

Abstract
Recently, a new model for glutamate uptake by glutamate transporters was proposed based on crystal structures of the bacterial glutamate transporter homolog GltPh. It was proposed that hairpin two (HP2) functions as the extracellular gate and that Na+ and glutamate binding closes HP2, thereby allowing for the translocation of the glutamate binding pocket across the membrane. However, the conformation of HP2 in the apo state and the Na+ bound state is unknown. We here use double site-directed spin-labeling electron paramagnetic resonance spectroscopy on the bacterial transporter GltPh from Pyrococcus horikoshi to examine conformational changes in HP2. Surprisingly, the cotransported substrates Na+ and aspartate induce opposite movements of HP2. We find that in the apo state, HP2 is in a similar conformation as in the aspartate-bound closed state. Na+ binding to the apo state opens HP2, whereas the subsequent binding of aspartate closes HP2. Our findings show that Na+ binding opens and stabilizes the extracellular gate, thereby allowing for amino acid substrate binding. In contrast, in the absence of Na+ and aspartate, HP2 closes, suggesting a potential mechanism for the translocation of the empty binding pocket necessary to complete the transport cycle. The finding that physiological Na+ concentrations stabilize the open HP2 state would ensure that the outward-facing conformation of the transporter is maintained in physiological solutions and that glutamate transporters are ready to quickly bind glutamate released from glutamatergic synapses.
Introduction
Mammalian excitatory amino acid transporters (EAATs) are essential for terminating synaptic excitation and for maintaining extracellular glutamate concentrations below neurotoxic levels (Zerangue and Kavanaugh, 1996). EAATs couple neurotransmitter uptake to the cotransport and countertransport of Na+ and K+, respectively (Zerangue and Kavanaugh, 1996). How this is structurally accomplished by EAATs is unknown. Structures of outward-facing conformations of GltPh, a bacterial aspartate transporter and EAAT homolog, suggest that hairpin (HP) 2 functions as the extracellular gate (Yernool et al., 2004; Boudker et al., 2007), because differences in crystal structures obtained in the presence of the transportable substrate l-aspartate or the nontransportable inhibitor dl-threo-β-benzyloxyaspartic acid (dl-TBOA) were localized to the tip of HP2. Recently, Reyes and colleagues (2009) presented a new crystal structure of GltPh with a cysteine in HP2 cross-linked to a cysteine in transmembrane segment two in what is thought to be an inward-facing conformation of the transporter. A model was proposed in which the binding of Na+ and aspartate closes HP2, thereby allowing the binding pocket to be translocated across the membrane. In the apo state, in absence of Na+ and aspartate, it was hypothesized that the external gate, HP2, is open (Reyes et al., 2009). Here, we directly measure conformations of HP2 for a transporter in a membrane environment in a number of different states, some of which have not been previously determined by x-ray crystallography, to further determine the conformational changes underlying the complete transport cycle.
We used double site-directed spin-labeling (DSDSL) electron paramagnetic resonance (EPR) spectroscopy to detect conformational changes in HP2 induced by the binding of Na+ and by the binding of the substrate l-aspartate or inhibitor dl-TBOA. Importantly, there is no crystallography data for the Na+ bound state of the transporter, and inconclusive crystallography data for the apo state (Boudker et al., 2007), due to the fragility of crystals in the absence of Na+. Previous mutagenesis studies have shown that the formation of disulfide bonds between cysteines inserted in HP2 and surrounding domains is modulated by substrates and inhibitors. However, these studies could not unequivocally show that HP2 moves upon substrate binding (Brocke et al., 2002; Leighton et al., 2006; Qu and Kanner, 2008). Substrate binding may restrict access to these cysteines, cause movement of other parts of the protein, or alter bond angles between cysteine side chains, thereby preventing disulfide bond formations without altering distances between the cysteines. DSDSL-EPR provides us the ability to measure the extent and directionality of conformational changes in HP2 because two paramagnetic spins in close proximity interact via dipole–dipole interactions depending on the distance, r, between spins to the third power (r3) (Liu et al., 2001). Importantly, DSDSL-EPR experiments can be performed on transporters in a lipid bilayer. Our results suggest a mechanism by which Na+ transport is strictly coupled to glutamate uptake and suggest a novel role for Na+ binding at glutamatergic synapses.
Materials and Methods
Site-directed mutagenesis.
Site-directed mutagenesis was performed on GltPh from Pyrococcus horikoshi using the QuikChange Kit (Stratagene). The one endogenous cysteine at position 321 was replaced by an alanine. The double cysteine mutations were made in this cysteineless C321A background (called GltPh-A).
Expression and purification of GltPh.
GltPh (GltPh-A and GltPh-A cysteine-introduced mutants) was expressed as His7 fusion proteins using the pBCH/G4-7CATS vector and Escherichia coli DH5α or TOP10 cells. Proteins were purified essentially as described previously (Yernool et al., 2004), with the following modifications. Membranes were solubilized with N-dodecyl-β-d-maltopyranoside (C12M, final concentration 40 mm; Anatrace) at room temperature for 2 h with gentle agitation. After solubilization, the mixture was diluted to a total volume of ∼30 ml (C12M concentration of 10–15 mm) in buffer #10 (20 mm HEPES, pH 7.6, 0.2 m NaCl, and 5 mm glutamate) and centrifuged at 150,000 g for 1 h. Three milliliters of Ni-NTA superflow (Qiagen) was then added to the supernatant and the mixture was incubated at 4°C overnight. The following day, the Ni-NTA superflow was washed once with buffer #10 followed by protein purification on a BioLogic LP system (Bio-Rad). Buffer #10 containing 40 mm imidizole was used to wash the column to remove nonspecific protein binding, then pure GltPh was eluted with buffer #10 containing 200 mm imidizole (Sigma). Protein was either concentrated at this step using an Amicon Ultra concentrator (100 kDa cutoff; Fisher Scientific) and stored at −80°C until future use or immediately spin labeled, as described below.
Site-directed spin labeling protocol and isolation of trimeric transporter state.
