

Abstract
Type I interferon (IFN)-dependent STAT1 and STAT2 activation requires specific tyrosine residues (337Y and 512Y) located in the cytoplasmic domain of IFNAR-2c, the β-subunit of the human type I IFN receptor. To identify STAT activation-independent induction of ISGs, we used a mutant cell line in which both 337Y and 512Y were substituted with phenylalanine (337F512F or FF mutant). In these cells, type I IFN failed to activate STAT1, STAT2, and STAT3 did not induce well-characterized ISGs and did not exert antiviral or antiproliferative effects. Using Oligonucleotide array (Affymetrix™) analysis, we showed that interferon regulatory factor-9 (IRF-9) was the only gene induced by IFN-β in FF cells. Transient transfection analysis using an IRF-9 promoter–reporter luciferase construct in FF cells confirmed induction of the IRF-9 transcription unit by IFN-β. EMSA analysis using an IFN-stimulated response element (ISRE)-like sequence on the IRF-9 promoter detected 2 novel DNA-binding complexes induced in nuclear extracts of IFN-β-treated FF cells. Supershift experiments identified the proteins IRF-1 and C/EBP-β in the complex. These studies provide the first evidence that signaling pathways leading to gene transcription are activated by IFN-β independent of STAT phosphorylation.
Introduction
The JAK-STAT pathways are now the major regulators of the transcription of the interferon (IFN)-stimulated genes (ISGs), whose protein products mediate the multiple biological responses to IFNs (Darnell and others 1994; Borden and others 2007). Type I IFN-dependent JAK-STAT signaling requires both the type I IFN receptor chains, IFNAR-1 and IFNAR-2c, and the 2 JAK kinases, JAK1 and TYK2 (Uze and others 1990; Novick and others 1994; Lutfalla and others 1995; Domanski and others 1998). IFN-α/β treatment activates the formation of trimetric transcription factor complex, ISGF3, comprised of STAT1, STAT2, and interferon regulatory factor-9 (IRF-9), which binds to the ISRE of many ISG promoters to stimulate their transcription (Darnell and others 1994; Stark and others 1998).
Binding of type I IFNs induces the aggregation of the receptor chains, leading to the phosphorylation of tyrosine (Y) residues located in the intracellular domain of each receptor chain. IFN-induced phosphorylation of the Y466 and Y481 on IFNAR-1 is required for the docking of STAT2 (Yan and others 1996). No human cells that lack IFNAR-1 exist. However, the role of IFNAR-2c in type I IFN signaling has been studied by expressing IFNAR-2c mutants in U5A cells (Russell-Harde and others 2000; Wagner and others 2002). These cells that express IFNAR-1 but lack IFNAR-2c fail to respond to type I IFN confirming the requirement of this receptor chain for IFN signaling (Lutfalla and others 1995). A mutant IFNAR-2c, with phenylalanines in place of the 7 tyrosines of cytoplasmic tail (7F mutant), failed to support type I IFN-dependent STAT activation, gene expression, antiproliferative and antiviral effects. However, JAK1 phosphorylation could still be detected in these cells (Russell-Harde and others 2000). In complementary studies, individual tyrosines were introduced into the 7F backbone. Surprisingly, presence of a single tyrosine at position either 337 or 512 was sufficient to restore a complete IFN response, equivalent to that observed in U5A cells rescued with expression of full-length IFNAR-2c (Wagner and others 2002).
The majority of type I IFN-induced ISGs requires only STAT proteins for their transcriptional induction. However, our recent work has focused on the identification and characterization of genes that require accessory signaling components in addition to the JAK-STAT signals in response to IFN-β (Rani and Ransohoff 2005). Using an IFNAR-2c mutant cell line (337F512F mutant, Fig. 1A), we report that the IRF-9 gene is induced in response to IFN-β independently of STAT1, STAT2, and STAT3 phosphorylation, indicating the existence of a novel IFN-induced signaling pathway.
FIG. 1. .
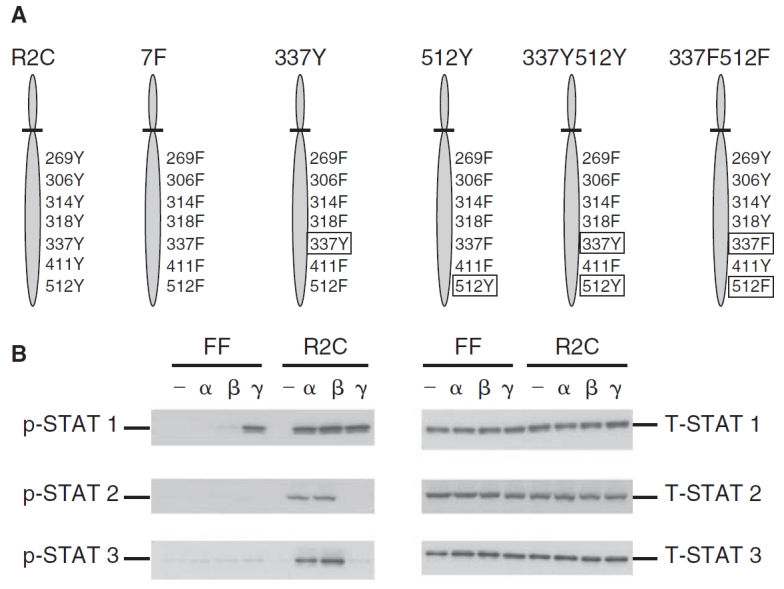
Phosphorylation of STAT1, STAT2, and STAT3 in the mutant IFNAR-2c (FF) and wild-type IFNAR-2c (R2C) cells. (A) A schematic representation of the tyrosine residues in the cytoplasmic domain of IFNAR-2c expressed in U5A cells. U5A cells expressing the wild-type IFNAR-2c with all 7 intact cytoplasmic tyrosine residues at 269, 306, 314, 318, 337, 411, and 512 (R2C); mutant IFNAR-2c in which all the above 7 tyrosines have been replaced with phenylalanine (7F); mutant IFNAR-2C with a single tyrosine at 337 (337Y); mutant IFNAR-2c with a single tyrosine at 512 (512Y); mutant IFNAR-2c with 2 tyrosines at 337 and 512 (337Y512Y) and mutant IFNAR-2c with 2 phenylalanines at 337 and 512 (337F512F) are shown. (B) FF and R2C cells were left untreated (—), or treated with interferon (IFN)-α2 (α) or IFN-β (β) (5,000 U/mL) or IFN-γ (γ) (1,000 U/mL) for 15 min and whole cell extracts prepared. Cell lysates were resolved by SDS-PAGE, blotted onto PVDF membranes, and subjected to immunoblotting using anti-phospho-STAT1, STAT2, or STAT3 (left set of 3 panels). Blots were stripped and re-probed with anti-STAT1, STAT2, or STAT3 (right set of panels) demonstrating that STAT1, STAT2, and STAT3 proteins were expressed at equivalent levels in both cell lines.
