section assessed. Three histological sections were sampled from each of 3 animals used per genotype. The design could thus be summarized as 15 fields ×2 ventricular territories ×3 sections ×3 animals ×2 genotypes. Images for counting arterial profiles were captured using a ×40 1.3 NA oil immersion objective on a Leica DML microscope as above. Images were projected on the Sony Triniton video screen with an overlaid Weibel grid and point counts were made directly from the screen.
2.6. Quantification of coronary arterial perfusion
The aorta of HF-1b knockout and wildtype hearts (six per genotype) were cannulated and the coronary arterial bed of these hearts were perfused via the aorta with Tyrode solution at a flow rate sufficient to maintain a perfusion pressure of 125 mm Hg. After 5 min of perfusion, 15 μm diameter fluorescent beads were injected into the aortic root at 250 beads/g of body weight—beads of this diameter are standard for assessing regional tissue perfusion [17]. Following a 5-min post-injection flush, the hearts were fixed with 4% paraformaldehyde. Imaging for bead counts was undertaken on a Lecia SP TCS LSCM using a ×20 objective using constant zoom, aperture and gain settings. A projected volume of bead-infused ventricle was generated from a series of 40 7.5 μm-spaced optical sections. Twenty such optical section series (10 each in the basal and apical halves of each ventricle) were imaged starting from the posterior epicardial surface. The bead counts per projection were normalized to myocardial volume estimates derived from background autofluorescence of the paraformaldehyde-fixed tissue. The design could thus be summarized as 10 confocal projections ×2 ventricular territories ×6 animals ×2 genotypes.
2.7. Optical mapping of activation
Optical recordings were made using a voltage-sensitive dye (di-4-ANEPPS) to compare the activation sequence on the posterior epicardial surface of HF-1b knockout and wildtype hearts (5–6 per genotype). The methods used have been previously described by others and us [6,18–20]. In brief, hearts were removed from the chest and immediately perfused through the coronary circulation via the aorta with oxygenated Tyrode solution at 125 mm Hg. The hearts were placed in a tissue bath mounted on a Leica FLZ III dissecting microscope. The temperature of the tissue bath and perfusate were maintained at 37± 0.5 °C. Hearts were stained via superfusion and perfusion with voltage-sensitive dye (di-4-ANEPPS) for 10 min. Excitation light was provided by 100 W mercury vapor light source and green filter. Hearts were paced from a stimulus electrode (frequency 5–6.7 Hz, 1 ms duration, 2× threshold voltage) placed on the epicardial surface at the ventricular base. A high speed SciMeasure CCD camera (RedShirtImaging, Fairfield CT) with 80×80 pixel array was used to capture epicardial voltage transients propagating from the stimulus electrode at 1–2000 frames/s. Cytochalasin D (10 μmol/L) was used to minimize the motion artifact. Spatial and temporal filtering was utilized in data post-processing (Cardioplex, RedShirtImaging). Isochronal maps of activation spread and conduction velocity measurements were derived by Cardioplex software. Cx43 immunolabelings and confocal imaging of mapped hearts were carried out as described above.
2.8. Statistical analysis
Data are expressed throughout as mean± SD (standard deviation). Except where otherwise noted, a one-way analysis of variance was performed on all data sets (p values of <0.05 were considered significant). Bartlett’s test for homogeneity of variance indicated no significant inhomogeneities of variance within the data. Normality was assessed by the univariate normality procedure prior to ANOVA and Cx43 content per tissue area data was log transformed to conform to a normal distribution. When appropriate, pair-wise comparisons were made using Student’s t-test. p values of <0.05 were considered significant.
3. Results
3.1. Cx43 levels are reduced and heterogeneous in the apical region of the HF-1b knockout ventricle
In the HF-1b knockout ventricle, the expression of the gap junction protein Cx40 is disrupted in distal Purkinje fibers, i.e. cells localized in the apical half of the ventricle [3,5]. This localization led to ask the question: Are there similar spatial variations in other structural elements relevant to electrical activation in the knockout heart? We initially focused on base-to-apex variations in the level and organization of Cx43. As HF-1b null heterozygotes (+/−) showed no significant difference from wildtype littermates in any parameter examined (e.g., see Table 1), the following description is mainly confined to comparisons between the knockout and wildtype genotypes.
Table 1.
Cx43 gap junction (GJ) content (Cx43 μm2/tissue μm2) and percent Cx43 area localized at intercalated disks overall and in the basal (base) or apical (apex) regions of the ventricle of HF-1b wildtype, heterozygous null knockout littermates
| Genotype/vent region | Cx43 μm2/tissue μm2 | % Cx43 area at intercalated disks |
|---|---|---|
| +/+ overall | 0.016± 0.005 | 45.1± 9.4 |
| +/− overall | 0.015± 0.004 | 34.6± 7.9 |
| −/− overall | 0.012± 0.006† | 25.7± 14.2† |
| +/+ base | 0.015± 0.005 | 42.9± 8.8 |
| +/− base | 0.015± 0.004 | 38.9± 6.9 |
| −/− base | 0.013± 0.007 | 25.4± 11.2† |
| +/+ apex | 0.017± 0.004 | 47.0± 9.7 |
| +/− apex | 0.014± 0.004 | 30.3± 8.8† |
| −/− apex | 0.010± 0.004† | 26.0± 16.7† |
Significant difference (p <0.001) between wildtype (+/+) and HF-1b knockout (−/−) groups.
