

Abstract
X-ray absorption and resonance Raman spectroscopies show that CmlA, the β-hydroxylase of the chloramphenicol biosynthetic pathway, contains a (μ-oxo)-(μ-1,3-carboxylato)diiron(III) cluster with 6-coordinate iron centers and 3 – 4 His ligands. This active site is found within a unique β-lactamase fold and is distinct from those of soluble methane monooxygenase and related enzymes that utilize a highly conserved diiron cluster with a 2-His-4-carboxylate ligand set within a 4-helix bundle motif. These structural differences may have an impact on the nature of the activated oxygen species of the reaction cycle.
Amino acid β-hydroxylation occurs during the biosynthesis of many natural products in nonribosomal peptide synthetase (NRPS)-based pathways.1 The products include many pharmaceutically important antibiotic and chemotherapeutic drugs.2–4 The newly introduced hydroxyl groups serve as sites for further modifications including glycosylation, oxidation, and macrocycle formation.1 These tailoring reactions are usually required for pharmacological activity.
While β-hydroxylation in NRPS biosynthetic pathways is typically catalyzed by cytochrome P450s5 and alpha-ketoglutarate-dependent nonheme iron-containing enzymes,6, 7 we have recently characterized a large new family of tailoring enzymes that contain a nonheme oxo-bridged diiron active site.8 The first member of this family to be isolated was CmlA from the chloramphenicol biosynthetic pathway. CmlA catalyzes β-hydroxylation of p-amino phenylalanine linked by a thioester bond to the thiolation domain of the NRPS CmlP (Scheme 1).8 Other enzymes from this family catalyze β-hydroxylation in the biosynthesis of a wide range of antibiotic and cytostatic agents, including bleomycin and the planin family of antibiotics. Unlike bacterial multicomponent monooxygenases (BMMs), which utilize a canonical four helix bundle (α4) protein fold to provide the carboxylate and imidazole ligands to the diiron center,9 members of the CmlA family of monooxygenases employ a metallo β-lactamase (αββα) fold to bind the cluster. While a few other diiron enzymes have been shown to utilize a lactamase fold,10 CmlA is the first example shown to catalyze substrate hydroxylation.8 As such, a detailed description of the CmlA active-site is warranted in order to understand how this unique enzyme class catalyzes hydroxylation at the dinuclear center. To date, no X-ray crystal structure of an enzyme from this family has been reported. In this communication, we utilize resonance Raman and X-ray absorption spectroscopies to structurally characterize the CmlA active site, including the bridging structure of the diiron center and elucidation of the ligand environment.
Scheme 1.
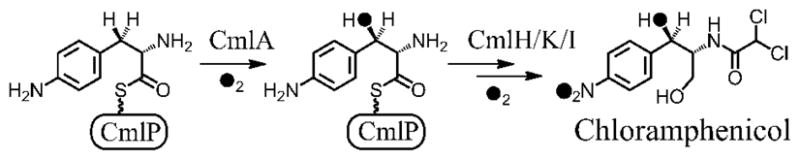
The biosynthetic pathway of chloramphenicol
Oxidized CmlA has a chromophore at ~340 nm, which typically originates from an oxo-to-iron(III) charge transfer band associated with the bridging oxygen of the diiron cluster.8 Resonance Raman (rR) spectroscopy provides a direct probe for the nature of this chromophore, including the precise bridging structure of the CmlA diiron center. Figure 1 shows the rR spectrum of as isolated, diferric CmlA in H216O, H218O, and D2O. The spectrum of the sample in H216O has a peak at 481 cm−1 (top trace), which downshifts to 464 cm−1 in H218O buffer (middle trace) but is unaffected in D2O buffer (bottom trace). The oxygen-isotopic sensitivity of this band clearly indicates that the 481 cm−1 peak arises from an oxygen-linked vibration. Indeed, the 481 cm−1 peak and its 17 cm−1 isotopic downshift values are consistent with a (μ-oxo)diiron cluster, as found in other diiron enzymes and model systems,11 but not with a μ-hydroxo or bis(μ-oxo) cluster. Previous results have demonstrated a strong correlation between the observed νs(Fe-O-Fe) and ∠Fe-O-Fe values;11 from this correlation, an Fe-μ-O-Fe angle of ~130° can be deduced for CmlA.
