

Summary
Study of the development of distinct CD4+ T-cell subsets from naive precursors continues to provide excellent opportunities for dissection of mechanisms that control lineage-specific gene expression or repression. Whereas it had been thought that the induction of transcription networks that control T-lineage commitment were highly stable, reinforced by epigenetic processes that confer heritability of functional phenotypes by the progeny of mature T cells, recent findings support a more dynamic view of T-lineage commitment. Here, we highlight advances in the mapping and functional characterization of cis elements in the Ifng locus that have provided new insights into the control of the chromatin structure and transcriptional activity of this signature T-helper 1 cell gene. We also examine epigenetic features of the Ifng locus that have evolved to enable its re-programming for expression by other T-cell subsets, particularly T-helper 17 cells, and contrast features of the Ifng locus with those of the Il17a–Il17f locus, which appears less promiscuous.
Keywords: cytokines, gene transcription, epigenetics, transcription factors, immunoregulation
Introduction
The differentiation of multipotent naive CD4+ T cells into distinct effector and regulatory lineages provides a central mechanism for linking the innate and adaptive arms of immunity. The pattern of antigen-driven CD4+ T-cell differentiation is controlled by cytokine cues that emanate from microbe-sensing innate immune cells and defines the course of the adaptive immune response, in large part through the release of effector or regulatory cytokines that are characteristic of distinct T-cell lineages. The recognition that clonal populations of mature CD4+ T cells produce distinct cytokine profiles formed the basis for the T helper 1 (Th1)–Th2 cell paradigm nearly 25 years ago (1) and has been expanded in the intervening years by discovery of additional T-cell subsets, including regulatory T cells (Tregs) and a third major effector lineage, Th17, which produce its own characteristic profile of cytokines (Reviewed in 2–4). Additional CD4+ T-cell subsets, including Th9 and T-follicular helper (Tfh) cells (5–8), have also been described, although they remain less well characterized.
Findings over the past two decades have elucidated contributions of cytokines and the lineage–specifying transcription factors they induce in the generation of helper CD4+ T cell diversity (Fig. 1). Although the complexity of CD4+ T-cell subsets continues to grow, a common theme is that a limited set of transcription factors induced early during T-cell activation guides the differentiation of individual effector or regulatory lineages in the context of rapid cell cycling that accompanies clonal expansion. Notably, each of the lineages described to date is linked to one or more members of the signal transducer and activator of transcription (STAT) family of transcription factors, which, as their name implies, act both as proximal signal transducers activated by different cytokine receptors and DNA-binding proteins that regulate transcriptional activation or repression of target genes in differentiating CD4+ T cells. The Th1 pathway is thus linked to IFN-α, β or γ−STAT1, IL-12–STAT4 and T-bet; the Th2 pathway is linked to IL-4–STAT6 and GATA3. The Th17 pathway is linked to TGF-β plus IL-6/IL-21/IL-23–STAT3 and RORγt/RORα. STAT5 and FoxP3 contribute to Treg differentiation, and the Th9 and T follicular helper (TFH) subsets have been recently linked to STAT6/PU.1 and STAT3/Bcl6, respectively (Fig. 1). Whereas other transcription factors also contribute to these lineages, including those already defined and those yet to be discovered, the factors listed above have been best characterized and appear essential to each of their respective lineages.
Fig. 1. Developmental pathways of effector CD4+ T-cell subsets that can lead to Ifng expression.
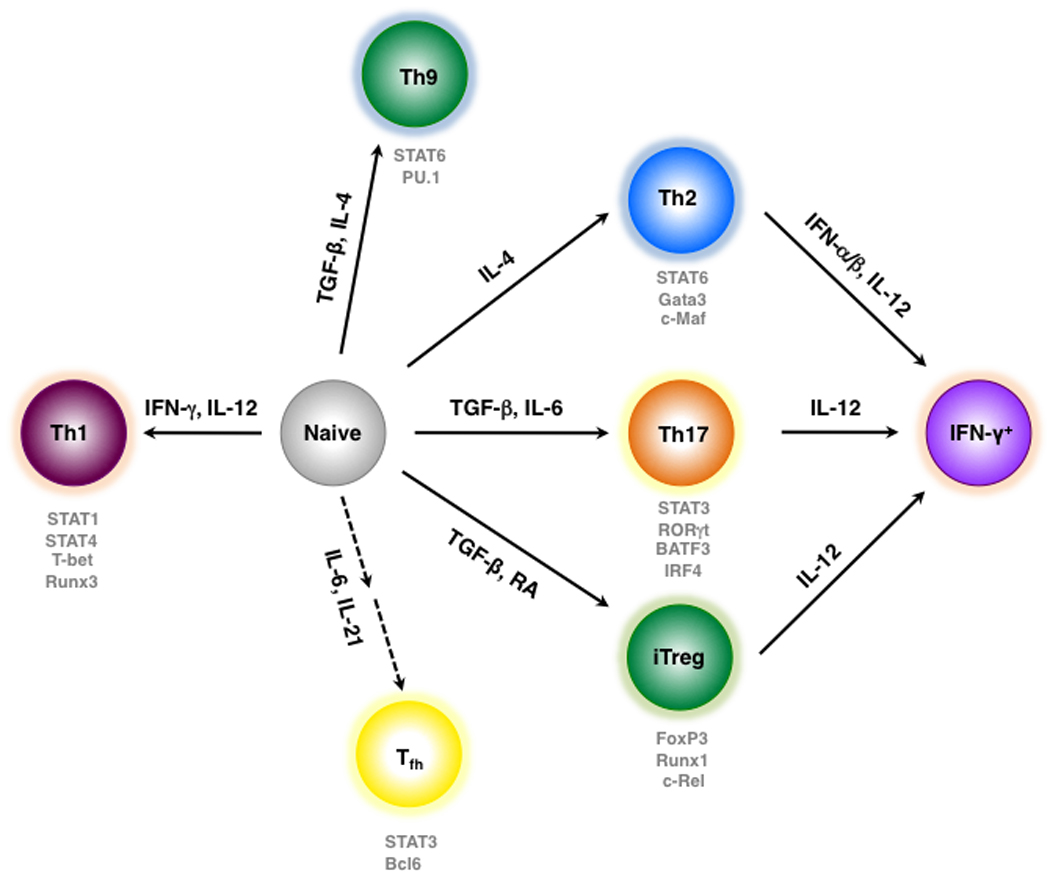
Following exposure to foreign antigen, naive CD4+ T cells are directed towards distinct developmental programs by cytokines that differentially induce lineage-specifying transcription factors (see main text for details). In addition to classical Th1 cells, Th17, iTreg, and Th2 cells can be induced to express IFN-γ under certain conditions. The broken lines associated with the Tfh pathway represent the possibility that these cells branch from developing Th1, Th2, and Th17 precursors rather than directly from naive precursors (123).
A key cell-intrinsic mechanism by which transcription factor networks stabilize T-lineage commitment is through epigenetic effects on target genes (9). Through histone or DNA modifications that activate or repress cis elements in target gene loci, T-lineage differentiation is accompanied by epigenetic imprinting such that the transcriptomes of mature T cells become stabilized and their functional phenotypes transmitted to their progeny. The identification of key trans-acting factors that specify T-cell fate decisions has enabled delineation of the cis elements with which these factors interact to coordinate lineage–specific regulation of multiple gene loci. With the advent of post-genomic technologies for more efficiently mapping of cis elements, our understanding of the regulatory complexities of cytokine genes has accelerated.
In this review, we will focus on the Ifng locus as a model for T-lineage-specific control of cytokine genes. Several excellent reviews have covered the identification of distal elements that regulate Ifng transcription and the importance of differentiation-dependent modifications of the chromatin architecture of the Ifng locus in regulating transcriptional competence (9–11). Here we will examine recent advances in understanding the interactions between cis elements and trans-acting factors that regulate T-lineage-specific remodeling of the Ifng locus and the role of acute trans-acting factors in driving stimulus-dependent transcription of Ifng in differentiated T effectors. Additionally, we consider the basis for plasticity of cytokine expression phenotypes that has been the subject of recent reports of non-Th1 cells transitioning into IFN-γ-competent effectors (12–15).
Cytokine and transcription factor networks that regulate Th1 differentiation
The temporal development of Th1 cells has been well scrutinized, giving rise to a sequential model of cytokine signaling and transcription factor utilization in commitment to this lineage. At least three transcription factors — STAT1, STAT4 and T-bet — play essential roles in programming naïve CD4+ T cells into IFN-γ–competent Th1 effectors. STAT1 is activated downstream of the type I (IFN-α, β) and type II (IFN-γ) interferon receptors, and STAT4 is activated downstream of the IL-12 receptor. Although Type 1 IFNs appear to be important in Th1 development in humans, their role in mice is limited due to a minisatellite insertion in the Stat2 gene (16). Here, we will limit subsequent discussion to IFN-γ-induced STAT1 activation, which has been more extensively studied.
Naive CD4+ T cells express the constitutive component of the IL-12 receptor (IL-12Rβ1), but low or undetectable levels of the inducible component of the IL-12 receptor (IL-12Rβ2), conferring efficient responsiveness to IL-12 only after upregulation of IL-12Rβ2. Concurrently with TCR signaling, IFN-γ activation of STAT1 drives initial up-regulation of the Th1-specifying transcription factor, T-bet (encoded by Tbx21). T-bet, a member of the T-box family of transcription factors, was originally cloned as a repressor of IL-2, but it was subsequently demonstrated that ectopic expression of T-bet conferred Ifng expression and that CD4 T cells lacking T-bet had a profound impairment in their ability to differentiate into Ifng competent Th1 cells (17). The expression of T-bet induces transcription of Il12rb2, expression of which generates a functional IL-12 receptor composed of IL-12Rβ1 and IL-12Rβ2, thereby enabling IL-12 signaling via activation of STAT4 (18, 19).
Recent kinetic and mathematical modeling of Th1 development has substantiated this developmental progression, showing that differentiating Th1 cells pass through at least two phases: an early TCR–IFN-γ–STAT1 dependent programming, which induces low levels of T-bet and induction of IL-12Rβ2, followed by a late IL-12–STAT4–T-bet–dependent commitment driven by STAT4-enhanced T-bet expression and heightened Ifng gene expression (20). In addition to activating increased competency of the Ifng locus, T-bet and STAT4 activate a number of additional genes that contribute to the Th1 differentiation program. STAT4 and T-bet act coordinately to induce the Th1-specific transcription factors Hlx and Runx3 (21–23). Whereas STAT4 plays a significant role in the upregulation of Etv 5 (ERM), a member of the Ets family, it remains to be seen whether T-bet is involved in this process (24). Thus, Runx3, Hlx and Ets family members cooperate with STAT4 and T-bet to confer Th1 identity, albeit through mechanisms that are not yet well defined.
