

Abstract
Apoptosis-inducing factor (AIF) is a flavin adenine dinucleotide-containing, NADH-dependent oxidoreductase residing in the mitochondrial intermembrane space whose specific enzymatic activity remains unknown. Upon an apoptotic insult, AIF undergoes proteolysis and translocates to the nucleus, where it triggers chromatin condensation and large-scale DNA degradation in a caspase-independent manner. Besides playing a key role in execution of caspase-independent cell death, AIF has emerged as a protein critical for cell survival. Analysis of in vivo phenotypes associated with AIF deficiency and defects, and identification of its mitochondrial, cytoplasmic, and nuclear partners revealed the complexity and multilevel regulation of AIF-mediated signal transduction and suggested an important role of AIF in the maintenance of mitochondrial morphology and energy metabolism. The redox activity of AIF is essential for optimal oxidative phosphorylation. Additionally, the protein is proposed to regulate the respiratory chain indirectly, through assembly and/or stabilization of complexes I and III. This review discusses accumulated data with respect to the AIF structure and outlines evidence that supports the prevalent mechanistic view on the apoptogenic actions of the flavoprotein, as well as the emerging concept of AIF as a redox sensor capable of linking NAD(H)-dependent metabolic pathways to apoptosis. Antioxid. Redox Signal. 14, 2545–2579.
I. Introduction
Apoptosis is a highly regulated and energy-dependent programmed cell death (PCD) essential for early embryonic development and tissue homeostasis. Dysregulation of apoptosis may lead to various acute and chronic pathologies such as stroke, cancer, neurodegeneration, and autoimmune syndromes (217). In mammalian cells, several pro-apoptotic and cell damage pathways, which either do or do not require caspase activation, converge on mitochondria. Permeabilization of the organelle leads to release of several proteins from the intermembrane space (IMS) that participate in the organized cell demise [reviewed in (184)]. One of the soluble factors released from mouse mitochondria and capable of forcing isolated nuclei to adopt apoptotic morphology in a caspase-independent manner was discovered by Kroemer and coworkers in 1996 (214, 248). The protein was named apoptosis-inducing factor (AIF), as it could maintain the apoptogenic ability in the presence of a pan-caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (z-vad.fmk) (212). This group demonstrated also that AIF binds favin adenine dinucleotide (FAD) and possesses various NAD(P)H-dependent redox activities (155, 212), but its specific enzymatic function is presently unknown.
In addition to the unraveling of a caspase-independent pathway of PCD, major advances in AIF research include (a) discovery of several naturally occurring and functionally distinct forms and splice variants of AIF; (b) identification of homologous proteins in lower eukaryotes, prokaryotes, and archaea and demonstration of evolutionary conservation of the AIF-dependent cell death pathways in uni- and multicellular organisms; (c) determination of the crystal structures of different redox forms of AIF and gaining insights into its unique architecture and redox-linked conformational reorganization; (d) exploitation of natural and creation of de novo genetic models to pinpoint effects caused by AIF knockout, deficiency or defects; and (e) identification of a diverse group of proteins interacting with AIF in different cellular compartments that showed the complexity and multilevel regulation of the AIF-mediated signal transduction.
Numerous reviews have appeared in recent years, covering various aspects of mitochondrial and cellular physiology related to AIF (45, 86, 104, 157, 179). This article includes most recent findings on AIF and discusses the experimental data from a structural viewpoint, emphasizing the importance of folding for the structural and functional integrity of the flavoprotein. A brief overview of multiple forms, transcriptional regulation, and phylogenetic roots of AIF is followed by a detailed description of its catalytic and structural properties. Since AIF was originally discovered as an apoptosis-inducing factor and its role in PCD has been investigated more extensively and, overall, is better understood than the function in normal mitochondria, it will be discussed first. In addition to in vitro and in vivo analyses of the AIF-mediated apoptotic cell death, current views on the mechanism of AIF liberation, translocation to the nucleus and interaction with pro-life and pro-death cytoplasmic and nuclear partners are presented. Apoptogenic potential of AIF homologs from lower eukaryotes is also discussed as it helps to better understand structure/function relations and cellular roles of the flavoprotein. The following sections highlight vital functions of AIF, especially its involvement in regulation of mitochondrial respiration and cristae morphology. A hypothesis on the redox-signaling role of AIF and supporting evidence are outlined at the end of the review. Since most investigations were performed on human and murine AIF, which vary in length, the amino acid sequence numbering will be given for human AIF, with the corresponding fragments/residues for the murine protein specified when necessary.
II. Multiple Forms of AIF
A. AIF precursor
The AIF gene, also known as AIFM1 and PCDC8, was mapped to chromosome X region A6 in mice and Xq25–26 region in humans (48). Transcription and translation of the nuclear-encoded gene gives rise to a 67 kDa precursor molecule, residues 1–613 (1–612 in mice), containing the N-terminal mitochondrial leading sequence (MLS), two nuclear leading sequences (NLS), and the NAD- and FAD-binding motifs (Fig. 1A, B) (212). The precursor can be imported to mitochondria only in a non-native form, meaning that its folding in the cytoplasm is either prevented or delayed. When refolded, full-length AIF becomes apoptogenic independently on whether FAD is bound or not (212).
FIG. 1.

Major forms and splice variants of AIF. (A) Schematic representation of the human AIF gene. Exons are numbered; alternative exons giving splice variants are in gray. Translation initiation (ATG) and stop codons (TGA/TAA) are indicated. (B, E–G) Naturally occurring transcripts corresponding to the AIF precursor, AIFsh, AIFsh2, and AIFsh3, respectively. (C) Mature form of AIF produced upon mitochondrial processing. Depending on the usage of exons 2a and 2b, the ubiquitously expressed AIF1 or brain-specific AIF2 isoforms can be synthesized. (D) Truncated apoptogenic form produced in the intermembrane space upon proteolytic processing. The FAD-binding, NAD(H)-binding, and C-terminal (Ct) domains are indicated. AIF, apoptosis-inducing factor; MLS, mitochondrial leading sequence; NLS, nuclear leading sequence.
B. Membrane-tethered mature AIFΔ1–54
For quite some time, the soluble 57 kDa fragment of AIF consisting of residues 103–613 (102–612 in mice; Fig. 1D) was thought to represent the mature form residing in the IMS (212). Several lines of evidence indicated, however, that mitochondrial AIF is associated with the inner membrane. In particular, it was demonstrated that (a) in response to several apoptotic stimuli, release of AIF from the IMS occurs downstream of cytochrome c and requires caspase activation (10); (b) during hypotonic lysis of mitochondria, AIF remains associated with the mitoplast pellet and is released only after partial solubilization of the inner membrane (222); and (c) truncated Bid (tBid), an outer membrane-permeabilizing agent, induces AIF release from isolated mitochondria only in the presence of active calpain, a calcium-dependent protease (178). Altogether, these studies suggested that AIF is somehow attached to the inner membrane and may need to be cleaved to become a soluble and apoptogenic protein.
A major breakthrough was made by Otera et al. (168), who demonstrated that the human AIF precursor is cleaved by a mitochondrial matrix peptidase at Met54/Ala55, resulting in a mature form that is 48-residue longer (∼62 kDa; Δ1–54) than originally reported (212). Residues 67–85 were identified as the IMS sorting signal and a trans-membrane fragment through which AIFΔ1–54 (Δ1–53 in mice), the healthy and vital form of the flavoprotein, is tethered to the inner membrane (168). However, Yu et al. argue against the proposed trans-membrane topology and suggest that AIF represents a peripheral rather than integral inner membrane protein because it can be stripped from the membranes by alkaline treatment (245). Concordantly, both endogenous and transiently overexpressed AIF1 (the ubiquitous isoform) were reported to behave as loosely membrane-associated proteins (87).
There is evidence that transport of AIF from the mitochondrial matrix into the IMS proceeds through the inner membrane channel Tim23 protein (89). Since AIF isolated from mitochondria contains FAD and the precursor does not (212), proteolytic maturation and import into the IMS seem to be required for the protein folding and flavin incorporation.
C. Soluble apoptogenic AIFΔ1–102/118
The 96–120 segment of AIFΔ1–54 has two proteolytic sites, cleavage of which can lead to formation of soluble Δ1–102 or Δ1–118 fragments (murine AIF numbering, ∼57 kDa; Fig. 1D) (38, 168, 178). Proteolysis of the membrane-bound mature AIF can be mediated by mitochondrial or cytoplasmic proteases, as discussed in detail in section VII.B.1. Upon permeabilization of the outer mitochondrial membrane (OMM), the detached Δ1–102/118 fragments can translocate from the IMS to the nucleus to execute apoptosis and, therefore, represent the lethal form of the flavoprotein.
D. AIF associated with the outer mitochondrial membrane
This pool of the protein has been recently identified by Yu et al., who showed that in brain mitochondria nearly 30% of total endogenous mitochondrial AIF is loosely associated with the cytoplasmic side of the OMM (245). This OMM-associated fraction, presumably an unfolded AIF precursor, can also be lethal because under certain conditions it can rapidly relocate to the nucleus and initiate PCD (see section VII.B.3 for details).
E. Splice variants AIF2, AIFsh, AIFsh2, and AIFsh3
An alternatively spliced AIF-exB (AIF2) isoform was the first splice variant discovered in mice and humans (136). AIF2 utilizes exon 2b instead of 2a (Fig. 1A) and differs from AIF1, the originally described ubiquitous form (212), only in a short N-terminal amino acid stretch that gets cleaved during apoptogenic processing. Exons 2a and 2b are phylogenetically conserved among mammals and their usage has no effect on the mitochondrial transport of AIF. AIF2 is a brain-specific isoform whose expression depends on the neuronal cell maturation status (87).
The AIFshort (AIFsh) isoform results from an alternative transcriptional start site located at intron 9 and consists of aa. 353–613 (35 kDa; Fig. 1E) (53). AIFsh lacks MLS and represents a cytoplasmic protein that upon an apoptotic insult can transport to the nucleus and promote cell death. AIFsh2 and AIFsh3, in turn, are produced via alternative usage of exon 9b. They are comprised of residues 1–322 and 87–322, respectively, and have additional Asp-Ile at the C-terminus (Fig. 1F, G) (54). These splice variants lack the C-terminal domain and NLS2 and cannot translocate to the nucleus. AIFsh3 is also missing the N-terminal MLS and confined exclusively to the cytoplasm. Mice have an AIFsh2 homolog but no AIFsh3. Owing to existence of multiple isoforms, the AIF-mediated function and signaling can be regulated at both the transcriptional and post-translational levels.
III. Transcriptional Regulation
A single 2.4 Kb AIF mRNA transcript and ubiquitous expression of AIF1 have been detected in murine and human organs (48, 53, 212). On the contrary, AIF2 mRNA is absent in all human tissues except brain and retina (87). Although in most areas of the human adult brain AIF1 and AIF2 are coexpressed, there are brain cells that solely express AIF2 (e.g., part of the anterior olfactory nucleus). The AIF1 and AIF2 mRNA levels are similar in the fetal brain but the AIF1/AIF2 ratio decreases as brain cells differentiate (87). Expression of AIF1 in human skeletal muscle is also age-dependent and increases by 10%–25% from the age of 10–40 (172). AIFsh is expressed at lower levels and regulated independently from AIF1 (53), whereas expression of AIFsh2 is tissue specific and, on average, lower than AIF1 (54).
Transcriptional factors regulating AIF expression remain largely unknown. It was found though that the AIF gene is a transcriptional target of p53 (209). Using isogenic cell lines differing in the p53 status, Stambolsky et al. demonstrated that the absence of functional p53 significantly reduces AIF mRNA and protein levels. They also identified a p53-responsive element in the fourth intron of the AIF gene. Positive modulation of AIF expression by basal levels of p53 is cell type-specific and does not depend on DNA-damaging stress.
Conversely, downstream effectors of hepatocyte growth factor, hepatocyte growth factor receptor, and focal adhesion kinase suppress AIF expression (34). Bcl-2 19 kDa interacting protein, a pro-cell-death BH3-only member of the Bcl-2 family, is another transcriptional repressor that binds to a response element in the AIF promoter and downregulates expression (26). In highly malignant human gliomas, Bcl-2 19 kDa interacting protein 3 reduces AIF expression through interactions with PTB (polypyrimidine tract binding protein)-associating splicing factor and histone deacetylase 1 (26) and thus is acting as a pro-survival factor. AIF expression is decreased in many other tumor types, contributing to their chemoresistance (54, 96), but is upregulated in malignant colorectal epithelial cells (99).
IV. Phylogenetic Roots
Determination of multiple prokaryotic and eukaryotic genome sequences enabled analysis of phylogenetic origins of AIF and predictions on its function. Phylogenetic analysis reveals strong conservation among mammalian AIFs (∼90% identity in the whole sequence) and ubiquitous presence of the AIF-like proteins in bacteria, archaea, and eukaryotes (27, 137, 124). Clustering of eukaryotic AIFs with the archaeal orthologs suggests an existence of a universal common ancestor that was secondarily recruited for a specific mitochondrial function. Homologs from zebrafish (Danio rerio), fruit fly (Drosophila melanogaster), and slime mold (Dictyostelium discoideum) are closely related to mammalian AIFs (Figs. 2 and 3), whereas proteins from Caenorhabditis elegans and Saccharomyces cerevisiae, widely used as model organisms in cell biology research, are more distant on the evolutionary tree.
FIG. 2.

Phylogenetic tree of AIF-like proteins. Analysis of the phylogenetic relationships between the full-length molecular sequences was performed using the Phylogeny.fr online server (www.phylogeny.fr), which utilizes MUSCLE for sequence alignment and PhyML for phylogeny (55). GenBank or UniProt identification numbers are indicated.
FIG. 3.

Amino acid sequence alignment of AIF and AIF-like proteins from Homo sapiens (H. s.), Mus musculus (M. m.), Danio rerio (D. r.), Drosophila melanogaster (D. m.), Dictyostelium discoideum (D. d.), Caenorhabditis elegans (C. e.), Arabidopsis thaliana (A. t.), Pseudomonas sp. strain KKS102 (Ps.), and Saccharomyces cerevisiae (S. c.). Sequence alignment was performed with ClustalW2 (128). GenBank or UniProt accession numbers are given in Figure 2. Functionally important structural elements are highlighted in gray and indicated. The membrane-binding fragment in C. elegans is boxed.
Most of the AIF-like proteins contain the GXGXXA and V/IXGXGXXG motifs, characteristic for the NAD(P)- and FAD-binding domains adopting the Rossmann fold (119). The N-termini of AIF from mammals and lower eukaryotes contain MLS and a stretch of hydrophobic amino acids serving as a membrane-binding fragment (Fig. 3). The plant, yeast, and bacterial homologs have a shortened N-terminus and lack both MLS and a membrane tether. The C. elegans protein has the longest N-terminal part that in addition to MLS contains an extended and highly charged linker region. The most important difference, however, is the presence of two insertions in mammalian proteins (191–203 and 510–560), which, as will be emphasized throughout the review, may define specific functions and signal transduction cascades. On the basis of the presence of the FAD- and NAD(H)-binding motifs and strong sequence homology to plant and bacterial ascorbate and ferredoxin reductases, AIF was suggested to possess oxidoreductase activities (212). This prediction was later confirmed in vitro using recombinantly expressed proteins (38, 155).
V. Redox Properties of Recombinant AIF
A. Refolded murine AIFΔ1–120
Recombinant murine AIFΔ1–120 was the first to be biochemically characterized. Cloned with the affinity tags fused to both termini, AIFΔ1–120 was expressed in E. coli as an apoprotein and had to be refolded to incorporate the flavin (155). FAD-bound AIFΔ1–120 can be fully reduced with sodium dithionite or NAD(P)H without accumulation of the semiquinone intermediate. However, addition of stoichiometric amounts of NAD(P)H to refolded AIFΔ1–120 is not accompanied by formation of an FADH2-NAD(P) charge–transfer complex (CTC), normally produced by other flavoenzymes as a result of π-π charge–transfer interactions between the parallel-stacked isoalloxazine and nicotinamide. Nonetheless, refolded AIFΔ1–120 can oxidize NAD(P)H (20–2244 min−1) and catalyze NADH-dependent reduction of small electron acceptors and cytochrome c directly or via superoxide anion, with a turnover number ranging from 0.5 to 22 min−1 (155). On the basis of these properties and the reductase activity of the native mitochondrial protein, AIF was proposed to function in vivo as a superoxide producing NADH oxidase (155).
B. Refolded human AIFsh2
Refolded human AIFsh2 is redox active and catalyses NAD(P)H oxidation and O2−•-mediated reduction of small electron accepting molecules similar to refolded murine AIFΔ1–120 (54).
C. Naturally folded murine AIFΔ1–53 and Δ1–101
Our group showed that recombinantly expressed AIF can naturally fold and incorporate FAD if no affinity or immune-tags are placed at the N-terminus, close to the flavin binding site (38). When expressed in this way, murine AIFΔ1–53 and Δ1–101 have properties distinct from those of refolded AIFΔ1–120. In particular, they have a considerably lower redox potential [−355/ −349 mV vs. − 308 mV at pH 7.5 (155)] and higher preference for NADH over NADPH. Both proteins react slowly with either cofactor and upon reduction form tight and long-lived FADH2-NAD(P) CTC (38). Due to resistance of CTC to oxidation, the NADH oxidase activity of AIF is negligible (<0.2 min−1). Nevertheless, both AIF forms directly shuttle electrons from NADH to one- and two-electron acceptors (30–60 min−1) but do not reduce O2−•, H2O2, ascorbate free radical, or dehydroascorbate and, hence, cannot function as reactive oxygen species (ROS) scavengers. On the basis of the kinetic parameters, such as high KM (1–2 mM) and low Kd for NADH (150 nM), mitochondrial AIF was concluded to function as a low turnover oxidoreductase (38). Other important findings on the naturally folded AIF include its redox-linked dimerization accompanied by a conformational switch in the 509–559 insertion, and involvement of the N-terminal peptide in stabilization of the AIF-NAD(P)H complex (38).
In summary, in vitro studies showed that the manner of folding and FAD incorporation defines redox properties of AIF. Naturally folded AIF functions as a low-turnover NADH-dependent oxidoreductase whose reduced form is dimeric and resistant to oxidation by O2. Although the specific enzymatic activity could not be identified, the biochemical data ruled out the possibility that AIF is an antioxidant.
VI. AIF Structure
A. X-ray structures of murine and human AIFΔ1–120
The x-ray structure of the oxidized form of refolded murine AIFΔ1–121 (150) provided first insights into the protein architecture and active site environment, defined surface properties of AIF, and enabled suggestions on how it may function in the cell. AIF has a glutathione reductase (GR) fold with the closest structural homology to BphA4, a ferredoxin reductase from Pseudomonas sp. strain KKS102 (200). The protein has three functional modules: the FAD- (aa. 129–170, 203–262 and 400–479) and NAD(H)-binding domains (aa.171–202 and 263–399) with a classical Rossmann topology, and the C-terminal domain (aa. 480–610) comprised of five antiparallel β-strands and two α-helices (Fig. 4A). The FAD-binding domain is most conserved in the GR-like proteins. The Lys176-Glu313 charge–charge pair in AIF assists FAD binding and regulates catalytic efficiency (150), whereas Phe309 plays a role of a gatekeeper controlling access of the pyridine nucleotide cofactors to the active site. A unique feature of the AIF FAD-binding domain is the N-terminal insertion (aa. 190–202; Fig. 3) folded as a β-hairpin (Fig. 4A, dark cyan region). This peptide establishes multiple contacts with the main core via hydrophobic (Leu190, Phe192, Trp195) and polar residues (Gln194, Arg200).
FIG. 4.