Figure 1.

Resonance Raman spectra of CmlA in 16OH2, > 85 % 18OH2, and > 85 % D2O buffer solutions. All spectra were subjected to polynomial baseline correction and 2 points binomial smoothing. The sharp phenylalanine signal at 1005 cm−1 was used to align the data. Asterisks indicate the laser plasma lines.
The structure of the diferric CmlA active site was further probed by X-ray absorption spectroscopy (XAS). The XAS spectrum of CmlA exhibits a pre-edge feature with an area of 13.4(4) units. This value is much smaller than those found for (μ-oxo)diiron(III) clusters with 5- or 4-coordinate iron centers but falls in the range found for those with 6-coordinate iron centers.12–14 Unfiltered EXAFS and Fourier transformed (FT) spectra of CmlA are shown as black lines in Fig. 2, with corresponding best fits shown in blue lines. The best fit parameters are listed in Table 1 and compared with those of diiron-cluster containing proteins and related model complexes in Tables S2–S4. The data analysis shows that CmlA contains a (μ-oxo)diiron(III) cluster with Fe-μ-O and Fe···Fe distances of ca. 1.80 and 3.32 Å, respectively, the latter clearly associated with the peak at r′ = 3 Å (Fig. 2). The Fe-μ-O and Fe···Fe distances together require an ∠Fe-O-Fe angle of ca. 134°, which agrees with the Raman results in Fig. 1. Based on comparisons with synthetic diiron(III) complexes,11 the spectroscopic data for CmlA indicate the presence of a (μ-oxo)(μ-1,3-carboxylato)diiron core, as also found in the β subunit of Class Ia ribonucleotide reductases (R2).15
Figure 2.

Fourier transform of unfiltered EXAFS data (inset) of CmlA (black lines) and its best fit (blue lines). Fit parameters are provided in Table 1. Green boxes highlight the features that arise from outer-shell atoms of imidazole ligands.
Table 1.
Best-fit parameters to unfiltered EXAFS data of CmlA
| Scattering path | N | R (Å) | σ2 × 103 (Å2) | Moiety* |
|---|---|---|---|---|
| Fe-O | 1 | 1.80 | 4.41 | μ-oxo |
| Fe-N/O | 5 | 2.10 | 4.97 | Im/μ-Car/OHx |
| Fe···C/N | 2 | 3.09 | 1.19 | Im/μ-Car |
| Fe···Fe | 1 | 3.32 | 5.36 | Fe |
|
| ||||
| Fe···C | 1 | 2.53 | 1.09 | Bidentate |
| Fe···C (with MS)# | 1 | 3.93 | 5.13 | carboxylate |
| Fe···C/N (with MS)# | 3.9 | 4.36 | 3.80 | Im |
k range = 2 – 13 Å, resolution ~ 0.144 Å. N = coordination number. R = distance. σ2 = respective Debye-Waller factor.
Paths associated with multiple scattering (see SI for details).
Moiety used in the FEFF model (SI), with Im = imidazole, Car = carboxylate, and OHx = terminal water derived ligand.
EXAFS analysis provides further insight into the nature of the iron ligands of CmlA. The average Fe-O/N bond distance (excluding the oxo bridge) for CmlA is 2.10 Å, which reflects the relative number of histidines and oxyanion ligands. This distance is shorter than those for various forms of metHr (2.13–2.17 Å),16, 17 which has a total of five histidines and two carboxylate ligands for the diiron cluster,18 but longer than those for other diiron enzymes with four carboxylates and one or two His ligands (1.99–2.06).14, 16, 19 The average Fe-O/N distance found for CmlA suggests that its diiron active site should have more than 2 but less than 5 His ligands.