Both STAT4 and T-bet play non-redundant roles in Th1 specification (22). STAT4-deficient CD4+ and CD8+ T cells fail to respond to IL-12 and are unable to undergo Th1 and Tc1 differentiation, respectively (25, authors’ unpublished findings). In contrast, T-bet–deficient mice have profoundly impaired Th1 responses, yet CD8+ T cells that lack T-bet readily acquire Ifng competence in an IL-12–dependent, T-bet–independent manner (26). Studies to understand this differential requirement of T-bet led to the identification of another T-box family member, Eomesodermin (Eomes), which mediates T-bet–independent acquisition of Ifng competence (27). Despite the availability of mice carrying a floxed Eomes allele, a possible role for Eomes in Th1 differentiation has not been directly evaluated. However, CD8+ T cells that lack both T-bet and Eomes fail to express Ifng and aberrantly express IL-17 in response to viral challenge. Thus, while acquisition of Ifng competence in cytotoxic T cells may be T-bet–independent, at least one T-box family member (Eomes) is required (28).
More recent studies have also demonstrated an important role for Runx3 that accounts for the disparity in T-bet requirement between CD4+ and CD8+ T cells. Although acquisition of Ifng competence is dependent on Runx3 in both CD4+ and CD8+ T cells, the availability of Runx3 in these two cell types differs significantly (23, 29). While CD8+ T cells upregulate Runx3 during thymic selection, generation of CD4 single-positive (CD4SP) cells requires downregulation of Runx3, leading to different levels of this factor in naive CD4+ and CD8+ T cells.
While STAT4 activates Th1-specific induction of Etv5, and cooperates with Etv5 to induce IFN-γ expression in Th1 cells (24), it remains to be seen whether STAT4 or T-bet play important roles in induction of another key Ets family member, Ets1. STAT4 appears to cooperate with Ets1 to sustain T-bet expression. Consequently, Ets1−/− T cells that are cultured under Th1 polarizing conditions upregulate T-bet transiently, but fail to sustain its expression (30). Notably, over-expression of T-bet in Ets1−/− T cells fails to restore Ifng transcription, indicating that Ets1 also contributes to Th1 differentiation through mechanisms that are independent of its effects on maintenance of T-bet expression (30). While Ets-1 and T-bet may cooperate to activate Ifng directly, it is also plausible that their function in regulating Ifng transcription is indirect through the activation of Runx3 and Hlx1. Studies on the role of Ets1 in thymocyte development suggest that the latter is more likely (31).
An emerging theme in our understanding of peripheral regulation of T-cell fates is the appreciation of shared attributes between Th1 differentiation and the generation of CD8SP cells in the thymus. Ets-1 and Runx3 are pitted against Gata3 and ThPOK in the latter, while in IL-12 driven polarization of CD4+ T cells, STAT4 and T-bet upregulate Ets-1 and Runx3 to silence GATA-3 and confer Th1 fate. It remains to be examined whether ThPOK, which cooperates with GATA-3 to facilitate generation of CD4SP thymocytes, contributes to Th2 development as well (32–34). Thus, in addition to activating the Th1 developmental program, Th1 specific transcription factors block the expression of several genes expressed by alternate T cell lineages. Repression of GATA-3 and IL-4 in particular is essential for Th1 commitment; accordingly, Tbx21−/− T cells have higher levels of Gata3 transcripts (23, 35). In recent years, the roles of several other families of transcription factors involved in early hematopoietic development including Ikaros, Notch, IRF, and mediators of β-catenin signaling have been ascribed roles in T-cell subset development as well (36).
Mapping of cis regulatory elements in the Ifng locus and their epigenetic modifications during effector T-cell differentiation
Though multiple transcription factors have been identified that regulate Th1 cell specification and Ifng expression, we are early in our understanding of sites in the Ifng locus to which these factors are recruited, the additional factors with which they complex, and how their binding influences transcriptional activity. Mapping of cis elements in the Ifng and IFNG loci began about 20 years ago with identification of intronic DNase I hypersensitive (HS) sites contained within an 8.6 kb fragment of human IFNG gene (37–39). While these intronic HS sites behaved as enhancers in vitro, a transgene containing the 8.6kb fragment of the IFNG locus was insufficient to direct Th1/Tc1 specific expression in a transgenic mouse model system (40, 41). Similarly, transgenic mice that included a 3.4 kb fragment of the Ifng promoter also lacked appropriate lineage- and activation-dependent expression (42). In contrast, mice carrying a human IFNG BAC transgene that contained ∼90kb of upstream and downstream flanking sequence recapitulated lineage-specific expression of IFNG in effector T cells, indicating that distal cis elements were required to direct appropriate expression of IFNG (40). This correlates well with the recently identified boundary elements that indicate spans of ∼180 kb and ∼140 kb for the IFNG and Ifng loci, respectively (43, 44)(see below).
Regulatory DNA elements tend to be conserved across species, enabling database searches for homologous stretches of DNA in the Ifng locus. We and others have identified at least nine conserved non-coding sequences (CNSs) that contribute regulatory functions in the Ifng locus, and it is likely that additional elements will be identified. A CNS is defined as a non-coding region of at least 100 bp that retains sequence homology of 70% or greater across multiple species (45, 46). Initial studies that identified CNS1 and CNS2 (subsequently renamed CNS-6 and CNS+17, respectively) (47, 48) and later CNSs-22, −34 and −55 (49), relied on conventional DNase I mapping and chromatin immunoprecipitation (ChIP) analyses of histone modifications. Newer technologies that couple ChIP and DNase I hypersensitivity with massive parallel sequencing (ChIP-seq and DNase-seq) or microarray (ChIP-chip and DNase-chip) approaches allow for unbiased and more comprehensive mapping of chromatin structure (reviewed in 50–53) and have accelerated discovery of candidate cis elements. Using these techniques, we and others have demonstrated that in the Ifng locus both distal elements and proximal elements are subject to considerable lineage-specific epigenetic modifications that act to either facilitate or limit recruitment of key trans-acting factors.
In our own studies, DNase-chip was employed to compare accessibility of multiple gene loci in naïve CD4 T cells and Th1, Th2 and Th17 effector populations (54, 55). In general, DNase I HS sites coincided remarkably well with conserved non-coding sequences (CNSs). The Ifng locus was found to contain a surprising number of HS sites in naïve CD4+ T cells, although the number and sensitivity of sites was greatly increased upon Th1 differentiation and was diminished upon Th2 differentiation (Fig. 2). Intriguingly, with a few notable exceptions, the HS map of the Ifng locus in Th17 cells is remarkably similar to that of Th1 cells (55), consistent with the ability of Th17 cells to acquire high-level Ifng expression late in their development (discussed below).
Fig. 2. DNase I hypersensitivity profiles of CD4+ T cells.
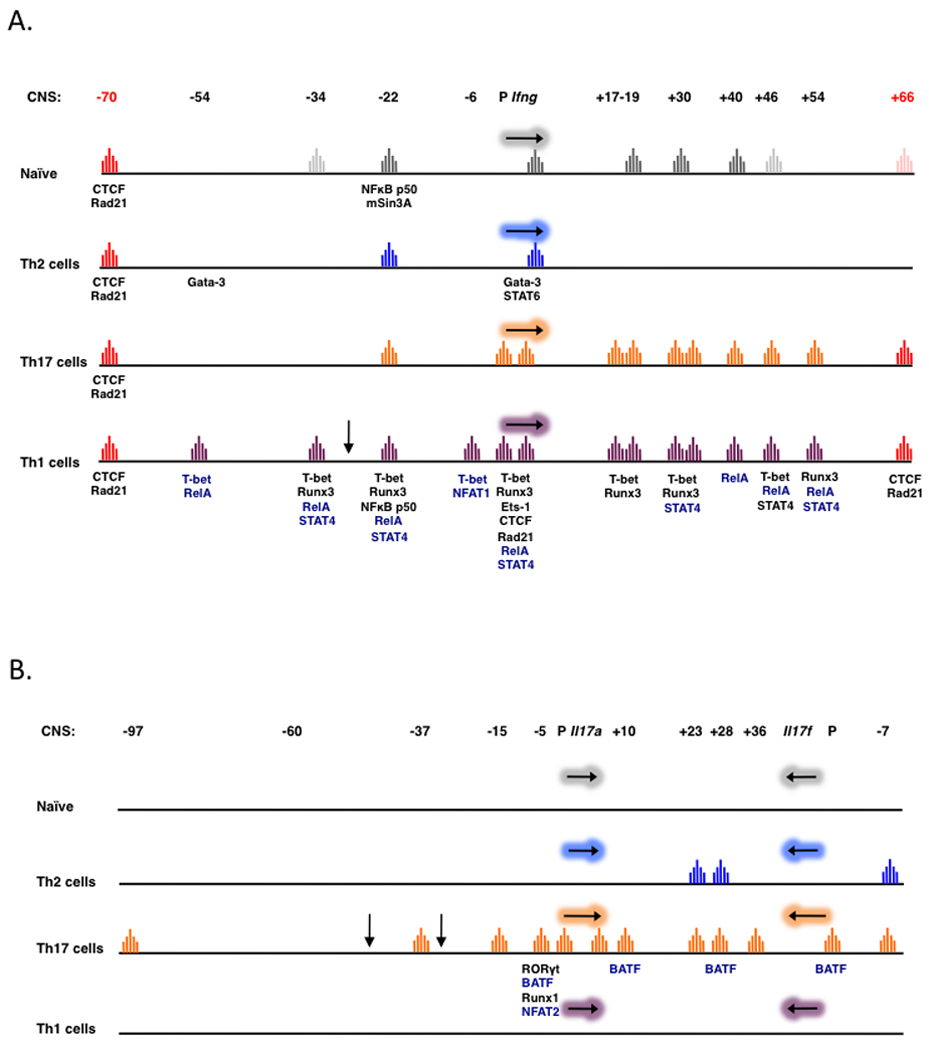
At the top of each figure a schematic displays the extended Ifng locus (A) or the Il17a–Il17f locus (B) composed of the gene(s), their associated promoter (P) and numeric descriptions (based on distance from the transcriptional start site) of identified conserved non-coding sequences (CNS). Regions of DNase I hypersensitivity, as determined by DNase-chip, are designated by colored peaks when corresponding to a CNS from resting naive or resting primary Th1, Th2, or Th17 cells. A downward arrow identifies a hypersensitive site that is not within a conserved region. CTCF-binding boundary elements are in red. Transcription factors that are recruited to the indicated conserved elements of each locus are listed. Paler peaks indicate activation-dependent HS sites within CNS elements (e.g. CNS-34 in Naive cells, A) or less intense HS peaks (e.g., +66 site in Naive cells, A)
The cis elements that show the most stringent Th1-specific hypersensitivity include the promoter, which is uniquely accessible in unstimulated Th1 cells, and the activation-induced proximal upstream element, CNS-6. The restriction of the promoter to unstimulated Th1 cells is consistent with global genome analyses that identify DNase I HS at proximal upstream elements of genes that are poised for transcriptional activity (56). In this regard, it is notable that the promoter and CNS-6 are not accessible in unstimulated Th17 cells, despite widespread HS at distal sites (55). In contrast to the restricted accessibility of the promoter and CNS-6, sites in CNS-22, the first intron (CNS+1.5), as well as the upstream boundary element at −70 kb (a non-conserved element), show lineage-unrestricted DNase I hypersensitivity, consistent with possible roles for these elements in early structural organization of the Ifng locus. Alternatively, these regions may be important in maintaining contacts with Th2 and Th17 loci to facilitate coordinated induction or silencing during effector differentiation (57). In Th1 cells, all of the Ifng CNSs characterized to date have been shown to interact with the Ifng gene using chromosome conformation capture (3C), suggesting that these distal elements are coordinately recruited to the promoter to regulate transcription of Ifng (44).