Fig. 1 indicates that 4-week postnate knockout mice show lower levels of localization of Cx43 particles/puncta at intercalated disks as compared to wildtype littermates (compare Fig. 1a with c—note inset in 1a showing disks labeled with rhodamine–phallodin). Consistent with this qualitative assessment, quantitative analysis of overall Cx43 immunolocalization patterns indicated that the percent of Cx43 area localized at intercalated disks was significantly lower (p <0.001) in the knockout ventricle (Table 1). This disruption to Cx43 pattern in the knockout was uniform within the knockout ventricle, showing no significant variation between the apical and basal halves (Table 1).
Overall, there was a highly significant decrease (p <0.001) in Cx43 content per tissue area in the 4-week-old knockouts as compared to control littermate ventricles (Table 1 and Fig. 2a). Western blotting confirmed that overall levels of ventricular Cx43 were reduced in the knockout (Fig. 1e). However, unlike percent of disk-localized Cx43, the reduction in Cx43 content showed territory specific variation within the ventricle. While there was no significant difference in Cx43 content between the basal halves of ventricles between genotypes, there was a highly significant (p <0.001) reduction in Cx43 content in the apical half of the ventricle of the knockout as compared with the wildtype (Table 1 and Fig. 2a).
Fig. 2.

Quantitative analyses of spatial heterogeneities in ventricular Cx43 in HF-1b knockout myocardium. (a) Cx43 content (μm2) per unit area of ventricular myocardium (μm2), shows an overall decrease in the HF-1b knockout ventricle compared to wildtype littermates. This reduction in Cx43 content is mainly localized to the apical territory of the ventricle (p <0.001). (b) Cumulative frequency distributions of Cx43 content measurements taken from the 90 optical sections sampled within wildtype and knockout apical myocardium. The leftward shift of the knockout plot demonstrates a population Cx43 immunolabeled optical sections with extremely low Cx43 content levels not found in the wildtype.
While moderate uniform reductions in Cx43 are not thought to slow or disrupt cardiac activation spread per se, evidence is mounting that spatial heterogeneities in Cx43 and other cardiac connexins can lead to profound conduction disturbance [21–24]. We thus undertook further analyses of spatial heterogeneities in Cx43 level within the apical territory of the HF-1b knockout ventricle. Fig. 2b provides cumulative frequency distributions of Cx43 content variation in the 45 optical sections that were sub-sampled within the apical territories of knockout and wildtype littermates. From this illustration, it can be ascertained that 8–10% of these optical section images had Cx43 content estimates that fell below a quarter of the value of the overall wildtype mean. By contrast, in the wildtype, no optical section had a Cx43 content that even fell below half the level of the overall wildtype mean (Fig. 2b). We concluded that there were sectors in the apical half of the knockout ventricle that displayed severe reductions in Cx43 level.
3.2. Myocyte size is reduced in the apical territory of the HF-1b knockout ventricle
WGA–FITC delineation of cell borders indicated that working ventricular myocytes of 4-week-old knockout mice were smaller and more irregularly shaped than those of wildtype littermates (compare Fig. 3a and b). This assessment was confirmed in a quantitative study (Fig. 3c). Moreover, it was determined that the dimensions of apically localized myocytes in the knockout showed a significant (p <0.05) decrease in size relative to apically located myocytes in wildtype littermates (Fig. 3c). In muscle fibers or neurons, conduction velocity varies as a square function of fiber diameter [25]. Thus, modest reductions in the myocyte diameter may have relatively large negative effects on conduction velocity.
Fig. 3.
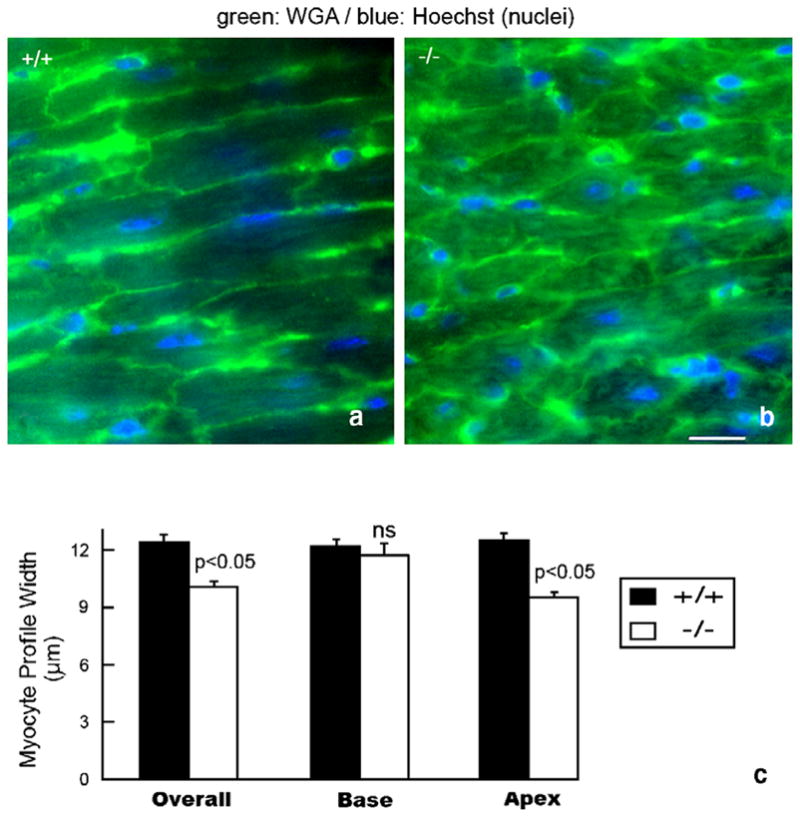
Diminished myocyte diameter and morphologic irregularity in the HF-1b knockout ventricular myocardium (a, b) WGA–FITC (green) and Hoechst (blue) staining delineate myocyte sarcolemma and nuclei in the apical myocardium of wildtype (a) and knockout (b) ventricles. (c) Plots of mean myocyte diameter overall in the ventricle and within the basal and apical territories. Significant differences (p <0.05) between knockout and wildtype measurements were found. Scale bar=25 μm.
