

Abstract
Apoptosis and the rapid clearance of apoptotic cells (ACs) by professional or nonprofessional phagocytes are normal and coordinated processes that ensure controlled cell growth and stress response with nonpathological outcomes. Uptake of ACs by phagocytes is thought to suppress autoimmune responses through the release of anti-inflammatory cytokines such as interleukin-10 (IL-10), transforming growth factor-β (TGF-β), and inhibition of proinflammatory cytokines. The production of pro- and anti-inflammatory cytokines by phagocytes is highly regulated as part of an intrinsic mechanism to prevent inflammatory and autoimmune reactions in a physiological state. Production of IL-10 by phagocytes during clearance of ACs is critical to ensuring cellular homeostasis and suppression of autoimmunity. The molecular mechanism whereby IL-10 production is induced by ACs is only beginning to be understood. This review summarizes our recent work in this aspect of an essential physiological and homeostatic process.
Phagocyte-Mediated Clearance of Apoptotic Cells
Apoptotic cell (AC) death is an essential process in the development of multicellular organisms (Morris and others 1984). Efficient removal of ACs helps sculpt organs, maintain homeostasis, and eliminate abnormal, nonfunctional, or harmful cells (Vaux and Korsmeyer 1999; Henson and Hume 2006). Moreover, removing ACs prevents harmful inflammatory and autoimmune responses owing to release of potentially dangerous contents. Inefficient engulfment of ACs, or degradation of apoptotic cell contents, results in chronic inflammatory and autoimmune diseases (Grigg and others 1991; Savill and others 1993; Cox and others 1995). In human systemic lupus erythematosis (SLE), impaired phagocytosis of apoptotic material by macrophages has been reported (Herrmann and others 1998; Baumann and others 2002), providing an explanation for increased levels of early ACs, DNA, and nucleosomes observed in the circulation of SLE patients (Raptis and Menard 1980; McCoubrey-Hoyer and others 1984; Steinman 1984; Perniok and others 1998). The impaired clearance of ACs resulting in an accumulation of late apoptotic and secondary necrotic cells including oligosomes might lead to an activation of autoreactive T and B cells (Voll and others 1997).
The process of removing dead cells is carried out by a wide variety of cell types. When apoptosis occurs at moderate rates such as during normal adult tissue turnover, neighboring cells such as fibroblasts can act as phagocytes in their ingestion and clearance. When apoptosis occurs on large scales such as during embryonic morphogenesis, ionizing radiation, and acute infections, macrophages are the major professional phagocytes that play important roles in the clearance of ACs. Macrophages are attracted to sites of high rates of apoptosis such as the thymus and the follicles of secondary lymphoid tissues in the immune system. The process of removing ACs involves multiple receptors (Savill and others 1993), such as scavenger receptors, oxidized low-density lipoprotein receptors, CD14, CD68, CD36, and vitronectin receptor. Animal studies have also identified some of the important nuclear, intracellular, and extracellular molecules in the clearance of potentially antigenic material from the circulation, such as DNAse I (Napirei and others 2000), serum amyloid P component (SAP) (Bickerstaff and others 1999), C1q (Teague and others 1999), and C-reactive protein (CRP) (Du Clos and others 1994).
The surface structure of ACs is altered during the death pathway so that they present patterns recognized by phagocytes as “altered self,” or sometimes referred to as AC-associated molecular patterns (ACAMPs). ACAMPs arise either from the exofacial exposure of endogenous molecules or the modification of pre-existing surface molecules. ACs exhibit numerous alterations of membrane lipid molecules and carbohydrates. There are 4 major phospholipids in the plasma membrane of many mammalian cells: phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and sphingomyelin. PS is the most-studied “eat-me” signal, exposed on the surface of dying cells (Fadok and others 1992; Williamson and Schlegel 2002). PS is maintained predominantly in the inner leaflet of the plasma membrane in viable cells through the action of ATP-dependent aminophospholipid translocases (Bratton and others 1997; Daleke and Lyles 2000; Vance and Steenbergen 2005). During apoptosis, the balance between translocase and scramblase activities that exchange PS between leaflets alters, and PS accumulates on the exoplasmic leaflet (Gardai and others 2006).
The exposed PS on ACs is recognized by several phagocyte receptors including a presumptive PS receptor (PSR) (Savill and others 1993). Ligation of this presumptive PSR has been proposed to be the primary mechanism through which these responses are initiated (Savill and others 1993), although experimental demonstration of such a receptor has been quite controversial (Williamson and Schlegel 2002). Very recently, several groups have identified receptors that both directly recognize PS and induce phagocytosis of ACs. These receptors include the brain-specific angiogenesis factor 1, the T-cell immunoglobulin domain and mucin domain 4, and stabilin-2 (Miyanishi and others 2007; Park and others, 2007, 2008).
Other “eat-me” signals expressed at the surface of ACs include modifications to the cell glycosylation profile such as altered oligosaccharides attached to cell-surface glycoproteins (Morris and others 1984), changes in the oxidation status of macromolecules such as low-density lipoprotein (Ramprasad and others 1995), exposure and redistribution of calreticulin, a 46-kDa soluble, highly charged protein that binds calcium ions (Gardai and others 2005).
Interactions Between Phagocytes and Apoptotic Cells
The process of AC clearance by phagocytes can be divided into a succession of steps, from the recognition of apoptosis-specific cues on the dying cells (“eat-me” signals), to internalization of the corpse, processing of the corpse as the phagocytic vacuole matures, and the consequences for the engulfing cell (Somersan and Bhardwaj 2001). Target selectivity in this phagocytic process is based on the specific cell–cell interaction between phagocytes and target cells undergoing apoptosis. Pattern-recognizing phagocytosis receptors present on the surface of phagocytes specifically bind, either directly or indirectly through bridging molecules, to ACAMPs and transmit signals to induce phagocytosis of bound ACs. Phagocytes often evoke subsequent actions, rather than simply digesting engulfed ACs, for a fine-tuning of tissue homeostasis.
Mechanisms for recognizing PS in mammals also involve secreted bridging molecules. Gas6 (growth arrest-specific gene 6) and protein S can bind PS, and are in turn recognized by Mer, a member of the family of tyrosine kinase receptors expressed at the phagocyte surface (Nagata and others 1996; Ishimoto and others 2000; Scott and others 2001). MFG-E8 (milk fat globule epidermal growth factor 8), another bridging molecule secreted by activated macrophages, also specifically bind PS (Borisenko and others 2004). MFG-E8-coated ACs are recognized by the vitronectin receptor, also called αv®3 integrin, then phagocytosed by macrophages (Hanayama and others 2002). Other bridging molecules include β2-glycoprotein I (Balasubramanian and others 1997), IgM (Kim and others 2002), and C1q (Païdassi and others 2008).
What are the mechanisms that lead to the engulfment of the apoptotic corpse into a phagosome? Reorganization of the cytoskeleton, as well as delivery of membrane to accompany the surface area increase of the phagocytic cup, is required. Maturation is the process by which particles internalized in phagosomes such as bacteria and ACs are trafficked into a series of increasingly acidified membrane-bound structures, leading to particle degradation (Kinchen and Ravichandran 2008). These phagosomes sequentially acquire different integral membrane and cytoplasmic proteins such as the Rab GTPases during maturation, and eventually fuse with lysosomes (Garin and others 2001; Stuart and others 2007). It is important to note that the phagocytosis of foreign particles and of ACs involves different phagocytic receptors that elicit different immune responses. Therefore, there may be specificity in the roles of particular Rabs in regulating AC clearance versus phagocytosis of other particles.