Support for this notion comes from an analysis of the outer shell features found in the 3.2 – 4.0 Å region of the FT data of CmlA (see green box in Fig. 2). These features can be attributed to the more distant atoms of imidazole-like ligands, the intensities of which are enhanced by multiple scattering pathways,20 and correspond to the double-humped feature at 4 Å−1 in the unfiltered k-space data (Fig. 2 inset). Similarly well defined double-humped features near 4.0 Å−1 can be found in unfiltered EXAFS data of metHr and model complexes that have 4–6 imidazole-like ligands per cluster.16, 17, 21 In contrast, such features are less well defined in the spectra of other diiron enzymes with only 1 – 2 His ligands per cluster.13, 14, 16, 19 Diiron complexes without imidazole-like ligands do not exhibit this feature.22, 23 Thus, the clearly defined double-humped feature in the EXAFS spectrum of CmlA suggests that there are more than 2 His ligands bound to its diiron active site.
To obtain a more quantitative analysis of the data, we have applied a multiple scattering approach to simulate the double-humped feature, as detailed in the SI. (Table S4 and Fig. S6). The best fit is shown in Fig. 2 and Table 1. The optimized numbers of scattering paths associated with the imidazole moiety require the presence of ~2 His ligands per iron, or ~4(±1) His ligands per cluster, given the ~25% uncertainty in the number of scatterers for a given shell determined from EXAFS analysis.24 However, the presence of 5 His ligands is highly unlikely on the basis of the average Fe-O/N distance of 2.10 Å found for CmlA, as discussed above.
The best fit of the EXAFS data also requires the inclusion of one low-Z scatterer per iron at 2.53 Å (Table S2). The small σ2 value associated with the scatterer excludes the possibility that it arises from the ligating atom of a weakly bound ligand and leads us to assign it to the carboxylate C atom (Ccar) of a symmetrically bidentate carboxylate ligand. Scatterers at similar distance have also been found in the EXAFS spectra of the terephthalate complex of protocatechuate 3,4-dioxygenase25 and the ternary complex of tyrosine hydroxylase with substrate and cofactor26 and are associated with the carbon atom of a bidentate carboxylate. The tyrosine hydroxylase result is corroborated by the crystal structure of the corresponding complex of the homologous phenylalanine hydroxylase.27 For CmlA, the Cα atom of the bidentate carboxylate can also be included in the best fit because of the collinearity of the Fe center and the carboxylate Ccar and Cα atoms; including the multiple scattering paths involving these atoms noticeably enhances the fit (Table S4). Taken together, the rR and XAS analyses described above show that CmlA contains a (μ-oxo)(μ-1,3-carboxylato)diiron(III) cluster with 6-coordinate iron centers, 2 symmetrically bidentate carboxylate ligands, and 3 – 4 His ligands (Fig. 3A).
Figure 3.

Comparison of the diiron(III) clusters of CmlA and related diiron proteins.
This structure is similar to that proposed in our previous study8 which was based on alignment of the sequences of CmlA and its homologs with the consensus sequence for metallo-β-lactamases. Sequence alignment predicted only 3 His ligands for CmlA,8 but the presence of an additional His ligand is also possible. Both the CmlA and metallo-β-lactamase sequences share the signature HxHxDH motif that provides one carboxylate and 3 His ligands for the metal cluster. Downstream of this sequence in CmlA, one Asp and two Glu residues are conserved and found at approximately the same positions as the other amino acid ligands of the dizinc cluster of the metallo-β-lactamases (2 His and 1 Cys/Ser). There is only one conserved His residue downstream of the signature sequence for CmlA (His378), and this could be the fourth His predicted in the current study. However, this His is adjacent to one of the postulated Glu ligands, and based on metallo-β-lactamases structures, replacement of this Glu by His would place three His ligands on one iron and one on the other. This would be expected to yield two quite different iron environments, which is not apparent in the Mössbauer spectrum of CmlA.8 It is possible that CmlA adopts a slightly different fold than a metallo-β-lactamase, changing the iron to which the additional His coordinates.
