

Abstract
The type III interferon (IFN) family elicits an antiviral response that is nearly identical to that evoked by IFN-α/β. However, these cytokines (known as IFN-λ1, 2, and 3) signal through a distinct receptor, and thus may be resistant to the evasion strategies used by some viruses to avoid the IFN-α/β response. Orthopoxviruses are highly resistant to IFN-α/β because they encode well-characterized immunomodulatory proteins that inhibit IFN activity. These include a secreted receptor (B18R) that neutralizes IFN-α/β, and a cytoplasmic protein (E3L) that blocks IFN-α/β effector functions in infected cells. We therefore determined the ability of these immunomodulators to abrogate the IFN-λ–induced antiviral response. We found that (i) vaccinia virus (VACV) replication is resistant to IFN-λ antiviral activity; (ii) neither VACV B18R nor the variola virus homolog B20R neutralizes IFN-λ; (iii) VACV E3L inhibits the IFN-λ–mediated antiviral response through a PKR-dependent pathway; (iv) VACV infection inhibits IFN-λR–mediated signal transduction and gene expression. These results demonstrate differential sensitivity of IFN-λ to multiple distinct evasion mechanisms employed by a single virus.
Introduction
Poxviruses are large double-stranded DNA viruses that replicate in the cytoplasm of infected cells. The genus Orthopoxvirus comprises a number of morphologically similar virus such as the variola virus (VARV; the etiological agent of smallpox), vaccinia virus (VACV), and monkeypox virus (Di Giulio and Eckburg 2004). Despite the fact that smallpox was eradicated 30 years ago, the use of poxviruses as agents of bioterrorism remains a potential threat. Because the smallpox vaccine has rare adverse effects including generalized infection (and even death) in some individuals, widespread immunization can be challenging in the current environment (Lane and others 1971; Fulginiti and others 2003). Hence, there is a need to develop new effective therapies against this family of viruses.
Although the cellular antiviral response elicited by interferon (IFN)-α/β or IFN-γ is a critical component of the host innate immune response to infection, poxviruses have evolved several strategies to evade this response (Seet and others 2003). First, they encode a number of proteins that function as soluble receptors that bind to the IFNs and neutralize their activity. Included in this category are proteins that bind to IFN-α/β (such as VACV B18R) and IFN-γ (VACV B8R) (Alcami and Smith 1995; Colamonici and others 1995; Symons and others 1995). VACV B18R is a secreted glycoprotein that belongs to the immunoglobulin superfamily and has homology to the type I IFN receptor α-subunit (Smith and Chan 1991; Colamonici and others 1995). B18R inhibits the antiviral response by preventing soluble IFN-α/β from binding to cellular receptors, and by binding to both infected and uninfected cells (Colamonici and others 1995; Symons and others 1995; Alcami and others 2000).
In addition to the secreted proteins, poxviruses also express cytoplasmic proteins that block the IFN response. The E3L protein binds to dsRNA (Chang and others 1992; Chang and Jacobs 1993), inhibits the activation of dsRNA-activated protein kinase (PKR; Chang and others 1992), and blocks IRF3 activation (Smith and others 2001; Xiang and others 2002). Numerous recent studies have shown the importance of PKR inactivation for E3L suppression of the IFN-α/β and IFN-γ antiviral response, at least in certain cell types (Arsenio and others 2008; Zhang and others 2008; Zhang and Samuel 2008; Zhang and others 2009). Interestingly, E3L was also recently shown to bind and inhibit the IFN-induced ubiquitin-like protein, ISG15 (Guerra and others 2008). There are 2 key functional domains in E3L: the dsRNA-binding domain at the C-terminus, which is necessary for IFN resistance, and the N-terminal Z-DNA-binding domain, which is required for in vivo pathogenesis (Chang and Jacobs 1993; Brandt and Jacobs 2001; Kim and others 2003). The importance of E3L in virus replication may also be evidenced by its high degree of similarity among different poxviruses [eg, 100% amino acid identity between VACV and VARV (Dave and others 2006)]. In addition to E3L, VACV also encodes a phosphatase (H1) that inhibits IFN-α/βR- and IFN-γR-mediated STAT tyrosine phosphorylation, and therefore blocks downstream antiviral gene expression (Najarro and others 2001; Mann and others 2008). Because they express these potent immunomodulatory factors, poxvirus replication is relatively resistant to the antiviral activity of IFN-α/β and IFN-γ (Haga and Bowie 2005).
A new family of IFN-related cytokines known as IFN-λ1, 2, and 3 (or IL-29, IL-28A, and IL-28B) was recently discovered (Kotenko and others 2003; Sheppard and others 2003), and designated type III IFN. Despite having very low sequence homology to the IFN-α/β family, these 2 families share a number of common characteristics. Both type I and type III IFNs are activated by virus infection and Toll-like receptor activation (Kotenko and others 2003; Sheppard and others 2003; Coccia and others 2004) through a common mechanism (Onoguchi and others 2007). However, the type III IFNs bind to a unique receptor containing the IL-10Rβ and IL-28Rα subunits, and do not bind to the IFN-α/β receptor (Kotenko and others 2003; Sheppard and others 2003; Donnelly and others 2004). Similar to the IFN-α/β receptor, activation of the type III IFN receptor activates STAT-1 and STAT-2, which subsequently results in formation of the IFN-stimulated gene factor 3 complex. Like IFN-α/β, the type III IFNs inhibit the replication of viruses such as encephalomyocarditis virus, vesicular stomatitis virus (VSV), cytomegalovirus, herpes simplex virus type 2, influenza A virus, hepatitis B virus, and hepatitis C virus (Kotenko and others 2003; Sheppard and others 2003; Brand and others 2005; Osterlund and others 2005; Robek and others 2005; Ank and others 2006; Marcello and others 2006a).
The interactions between type III IFN and the poxviruses are only beginning to be elucidated. Murine IFN-λ2 expressed from recombinant VACV decreases both virus replication and pathogenesis in mice (Bartlett and others 2005). However, this attenuation correlates with enhanced T-cell responses to the virus, and mouse IFN-λ2 does not inhibit virus replication in cultured mouse fibroblasts (Bartlett and others 2005). Furthermore, while a secreted glycoprotein produced by the primate yatapoxvirus Yaba-like disease virus inhibits IFN-λ activity, the VACV B18R protein does not, indicating that this pathway may be alternatively regulated by different viruses (Huang and others 2007).
In human poxvirus infections, IFN-λ may be an important determinant of virulence, depending on the ability of proteins such as B18R or E3L to blunt the IFN-λ–induced antiviral response. We therefore investigated the ability of the secreted and cytoplasmic immunomodulatory proteins to inhibit the antiviral activity of type III IFN. While the secreted IFN-α/β-binding proteins from VACV and VARV did not neutralize IFN-λ activity, the IFN-λ–mediated antiviral response was abrogated in VACV-infected cells in an E3L-dependent manner, and required PKR expression. These results indicate that the intracellular, but not extracellular, orthopoxvirus immunomodulatory proteins inhibit type III IFN activity.