3.3. Disruptions in electrical activation spread demonstrate a direct spatial association with Cx43 heterogeneities in the HF-1b knockout ventricle
The preceding analyses indicated base-to-apex heterogeneities in Cx43 level and myocyte size within the ventricle of the HF-1b knockout. Mathematical modeling has demonstrated that Cx43-mediated coupling and cell size are two of the most important determinants of conduction velocity in ventricular myocardium [26,27]. We next used optical mapping to address whether manifest alterations in electrical activation spread were detectable in the ventricle of the knockout. One key aspect of our approach was the use of a pacing electrode on the ventricular epicardial surface. This was done to eliminate complications to interpretation arising out of the previously described disruption to Cx40 in distal conduction cells in the knockout [3]. The stimulus electrode protocol ensured that the activation maps recorded reflected the conduction properties of the working ventricular myocardium alone, i.e., the tissue in which Cx43 and cell size heterogeneities were localized to in this study.
Epicardial electrical pacing of six 4-week-old wildtype mice indicated uniform anisotropic patterns of activation spread in all cases, whereby excitation propagated in the expected elliptical pattern [18] from the stimulus electrode (Fig. 4a, c). All knockout hearts exhibited strikingly more complex patterns of activation (Fig. 4b, d–g), although none demonstrated activation sequences consistent with an arrhythmia. In addition to areas of relatively normal conduction velocity, five of the six knockout hearts displayed areas in the middle or apical region of the ventricle of slow conduction (Fig. 4d–f). The remaining knockout heart displayed uniformly slow and thus abnormal conduction of excitation (Fig. 4g). Overall, average conduction velocities were significantly lower (p <0.05) in the HF-1b knockout hearts compared to wildtype littermates, as were minimal conduction velocities (Table 2). The ratio of maximum/minimum conduction velocity was also significantly greater in knockout hearts (p <0.05) compared to the wildtype (Table 2). These changes in activation parameters all appeared to reflect the discrete sectors of slowed conduction that occurred exclusively in HF-1b knockout ventricles.
Fig. 4.
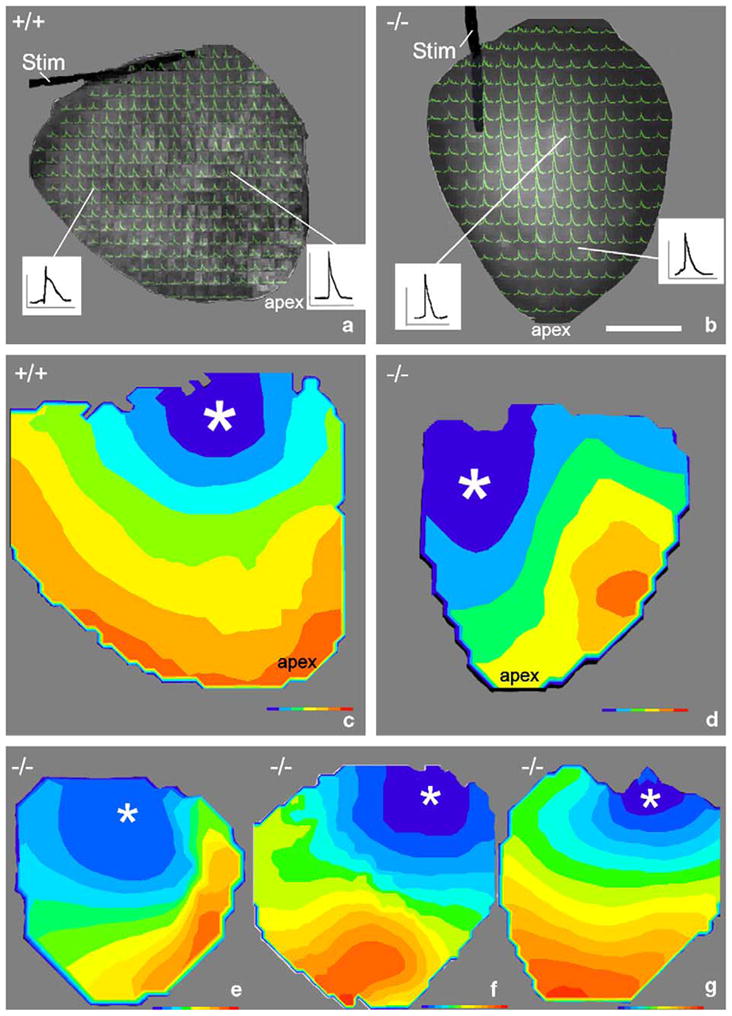
Abnormalities in ventricular activation pattern in HF-1b knockout ventricles (a) and (b) show action potentials superimposed on the posterior epicardial surfaces of knockout and wildtype hearts, respectively. The wildtype recording in (a) shows the apex in the upper right corner and the monopolar stimulus electrode (stim) at the ventricular base. Representative action potentials are shown in insets. Inset scales, vertical=50 mV and horizontal=100 ms. Isochronal maps of the epicardial activation were constructed from activation data in (a) and (b) and displayed in (c) and (d), respectively. Examples of maps from three further knockout hearts (apex down) are shown in e through g. Color isochrones on c–g=1 ms. Earliest isochrones are blue and latest red. Asterisks indicate site of the stimulus electrode at the ventricular base.
Table 2.
Conduction velocity measurements
| Genotype | Average CV | CVmax | CVmin | CVmax/CVmin |
|---|---|---|---|---|
| +/+ | 0.442± 0.26 | 0.858± 0.064 | 0.208± 0.041 | 4.30± 1.22 |
| −/− | 0.352± 0.23† | 0.709± 0.19 | 0.121± 0.33‡ | 6.06± 1.65† |
Velocity measurements are in millimeters per millisecond. For +/+ n =5 mice, −/− n =6 mice. Data are expressed as mean± SD.