Although a number of distal elements in the Ifng locus are hypersensitive to DNase I digestion in naive CD4+ T cells, only two distal elements, CNSs −22 and −34, appear to be in an ‘active’ state as assessed by histone methylation (H3K4 ) status. Although accessible in naive CD4+ T cells, distal elements downstream of the Ifng gene (e.g. CNSs +17, +40, +46) appear to be kept in check by a preponderance of EZH2–mediated H3K27 trimethylation (H3K27me3). Upon Th1 differentiation, permissive H3K4 methylation marks are detected at all Ifng CNSs. T-bet and STAT-4 are critical in inducing these changes. Further, removal of repressive H3K27me3 marks from the Ifng locus also appears to be dependent on T-bet. Although recruitment of histone-modifying enzymes is a conserved function of T-box factors, whether T-bet is directly responsible for mediating these changes is currently unclear (58, 59). While our studies support this hypothesis, it is also plausible that T-bet–dependent induction of other trans-factors, including Runx3 and Hlx, may indirectly regulate permissive remodeling of the Ifng locus, and will require further study.
Stat4−/− CD4+ T cells are resistant to IL-12–dependent Th1 polarization and therefore fail to up-regulate T-bet. In addition to impaired Th1 specific remodeling observed in the absence of T-bet, Stat4−/− cells also are deficient in long-range acetylation (60, authors’ unpublished findings}. In contrast, Tbx21−/− T cells cultured under Th1 conditions acquire acetylation marks across the extended Ifng locus, resembling those of cells derived from wild type mice (54). Although IL-12–dependent maturation of naïve CD4+ T cells leads to acquisition of both H4 hyperacetylation and H3K4 methylation across the extended locus, the relative contributions to accessibility of the Ifng locus remain to be defined. Nevertheless, IL-12–dependent differentiation of CD4+ and CD8+ T cells leads to extensive permissive remodeling of the Ifng locus, allowing these cells to efficiently transcribe Ifng upon reactivation.
Transcription factor recruitment to cis elements in the Ifng locus
As stated previously, CNSs tend to be regulatory in nature and this is primarily achieved by their ability to recruit DNA-binding proteins that either enhance or inhibit transcriptional activity of nearby genes when they are “accessible.” Each CNS in the Ifng locus that has been studied contains multiple transcription factor binding motifs that likely recruit various trans-factors, and nearly all CNSs have been shown to bind at least one transcription factor in Th1 cells (Fig. 2). Over the course of Th1 differentiation, the binding of trans-acting factors to each CNS is likely to be a dynamic process, although relatively little is known regarding the spatiotemporal utilization of these sites as Th1 development proceeds.
Our current understanding of the mechanisms that contribute to the transcriptional state of the Ifng locus have come from analyses of recruitment of three trans factors — STAT4, T-bet, and Runx3 (Fig. 2). Often these factors appear to be co-recruited, consistent with a role for cis elements in facilitating the assembly of multi-component transcriptional complexes that regulate gene expression (49, 60, 61). Based on their accessibility in naive CD4+ T cells, we speculate that CNSs −34 and −22 function as pioneering elements that initiate chromatin reorganization of the Ifng locus in developing Th1 and Tc1 cells, likely through the recruitment of both T-box (T-bet or Eomes) and STAT family members, for which each element contains consensus binding sites. Although multiple CNSs in the Ifng locus recruit STAT4 downstream of IL-12–driven acute activation of mature Th1 cells, constitutive association of STAT4 in resting Th1 cells is restricted to only two of these sites: CNSs −22 and +46 (54). T-bet binding, on the other hand, is readily detected at multiple Ifng CNSs prior to reactivation, including −34, −22, +30, +46 and the Ifng promoter (49). Recruitment of T-bet to CNS-6 is highly dependent on acute reactivation (47, 48). Although Runx3 recruitment to the Ifng locus has not been examined directly, recruitment of its partner CBFβ has provided insight into the role of Runx3 (61). Consistent with earlier predictions that Runx3 cooperates with T-bet, CBFβ recruitment closely overlaps with recruitment of T-bet, with the sole exception of CNS+46. Collectively, these studies support the view that CNSs −34, −22, +30 and +46 are key regulatory hubs that recruit multiple trans factors to facilitate Th1-specific remodeling of the Ifng locus.
While the roles of several key trans-acting factors, including STAT4, T-bet and Runx3, have been examined by gene-targeted deletion, few studies have interrogated the functions of cis elements through this approach. In the only such study of the Ifng locus, we evaluated the role of CNS-22, which serves as a recruitment site for STAT4, T-bet and Runx3, using a BAC-transgenic system (49). Deletion of CNS-22 from the transgenic locus resulted in marked ablation of transgene reporter expression in Th1 and Tc1 cells, as well as NK cells, suggesting that CNS-22 plays an important role in regulating Ifng gene transcription. As noted above, this element is highly DNaseI sensitive in naive T cells and all effector lineages, and in addition to its clustering of consensus binding sequences for several positive regulators of Ifng transcription, it also contains potential binding sites for negative regulators, including GATA-3 and STAT6 (55). Thus, although deletion of CNS-22 in this model did not induce aberrantly enhanced expression of the transgene that might result from loss of silencing in non-Th1 cells, the global deletion of both positive and negative regulatory functions within this element likely precluded this result. Additional studies in which selected mutation or deletion of specific trans-factor binding sites is performed will be necessary to parse out the functions of individual binding sites within this, and other, composite CNS elements. Other cis elements whose function would merit testing either by direct deletion or by careful transgenic analyses include CNSs −34 and +30 and +46.
Defining boundaries of the Ifng locus
The Ifng and IFNG genes in mouse and human share a common nearest gene neighbor downstream (Dyrk2/DYRK2) that is quite distant: ∼420KB and ∼500kb, respectively. They differ in their upstream neighbors due to an ancient insertion of repetitive elements in a common ancestor of the mouse that deleted an ancient IL26 homolog that is preserved in humans. In humans, the IL26 gene resides between ∼40–65 kb upstream of IFNG, whereas in mouse, the nearest upstream neighbor is Il22, which resides ∼245 kb upstream of Ifng in a position orthologous to the human IL22 gene. Because IL-22 and IL-26 are IL-10 family cytokines that are primarily expressed by Th17 cells, and not Th1 cells, an insulator or boundary element must separate the two loci to avoid regulatory crosstalk between these cytokines.
Insulator elements thought to define the boundaries of the Ifng and IFNG loci were identified recently based on the identification of recruitment of two molecules, the CCTC-binding factor (CTCF) and a cohesin component (Rad21) to these sites (43, 44). Although cohesins were originally identified as multi-component complexes that play an important role in sister chromatid cohesion during meiosis in yeast cells (62, 63), it was recently found that recruitment of cohesin analogs in mammals is restricted to DNase I HS sites (64, 65), the majority of which contain CTCF consensus sequence motifs (64). CTCF is a DNA-binding zinc-finger protein that typically binds to insulator elements that inhibit interactions between enhancers and promoters (66, 67). It has become apparent that CTCF can target cohesin complexes to both organize the topology of long-range intra-chromosomal interactions and serve as both insulator and architectural elements for organizing chromatin (43). These findings have also provided a new means to screen for insulator or boundary elements in mammalian genomes, through the identification by ChIP analysis of sites where CTCF and cohesin components (e.g. Rad21) co-localize.
Boundary elements thought to insulate Ifng from neighboring gene loci were identified based on recruitment of CTCF/Rad21 to sites −70 kb upstream and +66 kb downstream of Ifng (44). Homologous sites were identified in the human IFNG locus, where the upstream site resides within the IL26 gene (43, 44). Interestingly, conserved HS sites within a CNS within the Ifng and IFNG genes themselves serve as a third CTCF/cohesin recruitment site. Whereas recruitment of CTCF to site −70 was observed in naïve, Th1 and Th2 cells, CTCF recruitment to Ifng intron 1 and site +66 was restricted to Th1 cells (44). Our own data suggest that differential chromatin accessibility of these sites in distinct T lineages might be responsible for this disparity. While the CTCF recruitment site at −70 shows lineage-unrestricted DNase I hypersensitivity, both the intron 1 and +66 sites have prominent hypersensitivity only in Th1 cells (54). In fact, T-bet is recruited to HS sites in the first intron and at +66 of the Ifng locus in Th1 cells and appears to be responsible for chromatin reorganization that allows for CTCF-dependent recruitment of cohesins to these sites (44).
In Th1 cells, both the −70 and +66 boundary elements make contacts with the Ifng gene itself. It is thought that this looping is facilitated by interactions with the intronic CTCF binding site (44), although this remains to be directly tested. Of note, mapping of these interactions relied on chromosome conformation capture (3C), which does not discriminate between inter-chromosomal and intra-chromosomal interactions. Consequently, it is possible the two Ifng alleles interact with one another, which remains to be addressed. In any case, these studies suggest that the murine Ifng locus extends from about −70 kb to +66 kb relative to the transcription start site and that these boundary elements employ CTCF dependent recruitment of cohesins to both insulate transcriptional activity at the Ifng gene locus from neighboring gene loci and provide a mechanism to approximate the distal upstream and downstream cis elements to the Ifng promoter.