IRF-9 belongs to a family of structurally similar but genetically and functionally distinct DNA-binding proteins (Taniguchi and others 1995). IRF-1 and IRF-9 are activators of transcription, IRF-2 and IRF-8 are repressors, and IRF-3 and IRF-4 can both activate and repress transcription (Nguyen and others 1997). Gene-knockout studies have shown that IRF-9 plays an essential role in activation of ISGs and antiviral response (Holtschke and others 1996; Kimura and others 1996). As reported earlier, IRF-9 is a component of the transcription factor, IFN-stimulated gene factor 3 (ISGF3), which binds to the IFN-stimulated response element (ISRE) located in the promoters of ISGs to induce gene transcription (Darnell and others 1994). This is the first report of a gene induced by IFN-β in the absence of STAT1, STAT2, or STAT3 phosphorylation. We also show a unique complex consisting of IRF-1 and CCAAT/enhancer-binding protein-β to be important for regulating IRF-9 expression in response to IFN-β.
Materials and Methods
Cell culture and reagents
The 2fTGH fibrosarcoma cells, mutant U5A cells that lack the IFNAR-2c receptor, U5A cells transfected stably with the wild-type IFNAR-2c (R2C), and the mutant IFNAR-2c (FF) cells in which tyrosines at 337 and 512 are substituted with phenylalanine were maintained in Dulbeco’s modified Eagle’s medium supplemented with 10% calf serum (Russell-Harde and others 2000). Purified recombinant IFN-β-1b (2.3 × 107 IU/mg) was from Berlex Biosciences (Richmond, CA). LY294002, SB203580, and PD98059 were obtained from Biomol Research Labs (Plymouth Meeting, PA). Purified recombinant IFN-γ was obtained from Genentech Inc. (South San Francisco, CA). Total and anti-phosphotyrosine STAT1, STAT2, and STAT3 antibodies were obtained from Cell Signaling Technologies (Beverly, MA). Anti-IRF-1 was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Anti-C/EBP-β antibody and expression plasmid were described elsewhere (Roy and others 2002).
RNA isolation and RNase protection assay
Total RNA was prepared from IFN-treated cells using the TRIzol reagent (Invitrogen Inc., Carlsbad, CA). RNase protection assays were performed as described (Rani and others 1996). A 350 bp fragment of human IRF-9 cDNA was amplified by PCR using the primers, hIRF-9F1—CGCTTTCACTCCCCA GGTCCAGGCCCGCCC and hIRF-9R1—CATCCTGAGGCAGGAGTCAGC CCCAGCCT, and cloned into the pGEM-T vector (Promega Inc., Madison, MI) to generate the hIRF-9 plasmid. DNA was linearized with restriction enzyme BamHI and transcribed in vitro using T7 RNA polymerase to generate a 400 bp antisense probe. Probes corresponding to ISG 6-16 and an internal control γ-actin were described elsewhere (Ackrill and others 1991; Muller and others 1993). The IRF-9, 6-16, and γ-actin probes protect 350, 190, and 130 nucleotide long transcripts, respectively.
Transient transfection and luciferase assays
A −1,050 bp fragment corresponding to the upstream regulatory region of murine IRF-9 was amplified by PCR using genomic DNA as a template. The primers used for PCR were IRF-9F1—ATACGAGCTCCTT-GGTGGTCCTT and IRF-9R1—TTGGAAGATCTGATATATGAGAACTC with restriction sites SacI and BglII (underlined) for directional cloning of the DNA into the pGL3-basic vector (Promega Inc, Madison, MI) in which the expression of luciferase is controlled by the cloned IRF-9 promoter elements. A shorter construct of −340 bp IRF-9 DNA was also generated using the primers IRF-9F2-ATACGAGCTCGCTTGTGAATCTG and IRF-9R1 (sequence shown above). Transient transfection assays were done using the FuGENE reagent (Roche Applied Science, Nutley, NJ) according to manufacturer’s instructions. In brief, 1 µg of pGL3-IRF-9-1050 or pGL3-IRF-9-340 reporters were mixed along with 0.1 µg of Renilla luciferase control vector and transfected into cells in 24-well plates in serum-free media for 6 h at 37°C followed by the addition of serum (final concentration 10%) (Rani and others 2007). After over night incubation, cells were fed with fresh complete medium and were treated with IFN-β for 16 h. Cellular lysates were monitored for the luciferase activity using a commercially available dual luciferase assay kit (Promega Inc., Madison, MI). The firefly luciferase activity was normalized to that of Renilla luciferase and expressed as fold induction over control (Rani and others 2007).