Structure of AIF. (A) The x-ray model of refolded oxidized murine AIFΔ1–120 [PDB code 1GV4 (150)]. The FAD-binding, NAD(H)-binding, and C-terminal domains are depicted in beige, cyan, and purple, respectively; FAD is in CPK representation. The unique features of AIF are an extended N-terminus (shown in black), the N-terminal 190–202 insertion folded as a β-hairpin (deep cyan), and the C-terminal 509–559 insertion (red; proline residues are displayed). (B) Superposition of the crystallographic dimer of naturally folded oxidized murine AIFΔ1–77 [shown in gray, PDB code 3GD3 (202)] and biological dimer of the reduced NAD-bound AIFΔ1–101 [brown, PDB code 3GD4 (202)]. FAD is in CPK representation; NAD is in green. (C) A hydrogen-bonding network in the active site of reduced AIF. The nicotinamide group (green) is parallel stacked between Phe309 and the isoalloxazine of FAD (yellow) and establishes H-bonding network with the surrounding residues. One of these, His453, undergoes a large positional shift upon AIF reduction. (D, E) Views at the front side and the top of the reduced AIF dimer, respectively. The redox-sensitive 190–202 β-hairpin and Trp195 (in CPK representation) are shown in blue. NLS1 and NLS2 are in pink and magenta, respectively. NLS2, through which AIF is predominantly transported to the nucleus, comprises the dimer interface and, hence, becomes inaccessible upon AIF reduction. The α-helical portion of the regulatory peptide unwinds, and the 536–543 amino acid stretch transforms into the sixth strand of the C-terminal β-sheet (shown in red). The remaining residues of the regulatory peptide are not seen in the x-ray structure due to disorder (To see this illustration in color the reader is referred to the web version of this article at www.liebertonline.com/ars).
The C-terminal domains are most diverse in the GR-like flavoproteins and have structurally distinct peptides located on the surface. Similar to bacterial oxidoreductases, the C-terminal part of AIF lacks two long α-helices through which the GR family enzymes dimerize and catalyze disulfide reduction. Instead, AIF contains the 509–559 insertion (Figs. 3 and 4A), which makes its structure unique. The 509–559 peptide is organized as two short α-helices (aa. 517–523) and an extended loop (a red-colored region in Fig. 4A), whose open conformation is stabilized via crystal contacts. Together with the β-hairpin, the α-helical part of the C-terminal insertion blocks access to the active site.
The absence of canonical DNA-binding motifs in murine AIF was quite surprising, as the protein was known to directly associate with DNA. An explanation came later, when the 1.8 Å crystal structure of refolded human AIFΔ1–120 was determined (242). The human protein has an identical fold but due to different crystal packing, its 540–559 fragment was disordered. As a result, Arg584 and Lys590 became exposed, giving rise in a positive electrostatic surface potential. Together with Lys446, Arg449, Arg450, Arg451, and Lys593, these residues form a continuous positively charged patch, proposed to guide interactions with DNA (242).
B. X-ray structure of murine AIFΔ1–77
Lacking only the membrane tether, murine AIFΔ1–77 resembles the mature Δ1–53 protein in structure and function (38). In the crystal structure of naturally folded oxidized AIFΔ1–77, there are four molecules in the asymmetric unit, which are organized in two dimers topologically similar to that of refolded AIFΔ1–120 (202). The most notable difference between the two structures is in the FAD position (∼0.6 Å displacement) and conformation of the 438–453 peptide, connecting the active site with the crystallographic dimer interface (Fig. 5A). Due to conformational variations in the Arg448 side chain, the 438–450 fragment naturally folds into a 9-residue β-turn but remains as a loop upon AIF refolding. This alteration could be critical, as it may affect formation and stabilization of dimeric CTC (discussed below). The N-terminal and 538–559 peptides in all four AIFΔ1–77 molecules are structurally disordered. However, two 8- and 9-residue fragments, which could not be identified due to poor resolution and likely representing the missing N-termini, were modeled as antiparallel β-strands complementing the C-terminal β-sheets of two AIF monomers (Fig. 6).
FIG. 5.

Structural alterations in AIF caused by refolding. (A) Conformational differences in FAD and the 438–453 peptide, spanning from the crystallographic dimer interface to the active site. Due to conformational differences in the side chain of Arg448, forming a salt bridge with Asp414 in refolded AIF (gray) and an H-bond with the Val422 carbonyl oxygen in the naturally folded protein (black), the 438–540 fragment folds as a loop or β-turn, respectively. (B) By establishing an intersubunit salt bridge with Glu412, Arg448 may assist dimerization and stabilization of NADH-reduced AIF. Conformational alterations in the 438–453 peptide detected in refolded AIF (displayed in gray) could interfere with the dimerization process and be responsible for the perturbed redox properties of the protein.
FIG. 6.

Extension of the C-terminal antiparallel β-sheets in crystalline AIFΔ1–77. The N-termini of the neighboring molecules forming β-strands are shown in black and indicated by arrows.
C. X-ray structure of reduced NAD-bound murine AIFΔ1–101
The x-ray structure of reduced NAD-bound form of murine AIFΔ1–101 consists of one biological CTC dimer that resembles the crystallographic dimers of oxidized AIFΔ1–77 (Fig. 4B) (202). The CTC dimer interface has an intricate H-bonding network with two central Arg448-Glu412 intersubunit salt bridges (Fig. 5B). Since in refolded AIF Arg448 is locked in a conformation partially precluding charge–charge interactions with Glu442 and the CTC formation is perturbed (150, 155), the Arg448-Glu412 salt links are thought to promote AIF dimerization and stabilize CTC (202). Most importantly, Arg448 is part of NLS2 (445KLGRRRV451). This means that the NLS2 accessibility depends on the AIF redox/oligomeric state.
1. Redox-linked changes in the active site
Upon reduction, the flavin nucleotide portion of FAD shifts by 1.2 Å to optimize π-π charge–transfer interactions with the nicotinamide of NAD, parallel stacked between the isoalloxazine and Phe309 aromatic rings (Fig. 4C). The necessity of positional adjustments in FAD for optimization of the intercofactor charge transfer suggests that perturbations in the redox properties of refolded AIF may in part be due to an altered FAD conformation.
While the general mode of NAD(P) binding is conserved in the GR-like proteins, AIF has one distinctive feature in the active site: His453. Substituted by Leu or Ile in other flavoenzymes, His453 undergoes a large positional shift to establish an H-bond with the nicotinamide. This conformational switch is critical because it helps to orient the redox groups optimally for charge transfer and, most importantly, enables to transmit the redox signal from the active site to the surface via an adjacent 438–452 peptide (Fig. 5A). Thus, the redox-linked His453 movement could assist AIF dimerization and CTC stabilization. Drastic changes in the AIF redox activity caused by the H453L mutation (38) suggest that this residue also modulates the flavin redox potential and electron transfer to the acceptor molecules.
2. Reorganization in the C-terminal domain
The C-terminal domain undergoes most notable changes upon AIF reduction (202). Binding of NADH triggers rearrangement of a number of aromatic residues, whose side chains align in a tunnel spanning from the active site to the surface (Fig. 7). Tyr559 undergoes the largest movement (>9.0 Å) and shields the tunnel from the solvent. With the ring-to-ring distance of <4.0 Å, the aromatic cluster is perfectly suited for transfer/delocalization of electrons, which could extend the lifetime of the reduced species. Another important consequence of the redox-linked restructuring is dislocation of the entire 509–558 peptide, the α-helical portion of which unwinds and the 536–543 stretch becomes the sixth strand of the C-terminal β-sheet. In accord with the oxidized AIFΔ1–77 structure (Fig. 6) (202), the latter observation suggests that the surface-exposed 501–508 β-strand in AIF has a high affinity for the complementary peptides and may be predisposed for the β-sheet extension.
FIG. 7.

Redox-induced reorganization of aromatic residues in the C-terminal domain of murine AIFΔ1–101. Displacement of Phe481 and Phe309 by the nicotinamide group initiates the rearrangement and leads to formation of an aromatic tunnel that may serve as an electron delocalization site and prolong the lifetime of the reduced species.
3. Conformational changes in the 509–559 peptide
Conformational reorganization in the 509–559 peptide has far-reaching implications. One of the immediate effects is release of the 190–202 β-hairpin. Although the precise function(s) of this unique element are currently unknown, it has the potential to modulate redox properties and interaction of AIF with partnering proteins. Trp195, for instance, assists folding of the 509–538 peptide in oxidized AIF, modulates the FAD redox potential, and controls reactivity toward NADH and electron acceptors (202). A significantly prolonged lifetime of reduced AIF W195A suggests that Trp195 might be the site for electron exchange with oxygen and other acceptors. These features and an outward orientation make the hairpin suitable for binding to the membrane-associated or soluble partners. One protein predicted to interact with AIF through the 190–202-containing fragment is 70 kDa heat shock protein (Hsp70) (84, 196) (section VII.C.1.a). Severe and early onset mitochondrial encephalomyopathy linked to the AIF ΔR201 mutation in humans (81) (section VIII.C) also strongly argues for the functional importance of the 190–202 peptide.
Another redox-sensitive element that can modulate functioning of AIF is the 509–559 insertion. This fragment contains several regulatory elements (Fig. 8). Lysines 509 and 517 (510 and 518 in human AIF) are critical for the DNA binding and induction of apoptosis (242). It is possible that one or both of these surface lysines could undergo ubiquitination (42, 238). The proline/glutamic acid/serine/threonine-rich (PEST) sequence (aa. 528–559) is a proteolytic signal (148, 185) that, when flanked by arginine or lysine (Arg529 in human AIF; Fig. 8), correlates with the protein's short lifetime. Unstructured and easily accessible PEST-containing peptides can be cleaved by serine proteases and caspases, with the newly formed N- and C-termini acting as initiation sites promoting proteosomal degradation (19). The PEST sequence can also modulate the proteolytic action of μ-calpain (204), a protease involved in liberation of mitochondrial AIF (section VII.B.1). Further, biological activities of the PEST-containing proteins can be regulated through reversible phosphorylation, as the consensus sites of many kinases map to the PEST regions. The putative phosphorylation sites in human AIF include serines 518, 529, 531, and 538, and threonines 533 and 547 (Fig. 8). Two residues, Ser529 and Ser531, are predicted to be phosphorylated by casein kinase 2, implicated in a variety of cellular processes, including apoptosis, DNA repair, transcription, and cell cycle control (208).
FIG. 8.

Amino acid composition of the regulatory peptide in human and murine AIF (upper and lower sequences, respectively). The proline-rich motif within the PEST sequence is boxed; two lysines critical for DNA-binding are shown in bold italic. The putative phosphorylation sites were identified using the online NetPhos server (www.cbs.dtu.dk/services/NetPhos/) and are marked by asterisks. Human AIF has one additional phosphorylation site at Thr547, which is substituted by Ala in the murine protein. An arrow indicates the peptide transforming into the sixth strand of the C-terminal β-sheet upon AIF reduction.
The third functionally important element is a proline-rich sequence (aa. 543–554), a potential recognition and interaction site for the Src homology 3 (SH3) domain containing proteins [reviewed in (106)]. The human genome encodes ∼300 SH3 domains, some of which have been implicated in intracellular signaling, cytoskeletal rearrangements, cell growth and differentiation, protein and vesicle trafficking, and immune response (82, 112). T-cell ubiquitin ligand (TULA) is one SH3-domain containing protein identified as a cytoplasmic partner of apoptogenic AIF, although the TULA-AIF association may not require the SH3 module (42) (section VII.C.2.b). Histone H2AX, on the other hand, is a nuclear partner that lacks the SH3 domain but binds to the proline-rich region of AIF (13) (section VII.D.3.d). Thus, the regulatory peptide could mediate protein–protein interactions with or without involvement of the SH3 modules. Summarizing, it can be concluded that AIF has acquired several unique structural elements that help stabilize the NADH-reduced dimer and allow redox-controlled regulation of the AIF lifetime, posttranslational modification, and interaction with partnering proteins.
VII. Role of AIF in PCD
AIF is widely accepted as a caspase-independent PCD inducer although its contribution to apoptosis may depend on species, cell type, and death stimuli [reviewed in (104, 157)]. Since the manner of folding defines structural and functional integrity of AIF and the covalently attached affinity/epitope tags not only interfere with the protein folding but can also introduce artifactual effects (63, 90, 117), the AIF constructs used in some of the cited studies will be specified.
A. Apoptogenic effects of AIF in cell free systems and live cells
AIF was originally discovered as an IMS component capable of inducing chromatin condensation and DNA loss in the nuclei isolated from healthy cells (214, 215, 248). The protein was first proposed to act as a protease or protease activator that can be inhibited by a pan-caspase inhibitor z-vad.fmk (213). Later the same group demonstrated, however, that mitochondrial AIF is a flavoprotein whose apoptogenic activity is not affected by z-vad.fmk (211).
Like native AIF, the refolded precursor and AIFΔ1–120 trigger loss of DNA and peripheral chromatin condensation in purified nuclei (155, 210, 212). This process is rapid and accompanied by large-scale (∼50 Kb) DNA fragmentation which can be prevented by the divalent cation chelator EDTA. Induction of nuclear apoptosis does not require additional cytoplasmic factors but may involve nuclear protein(s), as AIF itself does not possess nuclease activity and has no effect on preheated nuclei. AIFsh induces similar effects in isolated nuclei, wherein the Δ353–613 mutant and AIFsh2 do not (53, 54). In cooperation with a heat-labile cytoplasmic factor, the precursor and AIFΔ1–120, but not the Δ1–351, Δ155–612 or Δ538–612 fragments, cause swelling of isolated mitochondria by increasing permeability of the OMM (212). Since AIF precursor becomes apoptogenic after refolding even in the absence of FAD (212) and the apoptogenic ability of poly-histidine (His)-tagged AIFΔ1–120 and Δ1–100 in cell-free systems is not affected by NAD(P)H or FAD modification (136, 155), the apoptogenic function of AIF was suggested to be defined by the protein conformation and require the C-terminal part but not the oxidoreductase activity.
The cell-death-inducing potency of recombinant AIF is abolished upon treatment with a thiol-reactive agent p-chloromercuriphenylsulfonate (pCMPS) (155). Among three cysteine residues present in AIF, only Cys441 (Cys440 in mice) is positioned close to the surface and is next to NLS2. Modification of Cys441 with pCMPS would introduce a negative charge near basic NLS2 and, hence, could interfere with the nuclear transport of AIF.
In live cells, the apoptogenic potency of AIF has been investigated by microinjection or transfection-enforced overexpression of the His- or green fluorescent protein-tagged precursor, Δ1–120, Δ1–100, and AIFsh fragments (53, 67, 136, 210, 212). The tagged Δ1–120 and Δ1–100 proteins diffusely distribute in the cytoplasm and nucleus, rapidly inducing chromatin condensation, loss of DNA, exposure of phosphatidylserine (PS) on the outer leaflet of the plasma membrane, and dissipation of the mitochondrial trans-membrane potential (ΔΨm). After liberation from the IMS, endogenous AIF increases mitochondrial permeability in a feedback loop (136, 212). The precursor is mainly targeted to mitochondria, but after prolonged cultivation, the cytoplasmic and nuclear distribution of AIF becomes predominant and nuclear apoptosis progresses (136). Lacking MLS, AIFsh diffusely distributes in the cytoplasm and then rapidly translocates to the nucleus, triggering large-scale DNA fragmentation in a caspase-independent manner (53).
When injected into the cytoplasm, FAD-free AIFΔ1–120 induces chromatin condensation and ΔΨm dissipation to the same degree as the FAD-bound protein, whereas the Δ1–351, Δ155–612, and Δ538–612 fragments fail to cause cell death (155, 212). To further test the role of the redox center, Loeffler et al. genetically modified 255CLIATG260 and 303TVIGGG308 motifs in the murine AIF precursor in attempt to preclude FAD and NADH binding (136). The apoptogenic potential of the 255CLIAAA260 and 303TAAAGG308 variants in live cells was similar to that of wild-type AIF (i.e., initial mitochondrial localization followed by spontaneous translocation and apoptosis). This was another argument to support the concept that the apoptogenic effect of AIF does not rely on the oxidoreductase activity, although no proof that the mutated proteins were incapable of binding the cofactors had been presented.
In live cells, the MLS-free Δ1–120, Δ1–100, and AIFsh proteins cause cell death more rapidly than the mitochondrion-targeted precursor, with the time difference varying from several hours to several days (53, 136). The same effects are observed in the presence of z-vad.fmk, indicating that when in the cytoplasm, AIF becomes a lethal factor leading to cell demise irrespective of whether caspases are activated or not. Contrarily, the proapoptotic action of Flag-tagged human AIFΔ1–480 was found to be mediated through cytochrome c release in a caspase 9-dependent manner (252). Most strikingly, enforced expression of tag-free human AIF1–613, Δ1–54, or Δ1–102 is nontoxic and even cytoprotective, as all three proteins reduce the basal and stimulated ROS levels (238). Thus, the cell death-triggering ability of AIF can be modulated by the attached tags and in some cases requires caspase activation.
Whether or not AIF is essential for cell death depends on the cell type and apoptotic stimuli. AIF−/y embryonic stem (ES) cells, for instance, display normal susceptibility to death in response to a variety of apoptosis-inducing agents, including staurosporine, etoposide, azide, tert-butylhydroperoxide, anisomycin, UV-radiation, and growth factor withdrawal, but fail to die in response to pro-apoptotic vitamin K3 in the presence of z-vad.fmk (105). Cells from AIF-deficient Harlequin (Hq) mice are more sensitive to induction of apoptosis (120), but neurons from Hq/Apaf−/− mice are impaired in both caspase-dependent and AIF-mediated cell death pathways (36). AIF gene silencing protects renal tubular epithelial cells against cisplatin-induced cell death (201), whereas AIF−/y human colon carcinoma cells are more sensitive to peroxide- and drug-induced apoptosis than the wild type (221).
Finally, the apoptotic machinery shares common pathways with autophagy, a self-destructive process involving lysosomes that could either link or polarize cellular responses [reviewed in (142)]. There are few studies where the functional relationship between caspase-independent cell death and autophagy has been explored. A regulatory crosstalk between AIF-mediated apoptosis and autophagy was observed in HCT116 cells treated with Lapatinib, an inhibitor of receptor tyrosine kinases (146), in malignant rhabdoid tumor cell line treated with the histone deacetylase inhibitor FK228 (235), and MCF-7 cells treated with crotoxin (241). In the presence of different inducers, cells undergo PCD that involves autophagy but not release of mitochondrial AIF (21, 80, 167). Thus, whether apoptosis and autophagy intersect depends on physiological settings.
In summary, AIF becomes a powerful lethal factor when released into the cytoplasm. Upon translocation to the nucleus, AIF triggers chromatin condensation and loss of DNA in a caspase-independent manner. The C-terminal portion of the protein but not its redox activity is responsible for the apoptogenic action. Contribution of AIF to PCD depends on the cell type and death triggers and, in some cases, may be amplified by caspases and autophagic factors, and modulated by the covalently attached tags.
B. Release of mitochondrial AIF
The mechanism regulating apoptogenic processing and translocation of mitochondrial AIF into the cytoplasm is complex and not fully understood. Here is a brief overview of the currently available data, schematically presented in Figure 9.
FIG. 9.