In our study of the Ifng BAC transgenic mouse model described above, while the inclusion of ∼160 kb of genomic sequence flanking the Ifng locus and conferred lineage–specific expression in Th1, Tc1 and NK cells (49, 68), expression was limited to ∼40–50% of IFN-γ+ cells, irrespective of transgene copy number or insertion site. Notably, although this transgene contained the +66 kb downstream CTCF element that was subsequently identified, it lacked the upstream CTCF element at HS-70, suggesting that the lack of absolute concordance of reporter and endogenous Ifng expression might be due to poor insulation of upstream cis-regulatory elements, deficient approximation of these upstream elements to the Ifng promoter, or both. Whether inclusion of another 10 kb of upstream sequence in the BAC reporter transgene would produce 100% concordance of reporter expression with the endogenous locus remains to be determined.
Actions of acute transcription factors at the Ifng locus
While lineage-specific transcription factors play essential roles in conferring transcriptional competence at cytokine gene loci, acute transcription factors that are often shared by multiple lineages play equally important roles in acute induction of gene transcription in mature T effectors. This is particularly true of trans-acting that are factors activated downstream of TCR signaling, which are largely conserved between T-cell subsets. Indeed, genes that encode signaling intermediates involved in the TCR signaling cascade are often expressed prior to expression of a functional TCR, and play important roles in positive and negative selection of developing thymocytes. Studies to understand the roles of acute transcription factors in Ifng production have focused on four families of transcription factors: the NFAT, AP-1, NF-κB and STAT families. Lineage-specific transcription factors such as T-bet often partner with these molecules both to determine cell fate and to govern stimulus-dependent acute gene transcription.
In addition to TCR-driven gene expression, Th1, Th2 and Th17 cells have retained TCR-independent pathways that induce effector cytokine production (69–71). These TCR-independent pathways are activated by the coordinated actions of two cytokine receptor pathways: one, an IL-1R family member that activates NF-κB (e.g. IL-1R for Th17 or IL-18R for Th1), and a second that activates a JAK-STAT signaling cascade (e.g. IL-23 for Th17 or IL-12 for Th1) (70). Through these TCR-independent pathways, mature effector T cell cytokine expression can be recruited in a manner analogous to that of innate immune cells (e.g., NK cells), which utilize the same pathways. Consequently, the cis regulatory elements of effector cytokine loci must have evolved to accommodate both TCR-dependent and -independent mechanisms that activate their transcription.
A critical role for NF-κB family transcription factors in activating Ifng
A role for NF-κB family members in activating Ifng transcription was initially recognized through the identification of NF-κB binding sites in the Ifng promoter (72, 73). Subsequent studies have extended the importance of NF-κB in Th1 differentiation and Ifng regulation, and have identified distinct roles for NF-κB family members. The NF-κB family is comprised of five members that bind DNA as homo- or heterodimers: p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), c-Rel (Rel) and RelB. In addition to an N-terminal Rel homology domain (RHD) that binds DNA, the three ‘Rel’ members of the NF-κB family also contain a C-terminal domain that enables these molecules to transactivate gene expression. In contrast, NF-κB p50 and p52 lack a transactivating domain and act as repressors of transcription unless heterodimerized with a family member that contains a transactivating domain (Reviewed in 74, 75).
The participation of NF-κB activation in both TCR- dependent and -independent induction of Ifng has led several groups to examine the role of individual NF-κB family members in Th1 differentiation and Ifng expression. CD4+ T cells that lack RelB, which in complex with p52 is activated via a “non-canonical” mode of NF-κB signaling, are severely impaired in their ability to undergo Th1 polarization. In addition to impaired proliferation, Relb−/− T cells show significant defects in upregulation of T-bet and phosphorylation of STAT4, indicating that RelB acts upstream of T-bet and STAT4 — and making difficult studies of the possible participation of RelB in acute transcription of Ifng (76). The importance of the canonical, or ‘classical’, mode of NF-κB activation was documented using a transgenic mouse that expressed a dominant negative form of IκB [(IκBα(ΔN)] under the control of the proximal Lck promoter, which restricts transgene expression to T lineages. IκBα(ΔN) is a mutant of IκBα that can be phosphorylated in response to activating classical NF-κB activating signals, but does not undergo ubiquitin-mediated degradation, thereby acting as a cytosolic sink that sequesters NF-κB dimers from nuclear translocation (77). In this model, both c-Rel– and RelA–mediated signaling are compromised. Under Th1-polarizing conditions, CD4+ T cells from IκBα(ΔN) mice displayed impaired antigen-driven proliferation and decreased IFN-γ production. Taken together, these studies established that both canonical and non-canonical activation of NF-κB could play important roles in Th1 differentiation and IFN-γ production.
Although studies using IκBα(ΔN) transgenic mice established the importance of canonical NF-κB activation in Ifng transcription, they provided little information on the specific contributions of c-Rel and RelA. To address a potential role for c-Rel in regulation of Ifng transcription, c-Rel–deficient mice were examined and were found to have impaired Th1 responses in vivo. However, this is primarily attributed to impaired IL-12 production by myeloid cells (78–80). Ex vivo, c-Rel-deficient CD4+ T cells cultured under Th1 polarizing conditions show impaired proliferation and Ifng production, both of which are rescued by addition of exogenous IL-2, indicating that c-Rel has less of a role in the induction of Ifng competence or transcription than in driving cell cycling required for Th1 cell differentiation (79, 81). Thus, analysis of c-Rel-deficient mice established a proximal role for NF-κB activation in T cell proliferation and suggested that acquisition of Ifng competence in Th1 cells might be independent of direct actions of c-Rel.
In aggregate, these studies hinted at an important role for RelA in acute Ifng induction, although this has been difficult to examine directly because Rela−/− mice die in utero (82). Generation of mice with T cell-specific deletion of Rela has recently allowed us to directly examine the role of RelA in Ifng transcription in both CD4+ and CD8+ T cells (54, 83). We find that in contrast to naïve c-Rel- and RelB-deficient T cells, naive RelA-deficient T cells proliferate well in response to antigen, but demonstrate profoundly impaired IFN-γ production in response to both TCR-dependent and -independent reactivation. These findings establish that RelA plays an indispensable role in Ifng transcription, leading us to define cis elements in the Ifng locus that might be targeted by RelA under conditions of TCR-dependent and -independent activation, and to assess the composition and mechanisms of recruitment of RelA-containing NF-κB complexes.
TCR-independent transcription of Ifng: roles for multiple distal elements that coordinately recruit STAT4 and RelA
Upregulation of the IL-18 receptor component IL-18R1 in developing Th1 cells is induced downstream of IL-12 signaling, conferring on mature Th1 cells the capacity for TCR-independent activation of Ifng transcription by concomitant IL-12 and IL-18 signaling. Originally described in Th1 cells, IL-12 plus IL-18–induced expression of Ifng was subsequently extended to NK cells, CD8+ T cells and NKT cells (69, 84–87). Like other IL-1 and Toll-like receptor family members, the IL-18 receptor is a potent activator of NF-κB, and critical roles for STAT4 and NF-κB in relaying signals downstream of the IL-12 and IL-18 receptors, respectively, were quickly recognized (69, 86, 88). A number of studies have explored mechanisms by which theses signals converge to activate Ifng gene transcription. This has led to several key observations, including the identification of de novo synthesis of GADD45β as a prerequisite for induction of IL-12 plus IL-18-dependent induction of Ifng (89). While NF-κB activation is a shared requirement for TCR-dependent and -independent activation of a number of cytokine genes, including Ifng, NFAT activation is dispensable for cytokine-induced Ifng expression (90). Accordingly, we recently reported that NF-κB response elements play an essential role in activating Ifng transcription in response to both these signals. In response to IL-12 plus IL-18, at least four distal elements — CNSs −34, −22, CNS+46 and CNS+54 — recruit RelA to drive Ifng transcription.
Although IL-12 is dispensable for NF-κB activation in Th1 cells, IL-18 driven Ifng transcription is quite limited in the absence of IL-12. In recent studies, we found that coordinate IL-12 signaling was necessary for optimal recruitment of RelA to all NF-κB targets in the Ifng locus (54). Further, it was shown that STAT4 and RelA are co-recruited to each of these elements, and that in the absence of IL-12, IL-18-induced activation of NF-κB was insufficient to induce RelA binding to these sites. Although previously it was assumed that STAT4 acted indirectly to augment IL-18 driven NF-κB activation, our findings indicate instead that STAT4 acts directly to enable binding of RelA-containing NF-κB complexes to multiple distal elements. Thus, the requirement for cooperative signaling downstream of IL-12 and IL-18 receptors appears due to a requirement for STAT4 to ‘chaperone’ binding of NF-κB to consensus sequences. Although our studies have been limited to Th1 and Tc1 cells, we believe that this may represent a highly conserved regulatory mechanism that will likely be extended to other Ifng-expressing cells, such as NK and NKT cells.
Generation of spliced Ifng transcripts in Th1 cells increases ∼100 fold within 2 hours of activation by IL-12 plus IL-18 (unpublished observations). Our findings indicate that it takes a complex consortium of trans-acting factors recruited to multiple distal and proximal regulatory elements to drive this remarkable increase in the rate of transcription. Although interactions between STAT and NF-κB family members have been described previously, the finding of a considerable multiplicity of cis elements that recruit cooperative binding of STAT4 and RelA suggests that this synergistic interaction might have acted as a driving force in the evolution of distal elements that regulate Ifng transcription, and might contribute to the high transcriptional rate that is characteristic of this gene. It remains to be determined whether STAT4 and RelA complexes further cooperate with GADD45β and other components of the p38MAPK cascade to augment IL-12 plus IL-18-driven Ifng activation.
Ifng transcription induced by antigen re-encounter
TCR signaling in Th1 cells initiates several cascades that ultimately activate the NFAT, AP-1, and NF-κB pathways, each of which appears important in the acute activation of Ifng transcription induced by antigen re-encounter. As is the case for Ifng transcription driven by IL-12 plus IL-18, TCR-driven transcription of Ifng is critically dependent on RelA, as its deletion results in profoundly impaired Ifng expression for which there is little or no compensation by other NF-κB family members (54). At least two distal elements (CNSs −34 and +40) recruit RelA in response to TCR-driven activation of Th1 cells (54). The recruitment of RelA to CNS+40 is uniquely induced by TCR-driven activation.