Electrophoretic mobility shift assay (EMSA)
Nuclear extracts were prepared from cells treated with IFN-β (5,000 U/mL) for various times as described earlier (Rani and others 2001). An oligonucleotide probe corresponding to the IFN-γ-activated transcriptional element (GATE) of IRF-9 gene was used for these assays. The reaction products were resolved on 8% non-denaturing polyacrylamide gels, which were dried and analyzed by autoradiography. For supershift assays, the extracts were incubated first with the antibodies for 20 min followed by the addition of the radiolabeled probe.
Western blots
FF cells were treated with IFN-β (5,000 U/mL) for various times and whole cell extracts prepared as described earlier (Rani and others 2002). Proteins were separated by SDS-PAGE and adsorbed to PVDF membranes and probed with the indicated primary and secondary antibodies. Immunoreactive bands were visualized using the ECL reagents (Amersham Biosciences, UK). Tyrosyl phosphorylation was monitored by Western blot analysis with anti-phosphotyrosine antibodies.
Microarray analysis
The human 6800 GeneChip array containing unique 20-mer oligonucleotide probes to >6,800 genes (Affymetrix™, Santa Clara, CA) was used for this analysis. FF cells were treated with recombinant IFN-β (1,000 U/mL) for 1, 3, or 6 h or reserved as controls. Total RNA was isolated and reverse-transcribed to cDNA using Superscript (Invitrogen Life Technologies, Carlsbad, CA), and subsequently converted to biotinylated cRNA using Enzo High Yield RNA Transcript labeling kit (Enzo Biochem, Farmingdale NY). After hybridization the gene chips were washed and stained with streptavidin-PE. Data analysis was performed using the Genespring 3.0 software (Agilent Technologies Inc., Santa Clara, CA). The data were normalized to the expression levels of actin and GAPDH. Genes induced ≥2-fold were selected for further analyses with PCR.
Results
FF cells lack IFN-β–induced phosphorylation of STAT1, STAT2, and STAT3
Phosphorylation of tyrosine residues on IFNAR-1 and IFNAR-2c is essential for the activation of STAT1 and STAT2 in response to type I IFNs. U5A cells lack IFNAR-2c (Lutfalla and others 1995). A schematic of the various mutants of IFNAR-2c is shown in Figure 1A. U5A cells reconstituted with wild-type IFNAR-2c receptor (R2C) and its functionally inactive mutants lacking either all 7 tyrosines (7F) or tyrosines at 337 and 512 (Y337F/Y512F or FF) were used for the impact on IFN-β inducible gene expression (Wagner and others 2002).
We first analyzed tyrosyl phosphorylation of STAT1, STAT2, and STAT3 in the FF mutant cells in response to IFN-α, IFN-β, or IFN-γ using Western blot analyses. None of the 3 STATs were phosphorylated in response to IFN-α or IFN-β, although all the STAT proteins were present in cells, suggesting a defective type I IFN signaling in these cells (Fig. 1B). However, STAT1 was phosphorylated in response to IFN-γ, demonstrating the intact type II IFN-induced signaling in FF cells (Fig. 1B). Cells expressing wild-type IFNAR-2c receptor (R2C) had an intact phosphorylation of STAT1, STAT2, and STAT3 in response to both type I IFNs (Fig. 1B). As predicted, STAT1 was phosphorylated only in response to IFN-γ in these cells (Fig. 1B). Unphosphorylated STAT1, STAT2, and STAT3 proteins were expressed at equivalent levels in both cell lines (Fig. 1B). The FF cells thus provided an ideal background for studying STAT phosphorylation-independent induction of ISG expression.
Gene expression by IFN-β in FF cells
In addition to JAK-STAT signaling, accessory signaling molecules can modulate the function of STATs or activate complementary signaling components (Rani and Ransohoff 2005). Induction of CXCL11 and TRAIL mRNAs by IFN-β not only required signaling involving phosphorylation of JAK-STATs but also other accessory signaling molecules like NF-κB and PI3K (Rani and Ransohoff 2005; Rani and others 2007). To identify genes induced in the absence of STAT phosphorylation by IFN-β, RNA was isolated from FF cells treated with and without IFN-β for 1 or 3 h. Oligonucleotide array analysis (Affymetrix™) identified a small subset of ISGs induced by IFN-β. However, real-time RT-PCR (results not shown) and RNase protection assays confirmed only IRF-9 induction by IFN-β in FF cells (Fig. 2A). IRF-9 was very weakly induced by IFN-β in 7F cells at 3 h, suggesting a requirement for a tyrosine residue in the cytoplasmic domain of IFNAR-2c for IFN-β–mediated induction of IRF-9 (Fig. 2A). A classical control ISG 6-16, whose expression required IFN-induced STAT1 and STAT2 phosphorylation and binding to the promoter was not induced in IFN-β-treated 7F or FF cells (Fig. 2A). In U5A cells expressing the full-length IFNAR-2c receptor (R2C) induction of IRF-9 and 6-16 mRNA occurred in response to IFN-β (Fig. 2A).These results indicate that optimal induction of IRF-9 by IFN-β requires phosphorylation of STAT1 and STAT2 plus accessory signaling. However, it is clear that accessory signaling components independent of STAT activation can mediate IRF-9 gene induction by IFN-β in FF cells.
FIG. 2. .