Indicates significantly different from +/+, p <0.05.
Indicates significantly different from +/+, p <0.02.
Mapped hearts were subsequently sectioned and immunolabeled to determine whether the apically located sectors of slow conduction in knockout ventricles corresponded to regions with reduced Cx43. Fig. 5a and b shows the isochronal maps for the same hearts illustrated in Fig. 4c and f, respectively. Arrowheads point to mid-ventricular and apical loci on these maps corresponding to the precise location of the four Cx43-immunolabeled tissue regions shown in Fig. 5c–f. A large reduction in Cx43 immunolabeling was observed at the precise location of slowed conduction in the knockout ventricle (Fig. 5b and f). Consistent with the data shown in Table 1, Cx43 levels in the basal region of the knockout was comparable to the levels in the basal and apical regions of the wildtype (compare Fig. 5e to c and d). The Cx43 content at the knockout apex in Fig. 5f is 12.5% of content estimate for the wildtype region shown in Fig. 5e. Danik et al. [28] have shown that reductions in Cx43 level below 18% of normal (via cre-mediated excision of Cx43) resulted in a significant reduction of conduction velocity in mouse ventricles. As such, we conclude that localized heterogeneities in Cx43 and myocyte size may be sufficient to explain the regions of slowed conduction in the ventricular myocardium of the HF-1b knockout mouse.
Fig. 5.
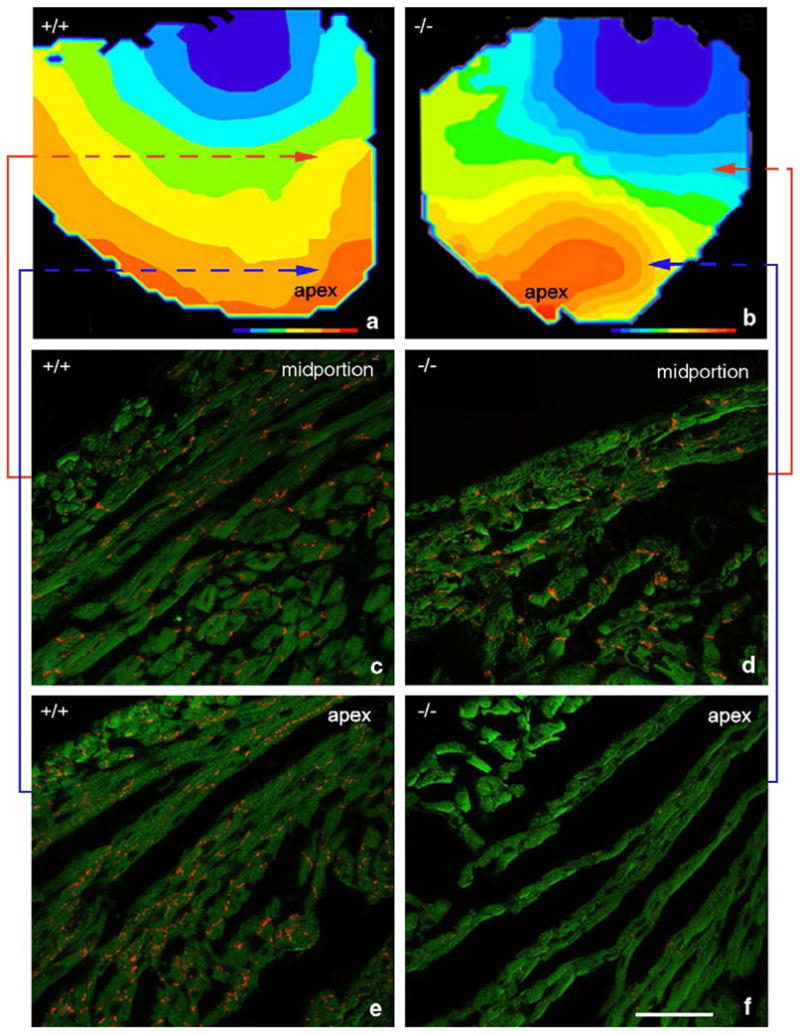
Direct co-localization of an apically located sector of disrupted and reduced Cx43 and a region of slow conduction in the HF-1b knockout ventricle. Isochronal maps of knockout and wildtype ventricles are shown in (a) and (b), respectively. (c–f) Cx43 immunolabeling (red) in ventricular regions indicated by shaded squares on the isochronal maps. Colored lines indicate shaded areas corresponding to areas of Cx43 immunolabeling. Note the reduction in Cx43 content in the apical region of the knockout ventricle (f), corresponding precisely with an area of slowed conduction in (b).
3.4. HF-1b knockout ventricles show base-to-apex deficits in coronary arterial structure and function
Stereomicroscopic examination of knockout hearts indicated coronary vascular abnormalities. While the main arterial branches seemed normal, collateral branching appeared reduced in the knockout. Fig. 6a and b show low magnification views of alpha-smooth muscle actin immunolabeled sections near the ventricular apex (see inset) of neonatal wildtype and knockout mice. Higher magnification details from these sections reveal irregularities in the smooth muscle coat of arterial profiles in knockout, not present in wildtype (compare Fig. 6c and d in particular the insets). It should be noted that only arteries are invested with smooth muscle in the neonatal mouse heart. It nonetheless could also not be excluded that there were also subtle vascular defects not revealed by smooth muscle actin immunolabeling in veins and capillaries.
Fig. 6.