Although the precise mechanisms by which TCR signaling activates RelA are not fully understood, an important role for CD28 co-signaling is apparent, as CD28-mediated costimulatory signals considerably enhance RelA recruitment to the Ifng locus (unpublished data). This is likely due, at least in part, to the phosphorylation of IκB by phospho-inositol (PI)-3-kinase. Because TCR and CD28 signaling do not activate STAT4, which is necessary for binding of RelA induced by IL-12 plus IL-18, TCR-dependent recruitment of RelA must occur through alternate mechanisms yet to be defined. It is not unlikely that activation of NFAT or AP-1 family members downstream of the TCR might cooperate with RelA to effect its binding to target cis elements, analogous to the requirement of STAT4 driven by IL-12 signaling. Although NFAT1−/− mice show significant impairment in Ifng transcription upon Th1 differentiation (91), it has not been determined whether NFAT1 is required for RelA binding. Further, while NFAT1 influences Ifng transcription, direct recruitment of NFAT has only been demonstrated to the proximal promoter and to one distal element, CNS-6 (47, 91, 92). Based on the presence of conserved NFAT binding consensus sequences in several sites of RelA recruitment, we speculate that co-recruitment of NFAT and NF-κB family members to these elements might be essential for maximal antigen-driven Ifng transcription.
The role of other signaling cascades activated by TCR signaling that contribute to the induction of Ifng transcription is also poorly defined. Mitogen-activated protein kinases (MAPKs) have been shown to function in both TCR and costimulatory signaling cascades (93) and the p38 MAPK and c-Jun amino terminal kinase-2 (JNK-2) pathways have received considerable attention for their roles in Th1 differentiation. Jnk2−/− CD4+ T cells are significantly impaired in their ability to secrete IFN-γ upon Th1 polarization (94). Pharmacological inhibitors of p38 MAPK specifically inhibited the generation of Th1 cells and acquisition of Ifng competence (95). Similarly, mice in which expression of a dominant negative p38 MAPK was restricted to T cells showed impaired Th1 function, and complementary analyses indicated that expression of a constitutively active form of p38 MAPK enhanced Ifng production (95). Several kinases that act upstream of p38 MAPK and JNK signaling cascades, including MKK3, GADD45β and GADD45γ, have also been implicated in the generation of Th1 cells (96–98). Establishing relationships between these signaling components and the downstream trans-activators they recruit to the Ifng locus will require more study. A re-evaluation of the impact of deletions of these factors on the Th1 transcriptome and epigenome would be invaluable to gaining a better understanding of interactions between these convergent signaling cascades that set in motion the Th1 differentiation program and activation of Ifng expression.
Promiscuity of the Ifng locus: implications for T-lineage plasticity
Recent reports have identified plasticity of some committed effector and regulatory T cells that has led to a reevaluation of the stability of T-cell lineages. Th17 cells and induced Treg (iTreg) cells in particular appear to have evolved mechanisms that permit their reprogramming in response to changing environmental cues, with important implications for host defense and autoimmunity (Reviewed in 99, 100). Whereas epigenetic modifications of developmentally regulated genes have been thought to reinforce transcription factor networks as a basis for T lineage specification (9), emerging data indicate that epigenetic modifications are more dynamic than previously appreciated, enabling shifts in expression or silencing of ‘signature’ genes that characterize distinct T-cell subsets.
While we are still in the early days of defining rules that regulate T-lineage transitions, a recent study that performed global ChIP-seq based analyses of H3K4 (permissive) and H3K27 (repressive) trimethylation in naive, Th1, Th2, and Th17 cells provided interesting insights (101). Whereas the promoters of cytokine genes tended to show highly constrained, lineage-specific histone modifications, promoters of genes that encode key lineage-determining factors showed a more complex pattern of epigenetic marks. Notably, while the promoters of Tbx21, Gata3, and Runx3 demonstrated a bivalent state (i.e. both permissive and repressive histone modifications often associated with genes poised for differential expression fates) in all lineages other than the canonical lineages with which they are linked, Rorc and Foxp3 completely lacked permissive modifications in Th1 and Th2 cells. These findings are consistent with reported transitions observed between iTreg and Th17 cells as well as the transition of Th17 cells towards Th1 cells but not the reverse.
Recent study of the Th17 to Th1 transition has been particularly informative, as it has provided a very tractable model for defining both the activation and repression of distinct effector cytokine gene loci: the Ifng locus and the Il17a–Il17f locus, respectively (55). Our group and others have demonstrated that Th17 cells derived ex vivo can retain low-level expression of the IL-12Rβ2 chain, resulting in responsiveness to IL-12 that induces transition into effectors that share many features with classical Th1 cells (15, 102, 103). The IL-12–induced transition of Th17 precursors involves the rapid extinction of the Th17 genes with reciprocal induction of genes associated with the Th1 differentiation program (15), and appears to be irreversible.
Akin to classical Th1 differentiation from naïve CD4+ T cells precursors, the Th17 to Th1 transition induced by IL-12 is dependent upon both STAT4 and T-bet. The importance of STAT4 in transcriptional competence of the Ifng gene is reinforced by the finding that over-expression of T-bet in the absence of IL-12 signaling is insufficient to induce this transition. As in classical Th1 cells (54), STAT4 recruitment to multiple sites in the Ifng locus of transitioning Th17 cells, including CNSs −34, −22 and +46, was rapidly detected following IL-12 signaling. Although, as noted above, the Ifng promoter is epigenetically repressed in Th17 cells, most of the remaining cis regulatory elements in the Ifng locus are DNase I hypersensitive and upon IL-12 signaling rapidly undergo histone modifications that closely resemble those of Th1 cells (55). A notable exception is CNS-6, which contains an activation-dependent HS site that is conspicuously absent and epigenetically repressed in Th17 cells prior to the IL-12-induced transition. Interestingly, although this cis element binds T-bet and NFAT downstream of TCR signaling, it does not bind STAT4 or RelA, yet appears to be particularly important for high-level Ifng transcription.
The finding that activated Th17 cells especially demonstrate a DNase I hypersensitivity pattern that is quite similar to that of activated Th1 cells, despite their development in the absence of Th1-specifying cytokines (e.g. IFN-γ or IL-12) or expression of T-bet, raises interesting questions as to the requirements for remodeling of the Ifng locus in immune cells. Indeed, a considerable fraction of IL-17–expressing Th17 cells maintained by continuous culture in TGF-β — and in the complete absence of Th1-specifying cytokines — acquire a progressive increase in Ifng co-expression, indicating that substantial Ifng transcription can result in the absence of typical Th1-promoting factors (15). While our studies of transitioned Th17 cells reaffirm the importance of STAT4 and T-bet in conferring high transcriptional competence on the Ifng locus, these factors are clearly not essential for much of the locus remodeling that accompanies effector T-cell differentiation. Indeed, given the number of HS sites that are already present in naive CD4+ T cells, and the loss of a number of these sites in Th2 cells, it would appear that considerable remodeling of the Ifng locus, and thus a highly ‘poised’ configuration, might be the default state of this locus following antigen-driven cell division unless it actively suppressed, as occurs in the context of Th2 differentiation. This ‘default’ remodeling of the Ifng locus may represent an unusual feature of the Ifng locus, as it is in stark contrast to the Il17a–Il17f locus in non-Th17 T cells or the Th2 cytokine gene cluster (Il4-Il13-Il5) in non-Th2 T cells. Indeed, in contrast to Th17 cells, naïve, Th1 and Th2 cells show little or no DNase I hypersensitivity across the extended Il17a–Il17f locus (55) (Fig. 2).
It was recently reported that Th2 cells transferred into naive recipients can transition into Gata-3+T-bet+ progenitors that co-express IL-4 and IFN-γ in response to LCMV infection: so-called ‘Th2+1’ cells (12). This transition was dependent on type I and II interferons and IL-12, and suggests an even greater capacity for re-programming of the Ifng locus than is evident from study of the Th17 to Th1 transition. In contrast to Th17 cells, however, where multiple cis elements that regulate Ifng transcription are found in a permissive state, the Ifng locus is largely silenced in Th2 cells (44, 54, 104, 105). Moreover, unlike Th17 cells, Th2 cells suppress expression of IL12Rβ2 early in their development (106), thereby extinguishing responsiveness to IL-12 and the downstream activation of STAT4 that is central to Ifng transcription. The mechanism by which Th2 cells can be induced to de-repress and activate the Ifng locus is unclear, but is consistent with the growing appreciation that no epigenetic state may be completely irreversible. The extent to which similar transitions occur during physiological immune responses and their importance to normal immune regulation remains to be determined.
Looking outside the Ifng locus: lessons from the Il17a–Il17f locus in Th17 cells, before and after Th1 transition
Compared to the rapidly emerging understanding of the functional organization of the Ifng locus, our understanding of the Il17a–Il17f locus is relatively nascent. While the precise extent of the Il17a–Il17f locus remains to be defined, DNase-chip analysis has identified Th17-specific HS sites spanning ∼185kb surrounding Il17a and IL17f genes, extending from the Pkhd1 gene on one flank to just downstream of the Mcm3 I gene on the other (55, authors’ unpublished data). Like the Ifng locus, most HS sites in the Il17a and IL17f locus correlate well with CNS elements, although a number of prominent CNSs in this locus are not hypersensitive in any of the T-cell lineages analyzed to date (55). Unlike the Ifng locus and the Th2 gene cluster, the Il17a–Il17f locus is essentially devoid of HS sites in naïve CD4+ T cells, and the acquisition of HS sites is largely Th17-specific (Fig. 2). A number of HS sites localize to CNSs in the intergenic region between Il17a and Il17f, consistent with a role in coordinating expression of both genes in developing Th17 cells.
While the functions of most of the HS sites in the locus remain to be determined, several of the sites between Il17a and Il17f undergo activation-induced epigenetic remodeling consisting with their function as acute enhancers (55), and at least two of these sites are also targeted for transcription factor binding very early in Th17 differentiation (107, authors’ unpublished data). Prompted by the discovery of a defect in Th17 development in mice with deletion of the AP-1 family factor BATF (108), intergenic CNS elements in the Il17a–Il17f locus targeted by this factor were identified (107) (Fig. 2). BATF recruitment is also detectable at the Il17f promoter and at CNS-5 upstream of the Il17a gene in activated Th17 cells, where it appears to bind as a heterodimer with Jun-B in proximity to putative RORγt and Runx1 binding sites (109). Interestingly, STAT3, which like BATF is required for Th17 development, appears to target the same two intergenic CNSs that bind BATF in Th17 cells (110), suggesting that these two elements (CNSs +10 and +28) are important cis regulatory nodes for the Il17a–Il17f locus.