Purified proteins were treated with 500 μm tris(2-carboxyethyl)phosphine (TCEP; Thermo Scientific) for 30 min at room temperature to reduce oxidized cysteines before being concentrated by an Amicon Ultra concentrator (100 kDa cutoff). Immediately following TCEP treatment, proteins were spin labeled with a 20:1 ratio of spin label:protein (mol/mol). Two separate labeling reactions were used to generate both maximally spin-labeled protein [100% (1-oxyl-2,2,5,5-tetramethyl-Æ3-pyrrolin-3-yl)methyl methanethiosulfonate (MTSL); Toronto Research Chemicals] and under-labeled protein (33% paramagnetic spin-label MTSL, 67% diamagnetic N-acetylated spin label analog; Toronto Research Chemicals) (Altenbach et al., 2001). Labeling was allowed to proceed for 30 min at room temperature before incubating overnight at 4°C. The following day, excess label was removed by FPLC (Superdex 200 10/300 GL; GE Healthcare), which simultaneously allowed the isolation of trimeric transporters and verified overall protein stability. All unlabeled and spin-labeled double mutants eluted in the same elution volume as sharp symmetrical peaks, superimposable on the elution peak from wild-type GltPh. This confirms that the mutants fold correctly and assemble as trimers. The extent of labeling was determined by whole mass electrospray mass spectrometry (OHSU Proteomics Shared Resource) and amino acid analysis (AAA Service Laboratory). We used amino acid analysis to obtain the precise concentration of spin-labeled protein in a given sample and compared the double-integrated EPR spectra from the sample and a linear dilution series of standard [MTSL and Cu(II)EDTA] to determine labeling efficiency.
GltPh proteoliposome preparation.
Pure protein was reconstituted into liposomes as previously described (Ryan and Mindell, 2007), with slight modifications. Briefly, a 3:1 ratio of Escherichia coli polar lipids and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids) was resuspended in either buffer (15 mm HEPES, pH 7.4, 100 mm NaCl, 1 mm MgCl2 and 2 mm KCl) containing 1 mm dl-TBOA (Tocris Bioscience) or 0.5 mm l-aspartate (Sigma), or buffer containing 100 mm Na+ or 100 mm K+ (or choline+) in the absence of any substrates. For protein reconstitution for EPR experiments, we typically used 4 mg of lipid for each condition and 250–500 μg of protein. Proteoliposomes were either concentrated by centrifugation at 135,000 g for 2 h at 4°C and resuspended in ∼ 8 μl of buffer containing either substrate or inhibitor, or subjected to several rounds of centrifugation and resuspension in Na+-free buffer after gramicidin-A incorporation (25 μg/ml) into liposomes to remove Na+, or resuspended in buffer containing 100 mm K+ (for uptake assays). Spin-labeled proteoliposomes were always used immediately for the EPR studies.
EPR spectroscopy.
Continuous-wave EPR spectra were obtained at room temperature on a Bruker E500 X-band EPR spectrometer equipped with a superX microwave bridge and an ER4123D dielectric resonator (Bruker Biospin). Approximately 4 μl of proteoliposome suspension at ∼100 μm were loaded into TPX capillaries (Bruker Biospin) and deoxygenated by continuous flushing of the cavity with N2. The microwave frequency was 9.78 GHz, the modulation frequency was 100 kHz, the modulation amplitude was 2 G, and the microwave power was 2 mW. Each spectrum corresponds to the accumulation of 10 scans.
EPR spectral analysis.
EPR spectra were baseline subtracted (first integration of spectra was zero at both extremes of spectra) and double integrated to obtain the number of spins present in each experiment. Each spectrum was normalized to the number of spins to enable comparison between under-labeled and fully labeled conditions (Altenbach et al., 2001). The ratios of the number of spins in under-labeled to fully labeled conditions were in the expected range of 0.3–0.4. Taking into account the measured labeling efficiency (70–100%), the single- and double-labeled spectra were calculated using simple linear algebra of the under-labeled and fully labeled spectra. In some cases, less than 1% of the EPR spectra was identified as coming from free label; this component was subtracted from the spectra. The single- and double-labeled spectra were deconvoluted to obtain the best fitted spin–spin distances and distance distributions using the program ShortDistances (Altenbach et al., 2001) run in LabView 2009 (National Instruments).
GltPh transport assay.
Frozen proteoliposomes were removed from −80°C and thawed on ice. For radioactive transport assays, proteoliposomes were previously loaded with internal buffer (15 mm HEPES, pH 7.4, 100 mm KCl, 0.8 mm MgCl2). The uptake reaction was initiated by diluting proteoliposomes into external buffer containing 100 mm NaCl, 2 mm KCl, 1 mm MgCl2, 5 mm HEPES, pH 7.4, and 400 nm l-14C-aspartate. Uptake was performed at 25°C for 6 min. Background was defined as the counts observed when the proteoliposomes were diluted into the buffer with which they were loaded.
SPA assay.
Cu2+ chelate YSi scintillation SPA beads (GE Healthcare) were used essentially as previously described (Quick and Javitch, 2007) and according to manufacturers instructions.
Results
Selection of GltPh residues for cysteine substitution
Using GltPh crystal structures (Yernool et al., 2004; Boudker et al., 2007), we selected pairs of residues that, when labeled with spin probes, would likely report on conformational changes measurable by DSDSL-EPR during HP2 movement. Specifically, in the crystal structure obtained in the presence of dl-TBOA, two residues on the tip of HP2 (Ala353 and Val355) are in close proximity with residues on TM4a (Pro129 and Leu130). However, in the crystal structure obtained in the presence of l-aspartate, these same two HP2 residues are located 6–10 Å further away from TM4a and are instead located closer to residues on HP1 (Ser279) (Fig. 1A,B). We used residues 353 or 355 at the tip of HP2 in all residue pairs, because these residues are predicted to move substantially and the side chains of these residues face away from the binding site (Fig. 1A,B). Other HP2 residues are predicted to either undergo minimal conformational changes during substrate binding or to face into the amino acid binding site (Fig. 1A,B).
Figure 1.

Strategy for measuring conformational changes with EPR. A, Models of GltPh crystal structures obtained in the presence of dl-TBOA and l-aspartate (ASP) (Boudker et al., 2007) showing the position of HP2 residues Val355 and Ala353 relative to TM4a residues Pro129 and Leu130. B, Models of GltPh crystal structures obtained in the presence of dl-TBOA and l-aspartate showing the position of HP2 residue Val355 relative to HP1 residue Ser279. A, B, Dashed lines indicate cysteine pairs that were used for EPR measurements. C, Distances between carbons for residue pairs 1, 2, and 3 in either dl-TBOA or l-aspartate in the outward-facing state (from A and B). D, Model of GltPh in the proposed inward-facing conformation (Reyes et al., 2009); residue pairs 353C–130C and 355C–129C are predicted to be too far from each other to detect spin–spin interactions. E, Structures of the paramagnetic (MTSL) and N-acetylated diamagnetic (Analog) labels used for EPR.