Factors modulating the cleavage and release of apoptogenic AIF from mitochondria. Proteolysis of the membrane tether in mature AIF can be mediated by local or cytoplasmic proteases entering the intermembrane space upon permeabilization of the outer membrane. Detached AIF can translocate to the cytoplasm with the involvement of the PTP complex or through pores formed by proapoptotic Bcl-2 family members Bax, Bak, and Bid. AIF associated with the outer leaflet of the OMM, likely a full-length precursor, can be released and transported to the nucleus in a PAR-dependent manner. ANT, adenine nucleotide translocase; PAR, poly(ADP-ribose); PTP, permeability transition pore; VDAC, voltage-dependent anion channel.
1. Proteolysis of mature AIF
Being tethered to the inner mitochondrial membrane, mature AIF must undergo N-terminal proteolysis to form an apoptogenic fragment that can be released into the cytoplasm (168). Studies conducted in cell-free systems demonstrated that mitochondrial μ-calpain, a Ca2+-dependent neutral cysteine protease, colocalizes with AIF in the IMS and can process it locally upon activation (77, 135, 156, 169, 170). An increase in cellular Ca2+ (162) and association with a protein–disulfide isomerase ERp57 (170) lead to activation of mitochondrial μ-calpain and AIF proteolysis. Conversely, an endogenous calpain inhibitor, calpastatin, can prevent AIF liberation (31, 227, 233). There is controversy, however, over whether calpastatin is present in mitochondria or not (108, 109, 169). Involvement of mitochondrial μ-calpain in AIF processing has also been questioned (101).
Studies on isolated mitochondria suggest that cytoplasmic μ-calpain and lysosomal Ca2+-independent cathepsins B, L, and S can gain access and process AIF after opening of the permeability transition pore (PTP) (31, 178, 246). Several lines of evidence disfavor this suggestion. First, there is no in cellulo experimental evidence demonstrating that the proteases can enter mitochondria during apoptosis. Second, lysosomal permeabilization would be required for the cathepsin-dependent AIF release, which is not always observed during apoptosis [reviewed in (164)]. Finally, in vitro cathepsins B and L cleave recombinant AIF nonspecifically (31), and cathepsin B knockout has no effect on AIF processing (159). Thus, it is still uncertain whether external enzymes are involved in proteolysis of mitochondrial AIF.
A single Leu103/Ser104 cleavage site for μ-calpain was identified in recombinant human AIFΔ1–54 (31). Mutagenesis screening revealed that only the L101G/L103G AIF variant is completely resistant to μ-calpain activity (31). On the basis of the proteolytic pattern of endogenous murine AIF, the existence of two μ-calpain-sensitive sites within the 96–120 fragment was predicted (178). This was later confirmed in vitro by our group (38). We found also that one site, Glu118-Gly119, is protected in reduced AIF (38). Further, NAD(P)(H) affected the μ-calpain-mediated cleavage of endogenous AIF in mitochondria treated with atractyloside or cBid (calpain activated Bid, the proapoptotic Bcl-2 family member) (38). The protective action of the reduced cofactors was more pronounced in mitochondria respiring on malate/glutamate than in succinate/rotenone media, and the NADP/NADPH pair had a stronger effect than NAD/NADH. This led us to suggest that the externally added pyridine nucleotides exert their action indirectly via modulation of mitochondrial permeability, morphology, and/or protein–protein interactions.
Contrary to our study (38), Norberg et al. reported that NAD(P)(H) has no effect on the Ca2+-stimulated AIF processing in isolated mitochondria in the presence of cyclosporin A (CsA) (163). Instead, Ca2+-induced production of ROS led to AIF carbonylation and accelerated cleavage by μ-calpain. The protective effect of NAD(P)(H), therefore, is not a general phenomenon and may depend on the apoptotic insult. tBid and atractyloside, for instance, cause drastic changes in the inner mitochondrial membrane topology (144) that can be inhibited by CsA (198).
That physical interaction and subsequent degradation of AIF by μ-calpains may be redox dependent follows also from the fact that recognition and proteolysis of protein targets by μ-calpain are expedited by PEST motifs (204, 229). If AIF's PEST sequence is indeed a calpain-targeting element, then AIF processing may be regulated via redox-linked reorganization in the regulatory peptide (section IV.C.3).
In summary, the membrane linker in AIF has at least one proteolytic site. Mitochondrial and cytoplasmic μ-calpains, as well as lysosomal cathepsins B, L, and S, can process AIF in cell-free systems, but whether these proteases participate in vivo is still a matter of debate. If the involvement of μ-calpain is confirmed, processing of AIF in the IMS may be redox controlled.
2. Release of truncated AIF into the cytoplasm
Permeabilization of mitochondria, irrespective of whether it occurs before or after AIF cleavage, is an obligatory event in the AIF-mediated apoptotic signaling. Currently, two different models have been proposed to explain how the apoptogenic IMS components are released from mitochondria. According to the first model, mitochondrial PTP, a conductance channel formed at the contact sites between the inner and outer membranes and consisting of the outer membrane voltage-dependent anion channel (VDAC), the inner membrane adenine nucleotide translocase (ANT), and associated with the matrix side of ANT cyclophilin D, is actively opened. This allows small molecules (<1.5 kDa) to enter the mitochondrial matrix, leading to swelling and rapture of the OMM. PTP opening is regulated by the mitochondrial membrane potential, ΔΨm, dissipation of which correlates with the cytoplasmic translocation of AIF (49, 129, 171). The ΔΨm/PTP-related liberation of AIF can be induced by Bid (127, 178), atractyloside (a competitive inhibitor of ANT), fatty and bile acids (54, 141, 159, 161, 207), α-eleostearic acid (123), and may require VDAC oligomerization (203). Conversely, bongkrekic acid, ROS scavengers, Bcl-2, and CsA inhibit translocation of mitochondrial AIF (129, 132, 163, 168, 171, 178, 212, 250). Pro-survival Bcl-2 prevents AIF liberation by sequestering proapoptotic Bax and Bak (discussed below) or via extramitochondrial mechanisms (20). The antibiotic bongkrekic acid acts as an ANT inhibitor, wherein CsA precludes PTP opening by forming complex with cyclophilin D on the matrix side of ANT (85). Intracellular Ca2+ dysregulation and mitochondrial Ca2+ overload facilitate AIF relocation by activating calpain, dissipating ΔΨm and inducing the OMM rupture (126, 131, 175), which could proceed in a caspase-independent manner (24, 227). In some cases, however, activation of caspases is required for the AIF release (9, 17, 201, 205).
Poly(ADP-ribose) (PAR) polymer, a byproduct of the reaction catalyzed by the activated nuclear DNA repair enzyme PAR polymerase-1 (PARP-1), triggers AIF translocation by dissipating ΔΨm, altering mitochondrial Ca2+ homeostasis, and activating calpain (39, 159, 227, 244). Additionally, PARP-1 activation leads to excessive consumption of cytoplasmic NAD and generation of AMP, which facilitates mitochondrial depolarization and AIF release (3, 69). PARP-1-dependent AIF efflux may also involve Bax (159). The nuclear PARP-2 isoform was recently demonstrated to substantially contribute to nuclear translocation of AIF, in part via PAR accumulation (133). Finally, intramitochondrial PARP-1 can directly provoke AIF liberation via interactions with the mitochondrial PTP (61, 186).
The second model suggests that mitochondrial apoptogenic factors can be released through the pores in the outer membrane formed by the pro-apoptotic Bcl-2 family members such as Bax, Bak, and Bid that do not affect ΔΨm. Bax and Bak are essential regulators of a diverse mitochondrial cell death pathways. Being in a latent state in healthy cells, they undergo conformational changes and homo- and hetero-oligomerization, forming channels in the OMM (6). While elimination of the PTP components has virtually no effect on the ability of cells to respond to apoptotic stimuli (15, 122, 194), Bax−/−/Bak−/− double-knockout abrogates apoptosis (134, 236). Release of AIF is also blocked in Bax−/− human colon cancer cells (182). Bax, Bak, and full-length and proteolytically activated Bid (cBid or tBid) can initiate mitochondrial demise and facilitate AIF translocation not only through the OMM pore formation (118, 121, 125, 188), but also by increasing mitochondrial fission and fragmentation (23, 110, 127) and altering cristae formation (73, 198). In addition, a pore that permits AIF efflux can be generated by Bax, VDAC, and ceramide (191). Certain types of Bax-related mitochondrial membrane injuries and AIF release are inhibited by Hsp70 (187, 189). Thus, liberation of AIF from the IMS can be regulated by a variety of factors that may or may not involve mitochondrial PTP and ΔΨm dissipation.
3. Release of AIF associated with the outer mitochondrial membrane
The AIF pool loosely associated with the cytoplasmic side of the OMM can be rapidly released during parthanatos (243, 245), a unique form of PARP-1-initiated cell death triggered by DNA damage. Parthanatos is characterized by accumulation of PAR, mitochondrial depolarization, early nuclear translocation of AIF, loss of cellular NAD and ATP, and late caspase activation [reviewed in (5, 50)]. Release of the OMM-associated AIF does not involve proteases and is mediated by pathogenic PAR polymers through an unknown mechanism (234).
PAR polymers are especially toxic for neurons (5) and their cytotoxicity increases with the dose, length, and complexity of the molecule but does not depend on the negative charge (5, 243). PAR binds covalently and noncovalently to a variety of proteins, including AIF, altering their function and conformation (74, 206). PAR binding/modification can be mediated by three protein motifs: (a) an amino acid sequence rich in positively charged residues with a consensus pattern -hxbxhhbbhhb-, where h, b, and x correspond to hydrophobic, basic, and any amino acids, respectively (177); (b) a macro domain, capable of recognizing monomeric and polymeric ADP-ribose (113); and (c) a PAR binding zinc finger motif (PBZ) (1). AIF does not have the consensus sequence or PBZ but contains two pyrophosphate/adenine binding pockets as part of the FAD- and NAD(H)-binding sites. It is possible, hence, that folded and FAD-bound oxidized AIF could associate with the ADP-ribose monomer through an unoccupied NAD(H)-binding pocket, wherein an unfolded FAD-free precursor may have affinity for both monomeric and branched ADP-ribose. Assuming that the OMM-associated fraction represents an unfolded and nonapoptogenic AIF precursor, one can speculate that PAR directly binds to the NAD- and FAD-binding sites and refolds the protein. This, in turn, could trigger detachment and translocation of the apoptogenic PAR-AIF complex to the nucleus.
C. Cytoplasmic interactions of apoptogenic AIF
Before relocating to the nucleus, apoptogenic AIF has a capacity to exert multiple and diverse effects through interactions with the cytoplasmic proteins acting as pro-survival or pro-death effectors (summarized in Fig. 10).
FIG. 10.

Cytoplasmic partners of apoptogenic AIF. After release into the cytoplasm, AIF can promote apoptosis by interacting with TULA, eIF3g, and phospholipid scramblase, the pro-death partners. Scythe facilitates cell death by regulating stability and lifetime of the cytoplasmic AIF precursor, wherein CypA assists cytonuclear translocation of apoptogenic AIF. Contrarily, Hsp70 retains AIF in the cytoplasm and, hence, can postpone or prevent initiation of the nuclear apoptosis. XIAP is another pro-life partner, which in co-operation with AIF reduces reactive oxygen species levels and promotes cell survival. CypA, cyclophilin A; eIF3g, eukaryotic translation initiation factor 3 subunit p44; Hsp70, 70 kDa heat shock protein; TULA, T-cell ubiquitin ligand; XIAP, X-linked inhibitor of apoptosis protein.
1. Pro-survival partners of AIF
a. Heat shock protein Hsp70
Inducible Hsp70 (70 kDa) was the first cytoplasmic partner of apoptogenic AIF to be identified (183). Hsp's are highly conserved molecular chaperones critical for cell viability, as they assist folding, thermal tolerance, and translocation of proteins across cellular membranes, reorganization of macromolecular complexes, degradation of misfolded proteins, and regulation of signaling pathways (65, 79, 88, 93, 115). Acting as monomers, Hsp70s contain the N-terminal ATPase domain and the C-terminal module comprised of the peptide-binding domain (PBD) and an EEVD motif required for the interaction with other chaperones and regulation of protein refolding and repair (72, 153). Hydrolysis of ATP allosterically modulates affinity and kinetics of substrate binding to PBD, which recognizes hydrophobic residues or unstructured backbone regions.
Hsp70-AIF interaction: In a cell-free system, Hsp70 antagonizes the apoptogenic effect of native and recombinant AIFΔ1–100 in an energy- and caspase-independent manner (183). Likewise, downregulation of the chaperone increases the cytotoxic potency of AIF (183). Direct Hsp70-AIF association was confirmed in vivo by several studies showing that cytoplasmic Hsp70 specifically interacts with exogenous and endogenous AIF and inhibits its relocation to the nuclear compartment (84, 139, 151, 189). Hsp70 also binds AIF precursor (189) but whether this interaction is related to the chaperone function of Hsp70 has not been elucidated yet.
Examination of physical and functional interactions between the tagged AIF and Hsp70 under conditions of normal cell growth revealed that the chaperone's PBD but not the ATPase activity is required for association and retention of AIF in the cytoplasm (183). In contrast, investigation of dynamic protein–protein interactions under conditions of physiological stress indicated that the Hsp70 ATPase domain is indispensable for interaction with native AIF (188). Again, whether these discrepancies are due to the tag attachment or/and differences in the cellular environment needs to be clarified.
Systematic deletion analysis identified the 150–228 peptide and, in particular, Arg192 and Lys194 in human AIF as critical for Hsp70 binding (Fig. 11) (84). In silico modeling, in turn, predicted that the protein–protein interface in the AIF-Hsp70 complex is comprised of the AIF 184–221 peptide and the 433–436, 469–473, and 534–541 fragments of the chaperone (196). The inability of Hsp70 to prevent nuclear apoptosis induced by AIFsh (53) and accelerated nuclear translocation of the R192A/K194A mutant of human AIF (242) support the notion that the N-terminal fragment containing the 190–202 β-hairpin mediates binding of the chaperone.
FIG. 11.