Induction of interferon regulatory factor-9 (IRF-9) in the mutant IFNAR-2c (FF) cells by interferon-β (IFN-β). (A) RNA was isolated from untreated cells (C) or cells treated with 1,000 IU/mL of recombinant IFN-β for 1 and 3 h, respectively. The following cell lines were examined: wild-type IFNAR-2c (R2C), FF, and 7F. RNase protection assay was performed using 20 µg of RNA and analyzed using probes for IRF-9 (protects 350 bp), 6-16 (protects 160 bp), and γ-actin (protects 110 bp). The figure shows an autoradiogram derived from one of three experiments. (B) Cells were treated with different doses of IFN-β for 6 h as indicated. RNase protection assay of total RNA (20 µg) was performed using probes for IRF-9, 6-16, and γ-actin. The figure shows an autoradiogram generated on a PhosphorImager, derived from one representative experiment (out of 3). (C) FF cells were treated with 1,000 IU/mL of IFN-β for periods of time indicated on the panel. The figure shows an autoradiogram derived from one representative experiment (out of 3).
We next addressed the kinetics and dose dependence of induction of IRF-9 by IFN-β (Fig. 2B). The induction of IRF-9 by IFN-β treatment was dose-dependent between 20 and 500 U/mL (Fig. 2B). For kinetic studies, FF cells were treated with IFN-β (1,000 IU/mL) for various periods of time (Fig. 2C). Maximal induction of IRF-9 mRNA was observed following a 6-h treatment with IFN-β, and IRF-9 mRNA accumulation was unchanged thereafter over 12 h of IFN-β exposure (Fig. 2C).
Transcriptional induction of IRF-9 by IFN-β in FF cells
To determine if induction of IRF-9 occurred at a transcriptional level, a 1,050 bp fragment of the murine IRF-9 gene upstream of the translation start site was cloned into the promoterless pGL3-basic vector (Promega Corp., Madison, WI) upstream of the firefly luciferase reporter gene to generate the plasmid IRF-9-1050 (Weihua and others 1997a). A schematic of the promoter of mIRF-9 is shown in Figure 3A. The sequence of the mIRF-9 promoter revealed the existence of potential regulatory elements, which included NF-κB, GAS, AP-1, GATE, Myc/Max, and SP-1 elements. Transient transfection assays in FF cells demonstrated a 9-fold increase in IRF-9 promoter luciferase activity after treatment with IFN-β (Fig. 3B). A reporter driven by the promoter of ISG-561 gene (a type I IFN-induced gene), which requires phosphorylation of STAT1 and STAT2, was not induced in the FF cells (results not shown). However, in the R2C cells, there was equivalent induction of IRF-9 promoter–reporter and 561 promoter–reporter luciferase activity by IFN-β (results not shown).
FIG. 3. .
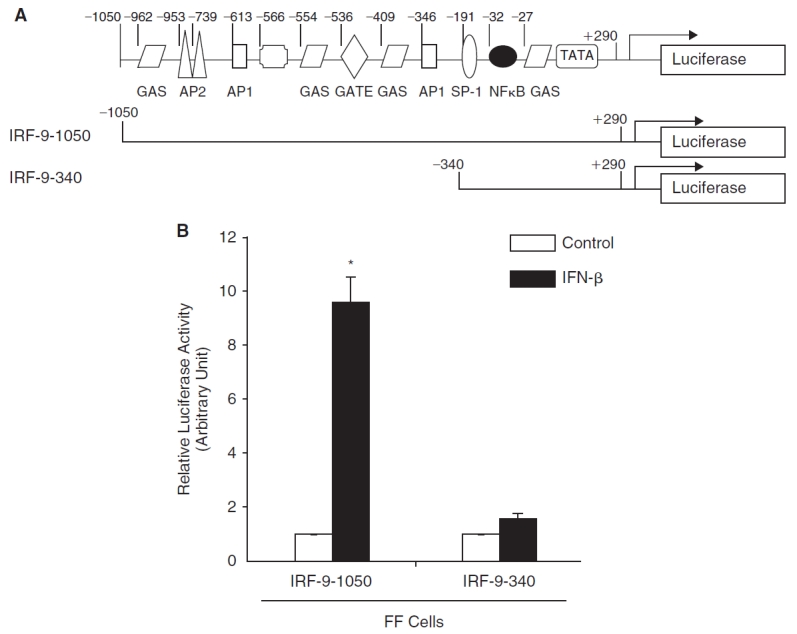
Induction of interferon regulatory factor-9 (IRF-9) by interferon-β (IFN-β) requires the upstream region between −1,050 bp and −340 bp. (A) A schematic of the IRF-9 promoter with its putative regulatory motifs. Also shown are 2 schematics of IRF-9 promoter–reporter luciferase constructs. (B) Mutant IFNAR-2c (FF) cells were transiently transfected overnight with a mIRF-9-1050 or mIRF-9-340 promoter–reporter luciferase construct together with Renilla luciferase control vector. The luciferase activity was determined and normalized to Renilla luciferase for transfection efficiency. The fold increase with IFN-β (filled bars) compared to untreated control (open bars) is shown for IRF-9-1050 and IRF-9-340. Results represent an average of 3 experiments with triplicate samples for each treatment.
We previously identified NF-κB as an accessory factor essential for the induction of CXCL11 and TRAIL by IFN-β (Rani and others 2001). The IRF-9 promoter contained a potential NF-κB element. To further define the role of this element in the IFN-β response, we created a deletion-mutation construct comprised of a 340 bp region of the promoter, which included the NF-κB, GAS, and SP-1 element (Fig. 3A). Transient transfection assays showed that the IRF-9-340 promoter–reporter construct was not induced by IFN-β in FF cells, indicating that the IFN-responsive region of the IRF-9 gene was located between −1050 and −340 (Fig. 3B). Further analyses revealed that this region contained an ISRE-like sequence.
