

Abstract
Although many signals are capable of activating MAPK signaling cascades in chondrocytes in vitro, the function of these pathways remains unclear in vivo. Here we report the phenotype of mice with a conditional deletion of TGF-β-activated kinase 1 (TAK1), a MAP3K family member, in cartilage using the collagen 2α promoter. These mice display chondrodysplasia characterized by neonatal-onset runting, delayed formation of secondary ossification centers, and defects in formation of the elbow and tarsal joints. This constellation of defects resembles the phenotype of mice deficient for receptors or ligands involved in signaling by BMP family members. Chondrocytes from these mice show evidence of defective BMP signaling in vivo and in vitro. Surprisingly, deletion of TAK1 seems to affect not only activation of the p38 MAPK signaling cascade, but also activation of the BMP-responsive Smad1/5/8. Biochemical analysis suggests that TAK1 can interact with Smad proteins and promote their activation through phosphorylation, revealing a previously unrecognized crosstalk between the MAPK and Smad arms of BMP signaling.
Keywords: BMP, cartilage, TAK1, TGF-β, Smad
Introduction
The bone morphogenic protein (BMP) family of ligands was originally identified as an active protein fraction in bone extracts that promoted ectopic bone and cartilage formation in vivo.1 Since then, BMPs have been found to be critical regulators of nearly every aspect of chondrocyte biology, including the formation of chondrocyte condensations in the early limb buds, the development and maintenance of synovial joints, and chondrocyte maturation in the chondroepiphysis of long bones.1–3,5 The BMP receptor complex signals through two broad downstream pathways, the Smad1/5/8 transcription factors and MAPK pathways. The activation of Smad1/5/8 occurs through direct phosphorylation by the type I BMP receptor kinase.6 The mechanisms for the activation of MAPK pathways are not well understood in chondrocytes.7 In other systems, BMPs activate the MAP3K TGF-β-activated kinase 1 (TAK1), leading to phosphorylation of MKK3/6 and activation of p38 isoforms.5
While mice with a conditional deletion of multiple Smad transcription factors in chondrocytes have been shown to have defects in the formation of chondrocyte condensations in the limb bud or in chondrocyte maturation in the chondroepiphysis, no equivalent data exist to identify the role of MAPK signaling downstream of BMPs in vivo. To this end, we generated mice with a conditional deletion of TAK1 in cartilage by crossing TAK1 floxed allele mice to a deleter strain with the expression of cre directed by the cartilage-specific collagen 2α promoter.8 These mice display chondrodysplasia with severe postnatal runting and defects in the development of the elbow and tarsal joints. This phenotype resembles the joint defects seen in mice with mutations in the secreted BMP ligand GDF5 or deletion of BMPR1b. Additionally, the reduced chondrocyte maturation and accompanying runting resemble the phenotype of mice with a deletion of multiple BMP ligands involved in limb outgrowth or the deletion of BMP-responsive Smads. The induction of BMP target genes, such as ID1, was defective in chondrocytes both in vivo and in vitro. Surprisingly, phosphorylation of Smad1/5/8 at the C-terminal serine phosphorylation site targeted by the BMP receptor was decreased in the absence of TAK1 both in vivo and in vitro. Further analysis demonstrated that TAK1 can interact with and phosphorylate Smad1/5/8 at the same site as the BMP receptor (S465/S468 for Smad1). These observations demonstrate a novel role for TAK1 in the development and maintenance of cartilage in vivo, and suggest that TAK1 is a critical mediator of crosstalk between the MAPK and Smad arms of BMP signaling.