Apical disruption of coronary artery structure in the HF-1b knockout. (a–d) Alpha-smooth muscle actin immunolabeling (green) with propidium iodide nuclear counterstaining (red) highlight coronary arterial distributions in apical regions of a neonatal HF-1b+/+ (a, c) and HF-1b+/+ (b, d) ventricles. Inset in (b) shows section level at the apex. Boxed areas from (a) and (b) are shown at higher magnification in (c) and (d), respectively. Although the distribution of the main branches is preserved, vessel walls show discontinuous smooth muscle in the knockout at high magnification (insets). In the 4-week-old, apical sections stained with anti-smooth muscle actin and propidium iodide in (e) and (f). Although the main branches appear normally deployed in the HF-1b−/− heart, there is a paucity of small caliber vessels (best seen in the higher magnification insets) compared to that of the wildtype. Small arrowheads point to these smaller arterial profiles in (e) and (f). Scales=1 mm (a,b) and 100 μm (c, d, e, f). (g) Stereological analyses indicate a reduction in the total volume density of coronary arteries in the HF-1b−/− ventricle, as compared to HF-1b+/+, **(p <0.01). Furthermore, this decrease is seen in the apical half of the ventricle (p <0.01), but not in the base.
At 4 weeks, normal arterial profiles were always found in the basal and apical territories of wildtype ventricles and in the basal region of knockout littermates. However, in the apical half of knockout ventricles, there was a paucity of small-caliber arteries (compare frequency of arrowheads in Fig. 6e and f, in particular the insets). Quantification of coronary arterial density confirmed that there were significantly lower densities of intramural arterialization in knockouts as compared to wildtype ventricles at 4 weeks of age (p <0.01— Fig. 6g). Consistent with the qualitative appraisal, this difference followed a base-to-apex variation by genotype. Specifically, while there was no difference in arterial density between the basal territories of wildtype and knockout ventricles (Fig. 6g—base), there was a significant (p <0.01) reduction in density in the apical territory of the knockout.
To determine whether loss of small resistance vessels in the knockout ventricle had effects on function of the vascular bed, we undertook a bead perfusion study. Fig. 7a and b shows isolated wildtype and knockout hearts from 4-week-old mice that were subject to retrograde perfusion of the aorta with fluorescent 15 μm beads under physiological conditions. A base-to-apex gradient in bead distribution can be discerned in the knockout (Fig. 7b). Overall, the quantitative analysis (Fig. 7c) indicated that the density of beads in the ventricles of knockouts was significantly lower than that of wildtype animals (p <0.05). Consistent with the pattern of loss of small resistance vessels, knockouts displayed a significant reduction (p <0.01) in bead density in the apical half of the ventricle. Bead perfusion of the basal territory (Fig. 7c) also appeared to be decreased in the knockout, though this reduction did not reach significance in comparison to the wildtype. In conclusion, our data in indicates that the apical territory of the HF-1b knockout has reduced densities of small resistance vessels and decreases arterial perfusion relative to wildtype littermates.
Fig. 7.
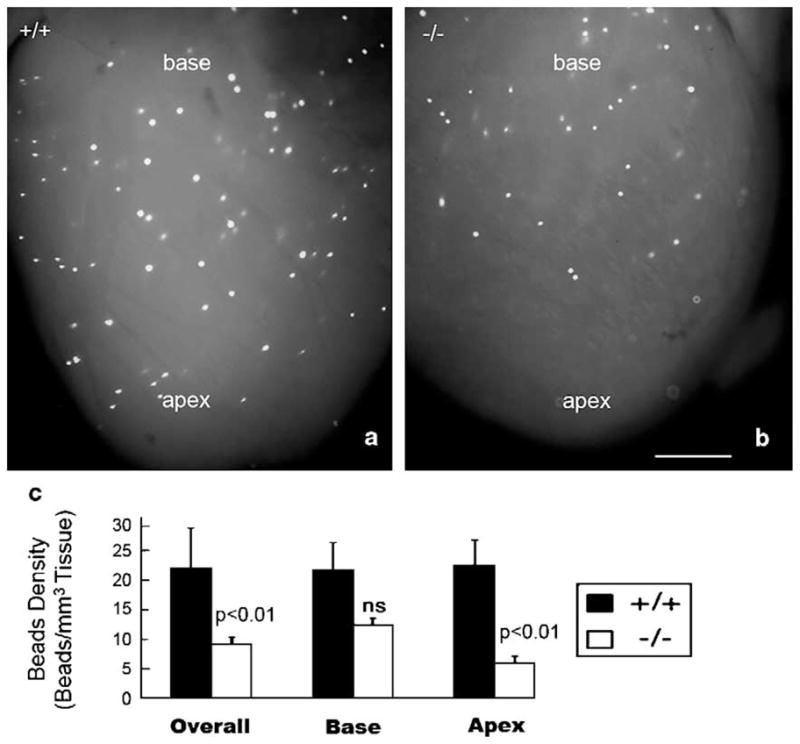
Injected fluorescent beads were distributed to a lesser level in the apical region of the HF-1b−/− heart when compared to the HF-1b+/+. Fluorescent beads (15 μm) were injected into the coronary circulation of six HF-1b−/− and six HF-1b+/+ hearts. (a) Typical bead distribution patterns of the wildtype (+/+) and (b) knockout hearts are seen in panels (a) and (b). Calibration bar=1 mm.