The two different structures of HP2 obtained in dl-TBOA and l-aspartate are referred to as the open and closed states of the extracellular gate, respectively (Boudker et al., 2007). To test these closed and open models of HP2, we generated three cysteine pairs using the residues mentioned above in a GltPh background where the only native cysteine (C321) was replaced with alanine. Distances between β-carbon atoms in the dl-TBOA-bound versus l-aspartate-bound states for these three cysteine pairs are in the range where spin labels attached to these residues would experience spin–spin interactions (Fig. 1C). In contrast, in the recently published inward state of GltPh (Reyes et al., 2009), none of these residue pairs (except 279–355) are close enough to experience spin–spin interactions (Fig. 1D).
Our strategy to measure conformational changes using DSDSL-EPR was to obtain EPR spectra for each cysteine pair in maximally labeled protein (two spin labels per pair of residues) and under-labeled protein (approximately one spin label per pair of residues). To ensure that maximally labeled and under-labeled proteins represented the same conformations, we used a mixture of 67% diamagnetic (N-acetylated MTSL analog) to 33% paramagnetic (MTSL) label for the under-labeled condition (Gross et al., 1999; Altenbach et al., 2001) (Fig. 1E). Spin–spin interactions were then calculated by deconvolution of EPR spectra measured from under-labeled and maximally labeled protein, normalized to the same number of spins (Altenbach et al., 2001). For interspin separations in the 8–20 Å range, spin–spin interactions result in a quantifiable distance-dependent decrease in the amplitude of the central peak and an overall broadening of the spectral line shape as spins come closer to one another (Altenbach et al., 2001).
Using protocols previously developed (Yernool et al., 2004), we obtained highly purified protein (Fig. 2A). The purified protein was labeled with the probes (100% MTSL or 67% N-acetylated MTSL analog and 33% MTSL) and then repurified using size-exclusion chromatography (Fig. 2B) to both isolate the trimeric protein and to remove any excess un-reacted label. No EPR signal was detected from cysteineless GltPh-A (Fig. 2C), having been exposed to the same labeling protocol as that of the double mutants. This verifies the purity of our proteins and shows the lack of any spin label contamination from other cysteines or from free un-reacted spin label. The labeling efficiency of our mutant proteins was determined by measuring the mass of labeled and unlabeled protein by mass spectrometry (Fig. 2D) and by amino acid analysis (see Materials and Methods, above). The maximum labeling efficiency ranged from 70 to 100% and was taken into consideration in the quantitative analysis of the EPR spectra (see Materials and Methods, above).
Figure 2.

Protein purification and spin-labeling efficiency. A, Representative example of SDS-PAGE analysis showing His-tagged GltPh purified using Ni-NTA superflow. Denaturing conditions result in the detection of GltPh monomer by Coomassie blue staining. B, Representative example of gel-filtration chromatography of GltPh used to simultaneously isolate functional trimeric transporters and remove excess spin label. C, No EPR signal is detected from the cysteineless mutant GltPh-A subjected to the same spin-labeling conditions as GltPh-A-353C–130C. EPR measurements were performed on purified, labeled proteins in N-dodecyl-β-d-maltopyranoside. D, Electrospray mass spectrometry data showing a characteristic 372 dalton shift in molecular weight indicating fully labeled double-mutant protein. Norm., Normalized.
Structural and functional properties of spin-labeled GltPh mutants
We next examined the functional properties of GltPh-A (cys-less) as well as the single cysteine mutants GltPh-A-353 and GltPh-A-355 using radioactive uptake assays on proteins incorporated into preformed liposomes. The single mutants were used in these uptake assays because either residues 353 or 355 were to be used in all EPR experiments. Therefore, the functionality of the transporter spin labeled at these residues was first examined. Although substantial transport was observed in the cys-less mutant (GltPh-A), spin-labeled GltPh-A-353 and GltPh-A-355 did not show any l-aspartate uptake over background levels (Fig. 3A). To ensure that the cysteine mutations themselves did not disrupt the structural integrity of the proteins, we examined uptake in all three of our unlabeled double cysteine mutants (Fig. 3B). Substantial uptake was observed for all three double-mutant proteins, indicating the functionality of the double-mutant proteins. Previous studies on mammalian transporters have shown that mutating and/or labeling of cysteines introduced in HP2 results in transporters that do not show significant uptake of radioactive substrates (Seal et al., 2001; Borre et al., 2002; Grunewald et al., 2002; Ryan and Vandenberg, 2002). However, these studies also demonstrated that the mutating and/or labeling of cysteines introduced into HP2 does not prevent substrate binding or extracellular conformational changes in mammalian transporters. In the new crystal structure in the presumed inward-facing state, residues 353 and 355 are buried inside the protein (Fig. 1D). Spin labels attached to 353 or 355 might sterically prevent the conformational change to the inward-facing state, which might explain the absence of aspartate transport when these residues are spin labeled. However, to minimize possible interference with the closing and opening of HP2 in the outward-facing conformations, we chose residue pairs that, in the two crystal structures of the outward-facing state, are facing away from the protein (Fig. 1A,B).
Figure 3.

Spin-labeling HP2 residues disrupts transport but not substrate and inhibitor binding. A, 14C-l-aspartic acid uptake in the presence of an inwardly directed Na+ gradient remains intact in GltPh-A proteoliposomes (squares), whereas spin labeling GltPh-A-353C (circles) and GltPh-A-355C (upward triangles) reduces uptake to background levels (downward triangles). Background was defined as the counts observed in the absence of a Na+ gradient (GltPh-A, 100 mm K+ on both sides of the membrane). B, 14C-l-aspartic acid uptake in the presence of an inwardly directed Na+ gradient remains intact in all three nonlabeled double-mutant proteins. Data are expressed as the percentage of maximum GltPh-A uptake to indicate the relative effect of the double cysteine mutations on uptake. All data points are background subtracted (uptake with 100 mm K+ on both sides of the membrane). GltPh-A-355C-279C (triangles), GltPh-A-353C-130C (squares), GltPh-A-355C-129C (circles). C, Scintillation proximity assay reveals 3H-l-aspartic acid binding and competitive inhibition by dl-TBOA remains intact in spin-labeled double-mutant transporters. Double mutant 355C–279C is shown as an example (n = 3).