Results
Tak1col2 mice show impaired growth of long bones and developmental defects in the tarsal and elbow joints
Immunohistochemical staining for TAK1 expression demonstrated that TAK1 is expressed in hypertrophic chondrocytes of long bones (data not shown). In TAK1 flox/flox Col2-cre mice (hereafter Tak1col2), where deletion of TAK1 is directed by Cre recombinase under the control of the collagen 2 promoter, staining for TAK1 was ablated in cartilage, confirming that TAK1 is efficiently deleted.8
Tak1col2 mice displayed a severe chondrodysplasia with runting as seen in alizarin red/alcian blue-stained skeletal preparations of p20 mice (Fig. 1A). E16.5 and E18.5 embryos were of comparable size to littermate controls, and p0 pups showed only a slight decrease in size, suggesting that the onset of the runting occurs between E18.5 and birth. Thereafter, the phenotype becomes progressively more dramatic until Tak1col2 mice die approximately 3 weeks after birth for unknown reasons. The proximal tibia showed nearly absent formation of secondary ossification centers by p20 in Tak1col2 mice (data not shown). The elbow of postnatal Tak1col2 mice shows dislocation of the proximal radius, a phenotype similar to that seen in GDF5 or BMPR1b-mutant mice.1,2,9 Given this phenotypic similarity and the known role of GDF5 in the developmental program of joint formation, we examined the elbow joint of E14.5 Tak1col2 mice (Fig. 1B). While control embryos showed clear separation between the distal humerus and the proximal radius (indicated with an arrow), Tak1col2 mice showed a complete failure to separate these skeletal elements. Histologic analysis of postnatal Tak1col2 mice showed a separation between the humerus and radius, suggesting that Tak1col2 mice display a delay in executing the program of joint cavitation downstream of GDF5. Analysis of the tarsal bones of Tak1col2 mice showed fusion of nearly all of the major tarsal elements in Tak1col2 mice, a feature also similar to the phenotype of mice with mutations in GDF5 and BMPR1b (Fig. 1C).
Figure 1.
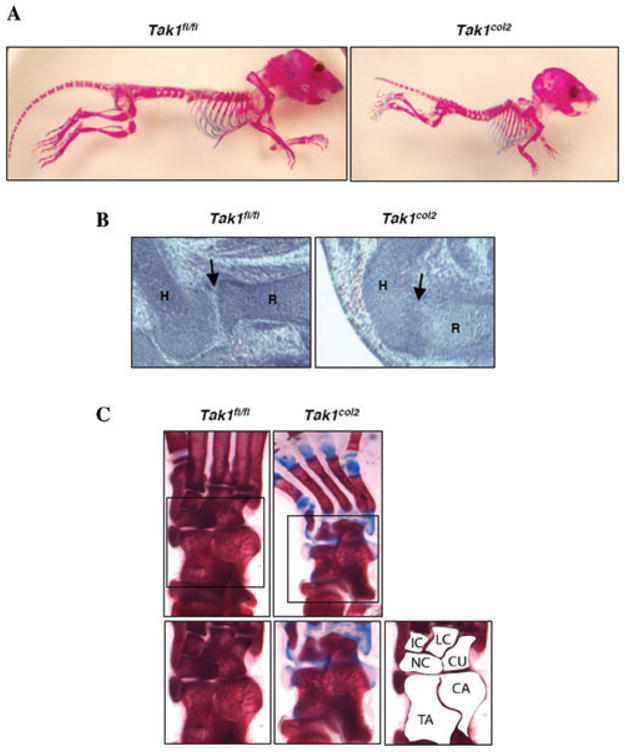
Phenotype of TAK1col2 mice. (A) Alizarin red/alcain blue-stained skeletal preparations of 3-week-old TAK1fl/fl and TAK1col2 mice. (B) Hematoxylin and eosin-stained sections of elbows of E14.5 Tak1fl/fl and Tak1col2 mice. Tak1col2 mice show failure to separate the cartilage elements of the humerus and the radius (arrow). The humerus (H) and radius (R) are labeled. (C) Alizarin red/alcian blue-stained skeletal preparations of the ankle of three week-old Tak1fl/fl and Tak1col2 mice, showing fusion of the tarsal bones. C, intermediate cuneiform; LC, lateral cuneiform; CU, cuboid; NC, navicular; CA, calcaneus; TA, talus.
Impaired BMP signaling in Tak1col2 mice in vivo
As shown above, Tak1col2 mice and mice with impaired GDF5 signaling show similar defects in the elbow and tarsal joints. Likewise, while many pathways contribute to the elongation of the long bones in the limbs, mice with mutations in multiple secreted BMP ligands display a reduction in limb size consistent with that seen in Tak1col2 mice. This suggests that the major function of TAK1 in cartilage in vivo is to transduce signals downstream of BMP ligands. To examine the status of BMP signaling in vivo, chondroepiphyses from Tak1col2 mice and littermate controls were immunohistochemically stained to determine the levels of phosphorylated Smad1/5/8. Phosphorylation of BMP-responsive R-Smad 1/5/8 at two C-terminal serines is necessary for their activation and transcriptional activity, and thus levels of phospho-Smad1/5/8 are a readout for the activation of the Smad arm of BMP signaling.10,11 Tak1col2 mice displayed a reduction in Smad1/5/8 phosphorylation in the hypertrophic/prehypertrophic region of the chondroepiphysis (Fig. 2A) that overlaps with the region of TAK1 expression.
Figure 2.