Role of IL-10 in Homeostatic Control of Inflammation and Immune Activation
Interleukin-10 (IL-10) is a pleiotropic cytokine produced by both T/B cells and macrophages and possesses both anti-inflammatory and immunosuppressive properties (Moore and others 1990). Extensive research showed that IL-10 is an inhibitor of a broad spectrum of monocyte/macrophage functions, including cytokine synthesis, nitric oxide production, and expression of MHC class II and co-stimulatory molecules such as CD80/CD86 (Moore and others 2001). Investigations in numerous inflammatory disease models including chronic enterocholitis, cutaneous inflammatory condition, endotoxic shock and Shwartzman reaction, and autoimmune encephalomyelitis in IL-10-deficient mice have yielded strong evidence that IL-10 plays a central role in vivo in restricting inflammatory responses (Fuss and others 2002). However, endogenous IL-10 production and systemic administration can also exacerbate macrophage and T-cell dysfunction, decrease T-cell apoptosis, blunt antimicrobial activity, and increase mortality in other less acute bacterial models of sepsis or after thermal injury (Oberholzer and others 2002). In addition, IL-10 also processes immunostimulatory effects that have not attracted sufficient attention. IL-10 is a potent growth factor for B lymphocytes. It promotes B-cell proliferation, antibody production, and class II expression (Schall and others 1990). IL-10 enhances, paradoxically, the development of cytotoxic T lymphocytes (CTL) (MacNeil and others 1990). It induces NK cytotoxicity against NK-resistant tumor cells in vitro and increases IL-2-induced NK cell proliferation (Carson and others 1995). It acts as a cofactor for colony formation by mast cell progenitors (Robinson and others 2003) and thymocytes (MacNeil and others 1990).
IL-10 Gene Expression in Microbially Stimulated Macrophages
IL-10 gene expression in macrophages is usually triggered by the same typical inflammatory stimuli such as lipopolysaccharides (LPSs) that induce the release of proinflammatory cytokines. However, the kinetics of its induction differs from those of the proinflammatory mediators (de Waal Malefyt and others 1991). Recent molecular analyses of the murine IL-10 promoter show that IL-10 transcription in macrophage cell types can be regulated by constitutive and ubiquitous transcription factors such as Sp1 and Sp3, suggesting that IL-10 may be produced at low levels constitutively to maintain certain level of control over “baseline” inflammation (Brightbill and others 2000). Another study provided evidence that posttranscriptional regulation of IL-10 gene expression through sequences in the 3′-untranslated region of the IL-10 mRNA contributes to its overall production as well (Alizadeh and others 2000). A critical role for Stat3 but not other Stat proteins in LPS-induced IL-10 transcription in a human B-cell line was reported by Benkhart and colleagues who demonstrated a direct interaction of Stat3 with the human IL-10 promoter at –120 (Benkhart and others 2000). Since Stat3 is also the mediator of IL-10 signaling via the IL-10 receptor (Moore and others 2001), this finding provides a mechanistic explanation for the noted autoregulation of IL-10. Cao and others showed that the NF-κB cis element on the mouse IL-10 proximal promoter was located to –55/–46, where p50 can homodimerize and form a complex with the transcriptional co-activator CREB-binding protein to activate transcription. The other Rel family members appeared to play a negligible role in IL-10 transcription (Cao and others 2006).
IL-10 Gene Expression During Phagocytosis of Apoptotic Cells
Resolution of inflammation depends not only on the removal of ACs but also on active suppression of inflammatory mediator production. Aberrations in either mechanism are associated with chronic inflammatory conditions and autoimmune disorders (Grigg and others 1991; Savill and others 1993; Cox and others 1995). Uptake of ACs by phagocytes is thought to suppress autoimmune responses through up-regulation of cell-surface expression of co-inhibitory molecules such as PD-L1 and ICOS-L, release of anti-inflammatory cytokines IL-10, transforming growth factor-β (TGF-β), platelet-activating factor (PAF), and prostaglandin E2 (PGE2), and inhibition of proinflammatory cytokines TNF-α, GM-CSF, IL-12, IL-1β, and IL-18 (Savill and others 1993; Voll and others 1997; Sun and Shi 2001; Kim and others 2004). However, little was known about the molecular mechanisms whereby the production of pro- and anti-inflammatory cytokines is regulated. We first reported that IL-10 production stimulated by ACs was regulated at the level of transcription in a manner dependent on p38 mitogen-activated protein kinase, partially on the scavenger receptor CD36, and required cell–cell contact but not phagocytosis (Chung and others 2007a). Using a reporter assay, we mapped the apoptotic cell response element (ACRE) in the human IL-10 promoter, and provided biochemical and physiological evidence that ACRE mediates the transcriptional activation of IL-10 via pre-B-cell leukemia transcription factor-1b (Pbx-1b) and another Hox cofactor Pbx-regulating protein 1 (Prep-1) in response to ACs (Chung and others 2007a). The Pbx homeoproteins are known to function as cofactors for the Hox family of homeodomain-containing transcription factors that pattern the embryonic body axes (Moens and Selleri 2006). Pbx-1a and Pbx-1b are the 2 isoforms of Pbx-1. Pbx-1a expression is restricted to neural tissues while Pbx-1b exhibits widespread expression patterns in the mouse embryo (Moens and Selleri 2006). Hox proteins and some orphan homeodomain proteins form complexes with either Pbx or Meis subclasses of homeodomain proteins. This interaction can increase the binding specificity and transcriptional effectiveness of the Hox partner (Moens and Selleri 2006). Our study on the induction of IL-10 during phagocytosis of ACs establishes a novel role of 2 developmentally critical factors in the regulation of homeostasis in the immune system. This study carries the prospect to open up a completely new area for future exploration at the intersection between cellular homeostasis and regulation of immune responses to exogenous pathogens as well as to endogenous danger signals.
IL-10 and SLE
Accumulating evidence suggests that IL-10 is a strong candidate gene in SLE susceptibility. First, it maps to human chromosome 1q31˜32, which is a susceptibility region for SLE (LOD = 3.79) (Johanneson and others 2002). It is also homologous to a murine SLE susceptibility region (Tsao and others 1997). Second, IL-10 is known to be an important immunoregulatory cytokine. It enhances B-cell survival, proliferation, differentiation, and autoantibodies production (Moore and others 2001), properties that could render IL-10 a causal factor for the hyperactivity of B cells in SLE. Third, high IL-10 production has been observed in B cells and macrophages from SLE patients in vitro (Llorente and others 1993), and elevated serum IL-10 levels were observed in SLE patients and have been shown to be associated with disease activity (Llorente and others 1997; Gröndal and others 2000). In NZB/W F1 lupus-prone mice, T-cell cytokine imbalance toward production of IFN-γ and IL-10 is associated with autoantibody levels and nephritis (Enghard and others 2006). Fourth, continuous administration of anti-IL-10 antibodies in this model delayed the onset of lupus-like autoimmunity and improved the survival rate from 10% to 80%, through up-regulation of endogenous TNF-α (Ishida and others 1994). Conversely, continuous administration of IL-10 accelerated the onset of autoimmunity in these mice. Collectively, these evidences suggest that elevated IL-10 levels may play a role in SLE pathogenesis by causing the hyperactivity of B cells and the autoantibody production.
IL-10 production is primarily controlled at the transcriptional level and is under strong genetic influence (Bienvenu and others 1995; Westendorp and others 1997). The 5′ flanking region of the IL-10 gene, which controls transcription, is polymorphic. Six single-nucleotide polymorphisms (SNPs) have been reported in the human IL-10 promoter to be associated with IL-10 production (Turner and others 1997; Eskdale and others 1999; D’Alfonso and others 2000; Gibson and others 2001). They are located at positions −3575 (T/A), −2849 (G/A), −2763 (C/A), −1082 (A/G), −819 (T/C), and −592 (A/C) from the transcription start site. The −3575 SNP lies within a putative Pit-1-binding site and −2763 SNP situates within putative lymphocyte-specific factor- and myeloid zinc-finger-binding sites (Gibson and others 2001). SNP at position −1082 is within a putative ETS-like transcription factor-binding site (Lazarus and others 1997). The SNP at −592 is located in a region that mediates negative regulatory function (Lazarus and others 1997), whereas the SNP at −819 may affect an estrogen receptor element (Kube and others 1995). Several studies showed that −1082, −819, and −592 combined to form 3 haplotypes (hts): GCC, ACC, and ATA that correlated with decreasing IL-10 expression levels (Turner and others 1997; Crawley and others 1999; Edwards-Smith and others 1999; Yilmaz and others 2005). In a study that involved 76 white patients with SLE and 119 controls, no significant change in the allele frequency of the 3 IL-10 gene promoter dimorphic polymorphisms in the SLE group compared with controls was found. However, when subgrouped according to autoantibody status and clinical features, −1082G, −819C, and −592C alleles were increased in patients possessing Ro autoantibodies and those with renal involvement (Lazarus and others 1997). These alleles are in preferential allelic association, namely GCC, ACC, and ATA haplotypes, and the GCC haplotype was increased in these patient subgroups. A larger scale study examining the association of IL-10 promoter SNPs (−3575T/A, −2849G/A, −2763C/A, −1082A/G, −819T/C, and −592A/C) with SLE in 554 Hong Kong Chinese SLE patients and 708 ethnically matched controls, and another study involving 119 Taiwanese Chinese SLE patients revealed that the homozygous genotype of high IL-10 production haplotypes was significantly increased in these SLE patients (Chong and others 2004; Zimmermann and others 2006). In addition, Gibson and others reported that the haplotype of −3575T and −2763C is associated with high IL-10 expression levels in African Americans (Gibson and others 2001). The allele frequency of −1082G in the Vietnamese SLE patients was significantly higher than that in the healthy controls (Khoa and others 2005). A study in a large population of Chinese patients with lupus nephritis (LN) showed a strong association of the −592A/C SNP with the disease activity and renal pathology of LN. Particularly, patients carrying the −592C allele had a higher risk of diffuse proliferative glomerulonephritis (Song and others 2005).