Using a scintillation proximity assay (Quick and Javitch, 2007), we verified that substrate and inhibitor binding remained intact in spin-labeled transporters (Fig. 3C). Note that, due to the estimated very high l-aspartate affinity (nm) of detergent solubilized GltPh in the presence of 100 mm Na+ (Boudker et al., 2007), it is not possible to directly measure the aspartate affinity with a scintillation proximity assay (or any other biochemical assay that requires micromolar concentration of protein), but it is only possible to show that l-aspartate binding occurs and that dl-TBOA can compete off l-aspartate (Fig. 3C). Due to the instability of detergent solubilized GltPh in the absence of Na+ (Boudker et al., 2007), Na+ binding was not tested with the scintillation proximity assay. However, Na+ binding (and l-aspartate and dl-TBOA binding) to GltPh incorporated in liposomes was further confirmed by line shape changes in the EPR spectra induced by these substrates (see below). In this study, we use to our advantage the fact that the spin-labeled transporters do not cycle continuously through the transport cycle in the presence of substrates. Because spin labeling did not abolish substrate/inhibitor binding, but prevented continuous transporter cycling, we were able to monitor in isolation the separate conformational changes of opening and closing of HP2 induced by the binding of Na+, l-aspartate, and dl-TBOA in the outward-facing conformations of GltPh.
Opposite GltPh HP2 movements in DL-TBOA compared with l-aspartate
We initially chose to measure spin–spin interactions under two conditions used for the published outward-facing crystal structures (Yernool et al., 2004; Boudker et al., 2007) in the presence of either inhibitor (dl-TBOA) or substrate (l-aspartate). GltPh protein has been reported to incorporate equally effectively into liposomes with the cytosolic part of the protein facing the outside or the inside of the liposome (Ryan et al., 2009). Therefore, to achieve a homogenous population of a specific conformational state for the EPR measurements, we reconstituted purified, labeled protein into preformed liposomes in the presence of either l-aspartate or dl-TBOA on both sides of the liposome membrane (Fig. 4).
Figure 4.

Experimental strategy to generate fully labeled and under-labeled proteoliposomes in the presence of dl-TBOA and l-aspartate (ASP). A–C, Double-cysteine mutant GltPh transporters were purified (A), labeled with MTSL or a mix of N-acetylated analog and MTSL (B), and repurified by size-exclusion FPLC (C). D, E, The spin-labeled GltPh protein was analyzed by mass spectroscopy to verify labeling efficiency (D) and reconstituted into liposomes in the presence of dl-TBOA or l-aspartate (E).
EPR spectra showed evidence of dipole–dipole interactions when comparing EPR spectra in under-labeled and maximally labeled transporters for all cysteine pairs (Fig. 5). This finding supports our assumption that the spin-labeled protein is in its outward-facing state because in the inward-facing state, the cysteines in two of the three cysteine pairs are not predicted to be close enough to each other for spin labels to experience dipole–dipole interactions (Fig. 1D). Comparing EPR spectra in under-labeled and maximally labeled transporters, residue pair 1 (V355C-S279C) on HP2 and HP1 displayed increased central peak heights in dl-TBOA compared with l-aspartate (Fig. 5). In contrast, a decrease in central peak height was observed in dl-TBOA compared with l-aspartate for residue pairs 2 and 3 (V355C-P129C and A353C-L130C, respectively) located on the tip of HP2 and TM4a (Fig. 5). Quantifying these changes using deconvolution of the EPR spectra (see Materials and Methods, above) (Altenbach et al., 2001) shows that spin labels are further apart in dl-TBOA than in l-aspartate for residue pair 1 (V355C-S279C), whereas for residue pairs 2 and 3 (A353C-L130C and V355C-P129C), spin labels are closer in dl-TBOA than in l-aspartate (Table 1). The pattern of changes in the spin–spin distances for all three pairs is best explained by a relative movement of the tip of HP2 away from TM4a and toward HP1 upon l-aspartate binding compared with the dl-TBOA-bound state.
Figure 5.
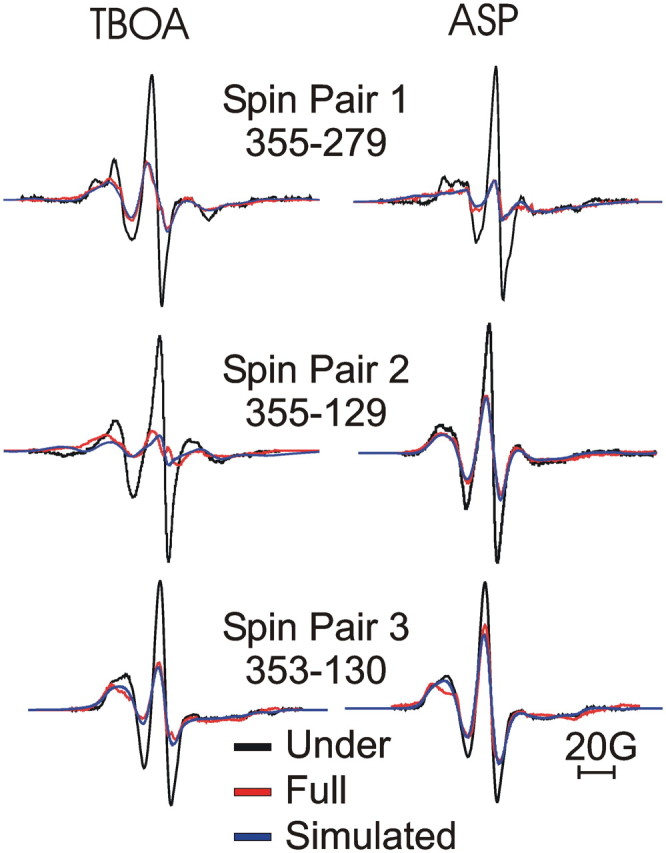
Changes in spin–spin interactions show movement of HP2. EPR spectra in the presence of dl-TBOA (left) and l-aspartate (ASP; right) for single-labeled (black lines) and double-labeled (red lines) cysteine pairs, normalized to the same number of spins (see Materials and Methods). Deconvolution of the single- and double-labeled spectra was used to obtain a best-fit simulated line shape (blue lines) to quantify the spin–spin interactions (see Materials and Methods) (Table 1).