An Hsp70 binding site identified by systematic deletion analysis and in silico modeling (84, 196). The 150–228 and, more precisely, 184–221 fragment (shown in dark gray and cartoon representation) and Arg192 and Lys194 in human AIF were identified as important for Hsp70 binding. Since the predicted site includes a redox-sensitive 191–203 β-hairpin, interaction between AIF and Hsp70 may be redox controlled.
In general, the Hcs70/Hsp70/DnaK (bacterial Hsp70 homolog) recognition motifs represent a stretch of seven residues with two or more bulky hydrophobic/aromatic groups in alternating positions, flanked by basic amino acids and mapped to the secondary structure elements such as packed together β-strands or partially exposed α-helices (12, 70, 140). The primary sequence and the β-hairpin structure of the AIF 190–202 peptide satisfy these requirements (Figs. 3 and 11). If this element indeed serves as the Hsp70 binding site, the AIF-Hsp70 interaction could be redox controlled, as the accessibility of the hairpin changes upon AIF oxidoreduction (section VI.C.3). Another level of regulation of the AIF-Hsp70 interaction could be via redox-dependent Hsp70 expression (71) and S-glutathionylation of a key redox-sensitive cysteine residue in the chaperone (94).
In accord with the structural prediction, our in vitro experiments showed that Hsp70 does not perturb the DNA-binding ability of oxidized AIF but attenuates the inhibiting effect of NADH on the AIF-DNA interaction (38). This could result from the preferential chaperone binding to the reduced AIF dimer. Selective AIF-Hsp70 association may be physiologically beneficial because when the NADH levels are sufficiently high, cytoplasmic retention of reduced AIF could slow down the apoptotic cascade and give the damaged cell a chance to recover. Depletion of the NAD(P)(H) pools and oxidation/monomerization of AIF, in turn, would indicate drastic metabolic changes, and sequestering of apoptogenic AIF would be of little help.
As a powerful cytoprotective agent whose dysregulation either facilitates apoptosis and kills cells or contributes to carcinogenic transformation, Hsp70 represents an attractive therapeutic target. Since cytoplasmic retention of apoptogenic AIF is pro-carcinogenic, designing of noncytotoxic Hsp70-binding peptides based on the AIF structure can be one of the anticancer strategies (196). One engineered peptide, ADD70 (AIF-derived decoy for Hsp70), consists of residues 101–366, 467–566, and 610–613 of human AIF (196). ADD70 sensitizes cells to apoptosis triggered by a number of PCD inducers (DNA damaging agents, serum depletion, staurosporine, etc.) through binding and neutralization of Hsp70. An analogous fragment with the Hsp70 binding site omitted (Δ150–228) has no such effect (196). The chemosensitizing potential of ADD70 is observed in multiple cancer lines but not in cells lacking Hsp70, which confirms that the engineered fragment acts through Hsp70 neutralization. In syngeneic animals, expression of ADD70 delays growth and reduces metastatic potential of tumor cells and sensitizes them to the anticancer drug cisplatin (195). Another approach for anticancer therapy that is currently being developed includes a combination of Hsp70 silencing and delivery of AIFΔ1–120, which enhances chemotherapy and drug-induced cell death in tumors (254).
b. X-linked inhibitor of apoptosis protein
X-linked inhibitor of apoptosis protein (XIAP) belongs to a family of the IAPs that selectively bind and inhibit caspases 3, 7, and 9 [reviewed in (62, 192)]. Mammalian XIAP is the most potent and broad suppressor of apoptosis known to date, as it directly and with high affinity inhibits enzymatic activities of both initiator and executioner caspases (63). Human XIAP is a multifunctional cytoplasmic protein consisting of three N-terminal baculoviral IAP repeat (BIR) domains and the C-terminal zinc-binding domain, also known as a RING finger. The BIR2 and BIR3 domains inhibit caspases 3/7 and 9, respectively (59), whereas the RING finger possesses E3 ubiquitin ligase activity and enables XIAP to remove caspases and other proteins from the cell via ubiquitination and proteasome degradation (95). In addition to the apoptotic signaling, XIAP is involved in the NF-κB, TGF-β, and c-Jun-N-terminal kinase pathways and copper homeostasis (92, 160, 190). Downregulation of XIAP can lead to cancer, neurodegenerative disorders, and autoimmunity, which makes the protein a potential drug target (192, 193).
XIAP-AIF interaction: Using a tandem affinity purification technique and a D148A/W310A variant of XIAP as a bait, Wilkinson et al. identified AIF as one of the XIAP-associated proteins in HEK 293 cell lysates (238). The AIF binding site was mapped to the XIAP BIR2 module. Further, it was demonstrated that AIF could be ubiquitinated by XIAP or alternative E3 ligases. This modification plays a regulatory role, as transient XIAP-AIF complexes formed in the cytoplasm decay quickly upon AIF ubiquitination but become more long-lived if the E3 ubiquitine ligase activity of XIAP is eliminated (238). Thus, although ubiquitination does not target AIF to the proteosome, it may modulate the apoptogenic potency of the flavoprotein.
Two lines of evidence suggest that the AIF-XIAP association plays a pro-survival role. First, AIF inhibits interaction of XIAP with Smac/DIABLO, another death-signaling protein released from mitochondria that prevents XIAP from binding to caspases (238). Second, coexpression of XIAP with either full-length, Δ1–54, or Δ1–102 AIF progressively decreases ROS levels in control and dying cells. Although the underlying mechanism is currently unknown, cooperation between AIF and XIAP is thought to affect caspase-independent functions of XIAP and promote cell survival by reducing cellular oxidative stress (238).
Using the docking program GRAMM (220), which performs a computational search of all possible configurations to find protein complexes with the highest surface complementarity, we compared preferable binding sites for the human XIAP BIR2 domain in the oxidized monomer and reduced dimer of murine AIF. In both types of computer-generated complexes (Fig. 12), the top ranking solutions for the BIR2 domain are clustered at two different sites that have similar binding energy (i.e., surface complementarity). This suggests that XIAP may bind the two redox forms of AIF with equal affinity. Independence of the XIAP-AIF association on the AIF redox state would be physiologically beneficial because it could increase the probability of the interprotein complex formation and promote cell survival.
FIG. 12.

Complexes between the BIR2 domain of human XIAP (PDB code 1I30) and the oxidized monomer and reduced dimer of AIF (A and B, respectively) generated with the program GRAMM (220). AIF molecules are in light gray and cartoon representation; BIR2 is in black and ribbon representation. In both types of complexes, the top ranking solutions for BIR2 are clustered at two docking sites that have similar binding energy. This suggests that association of AIF with XIAP may be redox independent.
2. Pro-death partners of AIF
a. Eukaryotic translation initiation factor 3 subunit p44
Eukaryotic translation initiation factor 3 (eIF3) is a 10–13 subunit complex that promotes binding of the 40S ribosomal subunit to mRNA (33). Although evolutionary conserved, the eIF3 subunit p44 (eIF3g) is not essential for the formation of an active mammalian eIF3 (149). eIF3g was reported to associate with the eIF3a subunit, stabilize the eIF3i subunit, and bind to rRNA through the C-terminal RNA recognition domain (22, 149).
eIF3g-AIF interaction: The AIF-binding ability of eIF3g was first detected by the yeast two-hybrid screening method where the N-terminally LexA-fused human AIFΔ1–101 was used as a bait (116). Direct interaction between eIF3g and AIF was confirmed both in vitro and in vivo and required the N-terminal part of eIF3g and the C-terminal portion of AIF (116). By specifically interacting with eIF3g, AIFΔ1–101 and Δ1–439 inhibit de novo protein synthesis in live cells and cell-free extracts, whereas the full-length precursor and 102–439 fragments have no such effect. Thus, the inhibitory properties of AIF could in part be defined by its folding. In MCF-7 cells undergoing cisplatin-induced apoptosis, AIFΔ1–101 not only inhibits the eIF3 machinery and protein synthesis but also amplifies apoptosis via caspase 7 activation and proteolytic cleavage of eIF3g, and colocalizes with eIF3g in apoptotic nuclei (116). This suggests that eIF3g and AIF may act synergistically in promoting cell death.
b. T-cell ubiquitin ligand
TULA is a cytoplasmic protein primarily expressed in T and B lymphocytes and other lymphoid cells where it acts as a negative regulator of c-Cbl-mediated inhibition of protein tyrosine kinases and epidermal growth factor receptor, induces c-Cbl degradation, and promotes cellular transformation (68). The N-terminal part of TULA represents a ubiquitin-associated domain (UBA), through which binding of ubiquitin and ubiquitinated proteins takes place. In addition, TULA has the central SH3 domain, the c-Cbl association site, and a region homologous to phosphoglyceromutases with unknown function (68).
TULA-AIF interaction: TULA was identified as a cytoplasmic partner of AIF by Collingwood et al., who showed that in HEK293T cell lysates AIF coimmunoprecipitates with the full-length and UBA/SH3-domain-containing 1–299 fragment of TULA (42). The UBA/SH3 module interacts with endogenous AIF stronger than the intact TULA, wherein the C-terminal portion (residues 300–623) is incapable of binding AIF. Despite a strict requirement of the N-terminal part of TULA for association with AIF, deletion of the SH3 or UBA domains or introduction of the W279L point mutation to inactivate the SH3 module have no effect on the protein–protein interaction. At the same time, both UBA and SH3 domains are indispensable for the apoptogenic action of TULA on T-cells (42). Further, in Jurkat T cells TULA plays a critical role in apoptosis induced by growth factor withdrawal that does not involve caspases or T-cell receptor/CD3 ligation but requires physical association with AIF liberated from mitochondria. On the basis of these findings, the TULA-AIF pair was proposed to amplify the cytoplasmic apoptotic cascade through cooperation with other factors. Whether this regulatory mechanism is specific for the malignant Jurkat cells (T-cell leukemia) or is more widespread remains to be established.
c. Cyclophilin A
Cyclophilins possess a peptidyl prolyl isomerase (PPI) activity and catalyze cis-trans isomerization of peptidylprolyl bonds. Cyclophilins have high affinity for CsA, an immunosuppressive drug and inhibitor of the PPI activity, and are implicated in protein folding, assembly and trafficking, immune response, and cell signaling [reviewed in (14, 154, 230)]. The proteins share a common fold but differ in surrounding structural elements that define subcellular compartmentalization and functional specialization of the individual family members (60). By recognizing and binding to a specific β-turn motif, cyclophilins can modulate functions of the protein targets and stabilize multiprotein complexes (98). Cyclophilin A (CypA) is the most abundant and ubiquitously expressed cyclophilin that, although not essential for cell viability (40), plays an important role in cellular biochemistry and pathogenesis of viral infection, cardiovascular disease, and cancer (165). A type VIb β-turn is one of the elements through which CypA selectively binds to its targets (98).
CypA-AIF interaction: CypA interacts with apoptogenic AIF in the cytoplasm (29) and cotranslocates to the nucleus (253). Formation of the CypA-AIF complex does not require any other factors or the PPI activity of CypA but may involve the AIF 367–399 fragment (Fig. 13A) and one of the helices (aa. 136–146) and part of the β-barrel of CypA (29). Binding kinetics between recombinant AIF (unspecified construct) and human CypA was investigated by surface plasmon resonance in the presence of NADPH (253). Assuming that under studied conditions the flavoprotein was predominantly reduced, the derived dissociation constant (1 μM) suggests that CypA has considerable affinity for the CTC dimer. Since no experimental data on the association of CypA with oxidized AIF had been reported, we utilized in silico modeling to test whether the AIF redox state affects CypA binding. CypA was found to preferably dock to the oxidized monomer at two low affinity sites, distinct from that predicted by Cande et al. (29), and clustered in the groove above the monomer–monomer interface in the CTC dimer (Fig. 13). While biological relevance of the predicted docking sites remains to be tested, the manner of the AIF-CypA interaction corroborates the kinetic data (253) and suggests that the cyclophilin has a higher affinity for the dimeric form of the flavoprotein. Since oxidized AIF has a tendency to oligomerize when present at high concentrations (38), it would be of interest to elucidate whether the binding preference of CypA for the reduced dimers is specific or the protein can associate as tightly with the oxidized AIF oligomers. In the latter case, cytoplasmic or nuclear accumulation/clustering of any form of AIF would facilitate CypA association and promote cell demise (see sections VII.D.3.b/c for additional information on the AIF-CypA interaction).
FIG. 13.

Complexes between CypA (PDB code 3KOM) and the oxidized monomer (A) and reduced dimer of AIF (B, C) generated with the program GRAMM (220). AIF molecules are depicted in light gray and carton representation. Computer modeling suggests that CypA (in ribbon representation) has a higher affinity for the dimeric form of AIF and preferably binds to the grove above the monomer–monomer interface. In the oxidized monomer, two favorable CypA docking positions are predicted, distinct from the site identified by Cande et al. [residues 367–399 (29); shown in black and indicated by an arrow]. Physiological relevance of the predicted CypA binding sites and whether the AIF-CypA interaction is redox dependent or simply requires clustering and oligomerization of AIF remain to be established.
d. Phospholipid scramblase
Scramblases are located in the cell membrane and belong to the family of trans-membrane lipid transporters, also known as flippases, that transport (scramble) negatively charged phospholipids from the inner- to outer-leaflet and vice versa. By translocating phospholipids against the concentration gradient, scramblases can establish or diminish asymmetrical distribution of trans-membrane lipids. Exposure of new lipid-head groups on the membrane leaflet, in turn, can serve as a specific signal for physiological modifications [reviewed in (58)]. Externalization of PS, normally distributed in the inner monolayer of the plasma membrane, is a hallmark event in mammalian apoptosis that serves as a signal for induction of phagocytosis (147). This process is promoted by AIF via an unknown mechanism (136, 212).
Transport of PS to the outer-leaflet of the plasma membrane also takes place during apoptosis in C. elegans and involves two proteins: scramblase SCRM-1, homologous to human scramblases, and the AIF homolog WAH-1 (231). WAH-1 specifically interacts and activates SCRM-1 in a Ca2+-independent manner. Further, genetic inactivation of WAH-1 diminishes PS exposure on the surface of apoptotic germ cells and compromises cell-corps engulfment. On the basis of these results, WAH-1 was proposed to mediate a mitochondrial-to-plasma-membrane signaling pathway that promotes changes on the surface of apoptotic cells (231). The 380–550 region in WAH-1 was identified as sufficient and Lys446 as critical for SCRM-1 binding (corresponding to the 270–440 fragment and Lys336 in human AIF; Fig. 14).
FIG. 14.
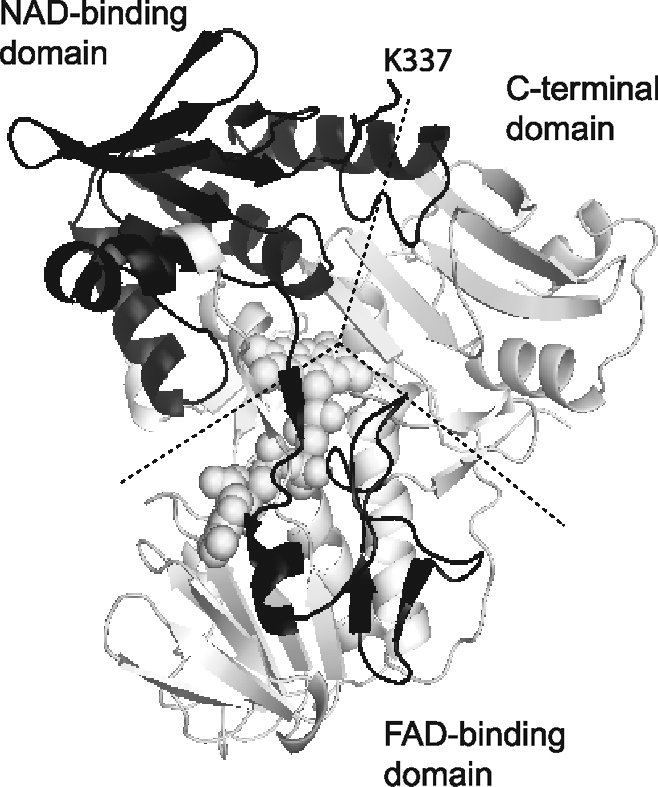
A potential binding site for phospholipid scramblase derived based on the sequence homology between human AIF and C. elegans WAH-1 (231). The 270–440 fragment and Lys337 in human AIF (highlighted in black) correspond to the WAH-1 380–550 peptide and Lys446, which are critical for interaction with SCRM-1, a worm homolog of human scramblase. The predicted scramblase-binding site includes residues comprising the NAD(H)- and FAD-binding domains.
The results obtained with C. elegans should be extrapolated to humans with caution. Although mammalian AIF promotes PS externalization (136, 212), direct interaction of AIF with the plasma membrane scramblase has never been reported. Besides, there are significant structural differences between human and worm proteins. The C. elegans SCRM-1, for instance, is lacking part of the N-terminal proline-rich domain and may have fewer, if any, SH3-domain containing partners than the human enzyme. WAH-1, on the other hand, is missing functionally important residues corresponding to Trp195, a Glu412-Arg448 salt bridge, the active site His453, and the regulatory 509–559 insertion (murine AIF numbering; see section VI.C for details). Instead, WAH-1 contains an extended and highly charged membrane linker whose function is currently unknown (Fig. 3). Owing to these dissimilarities, the human mechanism of PS exposure and AIF-mediated signal transduction could be distinct and more complex.
e. Scythe
Scythe, also known as BAT3 (HLA-B-associated transcript 3), is a 120 kDa protein that contains the N-terminal ubiquitin-like domain, a central proline-rich region, the C-terminal zinc finger motif, NLS, and the BAG-1(Bcl-2-associated gene 1)-like domain (16, 143, 219). Originally, Scythe had been identified as a regulator of apoptosis in Drosophila (218) and later was shown to play an important role during mammalian development. Inactivation of Scythe in mice leads to embryonic or perinatal lethality due to multiple developmental defects (56). Scythe−/− cells are defective in specific apoptotic signaling pathways and are resistant to apoptosis caused by thapsigargin (TG) or menadione, the endoplasmic reticulum stress inducers (56). An investigation of TG-related apoptosis in mouse ES cells revealed that (a) Scythe regulates expression and stability of the AIF precursor, (b) Scythe−/− cells become resistant to apoptosis because of decreased levels of AIF, and (c) expression of AIF can be regulated on a post-translational level and restored by reintroduction of Scythe into Scythe−/− cells (57). The Scythe-dependent regulation of AIF stability was suggested to take place in the cytoplasm via a mechanism that involves the proteosome and requires direct physical interaction of AIF with the N-terminal domain but not ubiquitin-like module of Scythe (57).
The cytoplasmic site for the functional association between AIF and Scythe proposed by Desmots et al. (57) contradicts the notion that Scythe is a nuclear protein that does not relocate during apoptosis (143). Another drawback of their study is utilization of the N-terminally green fluorescent protein-fused AIF precursor for investigating Scythe-mediated regulation of the AIF stability (54). The N-terminal tag could interfere with the mitochondrial sorting and artificially retain the AIF precursor in the cytoplasm, thus promoting interactions with the apoptotic machinery. Further studies are also necessary to clarify how the Scythe-mediated protection of a nonapoptogenic AIF precursor facilitates cell death.
In summary, upon translocation into the cytoplasm, apoptogenic AIF interacts with a group of structurally and functionally diverse proteins. These interactions are critical because they could promote, postpone, or even prevent cell death. To fully understand how the AIF-mediated signal transduction is regulated, more detailed investigations on the aforementioned partners and identification of other AIF-binding proteins are required.
D. Nuclear effects of apoptogenic AIF
Whether the release of mitochondrial AIF is a primary execution step or occurs downstream of caspase activation, AIF must enter the nucleus to promote cell death (35, 46, 210, 212, 242, 244). Relocation of apoptogenic AIF to the nuclear compartment is observed in different species during various but not all types of cell death.
1. Transport of AIF to the nucleus
Cytoplasmic proteins are imported to the nucleus by nuclear transport receptors that recognize an NLS, a surface peptide that generally contains a cluster of positively charged residues. On the basis of the sequence analysis, two NLS motifs were identified in murine AIF: NLS1 and NLS2, corresponding to the 277–301 and 445–451 fragments, respectively (Fig. 1) (212). Among the two, the C-terminal 445KLGRRRV451 peptide was proven to be the predominant NLS through which AIF is transported to the nucleus (84). The minor role of NLS1 is also evidenced by the inability of AIFsh2 and AIFsh3 to relocate from the cytoplasm to the nuclear compartment (54).
NLS2 is fully accessible in the oxidized monomer but becomes buried at the dimer interface upon AIF reduction (Fig. 4D, E; section VI.C). This means that the NLS2 accessibility to the nuclear transporters and the apoptogenic potency of AIF, in general, will depend on the cellular redox status. Besides NLS2, several positively charged surface residues collectively assist nuclear import of AIF (242). Cytoplasmic partners (Fig. 10) could also affect AIF trafficking. Accelerated relocation of human AIF R192A/K194A (242), for example, is likely caused by the perturbed association with Hsp70 (section VII.C.1.a) rather than nuclear transporters. Proteases degrading Hsp70 (32) or other AIF-binding proteins could be another potent regulatory factor. The importance of cytoplasmic interactions for the apoptogenic function of AIF is further evidenced by the ability of CypA to comigrate to the nucleus (216, 253) (Fig. 15) and mediate binding of AIF to DNA and nuclear components that promote chromatinolysis (13, 232) (sections VII.D.b/c).
FIG. 15.