4. Discussion
In the present study, the sudden cardiac death phenotype of the HF-1b knockout mouse is shown to be associated with multiple defects to ventricular structure and electrical activation. The most novel aspect of the study is that these ventricular defects demonstrate a base-to-apex axial gradation in severity. The study also supports and extends the concept that the HF-1b gene has effects ion channel and connexin expression in both working myocytes and conduction cells [2,3]. The abnormal expressions of Cx40 and minK have been noted previously [3]. Here, we report that the distribution of Cx43 gap junctions (GJs) is also disturbed in the HF-1b knockout heart, with increases in Cx43 immunolocalized to the lateral aspects of ventricular myocytes. Moreover, deletion of the HF-1b gene is associated with reductions in Cx43 content—particularly in the apical territory of the ventricle. The “lateralized” distribution of Cx43 observed in knockout ventricle resembles that found in the normal atria and at immature stages of postnatal development [29,30]. Non-disk-localized GJs and decreased Cx43 are characteristic of myocytes comprising the conduction system [31]. The pattern of alteration to Cx43 expression and cellular distribution seen in the HF-1b null ventricle is thus consistent with the previously suggested hypothesis of a “confused” identity” of myocyte phenotype in this mouse model [3].
The regions of slow conduction found in the apex of the HF-1b knockout heart were resolved following pacing of the epicardial surface of the ventricular base. As such, this abnormality of activation is not explained by the conduction system—e.g., resulting from disruption to Cx40 expression in Purkinje fibers. While it remains to be shown that these sectors lead to the lethal disturbances of conduction characterizing the HF-1b knockout heart, slow conduction has been mathematically modeled as a key property of propagation arrhythmias [24]. Importantly, reduced coupling by gap junctions supports very slow conduction in such models. Spach and others have also demonstrated that gap junctional coupling and cell size are significant factors in modeling conduction velocity in the working ventricular myocardium [26,27]. In this study, we show that the apical sectors of slow conduction present in the HF-1b knockout ventricle precisely co-localize with ventricular regions showing profound reduction in Cx43. Myocyte size also demonstrates reductions in the ventricle of the null mouse that are more pronounced in the apical territory. These localized changes in Cx43 level and cell size, together with the increases action potential duration and dispersion reported previously for working ventricular myocytes from the knockout [3], appear sufficient to explain conduction abnormalities present in the ventricle of HF-1b knockout mice. Furthermore, it is likely that these abnormalities in working myocardial structure, activation and refractoriness will contribute to the lethal cardiac arrhythmias that develop in this mouse model during postnatal life.
The mechanistic bases of the multiple changes in structure and function that are observed in the HF-1b knockout heart remain an outstanding question. One explanation could be that certain of the myocardial abnormalities of the knockout develop as a secondary consequence of the loss of coronary arterial structure and function possibly due to diminished growth factor(s) [32]. Proper function of the vascular bed is vital for the perfusion of cardiac muscle. It is well-established that gap junctional Cx43 in ventricular myocardium is exquisitely sensitive to even small changes in ionic concentrations, oxygen and pH [33,34]. The compromised perfusion of the apical territory of the ventricle in the knockout could alter such chemical gradients and in turn, such alterations may have local impacts on levels of Cx43 and other genes involved in normal conduction of activation. With respect to the occurrence of coronary arterial abnormalities in the knockout, it may be significant that HF-1b is expressed in neural, as well as cardiac tissues. Neural crest cells migrating from the neural tube are known to have important affects on cardiac morphogenesis [35], including coronary arterial development [15,36,37]. Most pertinently, in an apparent parallel to the effect of deletion of the HF-1b gene in mouse, ablation of neural crest in chick results in a reduction in coronary arterial density that is particularly marked in the apical territory of the ventricle [15,38].
Expression of HF-1b by neural tissues points to alternate potential explanations of the knockout phenotype. Coronary arteries are rich in intrinsic nerves and these nerves would presumably be attenuated in the knockout apex in association with the loss of arterial resistance vessels [39]. Additionally, this endogenous base–apex variation in norepinephrine is likely to be accentuated in the HF-1b knockout by ischemia in the apical region [40]. Furthermore, it is well understood that de facto denervation along with myocardial injury from diabetes is highly arrhythmogenic [41]. Vascular and neural trophic factors are expressed by cardiac cells and the loss of HF-1b function may attenuate the expression of such factors having wide range of biological actions [42,43]. It should also be considered that the loss of resistance vessels and their associated nerves in the knockout ventricle might be the primary defect in the myocardium rather than vice versa.
We conclude the HF-1b knockout mouse displays several defects in ventricular structure and function that may contribute to the development of lethal arrhythmias in this model. The ontogeny of these defects are likely to be multifactorial, with some of the contributing factors having possible cell autonomous origins in the ventricular myocardium, whilst others potentially result from disrupted function of cardiac neural and/or vascular tissues. There may also be complex interactions between myocardial and non-myocardial aspects of the phenotype that contribute to the eventual development of arrhythmias. Consequently, future work would best be directed at genetically dissecting the multiple input to the phenotype by undertaking tissue-specific knockout of HF-1b independently in ventricular myocardial and neural tissues.
Acknowledgments
This work was supported by NIH HL56728, HD39946, HL 36059 (to RGG), RR16434 (to DS) and HL-63714 (to SWK). The authors wish to thank Dr. Kenneth R. Chien for generously providing the HF-1b mouse for these experiments. The assistance of Jane Jourdan and Nan Fox are acknowledged with gratitude.