Materials and Methods
Cell lines, viruses, and reagents
Human hepatocellular carcinoma cells (Huh7), lung carcinoma cells (A549), cervical epithelial adenocarcinoma cells (HeLa), and epithelial kidney cells (HEK293T) were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 100 µg penicillin/mL, 100 U streptomycin/mL, 2 mM l-glutamine, 1× MEM nonessential amino acids (Invitrogen, Carlsbad, CA), 1 mM sodium pyruvate, 10 mM HEPES buffer, and 10% heat-inactivated fetal bovine serum (Invitrogen). An immortalized mouse hepatocyte cell line (provided by F. Chisari, The Scripps Research Institute) was propagated in RPMI 1640 medium containing 100 ng epidermal growth factor (BD Biosciences, Franklin Lakes, NJ)/mL, 16 ng insulin-like growth factor II/mL, and 10 µg insulin (Sigma Aldrich Inc, St. Louis, MO)/mL. VACV (strain WR) was propagated on HeLa S3 cells and recombinant vesicular stomatitis virus expressing GFP (VSV-EGFP; provided by J. Rose, Yale University) was propagated on BHK-21 cells. Viruses were titered and stored at −80°C prior to use. Human IFN-λ1 and murine IFN-λ2 were purchased from Peprotech (Rocky Hill, NJ), and the human IFN-α subtypes, universal IFN-α(A/D), IFN-β, and IFN-γ were purchased from PBL InterferonSource (Piscataway, NJ). The specific activities of IFN-α2A, IFN-α(A/D), and IFN-γ were 2 × 108 U/mg, 1.2 × 108 U/mg, and 1.6 × 107 U/mg, respectively. The VACVΔE3L viruses (strain WR and Copenhagen) were provided by B. Jacobs (University of Arizona), and the E3L-specific monoclonal antibody (Weaver and others 2007) was provided by S. Isaacs (University of Pennsylvania).
VACV B18R and VARV B20R expression vectors
Codon-optimized sequences of the VACV B18R (WR strain; accession number CAA01478) and VARV B20R (Sumatra 1970 strain; accession number ABF28175) open reading frames were chemically synthesized [GenScript Corporation (Piscataway, NJ)], and cloned into the pCMV-Tag4 expression vector (Stratagene, La Jolla, CA) to produce the plasmids pB18R-FLAG and pB20R-FLAG. Two million HEK293T cells were transfected with the expression vectors or with empty vector, and 4 mL of culture medium was collected 2 days post-transfection. Medium was concentrated 10-fold by centrifugation through Ambicon Ultra 30K cutoff separation membranes (Millipore, Billerica, CA). Protein expression was analyzed by Western blot analysis using a FLAG-specific antibody (Sigma), and concentrations were adjusted to normalize for protein expression level. Fifty microliters of sample (either undiluted or diluted as specified in the legend to Fig. 2) was incubated with the cytokines at the indicated concentrations. The experiments using the VARV polypeptide were approved by the Yale University Biological Safety Committee following review by the NIH Office of Biological Activities. These studies were also registered with the World Health Organization, and precautions outlined in the WHO Recommendations Concerning the Distribution, Handling, and Synthesis of Variola Virus DNA (May 2008) were followed.
FIG. 2. .

Vaccinia virus (VACV) B18R and VARV B20R both neutralize interferon (IFN)-α/β but not IFN-λ. DMEM or conditioned medium from HEK293T cells transfected with B18R-FLAG, B20R-FLAG, or empty expression vectors was incubated with the indicated concentrations of either (A) IFN-α2a or (B) IFN-λ1 for 30 min on ice before adding to Huh7 cells. (C) Conditioned medium diluted 1:10 was incubated with 5 ng of the indicated type I IFN. (D) Conditioned medium was diluted by the indicated ratio prior to Western blot analysis using a FLAG-specific antibody. Diluted conditioned media was incubated with 10 ng of IFN-α2a prior to addition to cells. (E) DMEM or conditioned medium from VACV-infected or -uninfected (uninf) HeLa S3 cells was incubated with either 0.5 ng/mL or 5 ng/mL IFN-α or IFN-λ1 for 30 min on ice prior to addition to Huh7 cells. For (A–E), induction of the representative IFN-stimulated gene MxA was measured 24 h after addition of IFN plus conditioned medium by quantitative RT-PCR. Expression is displayed as fold induction relative to untreated controls, and is normalized to GAPDH. All data are displayed as the mean of 3 independent experiments, and error bars represent SEM.
Quantitative real-time RT-PCR
Total RNA was harvested and prepared using the RNeasy Mini kit (Qiagen, Valencia, CA). Total RNA (1 µg) was reverse-transcribed with random hexamers using the TaqMan reverse transcription kit (Applied Biosystems, Foster City, CA). Quantitative PCR was performed using an Applied Biosystems 7500 real-time PCR system. The 25 µL PCRs contained 100 ng reverse-transcribed RNA, 200 nM sense and antisense primers (MxA, IFI27, or GAPDH), and 12.5 µL SYBR Green PCR master mix (Applied Biosystems). The primer sequences used were as follows: MxA, 5′-ACA GGA CCA TCG GAA TCT TG-3′ (sense), 5′-CCC TTC TTC AGG TGG AAC AC-3′ (antisense); IFI27, 5′-CCG TAG TTT TGC CCC TGG-3′ (sense), 5′-CGA GGC CAT TCC CGC CGC-3′ (antisense); GAPDH, 5′-AAG TAT GAC AAC AGC CTC AAG ATC-3′ (sense), 5′-CTG TGG TCA TGA GTC CTT C-3′ (antisense). After an initial incubation for 5 min at 95°C, PCR amplification was performed by cycling 50 times for 30 s at 95°C followed by 60 s at 60°C. Gene expression was quantified by the ΔΔCt method using the 7500 system Sequence Detection Software (Applied Biosystems).
STAT phosphorylation
Cells were washed once with PBS and lysed with 2× SDS sample buffer (125 mM Tris/HCl, 4% SDS, 20% glycerol, 0.02% bromophenol blue). Extracts were separated on a 10% SDS-polyacrylamide gel, transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA), and probed with antibodies specific for phospho-STAT-1 (Y701) and STAT-1 (Cell Signaling, Danvers, MA). Proteins were visualized using LumiGLO chemiluminescent reagents (Cell Signaling) and a Fuji LAS-3000 cooled CCD camera.
PKR siRNA transfections
Huh7 cells were seeded on a 24-well plate at 1 × 105 cells per well in 500 µL DMEM without antibiotics 1 day prior to transfection. Cells were transfected with 100 nM of siRNA specific to PKR 5′-GACGGAAAGACUUACGUUATT-3′ (sense) or Silencer Select Negative Control #1 (Ambion) using DharmaFECT 4 reagent (Dharmacon) in DMEM without antibiotics and incubated at 37°C overnight. Cells were treated with 2.5 ng/mL IFN-α or 250 ng/mL IFN-λ1 for 18 h, then infected at an MOI = 1 with VACVΔE3L. PKR silencing was confirmed by Western blot using a PKR-specific antibody (Cell Signaling).