Taken together, these studies strongly suggest that SNPs within the IL-10 gene promoter, which are associated with high IL-10 levels, may contribute significantly to the development of certain clinical features in SLE. We found that the homozygous GCC haplotype linked to greater SLE severity confers higher IL-10 gene transcriptional activity than the ATA haplotype in macrophages that encounter ACs, because of the differential DNA binding to the −592 SNP by a nuclear protein uniquely induced by ACs. Further, we identified this protein as poly(ADP-ribose) polymerase 1, confirmed its physiological role, and characterized its molecular properties, as a transcriptional repressor induced by ACs, in modulating IL-10 production during phagocytosis of ACs (Chung and others 2007b).
Poly(ADP-Ribose) Polymerase 1
PARP-1 is a nicotinamide adenine dinucleotide (NAD+)-dependent nuclear enzyme that detects and participates in DNA damage repair arising from genotoxic stress. It has also been linked to multiple events for transcriptional regulation in development (Tulin and others 2002; Ju and others 2004; Kim and others 2004; Pavri and others 2005). A large body of evidence demonstrates that PARP-1 is overactivated during the inflammatory response, depleting energy metabolism, and thus contributing greatly to tissue damage. Accordingly, pharmacological inhibition of PARP-1 is being actively investigated for therapeutic efficacy in animal models of inflammation such as ischemia–reperfusion (Szabó and Dawson 1998), chronic colitis (Jijon and others 2000), asthma (Virág and others 2004), experimental autoimmune encephalomyelitis (EAE) (Chiarugi 2002), diabetes mellitus (Akiyama and others 2001). Furthermore, PARP−/− mice are protected from endotoxic shock (Kühnle and others 1999; Oliver and others 1999).
PARP-1 is a 113-kDa protein composed of an N-terminal DNA-binding domain, containing 2 zinc finger motifs, a C-terminal catalytic domain, which can synthesize ADP-ribose polymers from the substrate NAD+, and an automodification site, which links the N- and C-terminal domains (de Murcia and others 1991). Typically catalytic activity is inhibited, but DNA binding to the N-terminal domain allosterically activates PARP polymerase function. There is evidence suggesting that PARP-1 can directly recognize specific DNA sequences. It has been shown that PARP-1 can directly interact with the promoter of Reg (regenerating gene) important for regeneration of pancreatic islet β-cells in the active transcriptional DNA/protein complex, and destabilize the complex by autopoly(ADP-ribosyl)ation of PARP (Akiyama and others 2001). PARP-1 binds in vitro and in vivo to specific sequences in a regulatory region of Bcl-6 (in the first intron) and inhibits Bcl-6 mRNA expression in B-cell lines (Ambrose and others 2007). PARP-1 has also been described as having a function in transcriptional regulation through their ability to modify chromatin-associated proteins and as a cofactor of different transcription factors, most notably NF-κB and AP-1 (Aguilar-Quesada and others 2007).
During apoptosis, PARP-1 is cleaved by caspase 3, resulting in separation of the N-terminal 24-kDa DNA-binding fragment from the C-terminal 89-kDa catalytic fragment. This cleavage is important for the regulation of inflammatory responses by PARP-1 (Pétrilli and others 2004). It has been shown that the 24-kDa fragment is an antagonist of full-length PARP-1 and it can inhibit DNA repair, ADP-ribose polymer formation, and damage-dependent up-regulation of transcription (D’Amours and others 2001; Yung and Satoh 2001).
The genetic loci located on chromosome 1 are associated with SLE in humans (Tsao 2000). Located in these loci are genes encoding TNFR2, complement component C1q, Fcγ receptors, TCR ζ chain, HRES-1 (an endogenous retrovirus), and interestingly, IL-10 and PARP-1 (Martens and others 2007). In patients with SLE, the highly ordered signal transduction cascade of apoptosis is disturbed. SLE patients show reduced PARP activity. PARP cleavage products are mainly found in association with either antinuclear or anti-dsDNA antibodies. Serum samples from SLE patients and other autoimmune diseases display anti-PAR and anti-PARP autoantibodies (Böhm 2006). In particular, autoantibodies to the catalytic fragment of PARP-1 were found in the sera of nearly 50% patients with SLE while they were not present in the sera of patients with rheumatoid arthritis, systemic sclerosis, or healthy donors (Lim and others 2002; Jeoung and others 2004). In the parent-into-F(1) model of chronic graft-versus-host disease (cGVHD) in which a lupus-like phenotype highly similar to human SLE is reliably induced in normal F(1) mice, PARP-1 was identified as a frequently targeted nuclear autoantigen modified by caspases during apoptosis (Grader-Beck and others 2007).
Septic Shock–Associated IL-10 −1082A>G Polymorphism
IL-10 attenuates the proinflammatory response in bacterial sepsis and reduces others (Steinhauser and others 1999). However, excess of IL-10 induces immunosuppression in sepsis (Steinhauser and others 1999) and increases mortality by impairing bacterial clearance in pneumococcal pneumonia (van der Poll and others 1996). A systemic infectious insult is often associated with subsequent hyporesponsiveness to endotoxin and an increased risk of late nosocomial infection in some patients. For example, immediately following cardiac surgery, many patients become relatively refractory to LPS stimulation. In humans, elevated circulating IL-10 has been associated with septic shock (Marchant and others 1994), severity of injury (Derkx and others 1995; Gómez-Jiménez and others 1995; Friedman and others 1997; Neidhardt and others 1997; Rodríguez-Gaspar and others 2001; Igonin and others 2004), and mortality (Monneret and others 2004; Simmons and others 2004). In acute respiratory distress syndrome (ARDS), the studies have been mixed. Lower levels of IL-10 were found in patients with ARDS compared with critically ill non-ARDS patients (Armstrong and Millar 1997). High plasma IL-10 but low bronchoalveolar lavage concentration of IL-10 correlated with increased mortality in ARDS (Donnelly and others 1996; Parsons and others 1997). Susceptibility for invasive pneumococcal disease has also been associated with the mannose-binding lectin gene, but no genetic linkage has been found for sepsis severity (Roy and others 2002).
Stanilova and others showed that the A allele of the −1082 polymorphism is associated with lower IL-10 production and sepsis susceptibility in patients, whereas G allele is associated with increased mortality in severe sepsis (Stanilova and others 2006). Another study demonstrated that the IL-10 intermediate/high producer genotype (−1082G-allele carrier) was associated with a lower risk of death among patients with acute renal failure who require dialysis (Jaber and others 2004). It was reported that individuals with genetic predisposition for increased IL-10 inducibility, as determined by the IL-10 −1082 polymorphism, have a higher risk of severe pneumococcal infection leading to septic shock (Schaaf and others 2003). A nested case–control epidemiology study in ARDS patients and controls who were admitted to an intensive care unit with sepsis, trauma, aspiration, or massive transfusions revealed that the high IL-10-producing −1082GG genotype is associated with variable odds for ARDS development depending on age, being associated with lower mortality and organ failure (Gong and others 2006). It was reported that in patients with acute pancreatitis, those who developed further into septic shock, showed a significantly higher prevalence of the −1082G allele than those without shock (Zhang and others 2005).