Novel IFN-β–induced DNA-binding complexes in FF cells
The GATE element in the promoter of murine IRF-9 is essential for its transcriptional induction by IFN-γ (Weihua and others 1997a). High concentrations of IFN-α/β were required for GATE-mediated IRF-9 induction in a human monocytic cell line (Xiao and others 2001). The GATE element shares some homology with the classical ISRE element that binds the transcription factor ISGF3 (Xiao and others 2001). The nucleotide sequences of the wtGATE and ISRE consensus are shown in Figure 4A. Two conserved nucleotides in the ISRE element are absent in the GATE element suggesting that ISGF3 would not be bound to the GATE element (Fig. 4A). To determine whether GATE formed complexes with proteins in response to IFN-β, we performed an EMSA. Two IFN-β-inducible DNA-binding complexes were detected in these assays. We termed these factors BBF-1 (β-IFN–induced DNA-binding factor-1) and BBF-2 (β-IFN–induced DNA-binding factor-2) (Fig. 4B). Untreated FF or R2C cells showed very low levels of BBF-1 but not BBF-2 (Fig. 4B). Both complexes were more robustly induced by IFN-β in R2C than in FF cells (Fig. 4B). It is important to note that these complexes were detectably induced in FF cells. The 2 complexes were competed out by 50-fold molar excess of unlabeled wtGATE oligonucleotides but not by a mutated mGATE oligonucleotide (Fig. 4B), suggesting the induced DNA–protein complexes bound to the GATE element in a sequence-specific fashion.
FIG. 4. .
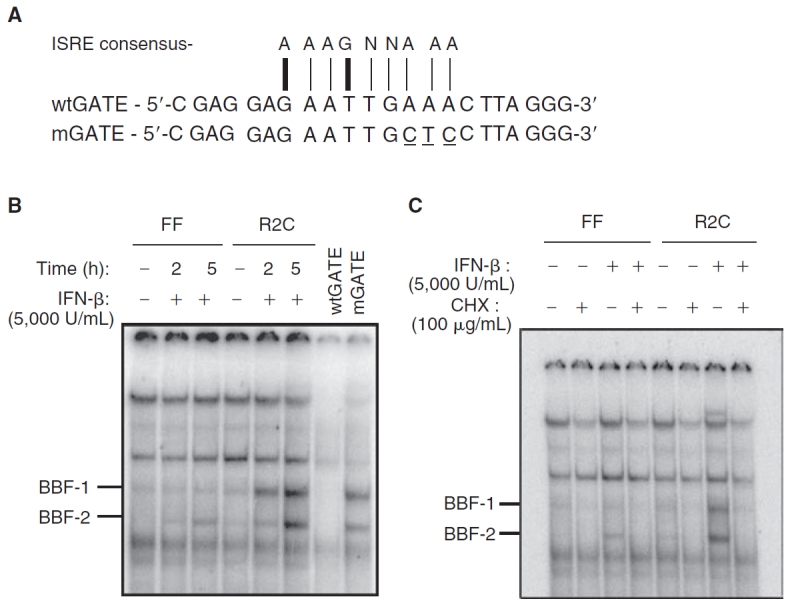
Novel DNA-binding complexes induced in mutant IFNAR-2c (FF) cells by interferon-β (IFN-β). (A) The nucleotide sequence of the wild-type GATE element (wtGATE), mutated GATE element (mGATE, mutated residues are shown underlined), and the consensus sequence for an ISRE are shown. The nucleotide residues not conserved between ISRE and GATE are indicated by thick vertical lines and thin vertical lines indicate consensus. (B) EMSA analysis of nuclear extracts from FF and wild-type IFNAR-2c (R2C) cells treated with recombinant IFN-β (5,000 U/mL) for 2 or 5 h or reserved as untreated controls (−) were assayed. The 32P-labeled GATE probe bound 2 complexes, BBF-1 and BBF-2. Results from one of three experiments are shown. Incubation with a 50-fold molar excess of unlabeled wild-type GATE oligonucleotide (wtGATE) competed out the complexes but not mutated GATE oligonucleotide (mGATE) prior to incubation with radiolabeled GATE probe. (C) IFN-β–induced DNA-binding complexes required de novo protein synthesis. FF and R2C cells were pretreated with cycloheximide (100 µg/mL) for 30 min followed by treatment with or without IFN-β (5,000 U/mL) for 6 h or reserved as untreated controls.
To address if new protein synthesis were required for the formation of BBF-1 and BBF-2 complexes, the FF and R2C cells were treated for 30 min with the protein synthesis inhibitor cycloheximide (100 µg/mL), prior to stimulation with IFN-β for 5 h. As shown in Figure 4C, the generation of both complexes required de novo protein synthesis in both FF and R2C cells. Also, basal expression of the complex BBF-1 was abrogated by cycloheximide treatment.
IRF-1 and C/EBP-β are components of BBF-1
To identify the factors that interact with GATE in response to IFN-β, antibodies against various members of IRF and STAT proteins involved in the IFN signal transduction pathway were used for supershifting the EMSA complexes. Nuclear extracts from R2C cells treated with IFN-β for 5 h were used in these assays, since they induced more abundant DNA-binding complexes than were observed in FF cells (Fig. 4B). A wide panel of antibodies against various STATs, p65, IRF-3, and c-myc had no effect on the mobility of BBF-1 and BBF-2 as shown in Figure 5A. Interestingly, anti-IRF-1 blocked the DNA binding to BBF-1 suggesting it was a component of the BBF-1 complex (Fig. 5A and 5B).
FIG. 5. .