Virus infectivity/replication assays
VSV-EGFP infectivity and replication were measured by 3 methods. First, the number of GFP(+) cells in infected cultures were quantified in a representative field by microscopic evaluation using an Olympus CK40 inverted microscope outfitted with an Olympus DP12 digital camera. Alternatively, flow cytometric analysis of GFP expression in infected cells was performed after fixation in 2% formaldehyde using a BD FACSCalibur. VSV-EGFP virus released into the media of infected cultures was titered using HeLa cells. VACV replication was assayed by titering virus from infected culture medium using BHK-21 or Huh7 cells. To measure levels of intracellular VACV replication, infected cells were harvested and lysed by 3 freeze-thaw cycles prior to titering.
Results
IFN-λ1 weakly inhibits VACV replication in cell culture
We first measured the sensitivity of VACV to the antiviral activity of human IFN-λ1. Human lung carcinoma A549 cells were either left untreated or were treated with increasing concentrations (0.025–250 ng/mL) of IFN-λ1 for 18 h, after which they were infected with VACV (strain WR) at a multiplicity of infection (MOI) of 1 or 0.1. A549 cells were used for these initial studies as the antiviral activity of IFN-λ is very well characterized in this cell line (Kotenko and others 2003; Meager and others 2005). Virus titers in the cell culture medium 48 h post-infection in IFN-λ1-treated cells were reduced only minimally compared to untreated controls (Fig. 1A). Although the inhibition of virus replication displayed a general dose-dependent trend, the reductions in virus titer reached a maximum of only 3-fold (MOI = 1) or 8-fold (MOI = 0.1) at the highest concentration of IFN-λ1 (250 ng/mL), and these levels of replication were not statistically different from untreated cells (P > 0.05 for all concentrations and MOI). In addition to the cell culture medium, we also measured the amount of virus in infected cell lysates, and found that intracellular replication was also not significantly reduced by IFN-λ1 treatment, even at low MOI (Fig. 1B). These decreases in virus replication did not differ substantially from the reductions observed when the cells were instead treated with similar concentrations of IFN-α2a (data not shown). Because we have previously shown that IFN-λ1 can function cooperatively with IFN-α or IFN-γ to inhibit viruses such as VSV and hepatitis C (Pagliaccetti and others 2008), we also treated the cells with combinations of IFN-λ1 plus IFN-α or IFN-γ. Like the single cytokines alone, the combinations of IFN-λ1 and IFN-α or IFN-γ failed to inhibit VACV-induced cytopathic effects (CPE) in infected cells (Fig. 1C). In contrast to VACV, VSV replication was blocked in A549 cells by IFN-λ1 (IC50 = 0.13 ng/mL) (data not shown), confirming that the cells do in fact respond to the cytokine. These results indicate that VACV is relatively resistant to the antiviral response induced by IFN-λ1, presumably due to the activity of the virus-encoded immunomodulatory proteins.
FIG. 1. .
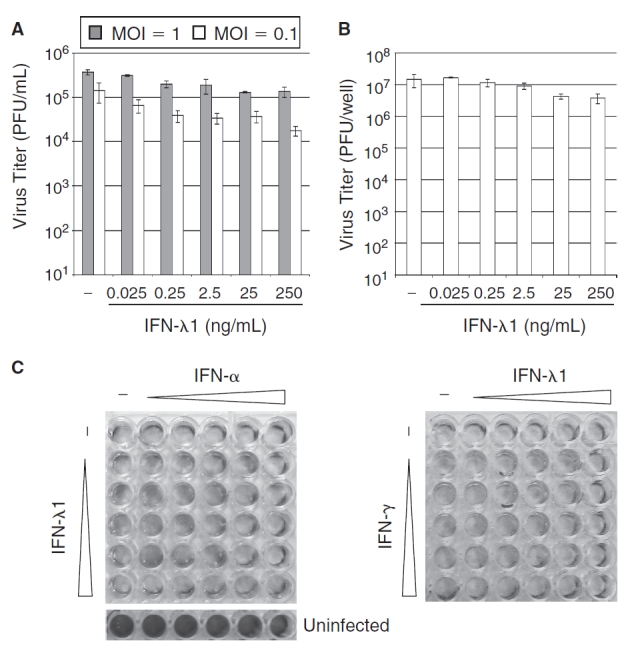
Interferon-λ (IFN-Δ) weakly inhibits vaccinia virus (VACV) replication. (A) A549 cells were treated with the indicated concentrations of IFN-λ1 for 18 h prior to infection with VACV at MOI = 1 or 0.1. Extracellular virus titers were measured in the cell culture medium 48 h post-infection. (B) A549 cells were treated with the indicated concentrations of IFN-λ1 for 18 h prior to infection with VACV at MOI = 0.1. Intracellular virus replication was measured 48 h post-infection. For both A and B, data are displayed as the mean of 2–3 independent experiments, and error bars represent standard error of the mean (SEM). (C) A549 cells were treated with increasing concentrations (0.025–250 ng/mL in 10× steps) of IFN-λ1 and IFN-α or IFN-γ alone or in combination for 18 h prior to infection with VACV at MOI = 1. Six untreated control wells were left uninfected. Cells were stained with 5% crystal violet 48 h post-infection to visualize virus-induced cytopathic effects.
Secreted IFN-α/β receptors B18R and B20R do not block IFN-λ–induced signal transduction and gene expression
We next determined the ability of 2 Orthopoxvirus secreted IFN-α/β-binding proteins (VACV B18R, VARV B20R) to inhibit IFN-λ1 activity. VARV B20R has 89% amino acid identity to VACV B18R, and therefore is presumed to possess similar activity, although this has not been formally demonstrated (Symons and others 1995). HEK293T cells were transfected with expression vectors encoding FLAG-tagged VACV B18R or VARV B20R, and conditioned medium was prepared as described in the Materials and Methods. Preincubation of human IFN-α2a over a wide range of concentrations (1–25 ng/mL) with conditioned media from VACV B18R- or VARV B20R-transfected cells efficiently blocked the induction of the representative IFN-stimulated gene MxA in Huh7 cells (Fig. 2A). In contrast, neither B18R nor B20R inhibited MxA expression induced by IFN-λ1 (Fig. 2B). To determine if this neutralization was specific to IFN-α2a, we also compared the ability of VACV B18R and VARV B20R to inhibit different type I IFNs (α-subtypes and IFN-β). All IFN-α/β types examined were efficiently neutralized by both B18R and B20R (Fig. 2C). Furthermore, we confirmed that our preparations of conditioned media contained similar levels of the recombinant proteins, and found that a 10-fold dilution of the media retained sufficient B18R or B20R to completely inhibit IFN-α2a activity (Fig. 2D). However, a 100-fold dilution only partially (20%–40%) blocked MxA induction, indicating that the undiluted preparations contain nearly 100× the EC50 for IFN-α2a neutralization, but still fail to neutralize IFN-λ1. These results demonstrate that the secreted IFN-α/β receptors encoded by VACV and VARV do not inhibit IFN-λ function, and that the VACV B18R and VARV B20R proteins have a similar capacity to neutralize IFN-α/β.