Several new insights derive from the proposed structure of the diiron cluster of CmlA. First, it is clear from the current studies that the diiron cluster of CmlA differs from those of the flavodiiron protein (FDP) family10 in which it was first shown that a diiron cluster can be supported in the metallo-β-lactamase αββα fold. FDPs are widespread among bacteria and archaea and function as reductases for O2 and NO rather than oxygenases. All crystal structures of FDPs reported thus far show that their diiron clusters are bridged by a water-derived ligand with an Fe-O distance of ~2.0 Å (Fig. 3B).10 This distance and the other spectroscopic features of the cluster indicate that this bridge is not a μ-oxo ligand as found here for CmlA, but rather a protonated form such as a hydroxo or an aqua ligand. Furthermore, the bridging atom appears to be hydrogen bonded to one of the terminal monodentate carboxylates. These structural differences between CmlA and FDPs may account for the contrasting reactivities, but additional structural and kinetic studies of both the FDPs and CmlA will be required to identify the molecular origin of these differences.
Second, despite having monooxygenase reactivity, the diiron core structure of CmlA differs from those of the clusters of all canonical oxygen-activating nonheme diiron enzymes characterized thus far.9 BMMs, desaturases, and R2 have a conserved 2-His-4-carboyxlate ligand set with 1 His per iron center (Fig. 3C). This ligand set promotes a generally accepted oxygen activation mechanism involving a (μ-1,2-peroxo)diiron(III) intermediate that converts to high-spin higher-valent diiron species responsible for substrate oxidation.9
Distinct oxygen activation mechanisms are postulated for some enzymes that have more than two His ligands bound to the diiron active site, including myo-inositol oxygenase (MIOX) with a total of 4 His ligands28 and AurF with a total of 3 His ligands29 (Fig. 3D). MIOX is unique in that it activates O2 with a mixed-valent diiron(II/III) cluster to form a C-H bond-cleaving (superoxo)diiron(III) intermediate that initiates the 4e-oxidation of substrate.30 AurF effects the conversion of aromatic amines to nitro groups via a diiron(III)-peroxo intermediate that differs from the canonical (μ-1,2-peroxo)diiron(III) intermediate discussed above.31
Like MIOX and AurF, CmlA has an additional His ligand on at least one and possibly both irons of its diiron cluster, but its molecular mechanism is unlikely to be similar to those of MIOX or AurF. In contrast to MIOX, we have shown that it is the diiron(II) state of CmlA that reacts with O2 to initiate catalysis.8 With respect to AurF, a similar peroxo intermediate may also be formed by CmlA, but this species is unlikely to be sufficiently reactive to attack the C-H bond on the substrate (BDE ~ 85 kcal/mol, Scheme 1). Accordingly, the peroxo intermediate of AurF has not been observed to attack C-H bonds of any substrate.
This analysis suggests that CmlA may generate some type of high valent diiron oxygen species in which the O-O bond of O2 has been broken as found for the canonical diiron monooxygenases such as methane monooxygenase (MMO). However, the additional His ligand(s) may alter the nature of this reactive species. There are many options for the species that could be formed as alternatives to the FeIV2O2 diamond core of MMO intermediate Q.32, 33 For example, our recent work has identified synthetic diiron(IV) complexes with nitrogen-rich ligand environments that are quite reactive towards C-H bonds.34 Although experimental evidence is needed to support the hypothesis for an alternative reactive species in CmlA, it is clear that variations to the canonical MMO reactive intermediate are emerging and may be relevant to catalysis by CmlA.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants GM37867 (to L.Q.), GM40466 (to J.D.L), and GM40466-S1 (to J.D.L.). V.V.V. was supported by fellowships from the Department of Chemistry and Graduate School at the University of Minnesota, and Vietnam Education Foundation. XAS data were collected at Beamline 7-3 of the Stanford Synchrotron Radiation Lightsource, which was supported by the U.S. Department of Energy and National Institutes of Health.
Footnotes
Supporting Information Available: Experimental details and XAS analysis. This material is free of charge via the Internet at http://pub.acs.org.