Table 1.
Spin–spin distances in aspartate and TBOA
| Pair | Mutants | TBOA | Asp |
|---|---|---|---|
| 1 | 355-279 | 16.0 ± 0.07 (15.6–16.4) | 8.9 ± 0.01 (8.3–9.7) |
| 2 | 355-129 | 8.6 ± 0.03 (8.0–9.2) | 18.6 ± 0.04 (17.9–19.3) |
| 3 | 353-130 | 12.5 ± 0.05 (11.5–13.5) | 14.9 ± 1.0 (13.0–16.8) |
Due to the mobility of the spin probes, the deconvolution results in a distribution of distances between the two spin labels. Therefore, spin–spin distances (in Å) are given as the center of the distance distributions with errors and the width of the distance distributions (in parentheses). The data was obtained by deconvolution of the single- and double-labeled spectra in Figure 5 (see Materials and Methods). Asp, l-aspartate.
HP2 movement upon Na+-binding to the apo state in GltPh
We next examined the pattern of dipole–dipole interaction between spin-labeled double mutants in the apo state (Na+- and l-aspartate-free) and in the presence of Na+ (l-aspartate-free). Spin-labeled proteins in detergent buffer containing 100 mm Na+ were incorporated into liposomes preloaded with buffer containing 100 mm Na+. The use of Na+-containing buffers served to ensure the stability of the trimeric protein during incorporation into liposomes (Boudker et al., 2007). Once the protein was in the more native environment of the liposomes, gramicidin A was incorporated into the liposomes and the 100 mm Na+ buffer was exchanged through several rounds of centrifugation and resuspension of liposomes in a 100 mm K+ buffer.
EPR spectra were recorded in this K+ buffer, which ensures that a majority of the transporters are in the apo state. Note that K+ does not interact with the bacterial GltPh transporters, in contrast to the mammalian EAATs (Ryan and Mindell, 2007; Ryan et al., 2009). We estimate that the free Na+ concentration is <10 μm after this buffer exchange, whereas the sodium affinity for GltPh in a lipid environment is estimated at 2–4 mm (Ryan et al., 2009). Na+ was then added to the samples at a final concentration of 100 mm and EPR spectra were recorded in the presence of Na+ but with no added l-aspartate. We estimate that the free l-aspartate concentration is negligible, as the protein is purified in the presence of 5 mm glutamate and l-glutamate concentrations are reduced to ≪1 μm after reconstitution and buffer exchange, whereas the l-aspartate and l-glutamate affinities for GltPh in a lipid environment is estimated at 0.12 and 100 μm, respectively (Boudker et al., 2007; Ryan et al., 2009), in the presence of 100 mm Na+. After the spectrum in Na+ was recorded, a spectrum in the presence of l-aspartate was recorded (Fig. 6). The line shapes of the EPR signal from under-labeled transporters were different in the absence of Na+ compared with in the presence of Na+, showing that Na+ induces a conformational change of the protein (Fig. 6A). The addition of l-aspartate further changed the lines shapes, showing that l-aspartate induces a different conformational change (Fig. 6A). Line shapes obtained in the presence of dl-TBOA (Fig. 5) are also shown in Figure 6A for further comparison. No change in central peak heights or line shapes were observed for any double mutant upon the addition of GABA to transporters in the presence of Na+ (Fig. 6B), demonstrating that the conformational changes induced by l-aspartate are specific for ligands that interact with the transporter. Additionally, central peak heights and line shapes were identical in the presence of K+ or choline+ in both under-labeled and maximally labeled transporters (Fig. 6C), further supporting the theory that K+ does not interact with GltPh (Ryan and Mindell, 2007; Ryan et al., 2009) and that the transporter is in the apo state in the presence of K+.
Figure 6.
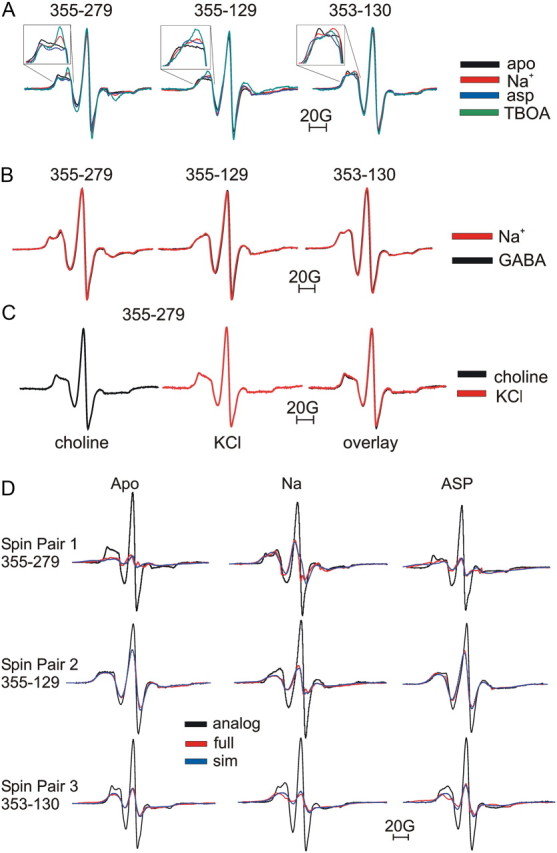
HP2 movement toward TM4 upon Na+-binding to the apo state. A, EPR line shapes obtained with under-labeled transporters in the apo (black), Na+-bound (red), aspartate-bound (asp; blue), or dl-TBOA-bound (green) states. Insets are shown to emphasize the differences in line shapes under different conditions. B, EPR line shapes obtained with under-labeled transporters in the Na+-bound state (red) and following the addition of GABA (black) to Na+-bound transporters. C, EPR line shapes obtained with under-labeled transporters in the presence of choline+ (absence of K+ and Na+) and after addition of 100 mm K+. Line shapes obtained in choline+ (black) and K+ (red) are overlaid for comparison. D, EPR spectra in the apo, Na+-bound, and l-aspartate-bound states for single-labeled (black lines) and double-labeled (red lines) cysteine pairs, normalized to the same number of spins (see Materials and Methods). Deconvolution of the single- and double-labeled spectra was used to obtain a best-fit simulated line shape (sim; blue lines) to quantify the spin–spin interactions (see Materials and Methods) (Table 2).