Nuclear effects of apoptogenic AIF. AIF is transported from the cytoplasm to the nucleus by nuclear transport receptors recognizing NLS1 and NLS2. CypA assists and Hsp70 prevents cytonuclear translocation of AIF. In the nucleus, AIF directly binds to DNA and in cooperation with other proteins causes peripheral chromatin condensation and, possibly, large-scale chromatin fragmentation. Histone H2AX and CypA cooperatively promote AIF-mediated nuclear apoptosis. Co-operation between AIF and endonuclease G was suggested but has not been proven thus far. Other unidentified proteins (indicated in the figure as triangles with a question mark) may interact with AIF as well, forming a DNA degrading complex, “degradosome.” Upon chromatinolysis, AIF might dissociate from oligonucleosomal DNA and translocate back to the cytoplasm.
2. Interaction of AIF with DNA
Genomic DNA degradation, a key feature of PCD, is a two-step process during which chromatin first condenses along the nuclear membrane and undergoes large-scale fragmentation (>50 kb; stage I), then DNA gets cleaved at the inter- and intranucleosomal sites, and apoptotic bodies are formed (stage II) (251). Both endogenous and recombinant AIF can trigger peripheral chromatin condensation and large-scale DNA fragmentation (36, 46, 75, 107, 173). There is also evidence suggesting that AIF is responsible only for stage I nuclear morphology but not large-scale DNA degradation (199, 247).
Lacking the nuclease activity, AIF is proposed to directly interact with DNA and disrupt/collapse chromatin structure by displacing chromatin-associated proteins and/or by recruiting proteases and nucleases to form DNA degrading complexes, degradosomes (210, 212). Colocalization of AIF with DNA during stage I chromatin condensation, as well as the inability of the K510A/K518A and K255A/R265A variants of human AIF to bind DNA in vitro and induce apoptosis in live cells are consistent with this hypothesis and indicate that direct AIF-DNA association is critical for execution of PCD (242). Concordantly, several RNA-binding proteins associating with AIF were identified in rat brain lysates and nuclear HeLa extracts (224). One of these, heterogeneous nuclear ribonucleoprotein, interacts with AIF via the RNA moiety.
Refolded His-tagged AIF binds to all kinds of nucleic acids in a sequence-independent manner but preferentially associates with single-stranded DNA (224). According to another in vitro study, recombinant AIF binds to double-stranded DNA but fails to interact with single-stranded oligonucleotides (242). Mg2+ ions promote AIF-DNA association, leading to thickening and shortening of DNA fragments and formation of intermolecular complexes via AIF-AIF contacts (224). NAD(P)H was reported to increase the mean size of the AIF-DNA complexes, NAD had no effect, and NADP promoted generation of high-order AIF-DNA and AIF-RNA agglomerates (224). These in vitro results led to the proposal that AIF invades chromatin at pre-existing DNA breakage sites and cooperatively propagates through the adjacent double-stranded DNA, causing chromatin condensation and aggregation into higher order structures.
In our study (38), naturally folded and tag-free AIF interacted with linearized double-stranded DNA quite differently. Formation of the AIF-DNA complexes was observed at considerably higher AIF concentrations [10–100 μg vs. 1 μg (224)] and higher AIF:DNA ratios [>40:1 vs. 20–30:1 (224)]. The DNA-binding ability of AIF was unaffected by Mg2+ and NADP but markedly inhibited by NAD(P)H. The latter finding is another supporting argument for the redox dependency of the apoptogenic action of AIF. The discrepancies in the manner of AIF-DNA interaction could result from structural aberrations in the refolded flavoprotein or/and attachment of the His-tag, which increases the AIF affinity for DNA by 10-fold (38).
In conclusion, AIF is transported from the cytoplasm to the nuclear compartment predominantly via NLS2 and with the assistance of CypA. In the nucleus AIF initiates chromatin condensation likely via direct interaction with DNA. Both nuclear transport and interaction with DNA may be redox controlled because the NLS2 accessibility and DNA-binding ability depend on the AIF redox state. Further clarification is needed on whether AIF participates in large-scale chromatin degradation or not.
3. Nuclear partners of AIF
a. Endonuclease G
Endonuclease G (EndoG) is an evolutionary conserved, nonspecific nuclease that cleaves single- and double-stranded DNA and RNA in an Mg2+/Mn2+-dependent fashion [reviewed in (237)]. Mammalian EndoG is a nuclear-encoded protein targeted to mitochondria, the 29 kDa mature form of which is compartmentalized in the IMS and implicated in mitochondrial DNA replication and apoptosis. Upon an apoptotic insult and release from the IMS, EndoG is activated and imported to the nucleus. In isolated nonapoptotic nuclei, EndoG first generates large fragments of DNA (>50kb), then cleaves at inter- and intranucleosomal sites.
In C. elegans, the EndoG homolog Csp-6 cooperates with WAH-1, the worm's homolog of AIF (Fig. 3; sections III and VII.C.2.d), in DNA degradation during apoptosis (232). The 196–700 fragment of WAH-1 (corresponding to human AIFΔ1–101) was shown to directly interact with Csp-6 and promote chromatinolysis in vitro and cell demise in vivo. On the basis of the similarity in cellular localization and apoptotic effects caused by AIF and EndoG and their worm homologs, the AIF/EndoG pair was proposed to define an evolutionary conserved DNA degradation pathway (232).
However, there is evidence that does not support this hypothesis. First, the WAH-1 deficient worms are vital although they grow slower and are smaller in size. Unlike mammalian AIFs, crucial for cell survival (section VIII), downregulation of WAH-1 expression by RNAi has little effect on the fate of the cells undergoing apoptosis but enhances the cell survival phenotypes of mutants that are partially or strongly defective in cell death. This indicates that WAH-1 normally promotes cell demise. Second, release of WAH-1 from mitochondria strongly depends on caspase activity and, thus, may be part of the caspase-dependent apoptotic cascade. The lack of a regulatory insertion in WAH-1 is another indication that signal transduction in the nematode may proceed via different pathways. Third, although WAH-1 is predicted to be a flavoprotein (Fig. 3, section III), the investigated 196–700 fragment does not contain FAD (232) and could be misfolded. Next, there is no direct evidence for cooperation between AIF and EndoG in DNA degradation during apoptosis in mammalian cells. Contrarily, one study showed that during caspase-independent noise-induced cell death in the inner ear, only EndoG translocates to the nucleus, whereas AIF is retained in the cytoplasm (240). Finally, EndoG null mice have no obvious defects in embryonic development and regulation of apoptosis (51), which also argues against the essential role of EndoG in PCD. Thus, the evolutionary conservation of the EndoG/AIF-mediated DNA degradation pathway in higher eukaryotes remains an open question.
b. Cyclophilin A
Presently, it is unclear whether cyclophilins possess or lack nucleolytic activities (145, 158). Nonetheless, recombinant CypA was demonstrated to cooperate with AIF in vitro in chromatinolysis and plasmid DNA degradation (29). That two proteins act synergistically is also evidenced by the facts that (a) CypA colocalizes with endogenous AIF in isolated nuclei during the initial stage of chromatin condensation, (b) transfection of CypA+/+ but not CypA−/− Jurkat cells with AIF increases frequency of staurosporine-induced apoptosis, and (c) the difference between CypA-sufficient and -deficient cells disappears when AIF expression is downregulated (29). The CypA- or DNA-binding domains of AIF (Δ269–442 and Δ263–399 or Δ473–552 and Δ567–609, respectively) but not the PPI activity of CypA are necessary for induction of apoptosis in CypA+/+ cells (29).
In our in vitro study, CypA had no effect on the DNA-binding ability of AIF, and the two proteins did not display any nuclease activity individually or in cooperation (38). It is unclear at the moment whether these functional dissimilarities are caused by aberrations in the AIF folding, covalent tag attachment, or contamination of CypA preparations with nucleases. Importance of the AIF-CypA interaction in triggering and promoting apoptosis is undermined also by the finding that CypA-deficient mice have no defects in cell death (41).
c. Histone H2AX
One possible role of CypA in the nucleus could be facilitation of AIF association with the proteins promoting chromatinolysis, in particular histone H2AX (13) (Fig. 15). As an integral component of the chromatin packaging, H2AX contributes to genome stability by participating in the assembly of the DNA damage repair foci [reviewed in (176)]. This H2AX function is regulated through post-translational modification. In response to treatments causing double-strand DNA breaks, the histone is rapidly phosphorylated at Ser139, which modifies chromatin and makes it accessible for repair. Phosphorylated H2AX (γH2AX) is known to regulate caspase-dependent apoptosis via the caspase-activated DNase pathway (138).
Artus et al. demonstrated that H2AX is also involved in caspase-independent necrosis via association with AIF (13). Interaction between H2AX and AIF is observed only in cells treated with the DNA alkylating agents that cause double-strand DNA breaks, such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (13). The proline-rich peptide of AIF (aa. 543–559) and phosphorylation of Ser139 in H2AX are required for their association. The AIF-H2AX interaction is essential for AIF-mediated chromatin condensation and degradation, as deletion of the proline-rich fragment in AIF and knockout of H2AX abolish chromatinolysis and nuclear apoptosis (13). The MNNG toxicity and all hallmarks of nuclear apoptosis are also eliminated when the CypA gene is silenced. This suggests that a synchronized action of AIF, H2AX, and CypA is necessary for initiation of nuclear DNA degradation. Downregulation of EndoG, on the other hand, has no effect on the AIF/H2AX-dependent chromatinolysis (13), which undermines once again the importance of the AIF/EndoG-mediated PCD pathway in mammalian cells.
It would be of interest to investigate whether the AIF redox state affects the AIF-H2AX interaction. If the proline-rich fragment is indeed a key element through which AIF binds to H2AX, association between the proteins should be redox controlled. Elucidation of the phosphorylation status of H2AX Tyr142 during AIF-mediated necrosis is also important because after DNA damage, the fate of the cell is determined by the Tyr142 but not Ser139 modification (43).
4. Relocation of AIF in late apoptosis
While association of apoptogenic AIF with the chromatin in cells undergoing early apoptosis is well documented (36, 46, 75, 107, 173, 247), little is known about what happens to the protein during the later stages of PCD. One of the first reports on AIF and a recent immunofluorescent study indicate that during stage II of nuclear apoptosis, when DNA is degraded to small oligonucleosomal fragments, colocalization of AIF with the chromatin is diminished (199, 242). It was observed also that during the late stages of etoposide- and actinomycin-induced apoptosis in HeLa cells, AIF relocates from the nuclei of karyorrhexic cells back to the cytoplasm and mitochondria (199). If confirmed, physiological relevance of this relocation should be investigated.
To summarize, AIF does not possess nuclease activities and can trigger DNA degradation only via co-operation with other proteins such as histone H2AX and CypA. EndoG is another potential partner but its involvement in AIF-mediated chromatinolysis in mammals has not been demonstrated yet.
E. Apoptogenic properties of the AIF homologs
As emphasized in sections VII.A–D, the mechanism of AIF-mediated apoptosis in mammalian cells is complex and, despite extensive research, is not fully understood. Comparative analysis of effects caused by downregulation of AIF in higher and lower eukaryotes helps better understand structure/function relations and cellular roles of the flavoprotein. In addition to the C. elegans WAH-1 (section VII.D.3.a), apoptogenic properties of four other AIF homologs have been investigated. The D. melanogaster and D. discoideum proteins are closely related to mammalian AIFs, whereas the Tetrahymena and S. cerevisiae counterparts are more distant on the evolutionary tree.
1. D. melanogaster
As in mammals, the PCD in D. melanogaster involves mitochondrial remodeling (83) that may lead to caspase activation or proceed in a caspase-independent manner (8, 152). The fly's homolog of AIF (DmAIF, 50% identity and 68% similarity) contains all structural elements characteristic for mammalian AIFs, including the N- and C-terminal insertions (Fig. 3). When transfected into live cells, the full-length DmAIF precursor is targeted to mitochondria, wherein the 178–674 fragment (corresponding to human AIFΔ1–120) is diffusely distributed in the cytoplasm (102). However, in the cells undergoing apoptosis triggered by UV/γ-irradiation or overproduction of the lethal protein GRIM, DmAIF remains compartmentalized in mitochondria (102). This raises doubts over the DmAIF ability to mediate PCD similarly to mammalian counterparts. One reason for the inability to transport from mitochondria could be the lack of a canonical PEST motif in DmAIF (Fig. 3). As discussed in section VII.B.1, PEST motifs regulate interaction of calpain proteases with protein targets (204, 229). Having a distinct C-terminal insertion, flies might be resistant to calpains.
The apoptogenic potential of extramitochondrial DmAIF was evaluated on transgenic flies that were expressing in the eye either full-length or Δ1–177 DmAIF fragments (102). Enforced expression of truncated DmAIF leads to a marked reduction in eye size and disruption in ommatidial architecture caused by ectopic cell death. This phenotype is also observed in the mutant fly lines deficient in caspases or caspase activators, and could be partially ameliorated by expression of caspase inhibitors Diap1 and Diap2 (Drosophila IAPs; homologous to human XIAP). On the contrary, coexpression of the baculoviral caspase inhibitor p35 enhanced the phenotypic changes induced by DmAIFΔ1–177 both in the eye and dorsal and thoracic bristles. Thus, extramitochondrial DmAIF can promote cell demise and alter tissue morphology in a manner that, in part, involves caspase proteases.
Drosophila thioredoxin-2 (DmTrx-2) was shown to be a natural mediator of DmAIFΔ1–177 cytotoxicity (102). Homologous to human thioredoxin, cytoplasmic DmTrx-2 participates in peroxide metabolism and redox signaling (18). Downregulation of DmTrx-2 abolishes the eye-damaging effects caused by DmAIFΔ1–177 overexpression and reverses to normal eye architecture. It was concluded therefore that the two proteins cooperate in vivo to facilitate PCD (102). That DmTrx-2 coimmunoprecipitates with endogenous murine AIF in ES cell lysates, as well as direct interaction between murine thioredoxin and AIF detected by yeast two-hybrid screening (102) support the interpretation that the thioredoxin-AIF partnership may be evolutionary conserved.
2. D. discoideum
Dictyostelium, also known as slime mold, undergoes a complex developmental process within its lifetime, transitioning from a collection of unicellular amoebae into a multicellular slug and a fruiting body. The cell death phenotype in D. discoideum is highly similar to that of multicellular animals and plants, and includes ΔΨm dissipation, plasma membrane PS exposure, intense vacuolization, large-scale DNA fragmentation, autophagy, and engulfment of apoptotic corpses by neighboring cells (11, 44). The slime mold homolog of AIF (DdAIF, 30% identity and 60% similarity) translocates from mitochondria to the nucleus during initiated or developmental cell death and triggers chromatin condensation and fragmentation in a caspase-independent fashion (11). DdAIF-containing cytosolic extracts induce condensation and partial degradation of DNA not only in healthy nuclei isolated from the slime mold but also in nuclei obtained from mammalian cells.
This similarity of action could arise from the common structural fold and functional elements shared by mammalian and Dictyostelium AIFs (Fig. 3). However, DdAIF does not contain a regulatory peptide but has a canonical DNA-binding motif (aa. 501–523; Fig. 16A) (11). In mammalian AIFs, the corresponding helix-turn-helix is surface-exposed and has the Asp-Gly-Glu cluster instead of basic Lys-Arg-Arg (Fig. 16B). This makes the surface more acidic and less suitable for DNA binding. An identical substitution is present in D. rerio, whereas in other species the turn between helices contains at least one basic residue (Fig. 16A). This suggests that the AIF ancestors were binding to DNA stronger. The lesser DNA-binding ability of mammalian AIFs may be beneficial as it enables to slow down and finely tune the apoptotic cascade.
FIG. 16.