However, these experiments did not rule out the possibility that a different VACV-encoded protein inhibits IFN-λ activity. To address this question, conditioned medium from VACV-infected cells was incubated with 2 different concentrations (0.5 or 5 ng/mL) of IFN-α or IFN-λ1 prior to addition to Huh7 cells. The IFN-mediated activation of STAT-1 phosphorylation or MxA mRNA expression was then assessed. As expected, conditioned medium from VACV-infected cells blocked the IFN-α–mediated phosphorylation of STAT-1 (data not shown) and induction of MxA (Fig. 2E). However, the conditioned medium did not neutralize STAT-1 phosphorylation (data not shown) or MxA expression (Fig. 2E) induced by IFN-λ1. Therefore, VACV does not appear to encode a secreted immunomodulatory protein distinct from B18R that blocks IFN-λ activity.
VACV infection rescues VSV from the antiviral activity of IFN-λ
It has been a longstanding observation that infection of cells with VACV rescues VSV from the antiviral activity of IFN-α/β (Thacore and Youngner 1973). Furthermore, the inhibition of the IFN-α response against VSV is primarily due to the activity of the cytoplasmic immunomodulator E3L (Shors and others 1998). We therefore determined if like IFN-α, VACV infection would rescue VSV from the antiviral activity of IFN-λ. For these experiments, we utilized a derivative of the mouse immortalized hepatocyte cell line MMHD3 (Amicone and others 1997), as in our hands these cells displayed the most complete inhibition of VSV replication after IFN-λ treatment (data not shown). In addition, murine IFN-λ2 was used for these experiments, because mice do not encode a functional IFN-λ1 (Lasfar and others 2006).
Mouse immortalized hepatocytes were treated for 18 h with either IFN-α or murine IFN-λ2 to activate gene expression and induce an antiviral state. The cells were then infected with VACV, and 2 h later were infected with VSV-EGFP. Twenty-four hours post-infection, intracellular VSV replication was monitored by microscopic evaluation of GFP expression (Fig. 3A), and extracellular VSV released into the culture medium was titered (Fig. 3B). Both IFN-α and IFN-λ efficiently blocked VSV replication, but pre-infection with VACV rescued VSV from the antiviral activity of both IFN-α (as expected) as well as IFN-λ. Therefore, VACV infection inhibits the IFN-λ–induced antiviral response.
FIG. 3. .
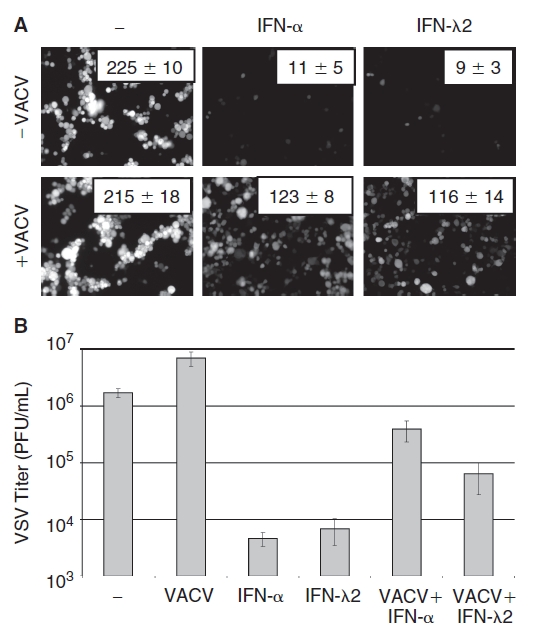
Vaccinia virus (VACV) infection rescues VSV from the antiviral activity of IFN-λ. (A) Immortalized mouse hepatocytes were treated with 2.5 ng IFN-α(A/D)/mL, 10 ng IFN-λ2/mL, or were left untreated for 24 h prior to infection with VACV (MOI = 1). Two hours later cells were infected with VSV-EGFP at MOI = 1 for 18 h. (A) Microscopic visualization of GFP+ cells. Numbers indicate mean ± SEM of GFP+ cells per field in 4 independent experiments. (B) Titer of VSV in infected culture medium displayed as plaque-forming units (PFU) per milliliter. Data are displayed as the mean of 4 independent experiments, and error bars represent SEM.
VACV E3L inhibits the antiviral activity of IFN-λ
We next used a recombinant VACV that lacks E3L (VACVΔE3L) to determine if the E3L protein is required to block the antiviral activity of IFN-λ (Beattie and others 1995). These experiments were performed identically to those described above except that (i) actinomycin D was added to the cultures 2 h post VACV infection to block VACV late gene expression, and thus abrogate potential differences in VACV and VACVΔE3L replication and (ii) VSV-EGFP expression was quantified by flow cytometry. Unlike pre-infection with VACV, infection with VACVΔE3L did not rescue VSV from IFN-λ antiviral activity (Fig. 4A). E3L expression (or lack of expression) in VACV- and VACVΔE3L-infected cells was confirmed by Western blot analysis using an E3L-specific antibody (Fig. 4B). Therefore, VACV infection inhibits IFN-λ antiviral activity against VSV in an E3L-dependent manner.
FIG. 4. .
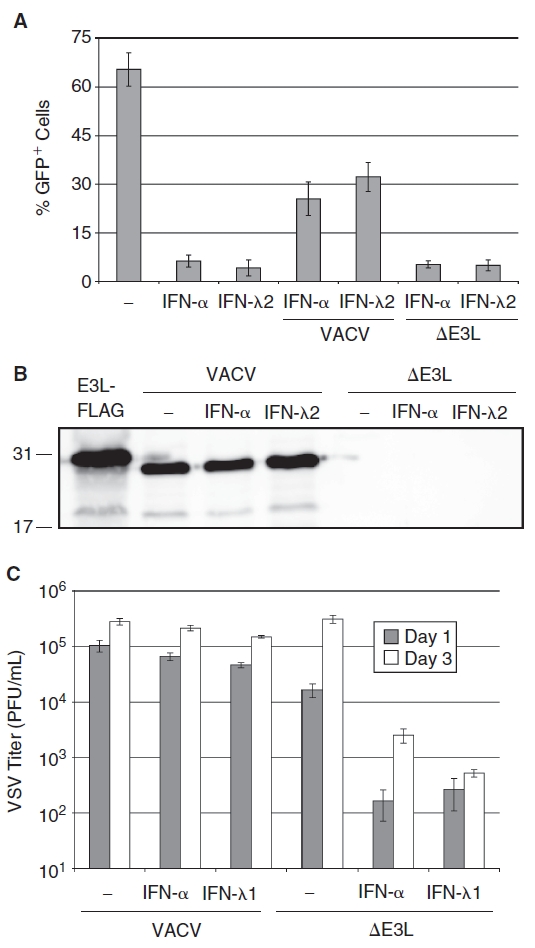
Inhibition of interferon-λ (IFN-Δ) antiviral activity requires vaccinia E3L. (A) Immortalized mouse hepatocytes were treated with 2.5 ng IFN-α(A/D)/mL, 10 ng IFN-λ2/mL, or were left untreated for 18 h prior to infection with wild-type vaccinia virus (VACV) or VACVΔE3L (MOI = 1). Two hours later cells were treated with 5 µg actinomycin D/mL to prevent late VACV gene expression, and were infected with VSV-EGFP at MOI = 1 for 18 h. GFP+ cells were quantified by flow cytometry. Data are displayed as the mean of 3 independent experiments, and error bars represent SEM. (B) Western blot analysis of E3L protein expression in wild-type VACV and VACVΔE3L-infected cells. Lysate from HEK293 cells transfected with a FLAG-tagged E3L expression vector is shown as a positive control. (C) VACVΔE3L is sensitive to the antiviral activity of IFN-λ. Huh7 cells were treated with 2.5 ng IFN-α/mL or 250 ng IFN-λ1/mL for 18 h prior to infection with VACV or VACVΔE3L (MOI = 1). Graph bars represent the mean extracellular viral titers 24 and 72 h post-infection from 3 independent experiments, and error bars indicate SEM.