Comparing all EPR spectra in under-labeled and maximally labeled transporters recorded under the apo, Na+-bound, and Na+ plus l-aspartate condition, a characteristic pattern emerged (Fig. 6D, Table 2). Residue pair 1 (355-279) displayed an increased central peak height in the presence of Na+ compared with the apo state (Fig. 6D). For residue pair 1, central peak height was substantially decreased compared with Na+ upon the addition of l-aspartate. Quantifying these changes using deconvolution of the EPR spectra shows that spin labels for residue pair 1 are further apart in the presence of Na+ compared with the apo and l-aspartate-bound states (Table 2). Residue pairs 2 and 3 (355-129 and 353-130, respectively) revealed the opposite pattern of interaction (Fig. 6D). Quantifying these changes using deconvolution shows that for residue pairs 2 and 3 (355-129 and 353-130, respectively), spins are closer in Na+ and further apart in the l-aspartate-bound and the apo states (Table 2). In Table 2, distances obtained in the presence of dl-TBOA (Fig. 5, Table 1) are included to show the comparison between Na+-bound and dl-TBOA-bound states. For each of the three residue pairs, both the distance estimates (Table 2) and the line shapes (Fig. 6A) were similar (but not identical) in dl-TBOA and Na+, as if the spin pairs occupy similar positions in dl-TBOA and Na+. In addition, for each of the three residue pairs, both the distance estimates (Table 2) and the line shapes (Fig. 6A) were similar (but not identical) in aspartate and the apo states, as if the spin pairs occupy similar positions in aspartate and apo states.
Table 2.
Spin–spin distances in apo, Na+, and in aspartate- and TBOA-bound states
| Pair | Mutants | Apo | Na+ | Asp | TBOA |
|---|---|---|---|---|---|
| 1 | 355-279 | 8.6 ± 0.04 (8.0–9.2) | 15.8 ± 0.06 (13.8–17.8) | 9.0 ± 0.1 (8.3–9.7) | 16.0 ± 0.07 (15.6–16.4) |
| 2 | 355-129 | 18.5 ± 0.01 (16.9–20.1) | 9.0 ± 0.03 (8.0–10.0) | 18.9 ± 0.05 (16.1–21.7) | 8.6 ± 0.03 (8.0–9.2) |
| 3 | 353-130 | 14.8 ± 0.04 (12.3–17.4) | 12.5 ± 0.07 (9.6–15.4) | 15.7 ± 1.0 (13.7–17.8) | 12.5 ± 0.05 (11.5–13.5) |
Spin–spin distances (in Å) given as the center of the distance distributions with errors and the width of the distance distributions (in parentheses). The data was obtained by deconvolution of the single- and double-labeled spectra in Figure 6 (see Materials and Methods). Asp, l-aspartate.
Discussion
The spin–spin interaction data and the line shapes from the apo, Na+-bound, dl-TBOA-bound, and l-aspartate-bound states led us to the following conclusions about the movement of HP2 in the bacterial GltPh transporter (Fig. 7). In the apo state, HP2 adopts a conformation more similar to that of the l-aspartate-bound closed HP2 state than the dl-TBOA-bound open HP2 state (Fig. 7A). Upon the binding of Na+ to the apo state of the transporter, HP2 undergoes a movement that brings the tip of HP2 further away from HP1 and into closer proximity with TM4a (Fig. 7A). This Na+-bound state resembles the dl-TBOA-bound open HP2 state (Fig. 7A). Finally, upon binding of l-aspartate to the Na+-bound state of the transporter, HP2 undergoes a movement that brings the tip of HP2 away from TM4a and into closer proximity with HP1, thereby closing the HP2 gate (Fig. 7A). Together, our EPR data show that Na+ and l-aspartate induce opposite movements of HP2 in the bacterial GltPh.
Figure 7.
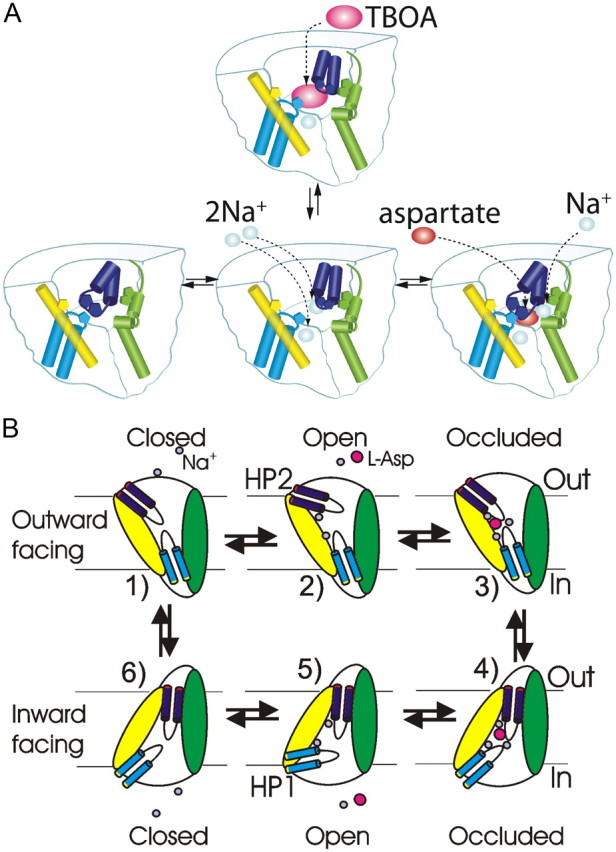
Model of HP2 positions illustrated for a single subunit. A, In the apo state, HP2 (dark blue) adopts a closed conformation, similar to the l-aspartate (asp)-bound state. The binding of Na+ to the apo state stabilizes HP2 in an open state, similar to that of the dl-TBOA-bound state. Green, TM4; yellow, TM2; light blue, HP1. B, Model of the whole transport cycle based on our data and the recent crystal structure of GltPh in the inward-facing conformation (Reyes et al., 2009). 1, In the outward-facing apo state, HP2 is closed. 2, The binding of Na+ stabilizes the open state of HP2 and creates the binding site for aspartate. 3, 4, Binding of aspartate and the last Na+ closes the HP2 gate (3), allowing for the translocation of the whole binding pocket according to the recent crystal structure (4) (Reyes et al., 2009). 5, 6, It is assumed that the subsequent intracellular release of aspartate and Na+ is accomplished by the reversed conformational changes of HP1 compared with the conformational changes of HP2 during the binding of aspartate and Na+ (1–3). In the inward-facing apo state, we assume that both HP1 and HP2 are closed, as in the outward-facing apo state. This would allow for the translocation of the empty binding pocket back to the outward-facing conformation by a similar mechanism as for the forward translocation of the substrate-bound binding pocket.