(A) The C-terminal sequence alignment of the AIF-like proteins and (B) the x-ray structure of human AIF. (A) A DNA-binding helix-turn-helix motif predicted in D. discoideum is boxed. Residues corresponding to the 512Lys-Arg-Arg514 cluster in D. discoideum are also indicated. (B) The 595–605 peptide corresponding to the DNA-binding motif in D. discoideum is displayed in black. Instead of the basic cluster predicted to comprise the turn between helices in D. discoideum, human AIF has 594Asp-Gly-Glu596, which makes the surface more acidic and less suitable for DNA binding.
3. Tetrahymena thermophila
T. thermophila are ciliates that have two functionally and morphologically distinct nuclei: a small diploid micronucleus and a large polyploid macronucleus responsible for reproduction and general cell regulation, respectively. Sexual reproduction in ciliates involves conjugation and programmed nuclear death, during which the parental macronucleus is eliminated (52). Degradation of the macronucleus proceeds in three stages that include large-scale DNA fragmentation (stage I), chromatin condensation and oligonucleosomal DNA fragmentation (stage II), and lysosomal autophagy (stage III).
The Tetrahymena homolog of AIF (TtAIF, 24% identity and 45% similarity) plays an important role in DNA degradation during early stages of programmed nuclear death (2). TtAIF contains MLS and NLS but lacks two insertions characteristic for mammalian AIFs. TtAIF is expressed continuously during conjugation and translocates to the parental macronucleus before nuclear differentiation. ΔAIF Tetrahymena strains have reduced growth rates and can initiate normal nuclear events during mating, although condensation and large-scale DNA fragmentation in the parental nuclei are delayed (2). Further, mitochondria isolated from ΔAIF cells display a considerably lower DNase activity relative to controls. These findings indicated that TtAIF is involved but not crucial for the initial stages of nuclear degradation, during which it may cooperate or activate a yet-unidentified mitochondrial DNase similar to EndoG. Unfortunately, it has not been investigated whether TtAIF mediates cell demise. Presently, Tetrahymena appear to employ TtAIF for a very specific task: elimination of a parental macronucleus, which requires precise temporal regulation of TtAIF expression, proteolysis, and translocation. It would be worthwhile to investigate how the TtAIF-mediated cascade is put into action. The mechanism by which two nuclei are distinguished is also intriguing.
4. S. cerevisiae
Aif1p, a yeast homolog of AIF (22% identity and 41% similarity), does not contain MLS and a membrane-binding fragment but associates with mitochondria and transports to the nucleus of yeast cells during H2O2-induced or physiologically induced apoptosis (i.e., chronological aging) (239). Aif1p is required for efficient apoptotic cell death in budding yeast. In synergy with mild oxidative stress, overexpression of Aif1p stimulates cell demise and leads to chromatin condensation and DNA fragmentation mostly in a caspase-dependent fashion. Further, recombinant Aif1p can degrade DNA in isolated nuclei. This activity does not require binding of FAD but may depend on other cofactors in vivo. The yeast homolog of human CypA is one of the proteins that promotes Aif1p-induced apoptosis (239). Similar to the human AIF-CypA pair (29), the cooperative apoptogenic action of yeast Aif1p and CypA does not require the cyclophilin's PPI activity.
In summary, a strong ability to promote PCD and high similarity of the apoptogenic effects exerted by the AIF-like proteins suggest that the mitochondrion-initiated apoptotic DNA degradation pathway involving AIF is phylogenetically old and conserved among eukaryotes (28). Similar to mammalian counterparts, the AIF-like proteins act in cooperation with the cytoplasmic and nuclear partners analogous to thioredoxin, CypA, EndoG, and possibly others. However, owing to structural and functional differences in AIFs, the PCD execution mechanisms may vary among species. During evolution, mammalian AIFs acquired the regulatory peptide and lost the DNA-binding motif. These features may be beneficial because they allow to slow down the apoptotic cascade and introduce additional regulatory steps through which the cell can more precisely regulate its demise.
VIII. Vital Functions of Mitochondrial AIF
Over the past years, compelling evidence has emerged to indicate that in addition to the apoptogenic function mitochondrial AIF has an essential pro-survival role. The precise role of the flavoprotein in the IMS, however, remains a puzzle. One obstacle to solving this problem is the difficulty in separating vital and lethal actions of AIF, as some of the cell-death-inducing effects may result from removal of the protein from the mitochondrial compartment. Analyses of phenotypes associated with the AIF deficiency and defects in cells, animal models and human patients strongly suggest that the flavoprotein plays an important role in regulation of mitochondrial morphology and energy metabolism.
A. Role of AIF in mitochondrial respiration
The most straightforward way to analyze function(s) of a protein would be its genetic inactivation through gene knockout. Homozygous AIF knockout in mice is embryonically lethal. The death of embryos was first reported to be the result of abolishment of the first wave of developmental cell death during early embryogenesis (105). This conclusion was later revised, as it was demonstrated that AIF is not required for cavitation and apoptotic cell death in early mouse embryos (25, 103). Moreover, the cavitation defects in AIF−/y ES cells were found to be secondary and caused by abnormalities in the function of respiratory complex I (66).
1. Hq mouse phenotype
An array of metabolic changes caused by AIF deficiency in vivo was first identified in Hq mice, a rodent strain that has 80% decreased AIF expression due to a retroviral insertion into the first intron of the AIF gene. Both Hq/Y males and Hq/Hq females are viable and do not exhibit obvious phenotypic alterations at early age. However, adult animals exhibit lack of hair and develop progressive neurodegeneration, ataxia, and blindness caused by retinal degeneration (120), but are resistant to a progressive weight gain and lipid accumulation during high-fat feeding (180). AIF-deficient neurons and cardiomyocytes are sensitized to peroxides and ROS-induced damage (120, 225). Further, AIF ablation leads terminally differentiated neurons to abortive cell cycle re-entry (120) and reduces oxidative phosphorylation (OXPHOS) in the brain and retina, but not in the liver and the heart, by diminishing expression of complex I subunits (223).
On the basis of strong correlation between AIF deficiency and oxidative stress, mitochondrial AIF was proposed to act as an antioxidant that plays a role in OXPHOS and assists free radical scavenging (120, 223, 225, 226). This conclusion contradicts in vitro data showing that AIF does not possess antioxidant activities (38) (section V.C). Concordantly, experiments on isolated brain mitochondria from wild-type and Hq mice demonstrated that AIF does not directly modulate ROS release, and the organelles can tolerate a 90% decrease in AIF and >40% in complex I without apparent changes in the mitochondrial ROS production (37). These findings indicated that oxidative stress in the AIF-deficient Hq brain is a consequence rather than a cause of mitochondrial dysfunction.
2. Tissue-specific AIF defects
Results obtained from genetic and RNAi knockdown models of AIF deficiency in various cells are inconsistent and suggest that downregulation of mitochondrial AIF elevates (7), decreases (180, 221), or has no effect on the cellular ROS levels (223). A general feature of cells lacking AIF is high lactate production and increased dependence on glycolytic ATP generation due to an altered oxidative metabolism (223). A 50% decrease in complex I activity is detected in both AIF−/y ES and AIF siRNA HeLa cells, whereas a proportional decrease in complex III activity is observed only when AIF expression is eliminated but not downregulated (223). Defects in the OXPHOS activity are caused in part by decreased expression of complex I subunits NDUFB7, NDUFS7, NDUFA9, and Grim19 and, to a lower extent, complex III subunits UQCRC1, UQCRC2, and UQCRFS1 (7, 223). Ablation of AIF also affects the NADH oxidase activity of complex I (223), indicating that there are defects both in the assembly and function of the enzyme. However, AIF-negative cells have no perturbations in expression of mitochondrial DNA-encoded respiratory chain subunits and functioning of complexes II, IV and V, PTP, or translocator of the outer membrane (223). Further, the AIF null cells have normal levels of oxidized cardiolipin, reduced glutathione, NAD(P)H, quinone-dependent, and superoxide dismutase activities (223). These findings are consistent with the interpretation that AIF knockout does not lead to oxidative stress.
Mice with cardiac- and skeletal-muscle-specific AIF knockout develop severe dilated cardiomyopathy and skeletal atrophy (103). A comprehensive metabolic analysis showed that the animals have reduced complex I-derived electron transfer and decreased ROS generation through increased coupling in the respiratory chain (180). Consequently, these functional changes improve insulin sensitivity and upregulate glucose uptake machinery, prevent diet-induced obesity and diabetes, and cause metabolic shifts in ATP, NAD, and AMP. Mice lacking AIF only in cortical neurons have defective cortical development and reduced neuronal survival due to defects in mitochondrial respiration (35). Depletion of AIF in the insulin-producing β-cells, on the other hand, results in an age-dependent mass decrease caused by misregulation of the cell cycle control and arrest in the G2 phase (197).
Despite in-depth in vivo analyses of metabolic changes caused by AIF ablation, the precise role of the mitochondrial flavoprotein remains elusive. A crucial role of the AIF redox activity for normal mitochondrial functioning is evidenced by the fact that only expression of full-length AIF can restore defects in complex I and the cell growth supportive function in AIF−/y cells (221, 223). Additionally, the flavoprotein was proposed to regulate the OXPHOS function and energy homeostasis indirectly, by acting as an assembly factor that assists biogenesis and/or stabilizes multisubunit complexes I and III (223). This hypothesis is mainly based on the findings that AIF does not tightly associate with any of the respiratory complexes and has no effect on their transcription at the mRNA level (223).
3. Role of AIF in neurodegeneration, neurogenesis, and neuroprotection
In vivo studies consistently demonstrate that AIF targeting has the largest effect on neurons. Having a high demand for energy and being unable to switch to anaerobic glycolysis when OXPHOS is limited, neurons are more dependent on mitochondrial metabolism than other types of cells. AIF deficiency in neurons leads to oxidative damage and, consequently, to neurodegeneration and blindness (35, 97, 103, 120, 225). What ROS molecules exert harmful effects is currently unknown.
Analysis of mouse embryos that had AIF deleted in the prospective midbrain and cerebellum at an early embryonic stage showed that AIF is critical for both postmitotic neuron survival and cerebellar development (97). Histological examination of the brains of these animals, which died soon after birth of unknown causes, revealed severe cerebellar hypoplasia with a marked reduction in the Purkinje and granule cell precursors. Deletion of AIF in the Purkinje cell precursors causes premature entry into S-phase and cell death via apoptosis, wherein the G1-S phase transition is disrupted in the precursors of granule cell. Thus, AIF is essential for neurogenesis and its loss causes cell cycle abnormalities in a neuron-specific manner. The mechanism underlying the abortive cell cycle re-entry in AIF-deficient neurons (97, 120) has not been established yet.
Predominant expression of AIF2 in the developing and adult central nervous system and retina correlates with decreased AIF-dependent neurotoxicity and, hence, may be neuroprotective (87). The trans-membrane fragment in brain-specific AIF2 is one residue longer and more hydrophobic than in AIF1, due to which AIF2 binds to the inner membrane tighter and is more difficult to extract. Upon MNNG treatment, AIF2 displays less relocation into the cytoplasm and, most importantly, retains AIF1 in the IMS, promoting neuronal survival (87).
In some cases, a decrease in the AIF levels can be neuroprotective as well. This effect is observed in neurons after acute injury, where AIF is the major factor mediating neuronal death [reviewed in (76)]. Further, compared to wild-type littermates, Hq mice display smaller infarct volumes and show dramatically reduced neuronal death in the ischemic zone after transient cerebral artery occlusion (47). The Hq mutation provides significant protection against photoreceptor apoptosis (91), whereas neurons carrying Hq and Apaf1−/− (apoptosis proteases-activating factor) mutations exhibit extended neuroprotection against DNA damage and glutamate-induced excitotoxicity (36). Downregulation of AIF by siRNA significantly reduces neuronal apoptosis induced by glutamate- and oxygen-glucose deprivation (47), and a neutralizing antibody to AIF prevents excitotoxic neuronal death (228). Thus, AIF can be beneficial or harmful for neurons, depending on physiological settings.
4. AIF deficiency in lower eukaryotes
Loss of zygotic expression of Drosophila DmAIF results in arrested larval growth, decreased viability, and OXPHOS defects mainly due to reduced enzymatic activities of complexes I and IV (102). Knockdown of C. elegans WAH-1 and Tetrahymena TtAIF, on the other hand, leads to a slower growth rate but has no affect on the viability of the mutants (2, 232). Compared to wild type, the ΔAif1p S. cerevisiae strain exhibits a significantly longer doubling time when grown on nonfermentable carbon sources, but not in glucose-rich media (223). These findings suggest that the AIF-like proteins may also be required for optimal OXPHOS.
Summarizing, AIF targeting mostly affects cells with high demand for energy, especially neurons. Analyses of metabolic changes caused by AIF deficiency showed that a yet unidentified redox activity of AIF is crucial for functioning of the respiratory chain and energy homeostasis. Additionally, the flavoprotein is thought to act as an assembly factor that regulates OXPHOS indirectly by assisting biogenesis/maintenance of complexes I and III.
B. AIF and mitochondrial morphology
Mitochondrial respiration and metabolism are spatially and temporarily regulated by the organelle's architecture, aberrations in which may lead to metabolic and energy failures. There is experimental evidence strongly suggesting that, solely or in cooperation with other proteins, AIF is involved in regulation of mitochondrial structure.
1. Mitochondrial abnormalities in telencephalon-specific AIFΔ mice
In an elegant study on the telencephalon-specific AIFΔ mice (tel AIFΔ), lacking the AIF gene only in cortical neurons, Cheung et al. were able to dissociate the physiological and apoptogenic roles of AIF (35). This group showed that translocation of AIF to the nucleus rather than depletion of the protein in mitochondria is the major event in apoptotic signaling. Most importantly, they found that tel AIFΔ neurons exhibit short and fragmented mitochondria with hyperpolarized membrane potential and abnormally dilated cristae. The altered cristae morphology and respiratory defects could be restored to normal by expression of AIF targeted to the outer leaflet of the inner mitochondrial membrane. It was suggested, therefore, that AIF regulates mitochondrial morphology and cristae formation, and that mitochondrial fragmentation could be the main cause of bioenergetic failure and reduced viability of the tel AIFΔ neurons (35).
2. Association of AIF with the optic atrophy 1 protein
Mitochondrial cristae are connected to the periphery of the inner membrane by narrow tubular junctions, which together with mitochondrial fusion and fission define the morphology of the organelle [reviewed in (144)]. The principal regulators of mitochondrial fusion and fission are the evolutionary conserved dynamin family GTPases (181). One of these, optic atrophy 1 (OPA1), is located on the outer leaflet of the inner membrane and directly involved in mitochondrial fusion and cristae remodeling (73, 130). In humans, mutations in OPA1 have been associated with familial autosomal dominant optic atrophy, characterized by progressive blindness due to loss of retinal ganglion cells (166).
Cells derived from the dominant optic atrophy patients display impaired complex I-dependent ATP synthesis, fragmented and balloon-like mitochondria, and 50% inhibition in mitochondrial fusion when grown in galactose medium (i.e., under forced oxidative metabolism), although expression and assembly of the respiratory complexes are normal (249). The same study found that AIF forms complexes with OPA1, even when complex I is partially assembled. This led to the hypothesis that AIF and OPA1 cooperatively regulate and stabilize the respiratory chain (249). The lack of difference in the OPA1-AIF interaction in the mutant and control cell lines was explained by the fact that all investigated mutations of OPA1 were located in the regions outside its coiled-coil domain, predicted to serve as a protein–protein interaction site (249).
Besides the dynamin family of enzymes, there are other inner membrane proteins shaping mitochondria. Mitofilin (100), SH3 domain containing endophilins (111), ATP synthase (174), and proteases (64) are some of the known cristae remodelers whose connection to AIF should be explored as well.
3. AIF isoform-specific cristae morphology
What AIF isoform predominates in the inner membrane may also define mitochondrial cristae morphology. Overexpression of AIF2 in human U2OS cells increases the curvature of cristae and forces them to adopt an onion-like shape, whereas overproduction of AIF1 leads to cristae rarefaction (87). Since AIF2 is expressed only in the central nervous system and the AIF1/AIF2 ratio depends on the brain cell maturation status (87), mitochondrial architecture in neurons is likely to be dynamic and affected by both the total and relative AIF1 and AIF2 contents.
In conclusion, several studies suggest that AIF may act as a regulator of mitochondrial cristae formation, and that mitochondrial fragmentation and abnormal cristae structure observed in AIF-deficient cells could be the primary cause of bioenergetic failure. Although the underlying mechanism is presently unknown, the AIF isoform content and AIF-OPA1 interaction are proposed to be the factors that could define mitochondrial morphology.
C. Human mitochondrial encephalomyopathy linked to the AIFΔ201 mutation
Only one mitochondrial disorder in humans directly linked to AIF has been described thus far (81). A 1601–1603 nucleotide deletion in exon 5 of the AIF gene was found in two related male infants. This mutation results in expression of AIF with the Arg201 deletion (AIFΔR210) leading to progressive mitochondrial encephalomyopathy characterized by axonal sensory and motor peripheral neuropathy, severe muscular atrophy, reduced OXPHOS activities, tissue-specific mitochondrial DNA depletion, and increased levels of plasma lactate. In contrast to the Hq mutation in mice, where residual levels of wild-type AIF are sufficient to provide normal embryonic development and support mitochondrial respiration in young offspring (120), the Arg201 deletion causes severe and early onset mitochondrial deficiency without affecting AIF expression levels.
Patient-derived fibroblasts have fragmented mitochondria and decreased complex I, III, and IV activities when grown under forced oxidative metabolism (81). Supplementation of AIFΔR201 cells with wild-type AIF recovers ∼30%–50% of complex III and IV activity, confirming a direct link between the AIF mutation and impairment of mitochondrial respiration. Deletion of Arg201 also increases the apoptogenic potency of AIF, as evidenced by increased frequency of staurosporine-induced apoptosis in AIFΔR201 fibroblasts relative to control, irrespective of whether z-vad.fmk was present or not.
Computer modeling provided some insights into why the ΔR201 mutation has such deleterious effects (81). Being part of the 191–203 β-hairpin (Figs. 3 and 4A), Arg201 (Arg200 in mice) establishes a salt link with Glu531 in oxidized AIF, thus, assisting folding of the regulatory peptide and limiting solvent access to the active site (Fig. 17A). By analogy with the murine protein (202), redox-induced restructuring of the regulatory peptide in human AIF is expected to release the hairpin, allowing Arg201 to form a hydrogen bond with the carbonyl oxygen of Phe205 (Fig. 17B). In silico modeling suggests that deletion of Arg201 shortens the hairpin and disrupts the β-turn structure. Since Arg201 defines the active site environment, as well as the length and positioning of the functionally important hairpin, the ΔR201 mutation could perturb both the folding and oxidoreductase function of AIF and, possibly, interaction with the partnering proteins.
FIG. 17.