A role for NFAT in driving transcription in the Il17a–Il17f locus has come from studies using mice deficient in Tec family tyrosine kinase, inducible T-cell kinase (Itk) (111). Itk regulates phosholipase C-γ (PLC-γ) activation, calcium mobilization and NFAT activation downstream of TCR signaling (112, 113). Consequently, mice lacking Itk have impaired calcium flux and dramatically impaired activation of NFAT signaling in response to TCR ligation. Itk−/− Th17 cells show a specific defect in transcription of Il17a, while expression of other Th17 lineage genes (Il17f, Il21 and Il22) remain unaffected (111). A conserved NFAT2 binding site ∼3kb upstream of the Il17a promoter was partially responsible for NFAT-driven activation of Il17a transcription (111), implicating a specific role for NFAT2 in Il17a transcription.
While roles for NFAT, STAT, Runx, and AP-1 family members in regulating expression of Th17 cytokines have become apparent, few studies have examined the role of NF-κB family members in Th17 cell gene expression. Based on the importance of IL-1 in Th17 differentiation and the TCR-independent induction of IL-17 in concert with IL-23 (71, 114, 115), it is likely that one or more NF-κB family members play essential roles in driving acute transcription of Th17 cytokines, analogous to their effects on Ifng transcription (114).
In contrast to the Ifng locus, which rapidly acquires additional permissive features in Th17 cells that transition to high Ifng expression, the Il17a–Il17f locus is equally rapidly repressed downstream of IL-12 signaling (55). And just as the activation of the Ifng locus requires STAT4 and T-bet, so too does the silencing of the Il17a–Il17f locus. While permissive H3K4 methylation marks accrue at multiple Ifng CNSs during the transition, there is corresponding acquisition of silencing H3K27me3 marks across the Il17a–Il17f locus downstream of IL-12 signaling. Similar epigenetic changes occur in the Rorc locus, and silencing of the Rorc gene may be the most crucial step underlying Il17a–Il17f locus repression, since retroviral over-expression of RORγt renders Th17 cells resistant to IL-12-induced reprogramming. While both STAT4 and T-bet cooperate to silence Rorc, RORγt induction in Th17 cells does not lead to effective silencing at either Tbx21 or Ifng gene loci (101). Notably, in addition to its effects to silence the Rorc locus, STAT4 binds at low levels to the same intergenic CNS elements in the Il17a–Il17f locus that are bound by BATF and STAT3, suggesting that STAT4 could directly suppress the Il17a–Il17f locus by interfering with positive trans-activating complexes that contain these factors.
Closing remarks and future directions
Twenty years since the identification of intronic hypersensitive sites in the Ifng gene, the Ifng locus has emerged as an important model for understanding T-lineage-specific gene regulation. Whereas mapping of important cis-elements in the Th2 cytokine cluster has long received considerable attention, the delineation of cis-elements that control the Ifng locus have made it useful as a counterpoint for defining principals of gene regulation during T-lineage differentiation. Study of the Ifng locus has helped establish CTCF-dependent recruitment of cohesins as a hallmark for the identification of chromatin structural elements that function in long-range chromatin interactions and as locus boundary elements (43, 44), has exposed the modular nature of cis-element utilization induced by differential modes of T cell activation and the interplay of STAT and NF-kB family members in this process (54), and has identified features of its epigenetic ‘promiscuity’ that render it particularly flexible for expression in the context of T-lineage plasticity (55, 102, 103). While a number of aspects of cis regulation of the Ifng locus remain elusive, including possible identities of silencer elements or a locus control region (LCR), the availability of more powerful tools for characterizing functional genomic elements promises rapid advances in this area. In this regard, bioinformatics-based analyses indicate that a number of cis elements are likely to have dual roles enhancers and silencers of Ifng transcription, as exemplified by CNS-22 (49). On the other hand, whether or not the Ifng locus utilizes a LCR is less clear. As previously alluded to, independent transgenic lines of mice that carried a BAC transgenic Ifng reporter showed no overt differences in expression despite significant differences in transgene copy numbers, raising speculation that the Ifng locus may not have a conventional LCR. Finally, while delineation of a surprisingly large number of distal elements that bind STAT-NF-κB complexes in the Ifng locus suggest considerable functional redundancy, whether there is a hierarchical utilization of these elements or whether they function to enable graded rates of gene transcription regulating Ifng transcription remains to be determined. Targeted deletion or mutation of these elements will be needed to definitively establish functional relevance of these elements, and here much work remains to be done.
It is anticipated that physical interactions between gene loci that encode cytokine genes and genes that encode key lineage-specific trans-acting factors will be an area of accelerated discovery in the near future. Until recently, mapping of interactions between gene loci relied almost entirely on the conventional 3C technique, whereas recent studies have employed more unbiased, high-throughput assays to examine chromosomal interactions (57, 116–119). These new approaches have corroborated the view that active and inactive genes are segregated into distinct nuclear compartments; active genes tend to co-localize with other active genes, while inactive genes tend to co-localize as well (119). The introduction of Hi-C (3C coupled to sequencing) and 4C (3C followed by hybridization on microarrays) to study of naive and differentiated T-cell populations is bound to provide invaluable insights into the role of nuclear sub-localization and formation of complex chromatin topologies in regulating T-lineage-specific transcription of genes. Our increased understanding of interactions between trans-acting factors and the cis elements they bind to effect lineage-specific remodeling at these gene loci will be immensely useful in interpreting the data generated from these experiments.
Given the propensity of differentiated T cells to transition in the course of an immune response, we anticipate that inter-chromosomal interactions will play an extremely important role in this process as well. In this respect, the Ifng locus is particularly fascinating, since multiple T lineages retain the ability to turn on Ifng transcription post-differentiation. Whether the Ifng locus is retained in an ‘active’ nuclear sub-domain upon Th2, Th17, and iTreg differentiation, or transition of these cells into IFN-γ+ T effectors is facilitated by relocation of the Ifng locus from an inactive to an active intranuclear compartment remains to be seen. We anticipate that key lineage-specific transcription factors act in concert with CTCF-driven cohesin recruitment to facilitate these inter-chromosomal interactions so as to confer distinct spatial compartmentalization of chromatin in distinct T effector subsets. In any case, chromosomal interaction studies to understand the role of three-dimensional chromatin architecture in regulating T-lineage decisions is likely to be a productive and exciting area in the near future, and should change considerably the way we perceive helper T-cell differentiation.
While the study of cis regulation at multiple gene loci has led to an appreciation of the importance of distal regulation, the precise mechanisms employed by cis elements to modulate transcription at the proximal promoter still remain poorly understood. Clearly, distal elements facilitate trans factor-mediated activation or repression of gene transcription and distal elements often directly interact with the proximal promoter to modulate transcription rates. However, there is relatively sparse information as to precisely how trans factor recruitment to a distal site impacts transcription at the proximal promoter. Since RNA Polymerase II (Pol II)-dependent transcription is ubiquitous in eukaryotic cells, there must be universal mechanisms that allow enhancers to control Pol II transcription rates. Inducible recruitment of p300 and CBP is often used to predict enhancer function, although how recruitment of these molecules confers enhancer activity remains to be understood (120). More importantly, not all enhancers are thought to act in a CBP/p300-dependent fashion. Studies of the γ-globin locus have fostered the idea that transcriptional activation complexes assembled at enhancers serve as chaperones that recruit Pol II to the proximal promoter (121). More recent studies to identify enhancers that are responsive to neuronal activity have established that at least a subset of enhancers recruit RNA Poll II and initiate transcription of non-coding transcripts at these enhancers (eRNA) (122). Indeed, Pol II recruitment to multiple enhancers that impact Ifng transcription has been observed (123, authors’ unpublished data), although whether or not this associated with initiation of eRNA synthesis remains to be seen. Enabled by the rapid advances in identification of multiple functional elements in the Ifng locus that are likely to contribute to its transcriptional regulation, we speculate that this locus will emerge not only as a model to understand cytokine regulation, but also to explore more universal mechanisms that regulate lineage- and activation-dependent transcription in eukaryotes.
Fig. 3. DNase I hypersensitivity profiles of IL-12–transitioned Th17 cells.
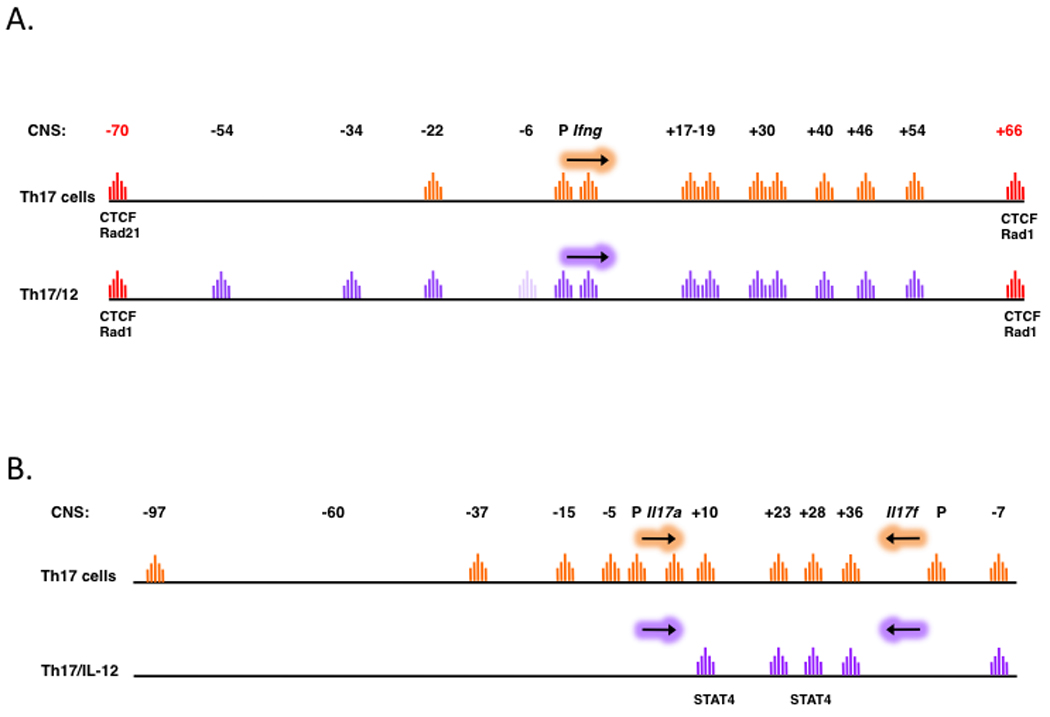
As in Fig. 2, DNase I hypersensitive sites are identified in the Ifng locus (A) and the IL17a–IL17f locus (B) in Th17 cells and in Th17 cells exposed to IL-12 (Th17/IL-12) that exhibit a Th1-like phenotype. Paler peaks indicate activation-dependent HS sites within CNS elements (e.g. CNS-6 in the Th17/IL-12 cells, A).