References
- 1.Chen H, Thomas MG, O’Connor SE, Hubbard BK, Burkart MD, Walsh CT. Biochemistry. 2001;40:11651–11659. doi: 10.1021/bi0115434. [DOI] [PubMed] [Google Scholar]
- 2.Ciabatti R, Kettenring JK, Winters G, Tuan G, Zerilli L, Cavalleri B. J Antibiotics. 1989;42:254–267. doi: 10.7164/antibiotics.42.254. [DOI] [PubMed] [Google Scholar]
- 3.Du L, Sanchez C, Chen M, Edwards DJ, Shen B. Chemistry & Biology. 2000;7:623–642. doi: 10.1016/s1074-5521(00)00011-9. [DOI] [PubMed] [Google Scholar]
- 4.Pootoolal J, Thomas MG, Marshall CG, Neu JM, Hubbard BK, Walsh CT, Wright GD. Proc Natl Acad Sci U S A. 2002;99:8962–8967. doi: 10.1073/pnas.102285099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen H, Walsh CT. Chemistry & Biology. 2001;8:301–312. doi: 10.1016/s1074-5521(01)00009-6. [DOI] [PubMed] [Google Scholar]
- 6.Neary JM, Powell A, Gordon L, Milne C, Flett F, Wilkinson B, Smith CP, Micklefield J. Microbiology. 2007;153:768–776. doi: 10.1099/mic.0.2006/002725-0. [DOI] [PubMed] [Google Scholar]
- 7.Strieker M, Nolan EM, Walsh CT, Marahiel MA. J Am Chem Soc. 2009;131:13523–13530. doi: 10.1021/ja9054417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makris TM, Chakrabarti M, Münck E, Lipscomb JD. Proc Natl Acad Sci U S A. 2010;107:15391–15396. doi: 10.1073/pnas.1007953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2657. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]; b) Kurtz DM., Jr J Biol Inorg Chem. 1997;2:159–167. [Google Scholar]; c) Sazinsky MH, Lippard SJ. Acc Chem Res. 2006;39:558–566. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 10.Kurtz DM., Jr Dalton Trans. 2007:4115–4121. [Google Scholar]
- 11.a) Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]; b) Zheng H, Zang Y, Dong Y, Young VG, Que L., Jr J Am Chem Soc. 1999;121:2226–2235. [Google Scholar]
- 12.a) Roe AL, Schneider DJ, Mayer RJ, Pyrz JW, Widom J, Que L., Jr J Am Chem Soc. 1984;106:1676–1681. [Google Scholar]; b) Westre TE, Kennepohl P, DeWitt JG, Hedman B, Hodgson KO, Solomon EI. J Am Chem Soc. 1997;119:6297–6314. [Google Scholar]
- 13.True AE, Scarrow RC, Randall CR, Holz RC, Que L., Jr J Am Chem Soc. 1993;115:4246–4255. [Google Scholar]
- 14.Shu L, Broadwater JA, Achim C, Fox BG, Münck E, Que L., Jr J Biol Inorg Chem. 1998;3:392–400. [Google Scholar]
- 15.a) Sjöberg BM, Loehr TM, Sanders-Loehr J. Biochemistry. 1982;21:96–102. doi: 10.1021/bi00530a017. [DOI] [PubMed] [Google Scholar]; b) Nordlund P, Eklund H. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 16.Scarrow RC, Maroney MJ, Palmer SM, Que L, Jr, Roe AL, Salowe SP, Stubbe J. J Am Chem Soc. 1987;109:7857–7864. [Google Scholar]
- 17.Zhang K, Stern EA, Ellis F, Sanders-Loehr J, Shiemke AK. Biochemistry. 1988;27:7470–7479. doi: 10.1021/bi00419a045. [DOI] [PubMed] [Google Scholar]
- 18.Stenkamp RE. Chem Rev. 1994;94:715–726. [Google Scholar]