Note that our measurements only show movement of HP2 relative to HP1 and TM4a. Another possible interpretation of our data would be that HP1 and TM4 move in concert relative to HP2 (e.g., in dl-TBOA, HP1 moves away from HP2 and TM4a moves toward HP2). However, due to the excellent agreement of our data in aspartate and dl-TBOA with the two available crystal structures in aspartate and dl-TBOA (Yernool et al., 2004; Boudker et al., 2007), we favor the interpretation that it is HP2 that moves relative to the comparatively static HP1 and TM4a (although small movements of HP1 and/ or TM4a could contribute to the distance changes measured).
In the apo state, our EPR data suggest that the position of HP2 is similar to the aspartate-bound state. Previous attempts to identify the apo state using crystallography were inconclusive (Boudker et al., 2007). Boudker et al. (2007) prepared Na+ and substrate-depleted crystals and examined threefold averaged electron density maps corresponding to the HP2 region. They found that HP2 could reside in an open conformation, essentially indistinguishable from that seen in the dl-TBOA-bound state. In unaveraged maps, however, they reported that subunits C and A showed density for HP2 in a more closed conformation. Our EPR analysis clearly suggests that in the apo state, HP2 resides in a position more closely resembling the l-aspartate-bound HP2 closed state (Fig. 6). We suggest that this HP2 conformation in the apo state might restrict access for l-aspartate, but not for Na+, to the binding pocket or distort the binding pocket so that l-aspartate cannot bind until Na+ has bound. Previous crystal structures of GltPh in which both substrate and ions were bound were deemed occluded by showing that the substrate was largely inaccessible to a 1.4 Å radius probe (Gouaux, 2009). Therefore, whereas HP2 in the substrate-bound occluded state effectively closes off extracellular access to bound substrate, a similar position of HP2 in the apo state would allow binding-site access to Na+ (crystal radius = 1.0 Å) while restricting access for the much larger l-aspartate. Consistent with this hypothesis, it has previously been shown by fluorescence and current measurements that aspartate/glutamate cannot bind to bacterial GltPh or mammalian EAATs transporters in the absence of Na+ (Kanner and Bendahan, 1982; Larsson et al., 2004; Boudker et al., 2007; Ryan et al., 2009).
In the Na+-bound state (Fig. 7A), another conformation of which there is no full crystal structures available (Boudker et al., 2007), our EPR data suggest a movement of HP2 that brings residues on HP2 (353 and 355) closer in proximity to residues on TM4a (129 and 130). The addition of Na+ to the apo state of the transporter resulted in increased interaction and decreased distance between spins for residue pairs 2 and 3 (353-130 and 355-129, respectively), but decreased interaction and increased distance between spins for residue pair 1 (355-279) (Fig. 6, Table 2). Our EPR analysis clearly suggests that, in the presence of Na+, HP2 resides in a position resembling the dl-TBOA-bound HP2 open state (Fig. 6, Table 2).
What roles do the different conformations of HP2 play in the function of the transporter? First, in a recent glutamate transporter model based on crystal structures of GltPh in the outward- and inward-facing conformations (Reyes et al., 2009), it is assumed that HP2 gate is open in the apo state and that binding of Na+ and aspartate closes the HP2 gate to allow for the translocation of the substrate-bound binding pocket to an inward-facing conformation. As pointed out by Reyes and colleagues (2009), this model with the HP2 gate open in the apo state does not explain how the transporter can reorient between the outward- and inward-facing conformations in the absence of substrate (apo state), which is necessary to complete the whole transporter cycle during substrate uptake or reverse transport. However, our finding that the HP2 gate adopts a more closed conformation in the apo state would allow for the empty binding pocket to be translocated across the membrane by a similar mechanism as the translocation of the substrate-bound binding pocket, thereby completing the whole transport cycle in GltPh in either uptake mode or reverse transport mode (Fig. 7B).
Second, upon the binding of Na+ to the apo state of the transporter, our EPR data on GltPh suggests that the tip of HP2 undergoes a movement that would open up the binding pocket, thereby allowing for amino acid substrate binding. Consistent with this, we recently have shown evidence that the backbone oxygen of residue 353 at the tip of HP2 actually contributes directly to one of the Na+ binding sites in the apo state (Larsson et al., 2010). Computer modeling further suggests that Na+ bound to this Na+-binding site contributes directly to the aspartate-binding site (Larsson et al., 2010). Additionally, in a recent study of the nonrelated neurotransmitter transporter homolog LeuT, it was also concluded that Na+ binding opens up the binding site to the extracellular solution (Claxton et al., 2010), suggesting that the coupling of Na+ to the opening of the binding site is a common theme among Na+-driven transporters. Together, these data support a model whereby a cation-specific conformational change of HP2 creates and stabilizes the substrate-binding site. This in turn could explain the strict coupling of Na+ transport to uptake of glutamate (Menaker et al., 2006). In addition, the stabilizing of the open HP2 state would ensure that the outward-facing conformation of the transporter is maintained in physiological solutions and ensure that the transporter is ready to quickly bind extracellular glutamate released from glutamatergic synapses.
Using DSDSL-EPR spectroscopy on the bacterial glutamate transporter homolog GltPh, we have expanded on the available crystal structures by showing that, in the apo state, the HP2 gate is in a conformation similar to a closed gate and that the initial binding of Na+ to the apo state results in a conformational change that opens the HP2 gate. Our results suggest that Na+ binding plays a role in stabilizing HP2 in an open to the outside conformation, thereby exposing the substrate binding site to the extracellular solution.