Next, we determined if the VACVΔE3L virus was sensitive to the antiviral activity of IFN-λ. VACV (Copenhagen strain) lacking E3L was recently shown to efficiently replicate in Huh7 cells, and replication of the E3L-deficient virus was inhibited by IFN-α or IFN-γ (Arsenio and others 2008). Huh7 cells were treated with 2.5 ng IFN-α/mL or 250 ng IFN-λ1/mL for 24 h prior to infection with VACV or VACVΔE3L at MOI = 1. Virus replication was then measured 1 and 3 days post-infection. Neither IFN-α nor IFN-λ significantly inhibited VACV replication (Fig. 4C), consistent with our previous result using VACV strain WR in A549 cells (Fig. 1A). In contrast, both IFN-α and IFN-λ reduced the replication of VACVΔE3L by ∼100-fold (Fig. 4C) and prevented CPE in VACVΔE3L-infected cells (data not shown). Therefore, E3L plays an important role in the resistance of VACV to the antiviral activity of IFN-λ.
Inhibition of VACVΔE3L by IFN-λ requires PKR
It has previously been shown that the sensitivity of VACVΔE3L to inhibition by type I and type II IFNs in Huh7 cells is mediated at least in part by PKR (Arsenio and others 2008; Zhang and others 2008). To determine if the IFN-λ–induced inhibition of VACVΔE3L also requires PKR activity, Huh7 cells were transfected with a negative control siRNA or a PKR-specific siRNA 24 h prior to treatment with 2.5 ng IFN-α/mL or 250 ng IFN-λ1/mL for 18 h, followed by infection with VACVΔE3L at an MOI = 1. PKR expression was measured 3 days post-infection, and extracellular virus titers and virus-induced CPE were determined at 1 and 3 days post-infection. Consistent with previously published results (Arsenio and others 2008), PKR expression was slightly decreased after VACVΔE3L infection, and was further substantially reduced by a PKR-specific siRNA (Fig. 5A). Virus replication was inhibited by IFN-α or IFN-λ in control siRNA-transfected cells but not in cells transfected with the PKR siRNA (Fig. 5B). In addition, in cells transfected with the PKR-specific siRNA, neither IFN-α nor IFN-λ protected cells from virus-induced CPE (Fig. 5C). These results indicate that PKR is required for IFN-λ–induced inhibition of VACVΔE3L replication in Huh7 cells.
FIG. 5. .
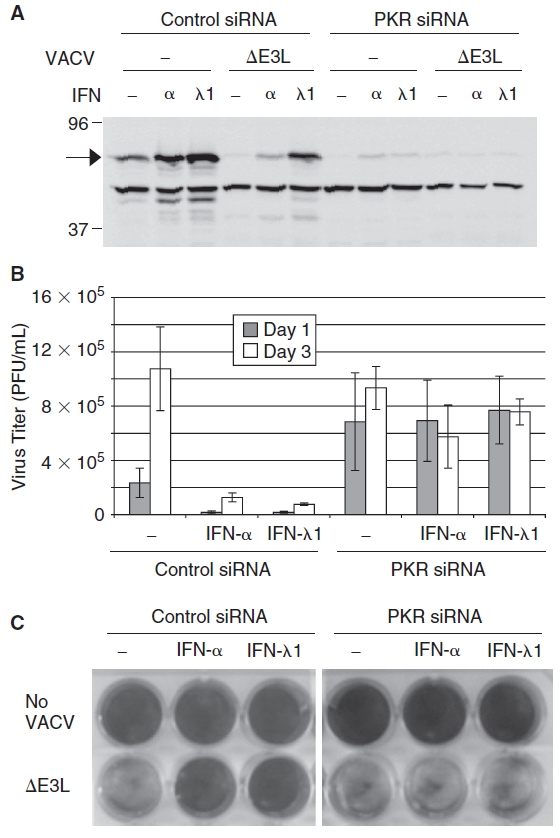
Inhibition of VACVΔE3L by interferon-λ (IFN-Δ) requires PKR. Huh7 cells were transfected with a nontargeting negative control or PKR-specific siRNA 24 h prior to treatment with 2.5 ng IFN-α/mL or 250 ng IFN-λ1/mL. Eighteen hours post-treatment with IFN, cells were infected with VACVΔE3L at an MOI = 1. (A) PKR siRNA reduces the amount of PKR (arrow) as compared to cells transfected with a negative control siRNA as shown by Western blot. (B) Titers of VACVΔE3L in culture media collected at 1 and 3 days post-infection, determined by plaque assay. Graph bars represent the mean of 3 independent experiments and error bars indicate the standard error of the mean. (C) PKR siRNA restores virus-induced CPE in VACVΔE3L-infected cells treated with IFN-α or IFN-λ1 as determined by crystal violet staining 72 h post-infection.
VACV infection inhibits IFN-λ–induced STAT activation and gene expression
In addition to neutralizing IFN-α/β extracellularly and blocking the downstream effector functions of antiviral proteins such as RNase L and PKR, VACV infection also inhibits IFN-α/β- and IFN-γ–mediated STAT phosphorylation through the activity of the virus-encoded H1 phosphatase (Najarro and others 2001; Mann and others 2008). We therefore determined if VACV infection also blocks IFN-λ–mediated signal transduction and gene expression. A549 cells were infected with VACV for 1 h prior to treatment with IFN-α or IFN-λ1, and cytokine-mediated signal transduction was measured by Western blot analysis of STAT-1 phosphorylation 1 and 4 h post-treatment (Fig. 6A) and quantitative RT-PCR analysis of IFN-stimulated gene expression 4 h post-treatment (Fig. 6B). As with IFN-α, VACV infection prior to IFN-λ treatment inhibited IFN-λ–mediated STAT-1 phosphorylation, as well as IFN-λ-stimulated gene expression.
FIG. 6. .

Vaccinia infection blocks interferon-λ (IFN-Δ)–mediated STAT phosphorylation and gene expression. A549 cells were infected with VACV for 1 h (MOI = 5) prior to treatment with 2.5 ng IFN-α/mL or 5 ng IFN-λ1/mL. Cells were harvested for (A) Western blot analysis of STAT-1 phosphorylation 1 and 4 h post-treatment or (B) quantitative RT-PCR analysis of IFN-stimulated gene expression 4 h post-treatment. Graph bars represent the mean of 3 independent experiments, and error bars indicate SEM.