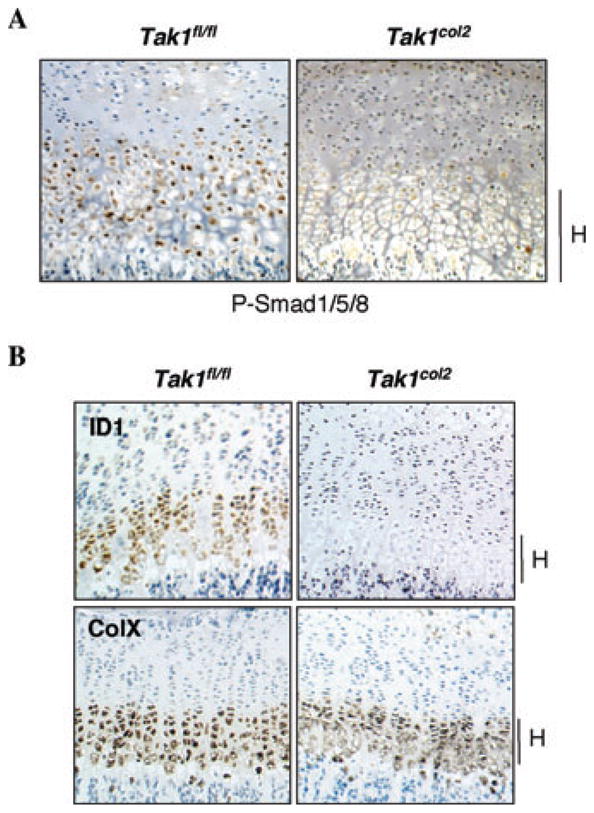
Reduced activation of BMP-responsive SMADs in TAK1col2 mice in vivo. (A) Immunohistochemically stained sections showing phosphorylation of BMP-responsive Smad proteins. Coronal sections of the proximal tibia of P0 Tak1fl/fl and Tak1col2 pups were stained with anti-phospho-Smad1/5/8 antibody. Hypertrophic chondrocytes in the terminal growth plate are indicated with an H. (B) In situ hybridization for ID1 and Collagen Xα (ColX). Coronal sections of the proximal tibia of 3-week-old Tak1fl/fl and Tak1col2 mice were probed for the expression of the indicated mR-NAs. The hypertrophic region of the growth plate is shown.
To further examine the functional activity of the BMP pathway, we performed in situ hybridization for a well-described BMP target gene, ID1. While ID1 showed robust expression in the prehypertrophic/hypertrophic region of the growth plate, ID1 transcript was nearly absent in this region of Tak1col2 mice (Fig. 2B). Taken together, these findings provide evidence for impaired BMP signaling in Tak1col2 mice in vivo.
TAK1 mediates C-terminal phosphorylation of Smad1/5/8
While TAK1 is known to be required for the activation of p38 downstream of BMP ligands in other cell types, the finding that phospho-Smad1/5/8 levels were reduced in Tak1col2 chondrocytes suggested the existence of a previously unrecognized crosstalk between the MAPK and Smad arms of BMP signaling. To analyze the biochemical mechanism of impaired BMP signaling in Tak1col2 mice, chondrocytes were isolated from the chondroepiphyses of Tak1col2 mice and grown in culture. When stimulated with BMP2/7, control chondrocytes showed a robust induction of Smad1/5/8 and p38 phosphorylation within 15 minutes (Fig. 3A). In contrast, Tak1col2 chondrocytes showed absent p38 phosphorylation and a moderate reduction in peak levels of Smad1/5/8 phosphorylation 15–30 min after stimulation, suggesting that TAK1 is responsible for amplifying the C-terminal phosphorylation of the BMP-responsive Smads. To determine whether the absence of TAK1 affected the stability of the BMP-R-Smad complex, the interaction between BMPR and R-Smads was examined in Tak1col2 and control chondrocytes by immunoprecipitation after stimulation with BMP2/7 (data not shown). This analysis showed normal levels of BMPR1a-associated p-Smad1/5/8, suggesting that the absence of TAK1 does not affect Smad phosphorylation by the BMP receptor complex.
Figure 3.

TAK1 mediates phosphorylation of BMP-responsive Smads. (A) Tak1fl/fl and Tak1col2 chondrocytes were serum-starved, stimulated with BMP2/7 (100 ng/mL) for the indicated times, and immunoblotted with antibodies specific to phospho-Smad1/5/8 or phospho-p38. (B) HEK293 cells were transfected with vector or HA-TAK1 (ΔN), and the immunoprecipitates were mixed with wt GST-Smad1 (WT) or mutant GST-Smad1 (AAVA).