The preponderance of clinical data on the association of the IL-10 −1082 SNP with bacterial septic shock and the sequelae is contrasted with an almost total lack of understanding of the molecular basis of the SNP-associated variability in IL-10 levels in different individuals. We recently reported that the −1082G>A allele in the promoter region of the human IL-10 gene physically interacts with PARP-1 in an haplotype-specific manner that results in different levels of IL-10 transcription. We showed that PARP-1 acts as a transcription repressor, and its DNA-binding activity is strongly regulated in macrophages that engulf ACs but not stimulated with LPS (Kang and others 2010). The critical difference between the −592 and −1082 SNPs, both of which interact with PARP-1 in response to ACs, is that PARP-1 binding is induced at the −592 site whereas it is inhibited at the −1082 site. In other words, the −592C/A alleles mediate PARP-1 inhibition of IL-10 transcription in response to ACs, whereas the −1082G/A alleles mediate AC-induced relief of the inhibition, both in an allele-specific manner.
Our studies (Chung and others 2007b; Kang and others 2010) unveil a novel role of PARP-1 in the regulation of IL-10 production in an allele-dependent way, which determines individual susceptibility to pathogen-induced inflammatory pathology and the immunological sequelae in physiological processes where clearance of infection-induced ACs by professional phagocytes triggers the cytokine synthesis. They provide clear molecular mechanisms whereby individual IL-10 promoter haplotypes confer differential IL-10 production. Further investigation and elucidation of the molecular nature and characteristics of PARP-1-mediated immunoregulation will likely benefit the design of effective therapeutic strategies against a wide range of inflammatory and pathological conditions such as SLE, arthritis, asthma, sepsis, cardiomyopathy, stroke, inflammatory bowel disease, diabetes, and cancer.
In summary of our work on the regulation of IL-10 production during phagocytosis of ACs by macrophages, we propose the model shown in Figure 1.
FIG. 1. .
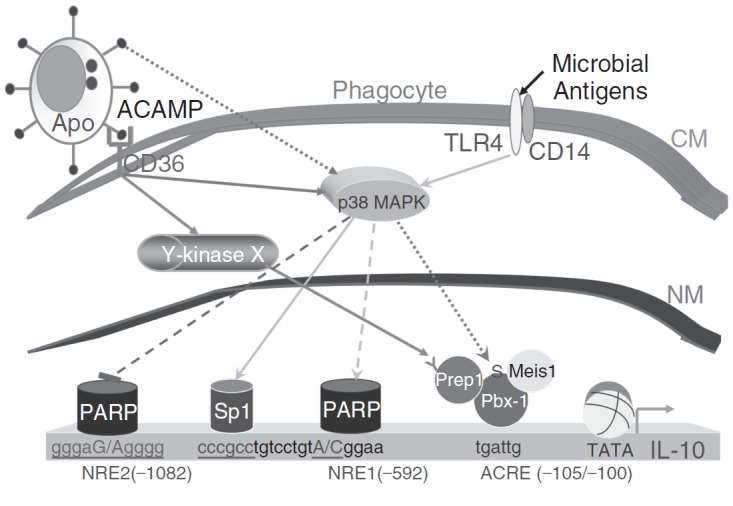
Model of regulation of interleukin-10 (IL-10) production in response to apoptotic cells (ACs). ACs, through not clearly defined AC-associated molecular pattern (ACAMP) interacting with phagocytic receptors, induce IL-10 production by macrophages. One of the phagocytic receptors is CD36, the engagement of which by ACs induces p38 MAPK phosphorylation and the activation of a yet to be identified protein tyrosine kinase (X). Activation of p38 MAPK leads to the assembly of a Hox complex, composed of Pbx-1b, Prep-1, and Meis1 as a minimum, on the IL-10 promoter via the apoptotic cell response element (ACRE) at −105/−100. Prep-1 tyrosine phosphorylation is presumably carried out by the hypothetical kinase X. The Hox complex is the main transcriptional factor driving IL-10 expression in response to ACs and it does not appear to be variable in human individuals. Two negative regulatory elements (NREs) exist: NRE1 at −592 and NRE2 at −1082, which also represent 2 major SNPs in human populations. Both NREs interact with PARP-1, which acts as a transcription repressor, but the modes of action are different. NRE1 does not bind PARP-1 in resting macrophages and the interaction with the 24-kDa cleaved fragment of PARP-1 is induced by ACs being ingested by the phagocyte. NRE2 binds the intact, 113-kDa PARP-1 in resting cells and the binding is reduced in macrophages in contact with ACs, resulting in inhibition of IL-10 transcription. It is possible that the cleavage product of PARP-1 is actually a transcriptional activator for IL-10, opposing the effect of its precursor. This possibility awaits formal confirmation. Both NREs regulate IL-10 transcription in an allele-specific manner, resulting in variable levels of IL-10 gene expression in different individuals in response to ACs. Note that p38 activation is also inducible via the TLR pathway by microbial pathogens, which represents an evolutionarily independent route to induce IL-10 production. This pathway leads to the binding of transcription factors such as Sp1 to the IL-10 promoter region. Abbreviations: CM, cell membrane; NM, nuclear membrane; TATA, TATA box; Y-kinase, tyrosine kinase; ACRE, apoptotic cell response element; NRE, negative regulatory element. Solid lines represent proven relationship while dashed lines designate hypothetical dependency.
Contributor Information
Yan Zhang, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York..
Ha-Jeong Kim, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York..
Soichiro Yamamoto, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York..
Xiaoyan Kang, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York..
Xiaojing Ma, Department of Microbiology and Immunology, Weill Medical College of Cornell University, New York.; Department of Pediatrics, Weill Medical College of Cornell University, New York. Program in Immunology and Microbial Pathogenesis, Weill Graduate School of Medical Sciences, Cornell University, New York, New York.