cEBP-β is a component of the interferon-β (IFN-β)–induced DNA complexes in mutant IFNAR-2c (FF) cells. (A) EMSA analysis was performed using nuclear extracts from wild-type IFNAR-2c (R2C) cells treated for 5 h with IFN-β (5,000 U/mL). Supershift assays were performed using anti-interferon regulatory factor (IRF)-3, 2 different antibodies to STAT1, anti-STAT2, anti-STAT3, anti-STAT5, anti-p65, anti-c-myc, and anti-IRF-1. (B) EMSA analysis was performed using the above extracts as explained in (A) and supershift experiments were done using anti-IRF-1 and anti-C/EBP-β. (C) Overexpression of cEBP-β enhanced IFN-β–induced transcription of IRF-9. FF cells were co-transfected with the wild-type IRF-9 promoter–reporter luciferase construct (wtIRF-9-1050) alone or in combination with wild-type cEBP-β expression vector. Cells were stimulated with and without IFN-β (1,000 U/mL for 16 h). IFN-β stimulation status are indicated as “+” for stimulated cells and “−” for nonstimulation. The luciferase activity was determined and normalized to Renilla luciferase for transfection efficiency. The fold induction with IFN-β compared to untreated control is shown. Results represent an average of 3 experiments with triplicate samples for each treatment.
The CCAAT/enhancer-binding protein-β (C/EBP-β) is an important component of GATE-dependent gene transcription (Roy and others 2002). Anti-C/EBP-β partially blocked the formation of the BBF-1 complex and shifted the mobility of the BBF-2 complex in nuclear extracts obtained from IFN-β-treated cells (Fig. 5B).
Nuclear extracts from IFN-β-treated R2C cells also contained a high-molecular-weight band (Fig. 5A), which was completely supershifted with STAT2 antibodies (Fig. 5A). Since this complex was absent in FF cells, it was not studied further.
To address if IRF-1 and C/EBP-β are capable of regulating IRF-9 transcription in response to IFN-β, we co-expressed vectors coding for these factors into FF cells. Co-transfection of a C/EBP-β expression plasmid enhanced luciferase transcription from the IRF-9 promoter–reporter in presence of IFN-β compared to a control transfected with an empty expression vector (Fig. 5C). These results further supported the involvement of C/EBP-β for IFN-β–mediated induction of IRF-9. In contrast, overexpression of IRF-1 expression plasmid suppressed induction of wtIRF-9 promoter–reporter luciferase activity by IFN-β (results not shown).
Discussion
The JAK-STAT pathways are the major signaling routes for IFN-induced transcriptional induction of ISGs (Stark and others 1998; Aaronson and Horvath 2002; Borden and others 2007). However, it has now become clear that the JAK-STAT pathway, which culminates in the formation of dimers of STAT proteins, each of which is phosphorylated on tyrosine, cannot by itself explain the full range of IFN-dependent biological effects in intact animals or different cell types (Ramana and others 2000a, 2000b; Pilz and others 2003; Wang and others 2003). Several lines of evidence support the concept that accessory signals generated by ligation of IFN receptors are biologically relevant and act in cell-type-specific fashion: for example, mice lacking type I IFN receptors are more susceptible to viral challenge than mice lacking STAT1, implying STAT-independent signaling (O’Shea and others 2002). Therefore, only a limited portion of the biological complexity of the IFN system is captured by examining the essential JAK-STAT components. Examination of the accessory signals that modulate the functions of STATs or activate complementary signaling components will provide insight into the mechanisms of IFN-induced expression of ISGs (Rani and Ransohoff 2005).
Here we describe IRF-9, an ISG induced by IFN-β in the absence of STAT phosphorylation. STAT1, STAT2, and STAT3 were not phosphorylated by IFN-β in the FF cells (which express a mutant IFNAR-2c that lack tyrosine residue at 337 and 512). IFN-induced activation of STAT4, STAT5, or STAT6 was also absent in these cells (results not shown). We used deletion mutagenesis and transient transfection assays to show functional importance of GATE, a positive regulatory element, for regulating IRF-9 expression. We demonstrated 2 sequence-specific GATE-binding complexes in nuclear extracts of FF cells treated with IFN-β. No STAT protein was part of this complex. Although GATE shows some homology to ISRE, proteins that bind to ISRE-like STAT1, STAT2, and IRF-9 did not bind to GATE. Supershift analysis revealed transcription factors IRF-1 and CAAT/enhancer-binding proteins (C/EBP-β) to be components of the GATE-binding complexes induced by IFN-β.
CAAT/enhancer-binding proteins (C/EBPs) belong to a family of leucine zipper transcription factors and are involved in cellular proliferation and differentiation in various tissues (Landschulz and others 1988; Poli 1998). The role of C/EBP-β in type I IFN-induced signal transduction pathways is unknown. However, C/EBP-β is essential for the induction of IRF-9 by type II IFN, IFN-γ (Weihua and others 1997a,b; Roy and others 2002; Roy and others 2005). Overexpression of C/EBP-β in FF cells enhanced transcriptional induction of IRF-9 luciferase by IFN-β. C/EBP-β binding to DNA requires phosphorylation on serine and threonine residues (Roy and others 2005).
The IFN-regulatory factors (IRFs) belong to a family of transcription factors with at least 10 members in humans and mice, each of which play distinct roles in biological processes as revealed by gene-targeting studies (Tamura and others 2008). They were initially characterized as transcriptional regulators of type I IFNs and IFN-inducible genes, which play a major role in adaptive and innate immune responses (Tamura and others 2008). The IRF-binding domain recognizes a DNA sequence corresponding to approximately IFN-stimulated response element (ISRE) half site (Darnell and others 1994). Our studies revealed that IRF-1 binds to the GATE element in the promoter of IRF-9 in response to IFN-β. However, it is not clear how it is regulated.