References
- 1.Zipes DP, Wellens HJ. Sudden cardiac death. Circulation. 1998;98:2334–51. doi: 10.1161/01.cir.98.21.2334. [DOI] [PubMed] [Google Scholar]
- 2.Chien KR. Genomic circuits and the integrative biology of cardiac diseases. Nature. 2000;407:227–32. doi: 10.1038/35025196. [DOI] [PubMed] [Google Scholar]
- 3.Nguyen-Tran VT, Kubalak SW, Minamisawa S, Fiset C, Wollert KC, Brown AB, et al. A novel genetic pathway for sudden cardiac death via defects in the transition between ventricular and conduction system cell lineages. Cell. 2000;102:671–82. doi: 10.1016/s0092-8674(00)00089-1. [DOI] [PubMed] [Google Scholar]
- 4.Supp DM, Witte DP, Branford WW, Smith EP, Potter SS. Sp4, a member of the Sp1-family of zinc finger transcription factors, is required for normal murine growth, viability, and male fertility. Dev Biol. 1996;176:284–99. doi: 10.1006/dbio.1996.0134. [DOI] [PubMed] [Google Scholar]
- 5.Miquerol L, Meysen S, Mangoni M, Bois P, van Rijen HV, Abran P, et al. Architectural and functional asymmetry of the His–Purkinje system of the murine heart. Cardiovasc Res. 2004;63:77–86. doi: 10.1016/j.cardiores.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 6.Litchenberg WH, Norman LW, Holwell AK, Martin KL, Hewett KW, Gourdie RG. The rate and anisotropy of impulse propagation in the postnatal terminal crest are correlated with remodeling of Cx43 gap junction pattern. Cardiovasc Res. 2000;45:379–87. doi: 10.1016/s0008-6363(99)00363-6. [DOI] [PubMed] [Google Scholar]
- 7.Coppen SR, Dupont E, Rothery S, Severs NJ. Connexin45 expression is preferentially associated with the ventricular conduction system in mouse and rat heart. Circ Res. 1998;82:232–43. doi: 10.1161/01.res.82.2.232. [DOI] [PubMed] [Google Scholar]
- 8.Severs NJ, Rothery S, Dupont E, Coppen SR, Yeh HI, Ko YS, et al. Immunocytochemical analysis of connexin expression in the healthy and diseased cardiovascular system. Microsc Res Tech. 2001;52:301–22. doi: 10.1002/1097-0029(20010201)52:3<301::AID-JEMT1015>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 9.Gourdie RG, Green CR, Severs NJ. Gap junction distribution in adult mammalian myocardium revealed by an anti-peptide antibody and laser scanning confocal microscopy. J Cell Sci. 1991;99:41–55. doi: 10.1242/jcs.99.1.41. [DOI] [PubMed] [Google Scholar]
- 10.Angst BD, Khan LU, Severs NJ, Whitely K, Rothery S, Thompson RP, et al. Dissociated spatial patterning of gap junctions and cell adhesion junctions during postnatal differentiation of ventricular myocardium. Circ Res. 1997;80:88–94. doi: 10.1161/01.res.80.1.88. [DOI] [PubMed] [Google Scholar]
- 11.Saitongdee P, Milner P, Becker DL, Knight GE, Burnstock G. Increased connexin43 gap junction protein in hamster cardiomyocytes during cold acclimatization and hibernation. Cardiovasc Res. 2000;47:108–15. doi: 10.1016/s0008-6363(00)00051-1. [DOI] [PubMed] [Google Scholar]
- 12.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol. 2000;278:H1662–70. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 13.Green CR, Peters NS, Gourdie RG, Rothery S, Severs NJ. Validation of immunohistochemical quantification in confocal scanning laser microscopy: a comparative assessment of gap junction size with confocal and ultrastructural techniques. J Histochem Cytochem. 1993;41:1339–49. doi: 10.1177/41.9.8354875. [DOI] [PubMed] [Google Scholar]
- 14.Sedmera D, Reckova M, deAlmeida A, Sedmerova M, Biermann M, Volejnik J, et al. Functional and morphological evidence for a ventricular conduction system in zebrafish and Xenopus hearts. Am J Physiol. 2003;284:H1152–60. doi: 10.1152/ajpheart.00870.2002. [Erratum in Am J Physiol 285:H919] [DOI] [PubMed] [Google Scholar]
- 15.Hyer J, Johansen M, Prasad A, Wessels A, Kirby ML, Gourdie RG, et al. Induction of Purkinje fiber differentiation by coronary arterialization. Proc Natl Acad Sci U S A. 1999;96:13214–8. doi: 10.1073/pnas.96.23.13214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weibel ER. Stereological methods. I. London, United Kingdom: Academic Press; 1979. pp. 301–11. [Google Scholar]
- 17.Decking UK, Pai VM, Bennett E, Taylor JL, Fingas CD, Zanger K, et al. High-resolution imaging reveals a limit in spatial resolution of blood flow measurements by microspheres. Am J Physiol Heart Circ Physiol. 2004;287:H1132–40. doi: 10.1152/ajpheart.00119.2004. [DOI] [PubMed] [Google Scholar]
- 18.Morley GE, Vaidya D, Samie FH, Lo C, Delmar M, Jalife J. Characterization of conduction in the ventricles of normal and heterozygous Cx43 knockout mice using optical mapping. J Cardiovasc Electrophysiol. 1999;10:1361–75. doi: 10.1111/j.1540-8167.1999.tb00192.x. [DOI] [PubMed] [Google Scholar]
- 19.Hall CE, Hurtado R, Hewett KW, Shulimovich M, Poma CP, Reckova M, et al. Hemodynamic-dependent patterning of endothelin converting enzyme 1 expression and differentiation of impulse-conducting Purkinje fibers in the embryonic heart. Development. 2004;131:581–92. doi: 10.1242/dev.00947. [DOI] [PubMed] [Google Scholar]
- 20.Efimov IR, Nikolski VP, Salama G. Optical imaging of the heart. Circ Res. 2004;95:21–33. doi: 10.1161/01.RES.0000130529.18016.35. [DOI] [PubMed] [Google Scholar]