Discussion
A recently characterized family of IFN-related cytokines (IFN-λ1, 2, and 3) are induced by viral infections and inhibit the replication of both RNA and DNA viruses (Kotenko and others 2003; Sheppard and others 2003; Robek and others 2005; Ank and others 2006; Doyle and others 2006a; Onoguchi and others 2007). The IFN-λs belong to a family of cytokines (class 2 α-helical) that also includes IFN-α/β and IFN-γ, as well as the IL-10 family proteins (Kotenko and others 2003; Sheppard and others 2003). Despite having a function that is nearly identical to IFN-α/β, IFN-λ is more similar to the IL-10 family of cytokines in terms of genomic structure and receptor usage. The IFN-λs have slightly less homology to IL-10 (11%–13%) than IFN-α (15%–19%), but like IL-10, the genes encoding the IFN-λs contain multiple exons, in contrast to the type I IFNs, which lack introns (Sheppard and others 2003). Furthermore, the IFN-λ receptor is composed of 2 subunits, one of which is the IL-10R2 subunit, and the other is unique to IFN-λ (IL-28R1) (Kotenko and others 2003). These structural and receptor usage differences distinguish the type I and type III IFN families.
Considering that IFN-α/β and IFN-λ utilize different host receptors and have low sequence homology, it might be predicted that immunomodulating factors such as B18R or B20R, which are IFN-α/β-specific, should be unable to block IFN-λ activity. Our data is consistent with another study that also demonstrated that VACV B18R does not block IFN-λ, despite the fact it inhibits multiple members of the type I IFN family (Huang and others 2007). Additionally, we found that the VARV homolog B20R, which is predicted to inhibit IFN-α/β but to our knowledge had not been directly shown to do so, does in fact block both IFN-α and IFN-β. However, as with VACV B18R, VARV B20R did not abrogate IFN-λ–induced signal transduction or gene expression. However, this is not the case for all poxviruses, as the IFN-α/β-binding protein of the Yaba-like disease virus, a Tanapoxvirus, inhibits both IFN-α/β and IFN-λ (Huang and others 2007). This protein, Y136, has low sequence homology to B18R, so its unique binding specificity is likely due to structural differences between these 2 factors (Huang and others 2007). Although differences in specificity for IFN-λ among these proteins may contribute to variations in virulence among poxviruses, it appears unlikely that the ability of B18R and B20R to neutralize IFN-λ accounts for the difference in virulence between VACV and VARV.
Despite differences in sequence and receptor usage, the downstream signaling that results from IFN-λ receptor activation is similar to the response induced by IFN-α/β. Both cytokines induce activation of STAT-1, -2, and -3, although IFN-λ induces slightly lower levels of phosphorylated STATs, and with different kinetics (Doyle and others 2006; Marcello and others 2006). Additionally, IFN-λ induces a similar gene expression profile as IFN-α/β with minor differences in magnitude and kinetics (Doyle and others 2006; Marcello and others 2006). Since VACV infection is known to inhibit IFN-α/β signal transduction and antiviral effector functions, we hypothesized that IFN-λ might also be blocked by the same factors.
We observed that STAT phosphorylation and ISG expression induced by either IFN-α/β or IFN-λ was inhibited by pre-infection with VACV. The VACV factor most likely responsible for this block is H1, which removes phosphates from activated STAT proteins, and prevents IFN-α and IFN-γ–mediated signal transduction and antiviral activity (Najarro and others 2001; Mann and others 2008). However, we cannot entirely rule out the possibility that another viral factor is instead responsible to blocking IFN-λ–mediated STAT phosphorylation, or that the kinetics of IFN-λ–induced STAT-1 phosphorylation are altered in infected cells.
Downstream of STAT phosphorylation, other poxvirus factors inhibit the antiviral effector proteins that are activated by IFN-α/β. VACV and VARV both encode an identical E3L protein, which can inhibit PKR, RNase L, and ISG15 function. Previous studies have shown that E3L expression is sufficient to prevent inhibition of VSV by IFN-α (Shors and others 1998). We found that unlike infection with wild-type VACV, pre-infection with VACVΔE3L did not rescue VSV from the antiviral effects of IFN-λ, supporting a role for E3L in blocking IFN-λ antiviral function. PKR plays an important role in blocking VSV replication by inhibiting translation of viral mRNAs (Balachandran and others 2000; Stojdl and others 2000). Because VSV replication was abrogated by both IFN-λ and IFN-α/β in VACVΔE3L-infected cells, this suggests that E3L inhibits antiviral effectors such as PKR that are activated by both cytokines.
We also found that the E3L-deficient VACV mutant is sensitive to the antiviral activity of IFN-λ (approximately 2-log inhibition at MOI = 1), in contrast to wild-type virus, which is relatively resistant to IFN-λ (approximately 2- to 3-fold inhibition at the same MOI). Recent studies have shown that inhibition of an E3L mutant VACV by IFN-α/β or IFN-γ can be suppressed by abrogation of PKR expression (Arsenio and others 2008; Zhang and others 2008). Similar to these findings, we found that the siRNA-mediated inhibition of PKR abolishes the antiviral activity of IFN-λ against VACV. In total, these results indicate that E3L plays an important role in the resistance of VACV to IFN-λ, and that PKR represents a common point of inhibition by this protein (Fig. 7). However, while these studies demonstrate PKR is necessary for the IFN-λ–mediated inhibition of VACV, they do not prove that it is sufficient, and other targets of E3L such as ISG15 also likely have important functions in this process.
FIG. 7. .

Summary of interferon-λ (IFN-Δ) neutralization by vaccinia virus (VACV). Although IFN-α and IFN-λ have low amino acid sequence homology and bind to distinct receptors, these receptors signal through pathways that are nearly identical. Therefore, the intracellular, but not extracellular, mechanisms used by VACV to block the IFN-α/β response also inhibit IFN-λ.
Although IFN-λ does not protect cells from VACV infection in vitro, IFN-λ may still inhibit VACV infection in vivo. Recent studies in mice indicate that IFN-λ functions not only to induce an intracellular antiviral state, but also to modulate the immune response (Bartlett and others 2005; Ank and others 2006a; Ank and others 2008). A modified VACV that expresses mouse IFN-λ2 displayed decreased infectivity levels compared to wild-type vaccinia (Bartlett and others 2005). In addition, these studies showed that infection with the modified virus was associated with increased lymphocyte recruitment and reduced lesion size and numbers. The results from our in vitro studies suggest that decreased viral infection rates might be attributed to the failure of B18R to inhibit extracellular IFN-λ from signaling to neighboring cells, as well as to induce an elevated immune response. This extracellular cytokine signal may also aid in prevention of virus spread resulting in fewer and smaller lesions.