- 19.a) DeWitt JG, Bentsen JG, Rosenzweig AC, Hedman B, Green J, Pilkington S, Papaefthymiou GC, Dalton H, Hodgson KO, Lippard SJ. J Am Chem Soc. 1991;113:9219–9235. [Google Scholar]; b) Shu L, Liu Y, Lipscomb JD, Que L., Jr J Biol Inorg Chem. 1996;1:297–304. [Google Scholar]; c) Rudd JD, Sazinsky MH, Merkx M, Lippard SJ, Hedman B, Hodgson KO. Inorg Chem. 2004;43:4579–4589. doi: 10.1021/ic049716b. [DOI] [PubMed] [Google Scholar]; d) Toussaint L, Cuypers M, Bertrand L, Hue L, Romão C, Saraiva L, Teixeira M, Meyer-Klaucke W, Feiters M, Crichton R. J Biol Inorg Chem. 2009;14:35–49. doi: 10.1007/s00775-008-0422-3. [DOI] [PubMed] [Google Scholar]; e) Bunker G, Petersson L, Sjöeberg BM, Sahlin M, Chance M, Chance B, Ehrenberg A. J Am Chem Soc. 1987;26:4708–4716. doi: 10.1021/bi00389a017. [DOI] [PubMed] [Google Scholar]
- 20.Riggs-Gelasco PJ, Stemmler TL, Penner-Hahn JE. Coord Chem Rev. 1995;144:245–286. [Google Scholar]
- 21.Brennan BA, Chen Q, Juarez-Garcia C, True AE, O’Connor CJ, Que L., Jr Inorg Chem. 1991;30:1937–1943. [Google Scholar]
- 22.Mizoguchi TJ, Kuzelka J, Spingler B, DuBois JL, Davydov RM, Hedman B, Hodgson KO, Lippard SJ. Inorg Chem. 2001;40:4662–4673. doi: 10.1021/ic010076b. [DOI] [PubMed] [Google Scholar]
- 23.Fiedler AT, Shan X, Mehn MP, Kaizer J, Torelli S, Frisch JR, Kodera M, Que L., Jr J Phys Chem A. 2008;112:13037–13044. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Penner-Hahn JE. Coord Chem Rev. 1999;190–192:1101–1123. [Google Scholar]
- 25.True AE, Orville AM, Pearce LL, Lipscomb JD, Que L., Jr Biochemistry. 1990;29:10847–10854. doi: 10.1021/bi00500a019. [DOI] [PubMed] [Google Scholar]
- 26.Chow MS, Eser BE, Wilson SA, Hodgson KO, Hedman B, Fitzpatrick PF, Solomon EI. J Am Chem Soc. 2009;131:7685–7698. doi: 10.1021/ja810080c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andersen OA, Stokka AJ, Flatmark T, Hough E. J Mol Biol. 2003;333:747–757. doi: 10.1016/j.jmb.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Brown PM, Caradoc-Davies TT, Dickson JMJ, Cooper GJS, Loomes KM, Baker EN. Proc Natl Acad Sci U S A. 2006;103:15032–15037. doi: 10.1073/pnas.0605143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi YS, Zhang H, Brunzelle JS, Nair SK, Zhao H. Proc Natl Acad Sci U S A. 2008;105:6858–6863. doi: 10.1073/pnas.0712073105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xing G, Diao Y, Hoffart LM, Barr EW, Prabhu KS, Arner RJ, Reddy CC, Krebs C, Bollinger JM. Proc Natl Acad Sci U S A. 2006;103:6130–6135. doi: 10.1073/pnas.0508473103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Korboukh VK, Li N, Barr EW, Bollinger JM, Krebs C. J Am Chem Soc. 2009;131:13608–13609. doi: 10.1021/ja9064969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shu L, Nesheim JC, Kauffmann K, Münck E, Lipscomb JD, Que L., Jr Science. 1997;275:515–518. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- 33.a) Dunietz BD, Beachy MD, Cao Y, Whittington DA, Lippard SJ, Friesner RA. J Am Chem Soc. 2000;122:2828–2839. [Google Scholar]; b) Rinaldo D, Philipp DM, Lippard SJ, Friesner RA. J Am Chem Soc. 2007;129:3135–3147. doi: 10.1021/ja0654074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.a) Xue G, Fiedler AT, Martinho M, Münck E, Que L., Jr Proc Natl Acad Sci USA. 2008;105:20615–20620. [Google Scholar]; b) Wang D, Farquhar ER, Stubna A, Münck E, Que L., Jr Nat Chem. 2009;1:145–150. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.