Functional importance of Arg201. (A) Superposition of the x-ray structures of oxidized human and murine AIF. By forming a salt bridge with Glu531, Arg201 assists folding of the β-hairpin and the helical part of the regulatory peptide (highlighted in black), which stabilizes FAD binding. (B) Superposition of the reduced forms of wild-type (light gray) and ΔR201 mutant (black) of human AIF, modeled based on the structure of the murine protein. In the wild-type protein, Arg201 forms an H-bond with the carbonyl oxygen of Phe204 and limits the flexibility of the β-hairpin. Deletion of Arg201 shortens the hairpin and disrupts the β-turn structure. In oxidized AIF this may lead to weaker FAD association, whereas in the reduced form, where binding of FAD is stabilized through charge–transfer and π-π stacking interactions with NAD, the Arg201 deletion and elimination of the H-bond with Phe204 may increase flexibility of the partially unstructured 191–203 peptide and, possibly, perturb protein–protein interactions. Through these changes, the ΔR201 mutation can affect both vital and apoptogenic functions of AIF.
Indeed, in vitro characterization showed that recombinant AIFΔR201 is structurally unstable and tends to lose FAD (81). Further, compared to wild type, the mutant reacts with NADH two-orders of magnitude faster, produces shorter-lived CTC, and has a higher capacity to bind DNA. The latter property and conformational distortions in the β-hairpin, predicted to serve as a Hsp70 binding site (84, 196) (section VII.C.1.a), explain the higher potency of the mutant to induce nuclear apoptosis. Thus, the available data suggest that the ΔR201 mutation alters structural and functional properties of AIF, ultimately leading to destabilization of the inner membrane, defects in the OXPHOS assembly/functioning, and reduction of mitochondrial DNA content. Conformational instability of the mutant, on the other hand, could facilitate the N-terminal proteolysis and promote caspase-independent cell death. The fact that AIFΔR201 and control cells have similar OPA1 content (81) does not exclude participation of AIF in mitochondrial morphogenesis.
It would certainly be worthwhile to investigate effects of Arg201 deletion in AIF in model animals and tissue-specific cells because this mutation creates unique physiological settings where expression levels and redox activity of AIF are preserved, but, nonetheless, mitochondrial morphology and OXPHOS are altered. Since one of the potential damaging effects of the ΔR201 mutation is disruption/perturbation of protein–protein interactions, the studies on the model systems might help to identify mitochondrial partners of AIF and clarify whether AIF is indeed an assembly factor or plays other roles in the IMS.
D. Involvement of AIF in regulation of cytoplasmic stress granules
Besides regulation of mitochondrial respiration and morphology, AIF controls cytoplasmic stress granule (SG) formation. SGs appear in the cytoplasm of mammalian cells under environmental stress and represent aggregates of stalled translation preinitiation complexes due to failed attempts to make proteins from mRNA [reviewed in (4, 114)]. SGs associate with the endoplasmic reticulum and are formed either to store/protect RNA from harmful conditions or serve as sites of mRNA triage by determining whether individual mRNAs are degraded or reinitiated. That AIF could act as an endogenous repressor of cytoplasmic SGs was suggested based on enhanced formation of SGs in AIF-deficient cells in response to arsenate (29, 30, 105). Since AIF does not coprecipitate with the cytoplasmic SG components, its inhibitory action on SG formation is thought to be indirect. AIF-mediated SG regulation requires the mitochondrial redox activity but not apoptogenic capacity of the flavoprotein. The Δ228–347 and Δ322–333 mutations, postulated to interfere with the FAD and NAD(H) binding (136), partially abolish the SG suppressing activity of AIF, whereas deletion of the C-terminal 567–609 fragment, indispensable for induction of apoptosis (196), has no effect (30). Therefore, it was suggested that mitochondrial AIF maintains a balance between the NAD(P)H and oxidized/reduced glutathione levels, which, in turn, directly control stress-induced SG formation (30).
IX. Possible Redox Sensing Role of AIF
As outlined throughout the review, a number of studies indicated that the apoptogenic activity of AIF is independent on its oxidoreductase function (53, 136, 155, 212, 242, 252). Indeed, the death-inducing potency of AIF is defined by the structural elements positioned on the surface remote from the redox center [e.g., NLS2, the N- and C-terminal insertions, lysines 510 and 518 (84, 195, 242)], and naturally occurring flavin-free AIFsh can be apoptogenic as well (53). However, there is sufficient experimental and structural evidence allowing to suggest that both vital and lethal functions of the major FAD-containing forms, AIF1, AIF2, and AIFΔ1–102/118, may be redox controlled (38, 202).
Ligand-induced monomer–oligomer transition is a key process in cell signaling. Reduction of AIF with NADH not only promotes protein dimerization but also leads to significant structural reorganization. As a result, AIF can exist in two functionally distinct forms (Fig. 4A, D): monomeric, where the active site is blocked by the β-hairpin and regulatory peptides, and the NLS2 and N-terminal proteolytic sites are accessible, and dimeric, where the active site and the β-hairpin are accessible but NLS2 and one of the N-terminal proteolytic sites are not. In normal mitochondria, where reduced cofactors are abundant, the reduced AIF dimer would be the predominant form. Although it is still unknown what exactly AIF does in the IMS and whether other proteins are needed to support its function(s), severe abnormalities in mitochondrial morphology caused by defects in AIF expression or structure/function (35, 81) favor the hypothesis that the redox active dimer is part of a multiprotein complex and its enzymatic activity is critical for biogenesis/restructuring of mitochondrial membranes and assembly/functioning of the OXPHOS system (38, 202). If interaction of AIF with the membrane-associated partners is mediated by the redox-sensitive β-hairpin and regulatory peptides, the IMS redox status could control the enzymatic function through both the oxidoreduction of AIF and AIF-mediated protein–protein interactions.
Depletion of NAD(P)(H) occurs early in apoptosis and together with other metabolic changes eventually leads to mitochondrial ΔΨm dissipation and PTP opening (78). Presently, little is known about pyridine nucleotide homeostasis in the IMS and its relevance to the AIF function. It is evident though that mitochondrial NAD(H) can directly regulate AIF monomer–dimer equilibrium. Small fluctuations in the NAD(H) levels could transiently affect the oligomeric state and association of AIF with the IMS partners. Depletion of NAD(H), in turn, would shift equilibrium toward the inactive monomeric form, more susceptible to the N-terminal proteolysis (38). It still needs to be determined whether dimeric AIF can undergo proteolysis and pass through the OMM openings. Even if it does, it would interact with the cytoplasmic partners and mediate signal transduction differently than the oxidized monomer. The lifetime, posttranslational modifications, and interactions of AIF with the cytoplasmic partners, for instance, could be differently regulated via the distinctly folded regulatory peptide and β-hairpin (Figs. 4A, D and 8). Transport to the nucleus, on the other hand, could be modulated by NADH through changes in NLS2 accessibility (202) (Fig. 4D, E). Reduced dimers may possess a lesser capacity, if any, for cytonuclear translocation owing to unreachable NLS2. This and the lower DNA-binding ability (38, 202) make reduced AIF less apoptogenic than the oxidized monomeric form.
Via the outlined mechanism (Fig. 18), both redox and apoptogenic functions of the flavoprotein, as well as AIF-mediated signal transduction could be controlled by NAD(H) at the mitochondrial, cytoplasmic, and nuclear levels. By undergoing conformational and oligomeric state changes, AIF could respond to fluctuations in the compartmental redox status and transmit the signal to the interacting proteins, thus linking the NAD(H)-dependent metabolic pathways to apoptosis. Existence of multilevel checkpoints in the AIF-mediated apoptotic cascade is highly beneficial, as it enables the cell to reverse, postpone, or promote its demise depending on the severity of metabolic changes.
FIG. 18.