Footnotes
This work was supported by a National Research Service Award predoctoral fellowship (P.F.) and by the National Institutes of Health (NIH) Grants R01-NS051169 and R01-HL095920 (H. P. L.). We thank E. Gouaux for helpful experimental insight and R. Duvoisin, A. Tzingounis, D. Farrens, J. Adelman, and C. Jahr for constructive criticism. We also thank L. Brown, S. Singh, C. Piscitelli, and D. Ortiz for help with technical aspects of the project. Mass spectrometry was conducted at the OHSU Proteomics Shared Resource (funded by the Oregon Opportunity and NIH Grants 5P30CA069533 and 5P30EY010572).
References
- Altenbach C, Oh KJ, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- Borre L, Kavanaugh MP, Kanner BI. Dynamic equilibrium between coupled and uncoupled modes of a neuronal glutamate transporter. J Biol Chem. 2002;277:13501–13507. doi: 10.1074/jbc.M110861200. [DOI] [PubMed] [Google Scholar]
- Boudker O, Ryan RM, Yernool D, Shimamoto K, Gouaux E. Coupling substrate and ion binding to extracellular gate of a sodium-dependent aspartate transporter. Nature. 2007;445:387–393. doi: 10.1038/nature05455. [DOI] [PubMed] [Google Scholar]
- Brocke L, Bendahan A, Grunewald M, Kanner BI. Proximity of two oppositely oriented reentrant loops in the glutamate transporter GLT-1 identified by paired cysteine mutagenesis. J Biol Chem. 2002;277:3985–3992. doi: 10.1074/jbc.M107735200. [DOI] [PubMed] [Google Scholar]
- Claxton DP, Quick M, Shi L, de Carvalho FD, Weinstein H, Javitch JA, McHaourab HS. Ion/substrate-dependent conformational dynamics of a bacterial homolog of neurotransmitter:sodium symporters. Nat Struct Mol Biol. 2010;17:822–829. doi: 10.1038/nsmb.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouaux E. Review. The molecular logic of sodium-coupled neurotransmitter transporters. Philos Trans R Soc Lond B Biol Sci. 2009;364:149–154. doi: 10.1098/rstb.2008.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross A, Columbus L, Hideg K, Altenbach C, Hubbell WL. Structure of the KcsA potassium channel from Streptomyces lividans: a site-directed spin labeling study of the second transmembrane segment. Biochemistry. 1999;38:10324–10335. doi: 10.1021/bi990856k. [DOI] [PubMed] [Google Scholar]
- Grunewald M, Menaker D, Kanner BI. Cysteine-scanning mutagenesis reveals a conformationally sensitive reentrant pore-loop in the glutamate transporter GLT-1. J Biol Chem. 2002;277:26074–26080. doi: 10.1074/jbc.M202248200. [DOI] [PubMed] [Google Scholar]
- Kanner BI, Bendahan A. Binding order of substrates to the sodium and potassium ion coupled L-glutamic acid transporter from rat brain. Biochemistry. 1982;21:6327–6330. doi: 10.1021/bi00267a044. [DOI] [PubMed] [Google Scholar]
- Larsson HP, Tzingounis AV, Koch HP, Kavanaugh MP. Fluorometric measurements of conformational changes in glutamate transporters. Proc Natl Acad Sci U S A. 2004;101:3951–3956. doi: 10.1073/pnas.0306737101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson HP, Wang X, Lev B, Baconguis I, Caplan DA, Vyleta NP, Koch HP, Diez-Sampedro A, Noskov SY. Evidence for a third sodium-binding site in glutamate transporters suggests an ion/substrate coupling model. Proc Natl Acad Sci U S A. 2010;107:13912–13917. doi: 10.1073/pnas.1006289107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton BH, Seal RP, Watts SD, Skyba MO, Amara SG. Structural rearrangements at the translocation pore of the human glutamate transporter, EAAT1. J Biol Chem. 2006;281:29788–29796. doi: 10.1074/jbc.M604991200. [DOI] [PubMed] [Google Scholar]
- Liu YS, Sompornpisut P, Perozo E. Structure of the KcsA channel intracellular gate in the open state. Nat Struct Biol. 2001;8:883–887. doi: 10.1038/nsb1001-883. [DOI] [PubMed] [Google Scholar]
- Menaker D, Bendahan A, Kanner BI. The substrate specificity of a neuronal glutamate transporter is determined by the nature of the coupling ion. J Neurochem. 2006;99:20–28. doi: 10.1111/j.1471-4159.2006.04003.x. [DOI] [PubMed] [Google Scholar]
- Qu S, Kanner BI. Substrates and non-transportable analogues induce structural rearrangements at the extracellular entrance of the glial glutamate transporter GLT-1/EAAT2. J Biol Chem. 2008;283:26391–26400. doi: 10.1074/jbc.M802401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick M, Javitch JA. Monitoring the function of membrane transport proteins in detergent-solubilized form. Proc Natl Acad Sci U S A. 2007;104:3603–3608. doi: 10.1073/pnas.0609573104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes N, Ginter C, Boudker O. Transport mechanism of a bacterial homologue of glutamate transporters. Nature. 2009;462:880–885. doi: 10.1038/nature08616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RM, Mindell JA. The uncoupled chloride conductance of a bacterial glutamate transporter homolog. Nat Struct Mol Biol. 2007;14:365–371. doi: 10.1038/nsmb1230. [DOI] [PubMed] [Google Scholar]
- Ryan RM, Vandenberg RJ. Distinct conformational states mediate the transport and anion channel properties of the glutamate transporter EAAT-1. J Biol Chem. 2002;277:13494–13500. doi: 10.1074/jbc.M109970200. [DOI] [PubMed] [Google Scholar]
- Ryan RM, Compton EL, Mindell JA. Functional characterization of a Na+-dependent aspartate transporter from Pyrococcus horikoshii. J Biol Chem. 2009;284:17540–17548. doi: 10.1074/jbc.M109.005926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal RP, Shigeri Y, Eliasof S, Leighton BH, Amara SG. Sulfhydryl modification of V449C in the glutamate transporter EAAT1 abolishes substrate transport but not the substrate-gated anion conductance. Proc Natl Acad Sci U S A. 2001;98:15324–15329. doi: 10.1073/pnas.011400198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yernool D, Boudker O, Jin Y, Gouaux E. Structure of a glutamate transporter homologue from Pyrococcus horikoshii. Nature. 2004;431:811–818. doi: 10.1038/nature03018. [DOI] [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Flux coupling in a neuronal glutamate transporter. Nature. 1996;383:634–637. doi: 10.1038/383634a0. [DOI] [PubMed] [Google Scholar]