A more complete understanding of the basic interactions between the innate immune response and viruses is necessary for the development of improved antiviral therapies. Successful anti-poxvirus therapies in animal models (ST-246 and cidofovir analogs) block virus formation or DNA polymerase activity, respectively (Buller and others 2004; Yang and others 2005; Nalca and others 2008; Parker and others 2008). A third line of defense might be to inhibit the activity of immunomodulating proteins, such as E3L and B18R, to allow the innate immune response to function properly and eliminate or control the viral infection. Based on our results, an antiviral agent [such as an siRNA (Dave and others 2006)] that inactivates or down-regulates E3L would sensitize the virus to IFN-λ as well as IFN-α. Additionally, IFN-λ may also be useful therapeutically to prevent virus spread, as it is not inhibited by B18R or B20R. In this regard, understanding the differential regulation of IFN-λ by poxvirus immunomodulating factors may lead to future therapeutic options.
Acknowledgments
Support for this work was provided by a Junior Faculty Career Development Award to M.D.R. from the Northeast Biodefense Center (U54 AI057158—Lipkin). N.E.P. was supported by PHS training grants T32 AI055403, T32 AI007640, and a fellowship from the Anna Fuller Fund. We thank Bertram Jacobs (University of Arizona), Stuart Isaacs (University of Pennsylvania), John Rose (Yale University), and Frank Chisari (The Scripps Research Institute) for providing reagents, and Ms. Minjung Han for technical assistance.
Contributor Information
Prasanthi Bandi, Department of Pathology, Yale University School of Medicine, New Haven, Connecticut..
Nicole E. Pagliaccetti, Department of Pathology, Yale University School of Medicine, New Haven, Connecticut..
Michael D. Robek, Department of Pathology, Yale University School of Medicine, New Haven, Connecticut.
Author Disclosure Statement
No competing financial interests exist.
References
- Alcami A, Smith GL. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J Virol. 1995;69((8)):4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcami A, Symons JA, Smith GL. The vaccinia virus soluble alpha/beta interferon (IFN) receptor binds to the cell surface and protects cells from the antiviral effects of IFN. J Virol. 2000;74((23)):11230–11239. doi: 10.1128/jvi.74.23.11230-11239.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amicone L, Spagnoli FM, Spath G, Giordano S, Tommasini C, Bernardini S, De Luca V, Della Rocca C, Weiss MC, Comoglio PM, Tripodi M. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 1997;16((3)):495–503. doi: 10.1093/emboj/16.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, Iversen MB, Bartholdy C, Staeheli P, Hartmann R, Jensen UB, Dagnaes-Hansen F, Thomsen AR, Chen Z, Haugen H, Klucher K, Paludan SR. An important role for type III interferon (IFN-lambda/IL-28) in TLR-induced antiviral activity. J Immunol. 2008;180((4)):2474–2485. doi: 10.4049/jimmunol.180.4.2474. [DOI] [PubMed] [Google Scholar]
- Ank N, West H, Bartholdy C, Eriksson K, Thomsen AR, Paludan SR. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo . J Virol. 2006a;80((9)):4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ank N, West H, Paludan SR. IFN-lambda: novel antiviral cytokines. J Interferon Cytokine Res. 2006b;26((6)):373–379. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- Arsenio J, Deschambault Y, Cao J. Antagonizing activity of vaccinia virus E3L against human interferons in Huh7 cells. Virology. 2008;377((1)):124–132. doi: 10.1016/j.virol.2008.04.014. [DOI] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13((1)):129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- Bartlett NW, Buttigieg K, Kotenko SV, Smith GL. Murine interferon lambdas (type III interferons) exhibit potent antiviral activity in vivo in a poxvirus infection model. J Gen Virol. 2005;86((Pt 6)):1589–1596. doi: 10.1099/vir.0.80904-0. [DOI] [PubMed] [Google Scholar]
- Beattie E, Denzler KL, Tartaglia J, Perkus ME, Paoletti E, Jacobs BL. Reversal of the interferon-sensitive phenotype of a vaccinia virus lacking E3L by expression of the reovirus S4 gene. J Virol. 1995;69((1)):499–505. doi: 10.1128/jvi.69.1.499-505.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand S, Zitzmann K, Dambacher J, Beigel F, Olszak T, Vlotides G, Eichhorst ST, Goke B, Diepolder H, Auernhammer CJ. SOCS-1 inhibits expression of the antiviral proteins 2′,5′-OAS and MxA induced by the novel interferon-lambdas IL-28A and IL-29. Biochem Biophys Res Commun. 2005;331((2)):543–548. doi: 10.1016/j.bbrc.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Brandt TA, Jacobs BL. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol. 2001;75((2)):850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller RM, Owens G, Schriewer J, Melman L, Beadle JR, Hostetler KY. Efficacy of oral active ether lipid analogs of cidofovir in a lethal mousepox model. Virology. 2004;318((2)):474–481. doi: 10.1016/j.virol.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Chang HW, Jacobs BL. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology. 1993;194((2)):537–547. doi: 10.1006/viro.1993.1292. [DOI] [PubMed] [Google Scholar]
- Chang HW, Watson JC, Jacobs BL. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc Natl Acad Sci USA. 1992;89((11)):4825–4829. doi: 10.1073/pnas.89.11.4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coccia EM, Severa M, Giacomini E, Monneron D, Remoli ME, Julkunen I, Cella M, Lande R, Uze G. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34((3)):796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- Colamonici OR, Domanski P, Sweitzer SM, Larner A, Buller RM. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J Biol Chem. 1995;270((27)):15974–15978. doi: 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- Dave RS, McGettigan JP, Qureshi T, Schnell MJ, Nunnari G, Pomerantz RJ. siRNA targeting vaccinia virus double-stranded RNA binding protein [E3L] exerts potent antiviral effects. Virology. 2006;348((2)):489–497. doi: 10.1016/j.virol.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Di Giulio DB, Eckburg PB. Human monkeypox: an emerging zoonosis. Lancet Infect Dis. 2004;4((1)):15–25. doi: 10.1016/S1473-3099(03)00856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly RP, Sheikh F, Kotenko SV, Dickensheets H. The expanded family of class II cytokines that share the IL-10 receptor-2 (IL-10R2) chain. J Leukoc Biol. 2004;76((2)):314–321. doi: 10.1189/jlb.0204117. [DOI] [PubMed] [Google Scholar]
- Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, Storey H, Yao L, Liu H, Barahmand-pour F, Sivakumar P, Chan C, Birks C, Foster D, Clegg CH, Wietzke-Braun P, Mihm S, Klucher KM. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44((4)):896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- Fulginiti VA, Papier A, Lane JM, Neff JM, Henderson DA. Smallpox vaccination: a review, part II. Adverse events. Clin Infect Dis. 2003;37((2)):251–271. doi: 10.1086/375825. [DOI] [PubMed] [Google Scholar]
- Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008;4((7)):e1000096. doi: 10.1371/journal.ppat.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga IR, Bowie AG. Evasion of innate immunity by vaccinia virus. Parasitology. 2005;130((Suppl)):S11–S25. doi: 10.1017/S0031182005008127. [DOI] [PubMed] [Google Scholar]