This raised the possibility that TAK1 was itself mediating phosphorylation of Smad1/5/8 at the same serines as the BMP receptor. To examine this directly, HA-TAK1 was expressed in 293 cells, and the ability of HA-TAK1 immunocomplexes to phosphorylate GST-Smad1 was examined. HA-TAK1 immunocomplexes showed a robust ability to phosphorylate GST-Smad1, and that activity was reduced nearly twofold in phosphorylating GST-Smad1 with a mutation in the C-terminal serines also phosphorylated by the BMP receptor (AAVA mutation of S465/S458, Fig. 3B). Immunoblotting analysis confirmed the absence of detectable BMP receptor complexes in the HA-TAK1 immunoprecipitate, arguing that contaminating BMPR1 proteins are not responsible for the kinase activity observed (data not shown). Taken together, these findings demonstrate that TAK1 mediates phosphorylation of Smad1 at multiple sites, one of which is the C-terminal S465/S468 site observed to show decreased phosphorylation levels in Tak1col2 mice in vivo. This argues that TAK1 is able to amplify Smad signaling downstream of BMP stimulation by promoting phosphorylation of the same site phosphorylated by the BMP receptor, a previously unrecognized point of crosstalk between MAPK and Smad signaling downstream of BMPs.
A model of TAK1 function downstream of BMPs
Previous studies have demonstrated that TAK1 can be recruited to the BMP receptor complex through two adaptor proteins XIAP and TAB1 (diagrammed in Fig. 4).12,13 From there it mediates activation of p38 via MKK3/6, as has been described in other systems. Surprisingly, we observed a reduction in Smad1/5/8 phosphorylation in chondrocytes in the absence of TAK1. Further analysis demonstrated that TAK1 is capable of phosphorylating Smad1 at the same C-terminal site phosphorylated by the BMP receptor. Thus, TAK1 is required for the activation of p38 MAPK and the early amplification of Smad1/5/8 phosphoryation downstream of BMP stimulation. The phenotypic similarities between Tak1col2 mice and mice deficient for secreted BMP family ligands along with the reduced levels of phospho-Smad1/5/8 and BMP target genes in vivo demonstrate that TAK1 is required for some aspects of BMP signaling in vivo.
Figure 4.
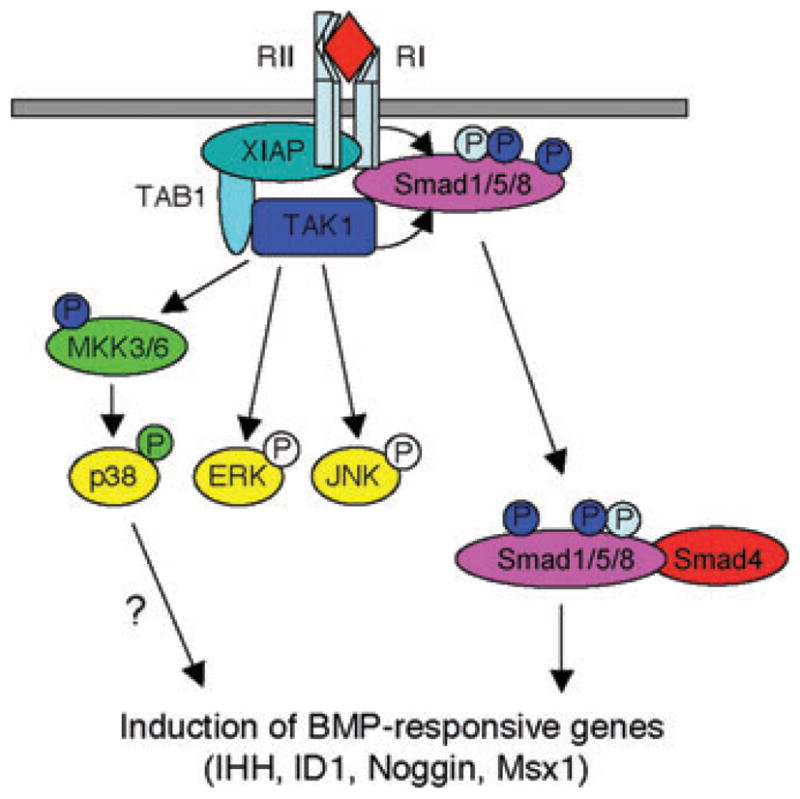
Model of the function of TAK1 in chondrocytes.