Acknowledgments
This work was supported by grants from the NIH (AI35783, AI77574 and DK64400) the Crohn’s and Colitis Foundation of America, and Daiichi-Sankyo Co., Ltd. We thank members of our lab for helpful comments.
References
- 1.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–2357. [PubMed] [Google Scholar]
- 2.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–944. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 3.Weaver CT, Murphy KM. T-cell subsets: the more the merrier. Curr Biol. 2007;17:R61–R63. doi: 10.1016/j.cub.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 4.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nurieva RI, et al. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnston RJ, et al. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science. 2009;325:1006–1110. doi: 10.1126/science.1175870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dardalhon V, et al. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Veldhoen M, et al. Transforming growth factor-beta 'reprograms' the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9:1341–1346. doi: 10.1038/ni.1659. [DOI] [PubMed] [Google Scholar]
- 9.Wilson CB, Rowell E, Sekimata M. Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol. 2009;9:91–105. doi: 10.1038/nri2487. [DOI] [PubMed] [Google Scholar]
- 10.Rowell E, Merkenschlager M, Wilson CB. Long-range regulation of cytokine gene expression. Curr Opin Immunol. 2008;20:272–280. doi: 10.1016/j.coi.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 12.Hegazy AN, et al. Interferons direct Th2 cell reprogramming to generate a stable GATA-3(+)T-bet(+) cell subset with combined Th2 and Th1 cell functions. Immunity. 2010;32:116–128. doi: 10.1016/j.immuni.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 13.Oldenhove G, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou X, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee YK, et al. Late developmental plasticity in the T helper 17 lineage. Immunity. 2009;30:92–107. doi: 10.1016/j.immuni.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000;1:65–69. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- 17.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 18.Afkarian M, et al. T-bet is a STAT1-induced regulator of IL-12R expression in naive CD4+ T cells. Nat Immunol. 2002;3:549–557. doi: 10.1038/ni794. [DOI] [PubMed] [Google Scholar]
- 19.Murphy KM, et al. T helper differentiation proceeds through Stat1-dependent, Stat4-dependent and Stat4-independent phases. Curr Top Microbiol Immunol. 1999;238:13–26. doi: 10.1007/978-3-662-09709-0_2. [DOI] [PubMed] [Google Scholar]
- 20.Schulz EG, Mariani L, Radbruch A, Hofer T. Sequential polarization and imprinting of type 1 T helper lymphocytes by interferon-gamma and interleukin-12. Immunity. 2009;30:673–683. doi: 10.1016/j.immuni.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 21.Mullen AC, et al. Hlx is induced by and genetically interacts with T-bet to promote heritable T(H)1 gene induction. Nat Immunol. 2002;3:652–658. doi: 10.1038/ni807. [DOI] [PubMed] [Google Scholar]
- 22.Thieu VT, et al. Signal Transducer and Activator of Transcription 4 Is Required for the Transcription Factor T-bet to Promote T Helper 1 Cell-Fate Determination. Immunity. 2008;29:679–690. doi: 10.1016/j.immuni.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 24.Ouyang W, et al. The Ets transcription factor ERM is Th1-specific and induced by IL-12 through a Stat4-dependent pathway. Proc Natl Acad Sci U S A. 1999;96:3888–3893. doi: 10.1073/pnas.96.7.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 26.Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science. 2002;295:338–342. doi: 10.1126/science.1065543. [DOI] [PubMed] [Google Scholar]
- 27.Pearce EL, et al. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science. 2003;302:1041–1043. doi: 10.1126/science.1090148. [DOI] [PubMed] [Google Scholar]
- 28.Intlekofer AM, et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science. 2008;321:408–411. doi: 10.1126/science.1159806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cruz-Guilloty F, et al. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J Exp Med. 2009;206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Grenningloh R, Kang BY, Ho IC. Ets-1, a functional cofactor of T-bet, is essential for Th1 inflammatory responses. J Exp Med. 2005;201:615–626. doi: 10.1084/jem.20041330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zamisch M, et al. The transcription factor Ets1 is important for CD4 repression and Runx3 up-regulation during CD8 T cell differentiation in the thymus. J Exp Med. 2009;206:2685–2699. doi: 10.1084/jem.20092024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Egawa T, Littman DR. ThPOK acts late in specification of the helper T cell lineage and suppresses Runx-mediated commitment to the cytotoxic T cell lineage. Nat Immunol. 2008;9:1131–1139. doi: 10.1038/ni.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Hamburg JP, de Bruijn MJ, Ribeiro de Almeida C, Dingjan GM, Hendriks RW. Gene expression profiling in mice with enforced Gata3 expression reveals putative targets of Gata3 in double positive thymocytes. Mol Immunol. 2009;46:3251–3260. doi: 10.1016/j.molimm.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 34.Wildt KF, et al. The transcription factor Zbtb7b promotes CD4 expression by antagonizing Runx-mediated activation of the CD4 silencer. J Immunol. 2007;179:4405–4414. doi: 10.4049/jimmunol.179.7.4405. [DOI] [PubMed] [Google Scholar]
- 35.Usui T, et al. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J Exp Med. 2006;203:755–766. doi: 10.1084/jem.20052165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka S, et al. The interleukin-4 enhancer CNS-2 is regulated by Notch signals and controls initial expression in NKT cells and memory-type CD4 T cells. Immunity. 2006;24:689–701. doi: 10.1016/j.immuni.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 37.Hardy KJ, Manger B, Newton M, Stobo JD. Molecular events involved in regulating human interferon-gamma gene expression during T cell activation. J Immunol. 1987;138:2353–2358. [PubMed] [Google Scholar]
- 38.Hardy KJ, Peterlin BM, Atchison RE, Stobo JD. Regulation of expression of the human interferon gamma gene. Proc Natl Acad Sci U S A. 1985;82:8173–8177. doi: 10.1073/pnas.82.23.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciccarone VC, Chrivia J, Hardy KJ, Young HA. Identification of enhancer-like elements in human IFN-gamma genomic DNA. J Immunol. 1990;144:725–730. [PubMed] [Google Scholar]
- 40.Soutto M, Zhou W, Aune TM. Cutting edge: distal regulatory elements are required to achieve selective expression of IFN-gamma in Th1/Tc1 effector cells. J Immunol. 2002;169:6664–6667. doi: 10.4049/jimmunol.169.12.6664. [DOI] [PubMed] [Google Scholar]
- 41.Young HA, et al. Expression of human IFN-gamma genomic DNA in transgenic mice. J Immunol. 1989;143:2389–2394. [PubMed] [Google Scholar]
- 42.Zhu H, et al. Unexpected characteristics of the IFN-gamma reporters in nontransformed T cells. J Immunol. 2001;167:855–865. doi: 10.4049/jimmunol.167.2.855. [DOI] [PubMed] [Google Scholar]
- 43.Hadjur S, et al. Cohesins form chromosomal cis-interactions at the developmentally regulated IFNG locus. Nature. 2009;460:410–413. doi: 10.1038/nature08079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sekimata M, et al. CCCTC-binding factor and the transcription factor T-bet orchestrate T helper 1 cell-specific structure and function at the interferon-gamma locus. Immunity. 2009;31:551–564. doi: 10.1016/j.immuni.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Loots GG, et al. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- 46.Li Q, Harju S, Peterson KR. Locus control regions: coming of age at a decade plus. Trends Genet. 1999;15:403–408. doi: 10.1016/s0168-9525(99)01780-1. [DOI] [PubMed] [Google Scholar]
- 47.Lee DU, Avni O, Chen L, Rao A. A distal enhancer in the interferon-gamma (IFN-gamma) locus revealed by genome sequence comparison. J Biol Chem. 2004;279:4802–4810. doi: 10.1074/jbc.M307904200. [DOI] [PubMed] [Google Scholar]
- 48.Shnyreva M, et al. Evolutionarily conserved sequence elements that positively regulate IFN-gamma expression in T cells. Proc Natl Acad Sci U S A. 2004;101:12622–12627. doi: 10.1073/pnas.0400849101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hatton RD, et al. A distal conserved sequence element controls Ifng gene expression by T cells and NK cells. Immunity. 2006;25:717–729. doi: 10.1016/j.immuni.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 50.Shibata Y, Crawford GE. Mapping regulatory elements by DNaseI hypersensitivity chip (DNase-Chip) Methods Mol Biol. 2009;556:177–190. doi: 10.1007/978-1-60327-192-9_13. [DOI] [PubMed] [Google Scholar]
- 51.Barski A, Zhao K. Genomic location analysis by ChIP-Seq. J Cell Biochem. 2009;107:11–18. doi: 10.1002/jcb.22077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. CSH Protoc. 2010 doi: 10.1101/pdb.prot5384. 2010:pdb prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weinmann AS. Novel ChIP-based strategies to uncover transcription factor target genes in the immune system. Nat Rev Immunol. 2004;4:381–386. doi: 10.1038/nri1353. [DOI] [PubMed] [Google Scholar]
- 54.Balasubramani A, Shibata Y, Crawford GE, Baldwin AS, Hatton RD, Weaver CT. Modular Utilization of Distal cis-Regulatory Elements Controls Ifng Gene Expression in T Cells Activated by Distinct Stimuli. Immunity. 2010;33:35–47. doi: 10.1016/j.immuni.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mukasa R, et al. Epigenetic instability of cytokine and transcription factor gene loci underlies plasticity of the T helper 17 cell lineage. Immunity. 2010;32:616–627. doi: 10.1016/j.immuni.2010.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyle AP, et al. High-resolution mapping and characterization of open chromatin across the genome. Cell. 2008;132:311–322. doi: 10.1016/j.cell.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spilianakis CG, Lalioti MD, Town T, Lee GR, Flavell RA. Interchromosomal associations between alternatively expressed loci. Nature. 2005;435:637–645. doi: 10.1038/nature03574. [DOI] [PubMed] [Google Scholar]
- 58.Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS. Coordinated but physically separable interaction with H3K27-demethylase and H3K4-methyltransferase activities are required for T-box protein-mediated activation of developmental gene expression. Genes Dev. 2008;22:2980–2993. doi: 10.1101/gad.1689708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis MD, Miller SA, Miazgowicz MM, Beima KM, Weinmann AS. T-bet's ability to regulate individual target genes requires the conserved T-box domain to recruit histone methyltransferase activity and a separate family member-specific transactivation domain. Mol Cell Biol. 2007;27:8510–8521. doi: 10.1128/MCB.01615-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang S, Aune TM. Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc Natl Acad Sci U S A. 2005;102:17095–17100. doi: 10.1073/pnas.0502129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yagi R, et al. The transcription factor GATA3 actively represses RUNX3 protein-regulated production of interferon-gamma. Immunity. 2010;32:507–517. doi: 10.1016/j.immuni.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Klein F, et al. A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell. 1999;98:91–103. doi: 10.1016/S0092-8674(00)80609-1. [DOI] [PubMed] [Google Scholar]