- Huang J, Smirnov SV, Lewis-Antes A, Balan M, Li W, Tang S, Silke GV, Putz MM, Smith GL, Kotenko SV. Inhibition of type I and type III interferons by a secreted glycoprotein from Yaba-like disease virus. Proc Natl Acad Sci USA. 2007;104((23)):9822–9827. doi: 10.1073/pnas.0610352104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YG, Muralinath M, Brandt T, Pearcy M, Hauns K, Lowenhaupt K, Jacobs BL, Rich A. A role for Z-DNA binding in vaccinia virus pathogenesis. Proc Natl Acad Sci USA. 2003;100((12)):6974–6979. doi: 10.1073/pnas.0431131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, Langer JA, Sheikh F, Dickensheets H, Donnelly RP. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4((1)):69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- Lane JM, Millar JD, Neff JM. Smallpox and smallpox vaccination policy. Annu Rev Med. 1971;22:251–272. doi: 10.1146/annurev.me.22.020171.001343. [DOI] [PubMed] [Google Scholar]
- Lasfar A, Lewis-Antes A, Smirnov SV, Anantha S, Abushahba W, Tian B, Reuhl K, Dickensheets H, Sheikh F, Donnelly RP, Raveche E, Kotenko SV. Characterization of the mouse IFN-lambda ligand-receptor system: IFN-lambdas exhibit antitumor activity against B16 melanoma. Cancer Res. 2006;66((8)):4468–4477. doi: 10.1158/0008-5472.CAN-05-3653. [DOI] [PubMed] [Google Scholar]
- Mann BA, Huang JH, Li P, Chang HC, Slee RB, O’Sullivan A, Anita M, Yeh N, Klemsz MJ, Brutkiewicz RR, Blum JS, Kaplan MH. Vaccinia virus blocks Stat1-dependent and Stat1-independent gene expression induced by type I and type II interferons. J Interferon Cytokine Res. 2008;28((6)):367–380. doi: 10.1089/jir.2007.0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcello T, Grakoui A, Barba-Spaeth G, Machlin ES, Kotenko SV, MacDonald MR, Rice CM. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131((6)):1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- Meager A, Visvalingam K, Dilger P, Bryan D, Wadhwa M. Biological activity of interleukins-28 and -29: comparison with type I interferons. Cytokine. 2005;31((2)):109–118. doi: 10.1016/j.cyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Najarro P, Traktman P, Lewis JA. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J Virol. 2001;75((7)):3185–3196. doi: 10.1128/JVI.75.7.3185-3196.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalca A, Hatkin JM, Garza NL, Nichols DK, Norris SW, Hruby DE, Jordan R. Evaluation of orally delivered ST-246 as postexposure prophylactic and antiviral therapeutic in an aerosolized rabbitpox rabbit model. Antiviral Res. 2008;79((2)):121–127. doi: 10.1016/j.antiviral.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Onoguchi K, Yoneyama M, Takemura A, Akira S, Taniguchi T, Namiki H, Fujita T. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282((10)):7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- Osterlund P, Veckman V, Siren J, Klucher KM, Hiscott J, Matikainen S, Julkunen I. Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J Virol. 2005;79((15)):9608–9617. doi: 10.1128/JVI.79.15.9608-9617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliaccetti NE, Eduardo R, Kleinstein SH, Mu XJ, Bandi P, Robek MD. Interleukin-29 functions cooperatively with interferon to induce antiviral gene expression and inhibit hepatitis C virus replication. J Biol Chem. 2008;283((44)):30079–30089. doi: 10.1074/jbc.M804296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker S, Touchette E, Oberle C, Almond M, Robertson A, Trost LC, Lampert B, Painter G, Buller RM. Efficacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir (CMX001) in a lethal mousepox model. Antiviral Res. 2008;77((1)):39–49. doi: 10.1016/j.antiviral.2007.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robek MD, Boyd BS, Chisari FV. Lambda interferon inhibits Hepatitis B and C virus replication. J Virol. 2005;79((6)):38851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seet BT, Johnston JB, Brunetti CR, Barrett JW, Everett H, Cameron C, Sypula J, Nazarian SH, Lucas A, McFadden G. Poxviruses and immune evasion. Annu Rev Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- Sheppard P, Kindsvogel W, Xu W, Henderson K, Schlutsmeyer S, Whitmore TE, Kuestner R, Garrigues U, Birks C, Roraback J, Ostrander C, Dong D, Shin J, Presnell S, Fox B, Haldeman B, Cooper E, Taft D, Gilbert T, Grant FJ, Tackett M, Krivan W, McKnight G, Clegg C, Foster D, Klucher KM. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4((1)):63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- Shors ST, Beattie E, Paoletti E, Tartaglia J, Jacobs BL. Role of the vaccinia virus E3L and K3L gene products in rescue of VSV and EMCV from the effects of IFN-alpha. J Interferon Cytokine Res. 1998;18((9)):721–729. doi: 10.1089/jir.1998.18.721. [DOI] [PubMed] [Google Scholar]
- Smith GL, Chan YS. Two vaccinia virus proteins structurally related to the interleukin-1 receptor and the immunoglobulin superfamily. J Gen Virol. 1991;72((Pt 3)):511–518. doi: 10.1099/0022-1317-72-3-511. [DOI] [PubMed] [Google Scholar]
- Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem. 2001;276((12)):8951–8957. doi: 10.1074/jbc.M008717200. [DOI] [PubMed] [Google Scholar]
- Stojdl DF, Abraham N, Knowles S, Marius R, Brasey A, Lichty BD, Brown EG, Sonenberg N, Bell JC. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol. 2000;74((20)):9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons JA, Alcami A, Smith GL. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell. 1995;81((4)):551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- Thacore HR, Youngner JS. Rescue of vesicular stomatitis virus from interferon-induced resistance by superinfection with vaccinia virus. I. Rescue in cell cultures from different species. Virology. 1973;56((2)):505–511. doi: 10.1016/0042-6822(73)90053-6. [DOI] [PubMed] [Google Scholar]
- Weaver JR, Shamim M, Alexander E, Davies DH, Felgner PL, Isaacs SN. The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res. 2007;130((1–2)):269–274. doi: 10.1016/j.virusres.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol. 2002;76((10)):5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pevear DC, Davies MH, Collett MS, Bailey T, Rippen S, Barone L, Burns C, Rhodes G, Tohan S, Huggins JW, Baker RO, Buller RL, Touchette E, Waller K, Schriewer J, Neyts J, DeClercq E, Jones K, Hruby D, Jordan R. An orally bioavailable antipoxvirus compound (ST-246) inhibits extracellular virus formation and protects mice from lethal orthopoxvirus Challenge. J Virol. 2005;79((20)):13139–13149. doi: 10.1128/JVI.79.20.13139-13149.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Jacobs BL, Samuel CE. Loss of protein kinase PKR expression in human HeLa cells complements the vaccinia virus E3L deletion mutant phenotype by restoration of viral protein synthesis. J Virol. 2008;82((2)):840–848. doi: 10.1128/JVI.01891-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Langland JO, Jacobs BL, Samuel CE. Protein kinase PKR-dependent activation of mitogen activated protein kinases occurs through mitochondrial adapter IPS-1 and is antagonized by vaccinia virus E3L. J Virol. 2009;83((11)):5718–5725. doi: 10.1128/JVI.00224-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Samuel CE. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J Biol Chem. 2008;283((50)):34580–34587. doi: 10.1074/jbc.M807029200. [DOI] [PMC free article] [PubMed] [Google Scholar]