- 21.Akar FG, Spragg DD, Tunin RS, Kass DA, Tomaselli GF. Mechanisms underlying conduction slowing and arrhythmogenesis in non-ischemic dilated cardiomyopathy. Circ Res. 2004;95:717–25. doi: 10.1161/01.RES.0000144125.61927.1c. [DOI] [PubMed] [Google Scholar]
- 22.Delmar M. Gap junction remodeling in the failing heart: different connexins—different message? J Cardiovasc Electrophysiol. 2003;14:1213–4. doi: 10.1046/j.1540-8167.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 23.Patel PM, Plotnikov A, Kanagaratnam P, Shvilkin A, Sheehan CT, Xiong W, et al. Altering ventricular activation remodels gap junction distribution in canine heart. J Cardiovasc Electrophysiol. 2001;12:570–7. doi: 10.1046/j.1540-8167.2001.00570.x. [DOI] [PubMed] [Google Scholar]
- 24.Kleber AG, Rudy Y. Basic mechanisms of cardiac impulse propagation and associated arrhythmias. Physiol Rev. 2004;84:431–88. doi: 10.1152/physrev.00025.2003. [DOI] [PubMed] [Google Scholar]
- 25.Hodgkin AL. A note on conduction velocity. J Physiol (Lond) 1954;125:221–4. doi: 10.1113/jphysiol.1954.sp005152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spach MS, Heidlage JF, Dolber PC, Barr RC. Electrophysiological effects of remodeling cardiac gap junctions and cell size: experimental and model studies of normal cardiac growth. Circ Res. 2000;86:302–11. doi: 10.1161/01.res.86.3.302. [DOI] [PubMed] [Google Scholar]
- 27.Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193–7. doi: 10.1161/01.res.86.12.1193. [DOI] [PubMed] [Google Scholar]
- 28.Danik SB, Liu F, Zhang J, Suk HJ, Morley GE, Fishman GI, et al. Modulation of cardiac gap junction expression and arrhythmic susceptibility. Circ Res. 2004;95:1035–41. doi: 10.1161/01.RES.0000148664.33695.2a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fromaget C, El Aoumari A, Gros D. Distribution pattern of connexin 43, a gap junctional protein, during the differentiation of mouse heart myocytes. Differentiation. 1992;51:9–20. doi: 10.1111/j.1432-0436.1992.tb00675.x. [DOI] [PubMed] [Google Scholar]
- 30.Gourdie RG, Green CR, Severs NJ, Thompson RP. Immunolabelling patterns of gap junction connexins in the developing and mature rat heart. Anat Embryol. 1992;185:363–78. doi: 10.1007/BF00188548. [DOI] [PubMed] [Google Scholar]
- 31.Gourdie RG, Harris BS, Bond J, Justus C, Hewett KW, O’Brien TX, et al. Development of the cardiac pacemaking and conduction system. Birth Defects Res. 2003;69:46–57. doi: 10.1002/bdrc.10008. [Part C] [DOI] [PubMed] [Google Scholar]
- 32.Tomanek RJ, Zheng W. Role of growth factors in coronary morphogenesis. Texas Heart Inst J. 2002;29:250–4. [PMC free article] [PubMed] [Google Scholar]
- 33.Matsushita T, Takamatsu T. Ischaemia-induced temporal expression of connexin43 in rat heart. Virchows Arch. 1997;431:453–8. doi: 10.1007/s004280050123. [DOI] [PubMed] [Google Scholar]
- 34.Beardslee MA, Lerner DL, Tadros PN, Laing JG, Beyer EC, Yamada KA, et al. Dephosphorylation and intracellular redistribution of ventricular connexin43 during electrical uncoupling induced by ischemia. Circ Res. 2000;87:56–662. doi: 10.1161/01.res.87.8.656. [DOI] [PubMed] [Google Scholar]
- 35.Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res. 2003;69:2–13. doi: 10.1002/bdrc.10002. [Part C] [DOI] [PubMed] [Google Scholar]
- 36.Hood LC, Rosenquist TH. Coronary artery development in the chick: origin and deployment of smooth muscle cells, and the effects of neural crest ablation. Anat Rec. 1992;234:291–300. doi: 10.1002/ar.1092340215. [DOI] [PubMed] [Google Scholar]
- 37.Ewart JL, Cohen MF, Meyer RA, Huang GY, Wessels A, Gourdie RG, et al. Heart and neural tube defects in transgenic mice overexpressing the Cx43 gap junction gene. Development. 1997;124:1281–92. doi: 10.1242/dev.124.7.1281. [DOI] [PubMed] [Google Scholar]
- 38.Li WE, Waldo K, Linask KL, Chen T, Wessels A, Parmacek MS, et al. An essential role for connexin43 gap junctions in mouse coronary artery development. Development. 2002;129:2031–42. doi: 10.1242/dev.129.8.2031. [DOI] [PubMed] [Google Scholar]
- 39.Kawano H, Okada R, Yano K. Histological study on the distribution of autonomic nerves in the human heart. Heart Vessels. 2003;18:32–9. doi: 10.1007/s003800300005. [DOI] [PubMed] [Google Scholar]
- 40.Li W, Knowlton D, Van Winkle DM, Habecker BA. Infarction alters both the distribution and noradrenergic properties of cardiac sympathetic neurons. Am J Physiol Heart Circ Physiol. 2004;286:H2229–22236. doi: 10.1152/ajpheart.00768.2003. [DOI] [PubMed] [Google Scholar]
- 41.Verrier RL, Antzelevitch C. Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr Opin Cardiol. 2004;19:2–11. doi: 10.1097/00001573-200401000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarama M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247–53. doi: 10.1002/path.878. [DOI] [PubMed] [Google Scholar]
- 43.Tammela T, Enholm B, Alitalo K, Paavonen K. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65:550–63. doi: 10.1016/j.cardiores.2004.12.002. [DOI] [PubMed] [Google Scholar]