Previous studies have demonstrated that IFNs can activate Ras-mitogen-activated protein kinase cascade in various cells (Berger and others 1997; Nishiya and others 1997; Sakatsume and others 1998). Our earlier work demonstrated a requirement of accessory signaling by PI3K and p38MAPK for the induction of ISGs-like β-R1 and TRAIL by IFN-β (Rani and others 2002; Rani and others 2007). Our studies using pharmacological inhibitors did not suggest a role for PI3K, p38 MAPK, or ERK kinase in the transcriptional induction of IRF-9 by IFN-β (results not shown).
Robust induction of IRF-9 by IFN-β was seen in the parental 2fTGH cells or R2C cells, suggesting the maximal induction of IRF-9 by IFN-β required the phosphorylation of the 2 STAT proteins STAT1 and STAT2. However, the induction of IRF-9 by IFN-β in FF cells that lacked phosphorylation of STAT1, STAT2, and STAT3 was clearly dependent on accessory signaling by IRF-1 and C/EBP-β involving non-STAT pathways. Defining the mechanism of activation of C/EBP-β, an essential and newly identified component in IFN-signaling pathway can help further understand how accessory signaling drives gene induction in the absence of STAT phosphorylation. We propose these studies will help define how the outcomes of accessory signals are integrated with and without activated STATs and are important for mediating cell-type-dependent selective transcriptional responses.
Acknowledgments
This research is supported by grant PO1 CA062220 from the National Cancer Institute. We are especially grateful to George R. Stark for his support and many helpful discussions.
Contributor Information
M.R. Sandhya Rani, Neuroinflammation Research Center, Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio..
Ed Croze, Department of Immunology, Bayer Healthcare Pharmaceuticals, Inc., Richmond, California..
Tao Wei, Neuroinflammation Research Center, Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio..
Jennifer Shrock, Neuroinflammation Research Center, Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio..
Anupama Josyula, Neuroinflammation Research Center, Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio..
Dhananjaya V. Kalvakolanu, Department of Microbiology and Immunology, University of Maryland School of Medicine, Baltimore, Maryland.
Richard M. Ransohoff, Neuroinflammation Research Center, Department of Neurosciences, Lerner Research Institute, Cleveland Clinic, Cleveland, Ohio.
References
- Aaronson DS, Horvath CM. A road map for those who know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- Ackrill AM, Reid LE, Gilbert CS, Gewert DR, Porter AC, Lewin AR, Stark GR. Differential response of the human 6-16 and 9-27 genes to alpha and gamma interferons. Nucleic Acids Res. 1991;19:591–598. doi: 10.1093/nar/19.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger LC, Tamir A, Ben-David Y, Hawley RG. Dose-dependent activation of p21ras by IFN-beta. J Interferon Cytokine Res. 1997;17:757–762. doi: 10.1089/jir.1997.17.757. [DOI] [PubMed] [Google Scholar]
- Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM, Foster GR, Stark GR. Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JE, Jr., Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. 1994;264:1415–1421. doi: 10.1126/science.8197455. [DOI] [PubMed] [Google Scholar]
- Domanski P, Nadeau OW, Platanias LC, Fish E, Kellum M, Pitha P, Colamonici OR. Differential use of the betaL subunit of the type I interferon (IFN) receptor determines signaling specificity for IFNalpha2 and IFNbeta. J Biol Chem. 1998;273:3144–3147. doi: 10.1074/jbc.273.6.3144. [DOI] [PubMed] [Google Scholar]
- Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, Lou J. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–317. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells. 1996;1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- Lutfalla G, Holland SJ, Cinato E, Monneron D, Reboul J, Rogers NC, Smith JM. Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J. 1995;14:5100–5108. doi: 10.1002/j.1460-2075.1995.tb00192.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller M, Laxton C, Briscoe J, Schindler C, Improta T, Darnell JE, Jr., Stark GR. Complementation of a mutant cell line: central role of the 91 kDa polypeptide of ISGF3 in the interferon-alpha and -gamma signal transduction pathways. EMBO J. 1993;12:4221–4228. doi: 10.1002/j.1460-2075.1993.tb06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Hiscott J, Pitha PM. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997;8:293–312. doi: 10.1016/s1359-6101(97)00019-1. [DOI] [PubMed] [Google Scholar]
- Nishiya T, Uehara T, Edamatsu H, Kaziro Y, Itoh H, Nomura Y. Activation of Stat1 and subsequent transcription of inducible nitric oxide synthase gene in C6 glioma cells is independent of interferon-gamma-induced MAPK activation that is mediated by p21ras. FEBS Lett. 1997;408:33–38. doi: 10.1016/s0014-5793(97)00383-9. [DOI] [PubMed] [Google Scholar]
- Novick D, Cohen B, Rubinstein M. The human interferon alpha/beta receptor: characterization and molecular cloning. Cell. 1994;77:391–400. doi: 10.1016/0092-8674(94)90154-6. [DOI] [PubMed] [Google Scholar]
- O’Shea JJ, Gadina M, Schreiber RD. Cytokine signaling in 2002: new surprises in the Jak/Stat pathway. Cell. 2002;109((Suppl)):S121–S131. doi: 10.1016/s0092-8674(02)00701-8. [DOI] [PubMed] [Google Scholar]