References
- Aguilar-Quesada R, Muñoz-Gámez JA, Martín-Oliva D, Peralta-Leal A, Quiles-Pérez R, Rodríguez-Vargas JM, Ruiz de Almodóvar M, Conde C, Ruiz-Extremera A, Oliver FJ. Modulation of transcription by PARP-1: consequences in carcinogenesis and inflammation. Curr Med Chem. 2007;14(11):1179–1187. doi: 10.2174/092986707780597998. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Takasawa S, Nata K, Kobayashi S, Abe M, Shervani NJ, Ikeda T, Nakagawa K, Unno M, Matsuno S, Okamoto H. Activation of Reg gene, a gene for insulin-producing beta-cell regeneration: poly(ADP-ribose) polymerase binds Reg promoter and regulates the transcription by autopoly(ADP-ribosyl)ation. Proc Natl Acad Sci USA. 2001;98(1):48–53. doi: 10.1073/pnas.240458597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, Powell JI, Yang L, Marti GE, Moore T, Hudson J, Jr, Lu L, Lewis DB, Tibshirani R, Sherlock G, Chan WC, Greiner TC, Weisenburger DD, Armitage JO, Warnke R, Levy R, Wilson W, Grever MR, Byrd JC, Botstein D, Brown PO, Staudt LM. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Ambrose HE, Papadopoulou V, Beswick RW, Wagner SD. Poly-(ADP-ribose) polymerase-1 (Parp-1) binds in a sequence-specific manner at the Bcl-6 locus and contributes to the regulation of Bcl-6 transcription. Oncogene. 2007;26(42):6244–6252. doi: 10.1038/sj.onc.1210434. [DOI] [PubMed] [Google Scholar]
- Armstrong L, Millar AB. Relative production of tumour necrosis factor alpha and interleukin 10 in adult respiratory distress syndrome. Thorax. 1997;52(5):442–446. doi: 10.1136/thx.52.5.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian K, Chandra J, Schroit AJ. Immune clearance of phosphatidylserine-expressing cells by phagocytes. The role of beta2-glycoprotein I in macrophage recognition. J Biol Chem. 1997;272(49):31113–31117. doi: 10.1074/jbc.272.49.31113. [DOI] [PubMed] [Google Scholar]
- Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46(1):191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Benkhart EM, Siedlar M, Wedel A, Werner T, Ziegler-Heitbrock HW. Role of Stat3 in lipopolysaccharide-induced IL-10 gene expression. J Immunol. 2000;165(3):1612–1617. doi: 10.4049/jimmunol.165.3.1612. [DOI] [PubMed] [Google Scholar]
- Bickerstaff MC, Botto M, Hutchinson WL, Herbert J, Tennent GA, Bybee A, Mitchell DA, Cook HT, Butler PJ, Walport MJ, Pepys MB. Serum amyloid P component controls chromatin degradation and prevents antinuclear autoimmunity. Nat Med. 1999;5(6):694–697. doi: 10.1038/9544. [DOI] [PubMed] [Google Scholar]
- Bienvenu J, Doche C, Gutowski MC, Lenoble M, Lepape A, Perdrix JP. Production of proinflammatory cytokines and cytokines involved in the TH1/TH2 balance is modulated by pentoxifylline. J Cardiovasc Pharmacol. 1995;25(Suppl. 2):S80–S84. doi: 10.1097/00005344-199500252-00017. [DOI] [PubMed] [Google Scholar]
- Böhm I. [The apoptosis marker enzyme poly-(ADP-ribose) polymerase (PARP) in systemic lupus erythematosus] Z Rheumatol. 2006;65(6):541–544. doi: 10.1007/s00393-006-0045-4. [DOI] [PubMed] [Google Scholar]
- Borisenko GG, Iverson SL, Ahlberg S, Kagan VE, Fadeel B. Milk fat globule epidermal growth factor 8 (MFG-E8) binds to oxidized phosphatidylserine: implications for macrophage clearance of apoptotic cells. Cell Death Differ. 2004;11(8):943–945. doi: 10.1038/sj.cdd.4401421. [DOI] [PubMed] [Google Scholar]
- Bratton DL, Fadok VA, Richter DA, Kailey JM, Guthrie LA, Henson PM. Appearance of phosphatidylserine on apoptotic cells requires calcium-mediated nonspecific flip-flop and is enhanced by loss of the aminophospholipid translocase. J Biol Chem. 1997;272(42):26159–26165. doi: 10.1074/jbc.272.42.26159. [DOI] [PubMed] [Google Scholar]
- Brightbill HD, Plevy SE, Modlin RL, Smale ST. A prominent role for Sp1 during lipopolysaccharide-mediated induction of the IL-10 promoter in macrophages. J Immunol. 2000;164(4):1940–1951. doi: 10.4049/jimmunol.164.4.1940. [DOI] [PubMed] [Google Scholar]
- Cao S, Zhang X, Edwards JP, Mosser DM. NF-kappaB1 (p50) homodimers differentially regulate pro- and anti-inflammatory cytokines in macrophages. J Biol Chem. 2006;281(36):26041–26050. doi: 10.1074/jbc.M602222200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson WE, Lindemann MJ, Baiocchi R, Linett M, Tan JC, Chou CC, Narula S, Caligiuri MA. The functional characterization of interleukin-10 receptor expression on human natural killer cells. Blood. 1995;85(12):3577–3585. [PubMed] [Google Scholar]
- Chiarugi A. Inhibitors of poly(ADP-ribose) polymerase-1 suppress transcriptional activation in lymphocytes and ameliorate autoimmune encephalomyelitis in rats. Br J Pharmacol. 2002;137(6):761–770. doi: 10.1038/sj.bjp.0704934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong WP, Ip WK, Wong WH, Lau CS, Chan TM, Lau YL. Association of interleukin-10 promoter polymorphisms with systemic lupus erythematosus. Genes Immun. 2004;5(6):484–492. doi: 10.1038/sj.gene.6364119. [DOI] [PubMed] [Google Scholar]
- Chung EY, Liu J, Homma Y, Zhang Y, Brendolan A, Saggese M, Han J, Silverstein R, Selleri L, Ma X. Interleukin-10 expression in macrophages during phagocytosis of apoptotic cells is mediated by homeodomain proteins Pbx1 and Prep-1. Immunity. 2007a;27(6):952–964. doi: 10.1016/j.immuni.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung EY, Liu J, Zhang Y, Ma X. Differential expression in lupus-associated IL-10 promoter single-nucleotide polymorphisms is mediated by poly(ADP-ribose) polymerase-1. Genes Immun. 2007b;8(7):577–589. doi: 10.1038/sj.gene.6364420. [DOI] [PubMed] [Google Scholar]
- Cox G, Crossley J, Xing Z. Macrophage engulfment of apoptotic neutrophils contributes to the resolution of acute pulmonary inflammation in vivo . Am J Respir Cell Mol Biol. 1995;12(2):232–237. doi: 10.1165/ajrcmb.12.2.7865221. [DOI] [PubMed] [Google Scholar]
- Crawley E, Kay R, Sillibourne J, Patel P, Hutchinson I, Woo P. Polymorphic haplotypes of the interleukin-10 5′ flanking region determine variable interleukin-10 transcription and are associated with particular phenotypes of juvenile rheumatoid arthritis. Arthritis Rheum. 1999;42(6):1101–1108. doi: 10.1002/1529-0131(199906)42:6<1101::AID-ANR6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- D’Alfonso S, Rampi M, Rolando V, Giordano M, Momigliano-Richiardi P. New polymorphisms in the IL-10 promoter region. Genes Immun. 2000;1(3):231–233. doi: 10.1038/sj.gene.6363666. [DOI] [PubMed] [Google Scholar]
- D’Amours D, Sallmann FR, Dixit VM, Poirier GG. Gain-of-function of poly(ADP-ribose) polymerase-1 upon cleavage by apoptotic proteases: implications for apoptosis. J Cell Sci. 2001;114(Pt 20):3771–3778. doi: 10.1242/jcs.114.20.3771. [DOI] [PubMed] [Google Scholar]
- Daleke DL, Lyles JV. Identification and purification of aminophospholipid flippases. Biochim Biophys Acta. 2000;1486(1):108–127. doi: 10.1016/s1388-1981(00)00052-4. [DOI] [PubMed] [Google Scholar]
- de Murcia G, Ménissier-de Murcia J, Schreiber V. Poly(ADP-ribose) polymerase: molecular biological aspects. Bioessays. 1991;13(9):455–462. doi: 10.1002/bies.950130905. [DOI] [PubMed] [Google Scholar]
- de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174(5):1209–1220. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkx B, Marchant A, Goldman M, Bijlmer R, van Deventer S. High levels of interleukin-10 during the initial phase of fulminant meningococcal septic shock. J Infect Dis. 1995;171(1):229–232. doi: 10.1093/infdis/171.1.229. [DOI] [PubMed] [Google Scholar]