- 63.Michaelis C, Ciosk R, Nasmyth K. Cohesins: chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997;91:35–45. doi: 10.1016/s0092-8674(01)80007-6. [DOI] [PubMed] [Google Scholar]
- 64.Parelho V, et al. Cohesins functionally associate with CTCF on mammalian chromosome arms. Cell. 2008;132:422–433. doi: 10.1016/j.cell.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 65.Wendt KS, et al. Cohesin mediates transcriptional insulation by CCCTC-binding factor. Nature. 2008;451:796–801. doi: 10.1038/nature06634. [DOI] [PubMed] [Google Scholar]
- 66.Ishihara K, Oshimura M, Nakao M. CTCF-dependent chromatin insulator is linked to epigenetic remodeling. Mol Cell. 2006;23:733–742. doi: 10.1016/j.molcel.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 67.Ishihara K, Sasaki H. An evolutionarily conserved putative insulator element near the 3' boundary of the imprinted Igf2/H19 domain. Hum Mol Genet. 2002;11:1627–1636. doi: 10.1093/hmg/11.14.1627. [DOI] [PubMed] [Google Scholar]
- 68.Harrington LE, Janowski KM, Oliver JR, Zajac AJ, Weaver CT. Memory CD4 T cells emerge from effector T-cell progenitors. Nature. 2008;452:356–360. doi: 10.1038/nature06672. [DOI] [PubMed] [Google Scholar]
- 69.Robinson D, et al. IGIF does not drive Th1 development but synergizes with IL-12 for interferon-gamma production and activates IRAK and NFkappaB. Immunity. 1997;7:571–581. doi: 10.1016/s1074-7613(00)80378-7. [DOI] [PubMed] [Google Scholar]
- 70.Guo L, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A. 2009;106:13463–13468. doi: 10.1073/pnas.0906988106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sica A, et al. Interaction of NF-kappaB and NFAT with the interferon-gamma promoter. J Biol Chem. 1997;272:30412–30420. doi: 10.1074/jbc.272.48.30412. [DOI] [PubMed] [Google Scholar]
- 73.Sica A, Tan TH, Rice N, Kretzschmar M, Ghosh P, Young HA. The c-rel protooncogene product c-Rel but not NF-kappa B binds to the intronic region of the human interferon-gamma gene at a site related to an interferon-stimulable response element. Proc Natl Acad Sci U S A. 1992;89:1740–1744. doi: 10.1073/pnas.89.5.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vallabhapurapu S, Karin M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol. 2009;27:693–733. doi: 10.1146/annurev.immunol.021908.132641. [DOI] [PubMed] [Google Scholar]
- 75.Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006;25:6758–6780. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- 76.Corn RA, Hunter C, Liou HC, Siebenlist U, Boothby MR. Opposing roles for RelB and Bcl-3 in regulation of T-box expressed in T cells, GATA-3, and Th effector differentiation. J Immunol. 2005;175:2102–2110. doi: 10.4049/jimmunol.175.4.2102. [DOI] [PubMed] [Google Scholar]
- 77.Boothby MR, Mora AL, Scherer DC, Brockman JA, Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185:1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST. Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci U S A. 2000;97:12705–12710. doi: 10.1073/pnas.230436397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML. c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol. 1999;11:361–371. doi: 10.1093/intimm/11.3.361. [DOI] [PubMed] [Google Scholar]
- 80.Mason NJ, Liou HC, Hunter CA. T cell-intrinsic expression of c-Rel regulates Th1 cell responses essential for resistance to Toxoplasma gondii. J Immunol. 2004;172:3704–3711. doi: 10.4049/jimmunol.172.6.3704. [DOI] [PubMed] [Google Scholar]
- 81.Kontgen F, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoral immunity, and interleukin-2 expression. Genes Dev. 1995;9:1965–1977. doi: 10.1101/gad.9.16.1965. [DOI] [PubMed] [Google Scholar]
- 82.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 83.Steinbrecher KA, Harmel-Laws E, Sitcheran R, Baldwin AS. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
- 84.Otani T, Nakamura S, Toki M, Motoda R, Kurimoto M, Orita K. Identification of IFN-gamma-producing cells in IL-12/IL-18-treated mice. Cell Immunol. 1999;198:111–119. doi: 10.1006/cimm.1999.1589. [DOI] [PubMed] [Google Scholar]
- 85.Leite-De-Moraes MC, et al. A distinct IL-18-induced pathway to fully activate NK T lymphocytes independently from TCR engagement. J Immunol. 1999;163:5871–5876. [PubMed] [Google Scholar]
- 86.Carter LL, Murphy KM. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon gamma production from CD4(+) versus CD8(+) T cells. J Exp Med. 1999;189:1355–1360. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang T, Kawakami K, Qureshi MH, Okamura H, Kurimoto M, Saito A. Interleukin-12 (IL-12) and IL-18 synergistically induce the fungicidal activity of murine peritoneal exudate cells against Cryptococcus neoformans through production of gamma interferon by natural killer cells. Infect Immun. 1997;65:3594–3599. doi: 10.1128/iai.65.9.3594-3599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berenson LS, Gavrieli M, Farrar JD, Murphy TL, Murphy KM. Distinct characteristics of murine STAT4 activation in response to IL-12 and IFN-alpha. J Immunol. 2006;177:5195–5203. doi: 10.4049/jimmunol.177.8.5195. [DOI] [PubMed] [Google Scholar]
- 89.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol. 2001;2:157–164. doi: 10.1038/84264. [DOI] [PubMed] [Google Scholar]
- 90.Yang J, Murphy TL, Ouyang W, Murphy KM. Induction of interferon-gamma production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur J Immunol. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 91.Kiani A, et al. Regulation of interferon-gamma gene expression by nuclear factor of activated T cells. Blood. 2001;98:1480–1488. doi: 10.1182/blood.v98.5.1480. [DOI] [PubMed] [Google Scholar]
- 92.Agarwal S, Avni O, Rao A. Cell-type-restricted binding of the transcription factor NFAT to a distal IL-4 enhancer in vivo. Immunity. 2000;12:643–652. doi: 10.1016/s1074-7613(00)80215-0. [DOI] [PubMed] [Google Scholar]
- 93.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 94.Yang DD, et al. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- 95.Rincon M, et al. Interferon-gamma expression by Th1 effector T cells mediated by the p38 MAP kinase signaling pathway. EMBO J. 1998;17:2817–2829. doi: 10.1093/emboj/17.10.2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lu B, et al. GADD45gamma mediates the activation of the p38 and JNK MAP kinase pathways and cytokine production in effector TH1 cells. Immunity. 2001;14:583–590. doi: 10.1016/s1074-7613(01)00141-8. [DOI] [PubMed] [Google Scholar]
- 97.Lu B, Ferrandino AF, Flavell RA. Gadd45beta is important for perpetuating cognate and inflammatory signals in T cells. Nat Immunol. 2004;5:38–44. doi: 10.1038/ni1020. [DOI] [PubMed] [Google Scholar]
- 98.Lu HT, et al. Defective IL-12 production in mitogen-activated protein (MAP) kinase kinase 3 (Mkk3)-deficient mice. EMBO J. 1999;18:1845–1857. doi: 10.1093/emboj/18.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 100.Roncarolo MG, Gregori S. Is FOXP3 a bona fide marker for human regulatory T cells? Eur J Immunol. 2008;38:925–927. doi: 10.1002/eji.200838168. [DOI] [PubMed] [Google Scholar]
- 101.Wei G, et al. Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity. 2009;30:155–167. doi: 10.1016/j.immuni.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lexberg MH, et al. Th memory for interleukin-17 expression is stable in vivo. Eur J Immunol. 2008;38:2654–2664. doi: 10.1002/eji.200838541. [DOI] [PubMed] [Google Scholar]
- 103.Bending D, et al. Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest. 2009 doi: 10.1172/JCI37865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schoenborn JR, et al. Comprehensive epigenetic profiling identifies multiple distal regulatory elements directing transcription of the gene encoding interferon-gamma. Nat Immunol. 2007;8:732–742. doi: 10.1038/ni1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chang S, Aune TM. Dynamic changes in histone-methylation 'marks' across the locus encoding interferon-gamma during the differentiation of T helper type 2 cells. Nat Immunol. 2007;8:723–731. doi: 10.1038/ni1473. [DOI] [PubMed] [Google Scholar]
- 106.Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–824. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schraml BU, et al. The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature. 2009;460:405–409. doi: 10.1038/nature08114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Williams KL, et al. Characterization of murine BATF: a negative regulator of activator protein-1 activity in the thymus. Eur J Immunol. 2001;31:1620–1627. doi: 10.1002/1521-4141(200105)31:5<1620::aid-immu1620>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 109.Zhang F, Meng G, Strober W. Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol. 2008;9:1297–1306. doi: 10.1038/ni.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Durant L, et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gomez-Rodriguez J, et al. Differential expression of interleukin-17A and −17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity. 2009;31:587–597. doi: 10.1016/j.immuni.2009.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Berg LJ, Finkelstein LD, Lucas JA, Schwartzberg PL. Tec family kinases in T lymphocyte development and function. Annu Rev Immunol. 2005;23:549–600. doi: 10.1146/annurev.immunol.22.012703.104743. [DOI] [PubMed] [Google Scholar]
- 113.Liao XC, Littman DR. Altered T cell receptor signaling and disrupted T cell development in mice lacking Itk. Immunity. 1995;3:757–769. doi: 10.1016/1074-7613(95)90065-9. [DOI] [PubMed] [Google Scholar]
- 114.Chung Y, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 116.Dostie J, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhao Z, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 118.Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 119.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Johnson KD, Grass JA, Park C, Im H, Choi K, Bresnick EH. Highly restricted localization of RNA polymerase II within a locus control region of a tissue-specific chromatin domain. Mol Cell Biol. 2003;23:6484–6493. doi: 10.1128/MCB.23.18.6484-6493.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kim TK, et al. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010;465:182–187. doi: 10.1038/nature09033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Collins PL, et al. Distal regions of the human IFNG locus direct cell type-specific expression. J Immunol. 2010;185:1492–1501. doi: 10.4049/jimmunol.1000124. [DOI] [PMC free article] [PubMed] [Google Scholar]