- Pilz A, Ramsauer K, Heidari H, Leitges M, Kovarik P, Decker T. Phosphorylation of the Stat1 transactivating domain is required for the response to type I interferons. EMBO Rep. 2003;4:368–373. doi: 10.1038/sj.embor.embor802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Chatterjee-Kishore M, Nguyen H, Stark GR. Complex roles of Stat1 in regulating gene expression. Oncogene. 2000a;19:2619–2627. doi: 10.1038/sj.onc.1203525. [DOI] [PubMed] [Google Scholar]
- Ramana CV, Grammatikakis N, Chernov M, Nguyen H, Goh KC, Williams BR, Stark GR. Regulation of c-myc expression by IFN-gamma through Stat1-dependent and -independent pathways. EMBO J. 2000b;19:263–272. doi: 10.1093/emboj/19.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani MR, Asthagiri AR, Singh A, Sizemore N, Sathe SS, Li X, DiDonato JD. A role for NF-kappa B in the induction of beta-R1 by interferon-beta. J Biol Chem. 2001;276:44365–44368. doi: 10.1074/jbc.C100417200. [DOI] [PubMed] [Google Scholar]
- Rani MR, Foster GR, Leung S, Leaman D, Stark GR, Ransohoff RM. Characterization of beta-R1, a gene that is selectively induced by interferon beta (IFN-beta) compared with IFN-alpha. J Biol Chem. 1996;271:22878–22884. doi: 10.1074/jbc.271.37.22878. [DOI] [PubMed] [Google Scholar]
- Rani MR, Hibbert L, Sizemore N, Stark GR, Ransohoff RM. Requirement of phosphoinositide 3-kinase and Akt for interferon-beta-mediated induction of the beta-R1 (SCYB11) gene. J Biol Chem. 2002;277:38456–38461. doi: 10.1074/jbc.M203204200. [DOI] [PubMed] [Google Scholar]
- Rani MR, Pandalai S, Shrock J, Almasan A, Ransohoff RM. Requirement of catalytically active Tyk2 and accessory signals for the induction of TRAIL mRNA by IFN-beta. J Interferon Cytokine Res. 2007;27:767–779. doi: 10.1089/jir.2007.0005. [DOI] [PubMed] [Google Scholar]
- Rani MR, Ransohoff RM. Alternative and accessory pathways in the regulation of IFN-beta-mediated gene expression. J Interferon Cytokine Res. 2005;25:788–798. doi: 10.1089/jir.2005.25.788. [DOI] [PubMed] [Google Scholar]
- Roy SK, Hu J, Meng Q, Xia Y, Shapiro PS, Reddy SP, Platanias LC. MEKK1 plays a critical role in activating the transcription factor C/EBP-beta-dependent gene expression in response to IFN-gamma. Proc Natl Acad Sci USA. 2002;99:7945–7950. doi: 10.1073/pnas.122075799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SK, Shuman JD, Platanias LC, Shapiro PS, Reddy SP, Johnson PF, Kalvakolanu DV. A role for mixed lineage kinases in regulating transcription factor CCAAT/enhancer-binding protein-{beta}-dependent gene expression in response to interferon-{gamma} J Biol Chem. 2005;280:24462–24471. doi: 10.1074/jbc.M413661200. [DOI] [PubMed] [Google Scholar]
- Russell-Harde D, Wagner TC, Rani MR, Vogel D, Colamonici O, Ransohoff RM, Majchrzak B. Role of the intracellular domain of the human type I interferon receptor 2 chain (IFNAR2c) in interferon signaling. Expression of IFNAR2c truncation mutants in U5A cells. J Biol Chem. 2000;275:23981–23985. doi: 10.1074/jbc.M002518200. [DOI] [PubMed] [Google Scholar]
- Sakatsume M, Stancato LF, David M, Silvennoinen O, Saharinen P, Pierce J, Larner AC. Interferon gamma activation of Raf-1 is Jak1-dependent and p21ras-independent. J Biol Chem. 1998;273:3021–3026. doi: 10.1074/jbc.273.5.3021. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–584. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Harada H, Lamphier M. Regulation of the interferon system and cell growth by the IRF transcription factors. J Cancer Res Clin Oncol. 1995;121:516–520. doi: 10.1007/BF01197763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uze G, Lutfalla G, Gresser I. Genetic transfer of a functional human interferon alpha receptor into mouse cells: cloning and expression of its cDNA. Cell. 1990;60:225–234. doi: 10.1016/0092-8674(90)90738-z. [DOI] [PubMed] [Google Scholar]
- Wagner TC, Velichko S, Vogel D, Rani MR, Leung S, Ransohoff RM, Stark GR. Interferon signaling is dependent on specific tyrosines located within the intracellular domain of IFNAR2c. Expression of IFNAR2c tyrosine mutants in U5A cells. J Biol Chem. 2002;277:1493–1499. doi: 10.1074/jbc.M108928200. [DOI] [PubMed] [Google Scholar]
- Wang J, Pham-Mitchell N, Schindler C, Campbell IL. Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-gamma in the brain. J Clin Invest. 2003;112:535–543. doi: 10.1172/JCI18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua X, Kolla V, Kalvakolanu DV. Interferon gamma-induced transcription of the murine ISGF3gamma (p48) gene is mediated by novel factors. Proc Natl Acad Sci USA. 1997a;94:103–108. doi: 10.1073/pnas.94.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihua X, Lindner DJ, Kalvakolanu DV. The interferon-inducible murine p48 (ISGF3gamma) gene is regulated by protooncogene c-myc. Proc Natl Acad Sci USA. 1997b;94:7227–7232. doi: 10.1073/pnas.94.14.7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Wang L, Yang X, Chen T, Hodge D, Johnson PF, Farrar W. CCAAT/enhancer-binding protein beta mediates interferon-gamma-induced p48 (ISGF3-gamma) gene transcription in human monocytic cells. J Biol Chem. 2001;276:23275–23281. doi: 10.1074/jbc.M010047200. [DOI] [PubMed] [Google Scholar]
- Yan H, Krishnan K, Greenlund AC, Gupta S, Lim JT, Schreiber RD, Schindler CW. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996;15:1064–1074. [PMC free article] [PubMed] [Google Scholar]