- Donnelly SC, Strieter RM, Reid PT, Kunkel SL, Burdick MD, Armstrong I, Mackenzie A, Haslett C. The association between mortality rates and decreased concentrations of interleukin-10 and interleukin-1 receptor antagonist in the lung fluids of patients with the adult respiratory distress syndrome. Ann Intern Med. 1996;125(3):191–196. doi: 10.7326/0003-4819-125-3-199608010-00005. [DOI] [PubMed] [Google Scholar]
- Du Clos TW, Zlock LT, Hicks PS, Mold C. Decreased autoantibody levels and enhanced survival of (NZB x NZW) F1 mice treated with C-reactive protein. Clin Immunol Immunopathol. 1994;70(1):22–27. doi: 10.1006/clin.1994.1005. [DOI] [PubMed] [Google Scholar]
- Edwards-Smith CJ, Jonsson JR, Purdie DM, Bansal A, Shorthouse C, Powell EE. Interleukin-10 promoter polymorphism predicts initial response of chronic hepatitis C to interferon alfa. Hepatology. 1999;30(2):526–530. doi: 10.1002/hep.510300207. [DOI] [PubMed] [Google Scholar]
- Enghard P, Langnickel D, Riemekasten G. T cell cytokine imbalance towards production of IFN-gamma and IL-10 in NZB/W F1 lupus-prone mice is associated with autoantibody levels and nephritis. Scand J Rheumatol. 2006;35(3):209–216. doi: 10.1080/03009740500417791. [DOI] [PubMed] [Google Scholar]
- Eskdale J, Keijsers V, Huizinga T, Gallagher G. Microsatellite alleles and single nucleotide polymorphisms (SNP) combine to form four major haplotype families at the human interleukin-10 (IL-10) locus. Genes Immun. 1999;1(2):151–155. doi: 10.1038/sj.gene.6363656. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148(7):2207–2216. [PubMed] [Google Scholar]
- Friedman G, Jankowski S, Marchant A, Goldman M, Kahn RJ, Vincent JL. Blood interleukin 10 levels parallel the severity of septic shock. J Crit Care. 1997;12(4):183–187. doi: 10.1016/s0883-9441(97)90030-7. [DOI] [PubMed] [Google Scholar]
- Fuss IJ, Boirivant M, Lacy B, Strober W. The interrelated roles of TGF-beta and IL-10 in the regulation of experimental colitis. J Immunol. 2002;168(2):900–908. doi: 10.4049/jimmunol.168.2.900. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: a perspective. J Leukoc Biol. 2006;79(5):896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, Bratton DL, Oldenborg PA, Michalak M, Henson PM. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- Garin J, Diez R, Kieffer S, Dermine JF, Duclos S, Gagnon E, Sadoul R, Rondeau C, Desjardins M. The phagosome proteome: insight into phagosome functions. J Cell Biol. 2001;152(1):165–180. doi: 10.1083/jcb.152.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson AW, Edberg JC, Wu J, Westendorp RG, Huizinga TW, Kimberly RP. Novel single nucleotide polymorphisms in the distal IL-10 promoter affect IL-10 production and enhance the risk of systemic lupus erythematosus. J Immunol. 2001;166(6):3915–3922. doi: 10.4049/jimmunol.166.6.3915. [DOI] [PubMed] [Google Scholar]
- Gómez-Jiménez J, Martín MC, Sauri R, Segura RM, Esteban F, Ruiz JC, Nuvials X, Bóveda JL, Peracaula R, Salgado A. Interleukin-10 and the monocyte/macrophage-induced inflammatory response in septic shock. J Infect Dis. 1995;171(2):472–475. doi: 10.1093/infdis/171.2.472. [DOI] [PubMed] [Google Scholar]
- Gong MN, Thompson BT, Williams PL, Zhou W, Wang MZ, Pothier L, Christiani DC. Interleukin-10 polymorphism in position -1082 and acute respiratory distress syndrome. Eur Respir J. 2006;27(4):674–681. doi: 10.1183/09031936.06.00046405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grader-Beck T, Casciola-Rosen L, Lang TJ, Puliaev R, Rosen A, Via CS. Apoptotic splenocytes drive the autoimmune response to poly(ADP-ribose) polymerase 1 in a murine model of lupus. J Immunol. 2007;178(1):95–102. doi: 10.4049/jimmunol.178.1.95. [DOI] [PubMed] [Google Scholar]
- Grigg JM, Savill JS, Sarraf C, Haslett C, Silverman M. Neutrophil apoptosis and clearance from neonatal lungs. Lancet. 1991;338(8769):720–722. doi: 10.1016/0140-6736(91)91443-x. [DOI] [PubMed] [Google Scholar]
- Gröndal G, Gunnarsson I, Rönnelid J, Rogberg S, Klareskog L, Lundberg I. Cytokine production, serum levels and disease activity in systemic lupus erythematosus. Clin Exp Rheumatol. 2000;18(5):565–570. [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, Nagata S. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417(6885):182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- Henson PM, Hume DA. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 2006;27(5):244–250. doi: 10.1016/j.it.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41(7):1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Igonin AA, Armstrong VW, Shipkova M, Lazareva NB, Kukes VG, Oellerich M. Circulating cytokines as markers of systemic inflammatory response in severe community-acquired pneumonia. Clin Biochem. 2004;37(3):204–209. doi: 10.1016/j.clinbiochem.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Ishida H, Muchamuel T, Sakaguchi S, Andrade S, Menon S, Howard M. Continuous administration of anti-interleukin 10 antibodies delays onset of autoimmunity in NZB/W F1 mice. J Exp Med. 1994;179(1):305–310. doi: 10.1084/jem.179.1.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto Y, Ohashi K, Mizuno K, Nakano T. Promotion of the uptake of PS liposomes and apoptotic cells by a product of growth arrest-specific gene, gas6. J Biochem. 2000;127(3):411–417. doi: 10.1093/oxfordjournals.jbchem.a022622. [DOI] [PubMed] [Google Scholar]
- Jaber BL, Rao M, Guo D, Balakrishnan VS, Perianayagam MC, Freeman RB, Pereira BJ. Cytokine gene promoter polymorphisms and mortality in acute renal failure. Cytokine. 2004;25(5):212–219. doi: 10.1016/j.cyto.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Jeoung D, Lim Y, Lee EB, Lee S, Kim HY, Lee H, Song YW. Identification of autoantibody against poly (ADP-ribose) polymerase (PARP) fragment as a serological marker in systemic lupus erythematosus. J Autoimmun. 2004;22(1):87–94. doi: 10.1016/j.jaut.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Jijon HB, Churchill T, Malfair D, Wessler A, Jewell LD, Parsons HG, Madsen KL. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279(3):G641–G651. doi: 10.1152/ajpgi.2000.279.3.G641. [DOI] [PubMed] [Google Scholar]
- Johanneson B, Lima G, von Salomé J, Alarcón-Segovia D, Alarcón-Riquelme ME, Collaborative Group on the Genetics of, The BIOMED II Collaboration on the Genetics of SLE and Sjögrens syndrome A major susceptibility locus for systemic lupus erythemathosus maps to chromosome 1q31. Am J Hum Genet. 2002;71(5):1060–1071. doi: 10.1086/344289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BG, Solum D, Song EJ, Lee KJ, Rose DW, Glass CK, Rosenfeld MG. Activating the PARP-1 sensor component of the groucho/ TLE1 corepressor complex mediates a CaMKinase IIdelta-dependent neurogenic gene activation pathway. Cell. 2004;119(6):815–829. doi: 10.1016/j.cell.2004.11.017. [DOI] [PubMed] [Google Scholar]
- Kang X, Kim H, Ramirez M, Salameh S, Ma X. The septic shock-associated IL-10 −1082 A>G polymorphism mediates allele-specific transcription via poly ADP-ribose polymerase 1 in macrophages engulfing apoptotic cells. J Immunol. 2010in press doi: 10.4049/jimmunol.0903613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoa PD, Sugiyama T, Yokochi T. Polymorphism of interleukin-10 promoter and tumor necrosis factor receptor II in Vietnamese patients with systemic lupus erythematosus. Clin Rheumatol. 2005;24(1):11–13. doi: 10.1007/s10067-004-0952-1. [DOI] [PubMed] [Google Scholar]
- Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21(5):643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196(5):655–665. doi: 10.1084/jem.20020542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9(10):781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kube D, Platzer C, von Knethen A, Straub H, Bohlen H, Hafner M, Tesch H. Isolation of the human interleukin 10 promoter. Characterization of the promoter activity in Burkitt’s lymphoma cell lines. Cytokine. 1995;7(1):1–7. doi: 10.1006/cyto.1995.1001. [DOI] [PubMed] [Google Scholar]
- Kühnle S, Nicotera P, Wendel A, Leist M. Prevention of endotoxin-induced lethality, but not of liver apoptosis in poly(ADP-ribose) polymerase-deficient mice. Biochem Biophys Res Commun. 1999;263(2):433–438. doi: 10.1006/bbrc.1999.1393. [DOI] [PubMed] [Google Scholar]
- Lazarus M, Hajeer AH, Turner D, Sinnott P, Worthington J, Ollier WE, Hutchinson IV. Genetic variation in the interleukin 10 gene promoter and systemic lupus erythematosus. J Rheumatol. 1997;24(12):2314–2317. [PubMed] [Google Scholar]
- Lim Y, Lee DY, Lee S, Park SY, Kim J, Cho B, Lee H, Kim HY, Lee E, Song YW, Jeoung DI. Identification of autoantibodies associated with systemic lupus erythematosus. Biochem Biophys Res Commun. 2002;295(1):119–124. doi: 10.1016/s0006-291x(02)00637-x. [DOI] [PubMed] [Google Scholar]
- Llorente L, Richaud-Patin Y, Couderc J, Alarcon-Segovia D, Ruiz-Soto R, Alcocer-Castillejos N, Alcocer-Varela J, Granados J, Bahena S, Galanaud P, Emilie D. Dysregulation of interleukin-10 production in relatives of patients with systemic lupus erythematosus. Arthritis Rheum. 1997;40(8):1429–1435. doi: 10.1002/art.1780400810. [DOI] [PubMed] [Google Scholar]
- Llorente L, Richaud-Patin Y, Wijdenes J, Alcocer-Varela J, Maillot MC, Durand-Gasselin I, Fourrier BM, Galanaud P, Emilie D. Spontaneous production of interleukin-10 by B lymphocytes and monocytes in systemic lupus erythematosus. Eur Cytokine Netw. 1993;4(6):421–427. [PubMed] [Google Scholar]
- MacNeil IA, Suda T, Moore KW, Mosmann TR, Zlotnik A. IL-10, a novel growth cofactor for mature and immature T cells. J Immunol. 1990;145(12):4167–4173. [PubMed] [Google Scholar]
- Marchant A, Devière J, Byl B, De Groote D, Vincent JL, Goldman M. Interleukin-10 production during septicaemia. Lancet. 1994;343(8899):707–708. doi: 10.1016/s0140-6736(94)91584-9. [DOI] [PubMed] [Google Scholar]
- Martens HA, Nolte IM, van der Steege G, Schipper M, Kallenberg CG, te Meerman GJ, Bijl M. Association of poly(ADP-ribose) polymerase 1 and a novel candidate locus, LOC127086, with systemic lupus erythematosus. Ann Rheum Dis. 2007;66(3):424–425. doi: 10.1136/ard.2006.065102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoubrey-Hoyer A, Okarma TB, Holman HR. Partial purification and characterization of plasma DNA and its relation to disease activity in systemic lupus erythematosus. Am J Med. 1984;77(1):23–34. doi: 10.1016/0002-9343(84)90431-5. [DOI] [PubMed] [Google Scholar]
- Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- Moens CB, Selleri L. Hox cofactors in vertebrate development. Dev Biol. 2006;291(2):193–206. doi: 10.1016/j.ydbio.2005.10.032. [DOI] [PubMed] [Google Scholar]
- Monneret G, Finck ME, Venet F, Debard AL, Bohé J, Bienvenu J, Lepape A. The anti-inflammatory response dominates after septic shock: association of low monocyte HLA-DR expression and high interleukin-10 concentration. Immunol Lett. 2004;95(2):193–198. doi: 10.1016/j.imlet.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- Moore KW, Vieira P, Fiorentino DF, Trounstine ML, Khan TA, Mosmann TR. Homology of cytokine synthesis inhibitory factor (IL-10) to the Epstein-Barr virus gene BCRFI. Science. 1990;248(4960):1230–1234. doi: 10.1126/science.2161559. [DOI] [PubMed] [Google Scholar]
- Morris RG, Hargreaves AD, Duvall E, Wyllie AH. Hormone-induced cell death. 2. Surface changes in thymocytes undergoing apoptosis. Am J Pathol. 1984;115(3):426–436. [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Ohashi K, Nakano T, Arita H, Zong C, Hanafusa H, Mizuno K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J Biol Chem. 1996;271(47):30022–30027. doi: 10.1074/jbc.271.47.30022. [DOI] [PubMed] [Google Scholar]
- Napirei M, Karsunky H, Zevnik B, Stephan H, Mannherz HG, Möröy T. Features of systemic lupus erythematosus in Dnase1-deficient mice. Nat Genet. 2000;25(2):177–181. doi: 10.1038/76032. [DOI] [PubMed] [Google Scholar]
- Neidhardt R, Keel M, Steckholzer U, Safret A, Ungethuem U, Trentz O, Ertel W. Relationship of interleukin-10 plasma levels to severity of injury and clinical outcome in injured patients. J Trauma. 1997;42(5):863–870. doi: 10.1097/00005373-199705000-00017. [DOI] [PubMed] [Google Scholar]
- Oberholzer A, Oberholzer C, Moldawer LL. Interleukin-10: A complex role in the pathogenesis of sepsis syndromes and its potential as an anti-inflammatory drug. Crit Care Med. 2002;30(1 Supp):S58–S63. [PubMed] [Google Scholar]
- Oliver FJ, Ménissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18(16):4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Païdassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, Arlaud GJ, Frachet P. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008;180(4):2329–2338. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- Parsons PE, Moss M, Vannice JL, Moore EE, Moore FA, Repine JE. Circulating IL-1ra and IL-10 levels are increased but do not predict the development of acute respiratory distress syndrome in at-risk patients. Am J Respir Crit Care Med. 1997;155(4):1469–1473. doi: 10.1164/ajrccm.155.4.9105096. [DOI] [PubMed] [Google Scholar]
- Pavri R, Lewis B, Kim TK, Dilworth FJ, Erdjument-Bromage H, Tempst P, de Murcia G, Evans R, Chambon P, Reinberg D. PARP-1 determines specificity in a retinoid signaling pathway via direct modulation of mediator. Mol Cell. 2005;18(1):83–96. doi: 10.1016/j.molcel.2005.02.034. [DOI] [PubMed] [Google Scholar]
- Perniok A, Wedekind F, Herrmann M, Specker C, Schneider M. High levels of circulating early apoptic peripheral blood mononuclear cells in systemic lupus erythematosus. Lupus. 1998;7(2):113–118. doi: 10.1191/096120398678919804. [DOI] [PubMed] [Google Scholar]
- Pétrilli V, Herceg Z, Hassa PO, Patel NS, Di Paola R, Cortes U, Dugo L, Filipe HM, Thiemermann C, Hottiger MO, Cuzzocrea S, Wang ZQ. Noncleavable poly(ADP-ribose) polymerase-1 regulates the inflammation response in mice. J Clin Invest. 2004;114(8):1072–1081. doi: 10.1172/JCI21854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramprasad MP, Fischer W, Witztum JL, Sambrano GR, Quehenberger O, Steinberg D. The 94- to 97-kDa mouse macrophage membrane protein that recognizes oxidized low density lipoprotein and phosphatidylserine-rich liposomes is identical to macrosialin, the mouse homologue of human CD68. Proc Natl Acad Sci USA. 1995;92(21):9580–9584. doi: 10.1073/pnas.92.21.9580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raptis L, Menard HA. Quantitation and characterization of plasma DNA in normals and patients with systemic lupus erythematosus. J Clin Invest. 1980;66(6):1391–1399. doi: 10.1172/JCI109992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SC, Scott KA, Wilson JL, Thompson RG, Proudfoot AE, Balkwill FR. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res. 2003;63(23):8360–8365. [PubMed] [Google Scholar]
- Rodríguez-Gaspar M, Santolaria F, Jarque-López A, González-Reimers E, Milena A, de la Vega MJ, Rodríguez-Rodríguez E, Gómez-Sirvent JL. Prognostic value of cytokines in SIRS general medical patients. Cytokine. 2001;15(4):232–236. doi: 10.1006/cyto.2001.0932. [DOI] [PubMed] [Google Scholar]