Notably, some level of Smad1/5/8 phosphorylation and activation is preserved in the absence of TAK1. As expected from this, the phenotype of Tak1col2 mice is less dramatic than that of mice with deletions of Smad1, Smad5, and Smad8 in cartilage. TAK1 was originally identified as a MAPKKK functioning downstream of TGF-β.14 Interestingly, previous studies have demonstrated that TGF-β functions in cartilage to pattern axial skeletal elements and suppress osteoarthritis.15,16 Since Tak1col2 mice display normal patterning of the axial skeleton and no joint abnormalities other than the defects in the elbow and tarsal joints discussed above, we suggest that TAK1 is functionally dispensable for TGF-β signaling in chondrocytes in vivo.
Summary and conclusions
Mice with a conditional ablation of TAK1 in chondrocytes develop several defects associated with impaired BMP signaling. They display runting and fusion of the elbow and tarsal joints. At the histologic level, chondrocyte hypertrophy is moderately impaired and the expression of BMP target genes such as IHH and ID1 is reduced. Surprisingly, ablation of TAK1 in chondrocytes results not only in impaired activation of p38 MAPK, but also in defective phosphorylation and activation of BMP-responsive Smads both in vivo and in vitro. Biochemical analysis showed that TAK1 can directly interact with the BMP-responsive Smads, and TAK1 immunocomplexes can directly phosphorylate Smad1/5/8 at the same C-terminal serines phosphorylated by the BMP receptor complex. Since phosphorylation at this site is essential for activity of Smads, TAK1 may play a previously unexpected role in amplifying the activity of BMP-responsive Smads in addition to its described role in activating p38 MAPK.
In summary, these results indicate that TAK1 is a critical mediator of BMP signaling in chondrocytes, driving chondrocyte hypertrophy and the cavitation step in the development of the elbow and tarsal joints. While this study was originally intended to address the role of MAPK signaling cascades in cartilage in vivo, the unexpected finding of crosstalk between TAK1 and Smad activation precludes a simple assignment of the in vivo phenotype observed to defective activation of p38 MAPK. Future studies should deconvolute the contribution of Smads versus p38 MAPK downstream of TAK1 through the use of mice with conditional ablation of p38 MAPK isoforms in cartilage.
Acknowledgments
We thank Drs. Marc Wein and Vicki Rosen for their thoughtful comments on the work, Dorothy Zhang, Michelle Schweitzer, and Pamela Okerholm for technical assistance, and Dallas Jones for helpful discussions. We also thank the many individuals who provided valuable reagents. This work was supported by National Institutes of Health Grant AI29673 and HD055601 (L.H.G.); J.S. was supported by an Arthritis Foundation postdoctoral fellowship.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
References
- 1.Wozney JM, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242:1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 2.Storm EE, et al. Limb alterations in brachypodism mice due to mutations in a new member of the TGF beta-superfamily. Nature. 1994;368:639–643. doi: 10.1038/368639a0. [DOI] [PubMed] [Google Scholar]
- 3.Yi SE, et al. The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development. 2000;127:621–630. doi: 10.1242/dev.127.3.621. [DOI] [PubMed] [Google Scholar]
- 4.Yoon BS, et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoon BS, Lyons KM. Multiple functions of BMPs in chondrogenesis. J Cell Biochem. 2004;93:93–103. doi: 10.1002/jcb.20211. [DOI] [PubMed] [Google Scholar]
- 6.Massague J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 7.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 8.Ovchinnikov DA, et al. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- 9.Bandyopadhyay A, et al. Genetic analysis of the roles of BMP2, BMP4, and BMP7 in limb patterning and skeletogenesis. PLoS Genet. 2006;2:e216. doi: 10.1371/journal.pgen.0020216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kretzschmar M, et al. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev. 1997;11:984–995. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- 11.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 12.Shibuya H, et al. Role of TAK1 and TAB1 in BMP signaling in early Xenopus development. EMBO J. 1998;17:1019–1028. doi: 10.1093/emboj/17.4.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamaguchi K, et al. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J. 1999;18:179–187. doi: 10.1093/emboj/18.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamaguchi K, et al. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–2011. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 15.Baffi MO, et al. Conditional deletion of the TGF-beta type II receptor in Col2a expressing cells results in defects in the axial skeleton without alterations in chondrocyte differentiation or embryonic development of long bones. Dev Biol. 2004;276:124–142. doi: 10.1016/j.ydbio.2004.08.027. [DOI] [PubMed] [Google Scholar]
- 16.Serra R, et al. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]