- Roy S, Knox K, Segal S, Griffiths D, Moore CE, Welsh KI, Smarason A, Day NP, McPheat WL, Crook DW, Hill AV, Oxford Pneumoccocal Surveillance Group MBL genotype and risk of invasive pneumococcal disease: a case-control study. Lancet. 2002;359(9317):1569–1573. doi: 10.1016/S0140-6736(02)08516-1. [DOI] [PubMed] [Google Scholar]
- Savill J, Fadok V, Henson P, Haslett C. Phagocyte recognition of cells undergoing apoptosis. Immunol Today. 1993;14(3):131–136. doi: 10.1016/0167-5699(93)90215-7. [DOI] [PubMed] [Google Scholar]
- Schaaf BM, Boehmke F, Esnaashari H, Seitzer U, Kothe H, Maass M, Zabel P, Dalhoff K. Pneumococcal septic shock is associated with the interleukin-10–1082 gene promoter polymorphism. Am J Respir Crit Care Med. 2003;168(4):476–480. doi: 10.1164/rccm.200210-1164OC. [DOI] [PubMed] [Google Scholar]
- Schall TJ, Bacon K, Toy KJ, Goeddel DV. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature. 1990;347(6294):669–671. doi: 10.1038/347669a0. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, Matsushima GK. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–211. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- Simmons EM, Himmelfarb J, Sezer MT, Chertow GM, Mehta RL, Paganini EP, Soroko S, Freedman S, Becker K, Spratt D, Shyr Y, Ikizler TA, PICARD Study Group Plasma cytokine levels predict mortality in patients with acute renal failure. Kidney Int. 2004;65(4):1357–1365. doi: 10.1111/j.1523-1755.2004.00512.x. [DOI] [PubMed] [Google Scholar]
- Somersan S, Bhardwaj N. Tethering and tickling: a new role for the phosphatidylserine receptor. J Cell Biol. 2001;155(4):501–504. doi: 10.1083/jcb.200110066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song E, Zhu P, Lee SK, Chowdhury D, Kussman S, Dykxhoorn DM, Feng Y, Palliser D, Weiner DB, Shankar P, Marasco WA, Lieberman J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat Biotechnol. 2005;23(6):709–717. doi: 10.1038/nbt1101. [DOI] [PubMed] [Google Scholar]
- Stanilova SA, Miteva LD, Karakolev ZT, Stefanov CS. Interleukin-10–1082 promoter polymorphism in association with cytokine production and sepsis susceptibility. Intensive Care Med. 2006;32(2):260–266. doi: 10.1007/s00134-005-0022-4. [DOI] [PubMed] [Google Scholar]
- Steinhauser ML, Hogaboam CM, Kunkel SL, Lukacs NW, Strieter RM, Standiford TJ. IL-10 is a major mediator of sepsis-induced impairment in lung antibacterial host defense. J Immunol. 1999;162(1):392–399. [PubMed] [Google Scholar]
- Steinman CR. Circulating DNA in systemic lupus erythematosus. Isolation and characterization. J Clin Invest. 1984;73(3):832–841. doi: 10.1172/JCI111278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart LM, Boulais J, Charriere GM, Hennessy EJ, Brunet S, Jutras I, Goyette G, Rondeau C, Letarte S, Huang H, Ye P, Morales F, Kocks C, Bader JS, Desjardins M, Ezekowitz RA. A systems biology analysis of the Drosophila phagosome. Nature. 2007;445(7123):95–101. doi: 10.1038/nature05380. [DOI] [PubMed] [Google Scholar]
- Sun EW, Shi YF. Apoptosis: the quiet death silences the immune system. Pharmacol Ther. 2001;92(2–3):135–145. doi: 10.1016/s0163-7258(01)00164-4. [DOI] [PubMed] [Google Scholar]
- Szabó C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19(7):287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- Teague TK, Hildeman D, Kedl RM, Mitchell T, Rees W, Schaefer BC, Bender J, Kappler J, Marrack P. Activation changes the spectrum but not the diversity of genes expressed by T cells. Proc Natl Acad Sci USA. 1999;96(22):12691–12696. doi: 10.1073/pnas.96.22.12691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao BP. Lupus susceptibility genes on human chromosome 1. Int Rev Immunol. 2000;19(4–5):319–334. doi: 10.3109/08830180009055502. [DOI] [PubMed] [Google Scholar]
- Tsao BP, Cantor RM, Kalunian KC, Chen CJ, Badsha H, Singh R, Wallace DJ, Kitridou RC, Chen SL, Shen N, Song YW, Isenberg DA, Yu CL, Hahn BH, Rotter JI. Evidence for linkage of a candidate chromosome 1 region to human systemic lupus erythematosus. J Clin Invest. 1997;99(4):725–731. doi: 10.1172/JCI119217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulin A, Stewart D, Spradling AC. The Drosophila heterochromatic gene encoding poly(ADP-ribose) polymerase (PARP) is required to modulate chromatin structure during development. Genes Dev. 2002;16(16):2108–2119. doi: 10.1101/gad.1003902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner DM, Williams DM, Sankaran D, Lazarus M, Sinnott PJ, Hutchinson IV. An investigation of polymorphism in the interleukin-10 gene promoter. Eur J Immunogenet. 1997;24(1):1–8. doi: 10.1111/j.1365-2370.1997.tb00001.x. [DOI] [PubMed] [Google Scholar]
- van der Poll T, Marchant A, Keogh CV, Goldman M, Lowry SF. Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J Infect Dis. 1996;174(5):994–1000. doi: 10.1093/infdis/174.5.994. [DOI] [PubMed] [Google Scholar]
- Vance JE, Steenbergen R. Metabolism and functions of phosphatidylserine. Prog Lipid Res. 2005;44(4):207–234. doi: 10.1016/j.plipres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96(2):245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Virág L, Bai P, Bak I, Pacher P, Mabley JG, Liaudet L, Bakondi E, Gergely P, Kollai M, Szabó C. Effects of poly(ADP-ribose) polymerase inhibition on inflammatory cell migration in a murine model of asthma. Med Sci Monit. 2004;10(3):BR77–BR83. [PubMed] [Google Scholar]
- Voll RE, Roth EA, Girkontaite I, Fehr H, Herrmann M, Lorenz HM, Kalden JR. Histone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody production. Arthritis Rheum. 1997;40(12):2162–2171. doi: 10.1002/art.1780401210. [DOI] [PubMed] [Google Scholar]
- Westendorp RG, Langermans JA, Huizinga TW, Elouali AH, Verweij CL, Boomsma DI, Vandenbroucke JP, Vandenbrouke JP. Genetic influence on cytokine production and fatal meningococcal disease. Lancet. 1997;349(9046):170–173. doi: 10.1016/s0140-6736(96)06413-6. [DOI] [PubMed] [Google Scholar]
- Williamson P, Schlegel RA. Transbilayer phospholipid movement and the clearance of apoptotic cells. Biochim Biophys Acta. 2002;1585(2–3):53–63. doi: 10.1016/s1388-1981(02)00324-4. [DOI] [PubMed] [Google Scholar]
- Yilmaz V, Yentür SP, Saruhan-Direskeneli G. IL-12 and IL-10 polymorphisms and their effects on cytokine production. Cytokine. 2005;30(4):188–194. doi: 10.1016/j.cyto.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Yung TM, Satoh MS. Functional competition between poly(ADP-ribose) polymerase and its 24-kDa apoptotic fragment in DNA repair and transcription. J Biol Chem. 2001;276(14):11279–11286. doi: 10.1074/jbc.M008044200. [DOI] [PubMed] [Google Scholar]
- Zhang DL, Zheng HM, Yu BJ, Jiang ZW, Li JS. Association of polymorphisms of IL and CD14 genes with acute severe pancreatitis and septic shock. World J Gastroenterol. 2005;11(28):4409–4413. doi: 10.3748/wjg.v11.i28.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann TS, Lee AC, Akinc A, Bramlage B, Bumcrot D, Fedoruk MN, Harborth J, Heyes JA, Jeffs LB, John M, Judge AD, Lam K, McClintock K, Nechev LV, Palmer LR, Racie T, Röhl I, Seiffert S, Shanmugam S, Sood V, Soutschek J, Toudjarska I, Wheat AJ, Yaworski E, Zedalis W, Koteliansky V, Manoharan M, Vornlocher HP, MacLachlan I. RNAi-mediated gene silencing in non-human primates. Nature. 2006;441(7089):111–114. doi: 10.1038/nature04688. [DOI] [PubMed] [Google Scholar]