Possible redox-sensing mechanism of AIF. In normal mitochondria, the reduced dimer is likely to be the predominant form of AIF and could assist cristae formation and OXPHOS functioning through a specific redox activity or/and protein–protein interactions. Fluctuations in the NAD(H) levels and aberrations in the inner membrane could lead to transient changes in monomer–dimer equilibrium and dissociation of the AIF-mediated protein complexes (preapoptotic state). Depletion of pyridine nucleotides and severe defects in the inner membrane coupled with the protease activation and OMM permeabilization would lead to AIF monomerization, proteolysis, and translocation into the cytoplasm. Owing to the distinctly folded β-hairpin and regulatory peptides (shown as bold lines) and differences in the accessibility of NLS2, the oxidized and reduced forms of AIF could recruit different cytoplasmic partners and, consequently, initiate different signaling pathways. The loss of redox active mitochondrial AIF, leading to OXPHOS failure and increased production of reactive oxygen species, may be another factor promoting cell death. If the reduced dimer does relocate to the nucleus, it will be less apoptogenic than the oxidized monomer due to the lower affinity for DNA. Existence of separate mitochondrial, cytoplasmic, and nuclear NAD(H) pools allows compartmental regulation of the AIF-mediated signal transduction and creates multilevel check points through which the cell can reverse, postpone, or promote its demise. OXPHOS, oxidative phosphorylation.
X. Concluding Remarks
Despite the tremendous progress that has been made over the past decade in AIF research, there are still some controversies, highlighted in this review, that need to be resolved. The major remaining challenge is elucidation of the vital activity of the flavoprotein and how it relates to OXPHOS functioning, ROS detoxification, mitochondrial morphology, and cell cycle regulation. Being structurally distinct from other NAD(P)H-dependent oxidoreductases, mammalian AIFs appear to operate via a unique redox mechanism that should be further explored. Biochemical characterization of the AIF homologs from lower eukaryotes might also be helpful in solving the AIF-related puzzles as it could provide valuable insights into the enzymatic mechanism, assist the identification of a specific redox activity, and relate structure to function. Development of new genetic animal models, on the other hand, particularly those with the AIFΔR201 mutation, may help to unravel the pathophysiological mechanism associated with AIF defects and pinpoint mitochondrial proteins dependent on the AIF activity. Identification of novel cytoplasmic and nuclear partners of AIF and detailed investigations on those already identified are also critical, as they could test the proposed redox-sensing mechanism and provide deeper insights into relations between the cellular redox status and signaling networks. Considering the wide range and importance of cellular processes regulated by AIF and its partners, there is little doubt that future studies will make a significant contribution to better understanding of normal development, degenerative diseases, and carcinogenesis.
Abbreviations Used
- ΔΨm
mitochondrial trans-membrane potential
- AIF
apoptosis-inducing factor
- ANT
adenine nucleotide translocase
- CsA
cyclosporin A
- CTC
charge–transfer complex
- CypA
cyclophilin A
- eIF3g
eukaryotic translation initiation factor 3 subunit p44
- EndoG
endonuclease G
- ES
embryonic stem
- GR
glutathione reductase
- Hq
Harlequin
- Hsp70
70 kDa heat shock protein
- IMS
intermembrane space
- MLS
mitochondrial leading sequence
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- NLS
nuclear leading sequence
- OMM
outer mitochondrial membrane
- OPA1
optic atrophy 1
- OXPHOS
oxidative phosphorylation
- PAR
poly(ADP-ribose)
- PARP
PAR polymerase
- PCD
programmed cell death
- PPI
peptidyl prolyl isomerase
- PS
phosphatidylserine
- PTP
permeability transition pore
- ROS
reactive oxygen species
- SG
stress granule
- SH3
Src homology 3
- TULA
T-cell ubiquitin ligand
- VDAC
voltage-dependent anion channel
- XIAP
X-linked inhibitor of apoptosis protein
- z-vad.fmk
benzyloxycarbonyl-Val-Ala-Asp (Ome) fluoromethylketone
Footnotes
Reviewing Editors: Ted M. Dawson, Douglas Ethell, John Mieyal, Vijayalakshmi Ravindranath, Stefan Ryter, Anna Ivana Scovassi, Massimo Zeviani, and Boris Zhivotovsky
Acknowledgments
This work was supported in part by the National Institutes of Health grant GM67637. Due to space limitations, some important references could not be cited and I apologize for these omissions.
References
- 1.Ahel I. Ahel D. Matsusaka T. Clark AJ. Pines J. Boulton SJ. West SC. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451:81–85. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 2.Akematsu T. Endoh H. Role of apoptosis-inducing factor (AIF) in programmed nuclear death during conjugation in Tetrahymena thermophila. BMC Cell Biol. 2010;11:13. doi: 10.1186/1471-2121-11-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alano CC. Garnier P. Ying W. Higashi Y. Kauppinen TM. Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson P. Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 5.Andrabi SA. Kim NS. Yu SW. Wang H. Koh DW. Sasaki M. Klaus JA. Otsuka T. Zhang Z. Koehler RC. Hurn PD. Poirier GG. Dawson VL. Dawson TM. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A. 2006;103:18308–18313. doi: 10.1073/pnas.0606526103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antignani A. Youle RJ. How do Bax and Bak lead to permeabilization of the outer mitochondrial membrane? Curr Opin Cell Biol. 2006;18:685–689. doi: 10.1016/j.ceb.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Apostolova N. Cervera AM. Victor VM. Cadenas S. Sanjuan-Pla A. Alvarez-Barrientos A. Esplugues JV. McCreath KJ. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2006;13:354–357. doi: 10.1038/sj.cdd.4401776. [DOI] [PubMed] [Google Scholar]
- 8.Arama E. Bader M. Srivastava M. Bergmann A. Steller H. The two Drosophila cytochrome c proteins can function in both respiration and caspase activation. EMBO J. 2006;25:232–243. doi: 10.1038/sj.emboj.7600920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arnoult D. Gaume B. Karbowski M. Sharpe JC. Cecconi F. Youle RJ. Mitochondrial release of AIF and EndoG requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J. 2003;22:4385–4399. doi: 10.1093/emboj/cdg423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arnoult D. Parone P. Martinou JC. Antonsson B. Estaquier J. Ameisen JC. Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol. 2002;159:923–929. doi: 10.1083/jcb.200207071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arnoult D. Tatischeff I. Estaquier J. Girard M. Sureau F. Tissier JP. Grodet A. Dellinger M. Traincard F. Kahn A. Ameisen JC. Petit PX. On the evolutionary conservation of the cell death pathway: mitochondrial release of an apoptosis-inducing factor during Dictyostelium discoideum cell death. Mol Biol Cell. 2001;12:3016–3030. doi: 10.1091/mbc.12.10.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Artigues A. Iriarte A. Martinez-Carrion M. Identification of Hsc70 binding sites in mitochondrial aspartate aminotransferase. Arch Biochem Biophys. 2006;450:30–38. doi: 10.1016/j.abb.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 13.Artus C. Boujrad H. Bouharrour A. Brunelle MN. Hoos S. Yuste VJ. Lenormand P. Rousselle JC. Namane A. England P. Lorenzo HK. Susin SA. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacting with histone H2AX. EMBO J. 2010;29:1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Avramut M. Achim CL. Immunophilins in nervous system degeneration and regeneration. Curr Top Med Chem. 2003;3:1376–1382. doi: 10.2174/1568026033451871. [DOI] [PubMed] [Google Scholar]
- 15.Baines CP. Kaiser RA. Sheiko T. Craigen WJ. Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banerji J. Sands J. Strominger JL. Spies T. A gene pair from the human major histocompatibility complex encodes large proline-rich proteins with multiple repeated motifs and a single ubiquitin-like domain. Proc Natl Acad Sci U S A. 1990;87:2374–2378. doi: 10.1073/pnas.87.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baud O. Li J. Zhang Y. Neve RL. Volpe JJ. Rosenberg PA. Nitric oxide-induced cell death in developing oligodendrocytes is associated with mitochondrial dysfunction and apoptosis-inducing factor translocation. Eur J Neurosci. 2004;20:1713–1726. doi: 10.1111/j.1460-9568.2004.03616.x. [DOI] [PubMed] [Google Scholar]
- 18.Bauer H. Kanzok SM. Schirmer RH. Thioredoxin-2 but not thioredoxin-1 is a substrate of thioredoxin peroxidase-1 from Drosophila melanogaster: isolation and characterization of a second thioredoxin in D. melanogaster and evidence for distinct biological functions of Trx-1 and Trx-2. J Biol Chem. 2002;277:17457–17463. doi: 10.1074/jbc.M200636200. [DOI] [PubMed] [Google Scholar]
- 19.Belizario JE. Alves J. Garay-Malpartida M. Occhiucci JM. Coupling caspase cleavage and proteasomal degradation of proteins carrying PEST motif. Curr Protein Pept Sci. 2008;9:210–220. doi: 10.2174/138920308784534023. [DOI] [PubMed] [Google Scholar]
- 20.Belizario JE. Alves J. Occhiucci JM. Garay-Malpartida M. Sesso A. A mechanistic view of mitochondrial death decision pores. Braz J Med Biol Res. 2007;40:1011–1024. doi: 10.1590/s0100-879x2006005000109. [DOI] [PubMed] [Google Scholar]
- 21.Billington RA. Genazzani AA. Travelli C. Condorelli F. NAD depletion by FK866 induces autophagy. Autophagy. 2008;4:385–387. doi: 10.4161/auto.5635. [DOI] [PubMed] [Google Scholar]
- 22.Block KL. Vornlocher HP. Hershey JW. Characterization of cDNAs encoding the p44 and p35 subunits of human translation initiation factor eIF3. J Biol Chem. 1998;273:31901–31908. doi: 10.1074/jbc.273.48.31901. [DOI] [PubMed] [Google Scholar]
- 23.Bossy-Wetzel E. Barsoum MJ. Godzik A. Schwarzenbacher R. Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–716. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 24.Braun JS. Sublett JE. Freyer D. Mitchell TJ. Cleveland JL. Tuomanen EI. Weber JR. Pneumococcal pneumolysin and H2O2 mediate brain cell apoptosis during meningitis. J Clin Invest. 2002;109:19–27. doi: 10.1172/JCI12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown D. Yu BD. Joza N. Benit P. Meneses J. Firpo M. Rustin P. Penninger JM. Martin GR. Loss of AIF function causes cell death in the mouse embryo, but the temporal progression of patterning is normal. Proc Natl Acad Sci U S A. 2006;103:9918–9923. doi: 10.1073/pnas.0603950103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burton TR. Eisenstat DD. Gibson SB. BNIP3 (Bcl-2 19 kDa interacting protein) acts as transcriptional repressor of apoptosis-inducing factor expression preventing cell death in human malignant gliomas. J Neurosci. 2009;29:4189–4199. doi: 10.1523/JNEUROSCI.5747-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cande C. Cecconi F. Dessen P. Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 28.Cande C. Cohen I. Daugas E. Ravagnan L. Larochette N. Zamzami N. Kroemer G. Apoptosis-inducing factor (AIF): a novel caspase-independent death effector released from mitochondria. Biochimie. 2002;84:215–222. doi: 10.1016/s0300-9084(02)01374-3. [DOI] [PubMed] [Google Scholar]
- 29.Cande C. Vahsen N. Kouranti I. Schmitt E. Daugas E. Spahr C. Luban J. Kroemer RT. Giordanetto F. Garrido C. Penninger JM. Kroemer G. AIF and cyclophilin A cooperate in apoptosis-associated chromatinolysis. Oncogene. 2004;23:1514–1521. doi: 10.1038/sj.onc.1207279. [DOI] [PubMed] [Google Scholar]
- 30.Cande C. Vahsen N. Metivier D. Tourriere H. Chebli K. Garrido C. Tazi J. Kroemer G. Regulation of cytoplasmic stress granules by apoptosis-inducing factor. J Cell Sci. 2004;117:4461–4468. doi: 10.1242/jcs.01356. [DOI] [PubMed] [Google Scholar]
- 31.Cao G. Xing J. Xiao X. Liou AK. Gao Y. Yin XM. Clark RS. Graham SH. Chen J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–9293. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chaitanya GV. Babu PP. Multiple apoptogenic proteins are involved in the nuclear translocation of Apoptosis Inducing Factor during transient focal cerebral ischemia in rat. Brain Res. 2008;1246:178–190. doi: 10.1016/j.brainres.2008.09.075. [DOI] [PubMed] [Google Scholar]
- 33.Chaudhuri J. Chowdhury D. Maitra U. Distinct functions of eukaryotic translation initiation factors eIF1A and eIF3 in the formation of the 40 S ribosomal preinitiation complex. J Biol Chem. 1999;274:17975–17980. doi: 10.1074/jbc.274.25.17975. [DOI] [PubMed] [Google Scholar]
- 34.Chen JT. Huang CY. Chiang YY. Chen WH. Chiou SH. Chen CY. Chow KC. HGF increases cisplatin resistance via downregulation of AIF in lung cancer cells. Am J Respir Cell Mol Biol. 2008;38:559–565. doi: 10.1165/rcmb.2007-0001OC. [DOI] [PubMed] [Google Scholar]
- 35.Cheung EC. Joza N. Steenaart NA. McClellan KA. Neuspiel M. McNamara S. MacLaurin JG. Rippstein P. Park DS. Shore GC. McBride HM. Penninger JM. Slack RS. Dissociating the dual roles of apoptosis-inducing factor in maintaining mitochondrial structure and apoptosis. EMBO J. 2006;25:4061–4073. doi: 10.1038/sj.emboj.7601276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheung EC. Melanson-Drapeau L. Cregan SP. Vanderluit JL. Ferguson KL. McIntosh WC. Park DS. Bennett SA. Slack RS. Apoptosis-inducing factor is a key factor in neuronal cell death propagated by BAX-dependent and BAX-independent mechanisms. J Neurosci. 2005;25:1324–1334. doi: 10.1523/JNEUROSCI.4261-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinta SJ. Rane A. Yadava N. Andersen JK. Nicholls DG. Polster BM. Reactive oxygen species regulation by AIF- and complex I-depleted brain mitochondria. Free Radic Biol Med. 2009;46:939–947. doi: 10.1016/j.freeradbiomed.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Churbanova IY. Sevrioukova IF. Redox-dependent changes in molecular properties of mitochondrial apoptosis-inducing factor. J Biol Chem. 2008;283:5622–5631. doi: 10.1074/jbc.M709147200. [DOI] [PubMed] [Google Scholar]
- 39.Cipriani G. Rapizzi E. Vannacci A. Rizzuto R. Moroni F. Chiarugi A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem. 2005;280:17227–17234. doi: 10.1074/jbc.M414526200. [DOI] [PubMed] [Google Scholar]
- 40.Colgan J. Asmal M. Luban J. Isolation, characterization and targeted disruption of mouse Ppia: cyclophilin A is not essential for mammalian cell viability. Genomics. 2000;68:167–178. doi: 10.1006/geno.2000.6295. [DOI] [PubMed] [Google Scholar]
- 41.Colgan J. Asmal M. Yu B. Luban J. Cyclophilin A-deficient mice are resistant to immunosuppression by cyclosporine. J Immunol. 2005;174:6030–6038. doi: 10.4049/jimmunol.174.10.6030. [DOI] [PubMed] [Google Scholar]
- 42.Collingwood TS. Smirnova EV. Bogush M. Carpino N. Annan RS. Tsygankov AY. T-cell ubiquitin ligand affects cell death through a functional interaction with apoptosis-inducing factor, a key factor of caspase-independent apoptosis. J Biol Chem. 2007;282:30920–30928. doi: 10.1074/jbc.M706870200. [DOI] [PubMed] [Google Scholar]
- 43.Cook PJ. Ju BG. Telese F. Wang X. Glass CK. Rosenfeld MG. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458:591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cornillon S. Foa C. Davoust J. Buonavista N. Gross JD. Golstein P. Programmed cell death in Dictyostelium. J Cell Sci. 1994;107((Pt 10)):2691–2704. doi: 10.1242/jcs.107.10.2691. [DOI] [PubMed] [Google Scholar]
- 45.Cregan SP. Dawson VL. Slack RS. Role of AIF in caspase-dependent and caspase-independent cell death. Oncogene. 2004;23:2785–2796. doi: 10.1038/sj.onc.1207517. [DOI] [PubMed] [Google Scholar]
- 46.Cregan SP. Fortin A. MacLaurin JG. Callaghan SM. Cecconi F. Yu SW. Dawson TM. Dawson VL. Park DS. Kroemer G. Slack RS. Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol. 2002;158:507–517. doi: 10.1083/jcb.200202130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Culmsee C. Zhu C. Landshamer S. Becattini B. Wagner E. Pellecchia M. Blomgren K. Plesnila N. Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J Neurosci. 2005;25:10262–10272. doi: 10.1523/JNEUROSCI.2818-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daugas E. Nochy D. Ravagnan L. Loeffler M. Susin SA. Zamzami N. Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett. 2000;476:118–123. doi: 10.1016/s0014-5793(00)01731-2. [DOI] [PubMed] [Google Scholar]
- 49.Daugas E. Susin SA. Zamzami N. Ferri KF. Irinopoulou T. Larochette N. Prevost MC. Leber B. Andrews D. Penninger J. Kroemer G. Mitochondrio-nuclear translocation of AIF in apoptosis and necrosis. FASEB J. 2000;14:729–739. [PubMed] [Google Scholar]
- 50.David KK. Andrabi SA. Dawson TM. Dawson VL. Parthanatos, a messenger of death. Front Biosci. 2009;14:1116–1128. doi: 10.2741/3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.David KK. Sasaki M. Yu SW. Dawson TM. Dawson VL. EndoG is dispensable in embryogenesis and apoptosis. Cell Death Differ. 2006;13:1147–1155. doi: 10.1038/sj.cdd.4401787. [DOI] [PubMed] [Google Scholar]
- 52.Davis MC. Ward JG. Herrick G. Allis CD. Programmed nuclear death: apoptotic-like degradation of specific nuclei in conjugating Tetrahymena. Dev Biol. 1992;154:419–432. doi: 10.1016/0012-1606(92)90080-z. [DOI] [PubMed] [Google Scholar]
- 53.Delettre C. Yuste VJ. Moubarak RS. Bras M. Lesbordes-Brion JC. Petres S. Bellalou J. Susin SA. AIFsh, a novel apoptosis-inducing factor (AIF) pro-apoptotic isoform with potential pathological relevance in human cancer. J Biol Chem. 2006;281:6413–6427. doi: 10.1074/jbc.M509884200. [DOI] [PubMed] [Google Scholar]
- 54.Delettre C. Yuste VJ. Moubarak RS. Bras M. Robert N. Susin SA. Identification and characterization of AIFsh2, a mitochondrial apoptosis-inducing factor (AIF) isoform with NADH oxidase activity. J Biol Chem. 2006;281:18507–18518. doi: 10.1074/jbc.M601751200. [DOI] [PubMed] [Google Scholar]
- 55.Dereeper A. Guignon V. Blanc G. Audic S. Buffet S. Chevenet F. Dufayard JF. Guindon S. Lefort V. Lescot M. Claverie JM. Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008;36:W465–W469. doi: 10.1093/nar/gkn180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Desmots F. Russell HR. Lee Y. Boyd K. McKinnon PJ. The reaper-binding protein scythe modulates apoptosis and proliferation during mammalian development. Mol Cell Biol. 2005;25:10329–10337. doi: 10.1128/MCB.25.23.10329-10337.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Desmots F. Russell HR. Michel D. McKinnon PJ. Scythe regulates apoptosis-inducing factor stability during endoplasmic reticulum stress-induced apoptosis. J Biol Chem. 2008;283:3264–3271. doi: 10.1074/jbc.M706419200. [DOI] [PubMed] [Google Scholar]
- 58.Devaux PF. Herrmann A. Ohlwein N. Kozlov MM. How lipid flippases can modulate membrane structure. Biochim Biophys Acta. 2008;1778:1591–1600. doi: 10.1016/j.bbamem.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 59.Deveraux QL. Reed JC. IAP family proteins—suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 60.Dornan J. Taylor P. Walkinshaw MD. Structures of immunophilins and their ligand complexes. Curr Top Med Chem. 2003;3:1392–1409. doi: 10.2174/1568026033451899. [DOI] [PubMed] [Google Scholar]
- 61.Du L. Zhang X. Han YY. Burke NA. Kochanek PM. Watkins SC. Graham SH. Carcillo JA. Szabo C. Clark RS. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J Biol Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 62.Dubrez-Daloz L. Dupoux A. Cartier J. IAPs: more than just inhibitors of apoptosis proteins. Cell Cycle. 2008;7:1036–1046. doi: 10.4161/cc.7.8.5783. [DOI] [PubMed] [Google Scholar]
- 63.Eckelman BP. Salvesen GS. Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Escobar-Henriques M. Langer T. Mitochondrial shaping cuts. Biochim Biophys Acta. 2006;1763:422–429. doi: 10.1016/j.bbamcr.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 65.Esser C. Alberti S. Hohfeld J. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta. 2004;1695:171–188. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 66.Feraud O. Debili N. Penninger JM. Kroemer G. Cavitation of embryoid bodies requires optimal oxidative phosphorylation and AIF. Cell Death Differ. 2007;14:385–387. doi: 10.1038/sj.cdd.4402041. [DOI] [PubMed] [Google Scholar]
- 67.Ferri KF. Jacotot E. Blanco J. Este JA. Zamzami N. Susin SA. Xie Z. Brothers G. Reed JC. Penninger JM. Kroemer G. Apoptosis control in syncytia induced by the HIV type 1-envelope glycoprotein complex: role of mitochondria and caspases. J Exp Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feshchenko EA. Smirnova EV. Swaminathan G. Teckchandani AM. Agrawal R. Band H. Zhang X. Annan RS. Carr SA. Tsygankov AY. TULA: an SH3- and UBA-containing protein that binds to c-Cbl and ubiquitin. Oncogene. 2004;23:4690–4706. doi: 10.1038/sj.onc.1207627. [DOI] [PubMed] [Google Scholar]
- 69.Formentini L. Macchiarulo A. Cipriani G. Camaioni E. Rapizzi E. Pellicciari R. Moroni F. Chiarugi A. Poly(ADP-ribose) catabolism triggers AMP-dependent mitochondrial energy failure. J Biol Chem. 2009;284:17668–17676. doi: 10.1074/jbc.M109.002931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fourie AM. Sambrook JF. Gething MJ. Common and divergent peptide binding specificities of hsp70 molecular chaperones. J Biol Chem. 1994;269:30470–30478. [PubMed] [Google Scholar]
- 71.Fratelli M. Goodwin LO. Orom UA. Lombardi S. Tonelli R. Mengozzi M. Ghezzi P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc Natl Acad Sci U S A. 2005;102:13998–14003. doi: 10.1073/pnas.0504398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Freeman BC. Myers MP. Schumacher R. Morimoto RI. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. EMBO J. 1995;14:2281–2292. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Frezza C. Cipolat S. Martins de Brito O. Micaroni M. Beznoussenko GV. Rudka T. Bartoli D. Polishuck RS. Danial NN. De Strooper B. Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 74.Gagne JP. Isabelle M. Lo KS. Bourassa S. Hendzel MJ. Dawson VL. Dawson TM. Poirier GG. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gallego MA. Joseph B. Hemstrom TH. Tamiji S. Mortier L. Kroemer G. Formstecher P. Zhivotovsky B. Marchetti P. Apoptosis-inducing factor determines the chemoresistance of non-small-cell lung carcinomas. Oncogene. 2004;23:6282–6291. doi: 10.1038/sj.onc.1207835. [DOI] [PubMed] [Google Scholar]
- 76.Galluzzi L. Morselli E. Kepp O. Kroemer G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim Biophys Acta. 2009;1787:402–413. doi: 10.1016/j.bbabio.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 77.Garcia M. Bondada V. Geddes JW. Mitochondrial localization of μ-calpain. Biochem Biophys Res Commun. 2005;338:1241–1247. doi: 10.1016/j.bbrc.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 78.Gendron MC. Schrantz N. Metivier D. Kroemer G. Maciorowska Z. Sureau F. Koester S. Petit PX. Oxidation of pyridine nucleotides during Fas- and ceramide-induced apoptosis in Jurkat cells: correlation with changes in mitochondria, glutathione depletion, intracellular acidification and caspase 3 activation. Biochem J. 2001;353:357–367. [PMC free article] [PubMed] [Google Scholar]
- 79.Georgopoulos C. Welch WJ. Role of the major heat shock proteins as molecular chaperones. Annu Rev Cell Biol. 1993;9:601–634. doi: 10.1146/annurev.cb.09.110193.003125. [DOI] [PubMed] [Google Scholar]
- 80.Ghavami S. Asoodeh A. Klonisch T. Halayko AJ. Kadkhoda K. Kroczak TJ. Gibson SB. Booy EP. Naderi-Manesh H. Los M. Brevinin-2R(1) semi-selectively kills cancer cells by a distinct mechanism, which involves the lysosomal-mitochondrial death pathway. J Cell Mol Med. 2008;12:1005–1022. doi: 10.1111/j.1582-4934.2008.00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ghezzi D. Sevrioukova I. Invernizzi F. Lamperti C. Mora M. D'Adamo P. Novara F. Zuffardi O. Uziel G. Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am J Hum Genet. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gmeiner WH. Horita DA. Implications of SH3 domain structure and dynamics for protein regulation and drug design. Cell Biochem Biophys. 2001;35:127–140. doi: 10.1385/CBB:35:2:127. [DOI] [PubMed] [Google Scholar]
- 83.Goyal G. Fell B. Sarin A. Youle RJ. Sriram V. Role of mitochondrial remodeling in programmed cell death in Drosophila melanogaster. Dev Cell. 2007;12:807–816. doi: 10.1016/j.devcel.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gurbuxani S. Schmitt E. Cande C. Parcellier A. Hammann A. Daugas E. Kouranti I. Spahr C. Pance A. Kroemer G. Garrido C. Heat shock protein 70 binding inhibits the nuclear import of apoptosis-inducing factor. Oncogene. 2003;22:6669–6678. doi: 10.1038/sj.onc.1206794. [DOI] [PubMed] [Google Scholar]
- 85.Halestrap AP. Connern CP. Griffiths EJ. Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–172. [PubMed] [Google Scholar]
- 86.Hangen E. Blomgren K. Benit P. Kroemer G. Modjtahedi N. Life with or without AIF. Trends Biochem Sci. 2010;35:278–287. doi: 10.1016/j.tibs.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 87.Hangen E. De Zio D. Bordi M. Zhu C. Dessen P. Caffin F. Lachkar S. Perfettini JL. Lazar V. Benard J. Fimia GM. Piacentini M. Harper F. Pierron G. Vicencio JM. Benit P. de Andrade A. Hoglinger G. Culmsee C. Rustin P. Blomgren K. Cecconi F. Kroemer G. Modjtahedi N. A brain-specific isoform of mitochondrial apoptosis-inducing factor: AIF2. Cell Death Differ. 2010;17:1155–1166. doi: 10.1038/cdd.2009.211. [DOI] [PubMed] [Google Scholar]
- 88.Hartl FU. Heat shock proteins in protein folding and membrane translocation. Semin Immunol. 1991;3:5–16. [PubMed] [Google Scholar]
- 89.Herrmann JM. Hell K. Chopped, trapped or tacked—protein translocation into the IMS of mitochondria. Trends Biochem Sci. 2005;30:205–211. doi: 10.1016/j.tibs.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 90.Hervey WJ., 4th Khalsa-Moyers G. Lankford PK. Owens ET. McKeown CK. Lu TY. Foote LJ. Asano KG. Morrell-Falvey JL. McDonald WH. Pelletier DA. Hurst GB. Evaluation of affinity-tagged protein expression strategies using local and global isotope ratio measurements. J Proteome Res. 2009;8:3675–3688. doi: 10.1021/pr801088f. [DOI] [PubMed] [Google Scholar]
- 91.Hisatomi T. Nakazawa T. Noda K. Almulki L. Miyahara S. Nakao S. Ito Y. She H. Kohno R. Michaud N. Ishibashi T. Hafezi-Moghadam A. Badley AD. Kroemer G. Miller JW. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008;118:2025–2038. doi: 10.1172/JCI34267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hofer-Warbinek R. Schmid JA. Stehlik C. Binder BR. Lipp J. de Martin R. Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J Biol Chem. 2000;275:22064–22068. doi: 10.1074/jbc.M910346199. [DOI] [PubMed] [Google Scholar]
- 93.Hohfeld J. Cyr DM. Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. EMBO Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoppe G. Chai YC. Crabb JW. Sears J. Protein S-glutathionylation in retinal pigment epithelium converts heat shock protein 70 to an active chaperone. Exp Eye Res. 2004;78:1085–1092. doi: 10.1016/j.exer.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 95.Huang H. Joazeiro CA. Bonfoco E. Kamada S. Leverson JD. Hunter T. The inhibitor of apoptosis, cIAP2, functions as a ubiquitin-protein ligase and promotes in vitro monoubiquitination of caspases 3 and 7. J Biol Chem. 2000;275:26661–26664. doi: 10.1074/jbc.C000199200. [DOI] [PubMed] [Google Scholar]
- 96.Huerta S. Heinzerling JH. Anguiano-Hernandez YM. Huerta-Yepez S. Lin J. Chen D. Bonavida B. Livingston EH. Modification of gene products involved in resistance to apoptosis in metastatic colon cancer cells: roles of Fas, Apaf-1, NFkappaB, IAPs, Smac/DIABLO, and AIF. J Surg Res. 2007;142:184–194. doi: 10.1016/j.jss.2006.12.551. [DOI] [PubMed] [Google Scholar]
- 97.Ishimura R. Martin GR. Ackerman SL. Loss of apoptosis-inducing factor results in cell-type-specific neurogenesis defects. J Neurosci. 2008;28:4938–4948. doi: 10.1523/JNEUROSCI.0229-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ivery MT. Immunophilins: switched on protein binding domains? Med Res Rev. 2000;20:452–484. doi: 10.1002/1098-1128(200011)20:6<452::aid-med2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 99.Jeong EG. Lee JW. Soung YH. Nam SW. Kim SH. Lee JY. Yoo NJ. Lee SH. Immunohistochemical and mutational analysis of apoptosis-inducing factor (AIF) in colorectal carcinomas. APMIS. 2006;114:867–873. doi: 10.1111/j.1600-0463.2006.apm_502.x. [DOI] [PubMed] [Google Scholar]
- 100.John GB. Shang Y. Li L. Renken C. Mannella CA. Selker JM. Rangell L. Bennett MJ. Zha J. The mitochondrial inner membrane protein mitofilin controls cristae morphology. Mol Biol Cell. 2005;16:1543–1554. doi: 10.1091/mbc.E04-08-0697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Joshi A. Bondada V. Geddes JW. Mitochondrial micro-calpain is not involved in the processing of apoptosis-inducing factor. Exp Neurol. 2009;218:221–227. doi: 10.1016/j.expneurol.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Joza N. Galindo K. Pospisilik JA. Benit P. Rangachari M. Kanitz EE. Nakashima Y. Neely GG. Rustin P. Abrams JM. Kroemer G. Penninger JM. The molecular archaeology of a mitochondrial death effector: AIF in Drosophila. Cell Death Differ. 2008;15:1009–1018. doi: 10.1038/cdd.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Joza N. Oudit GY. Brown D. Benit P. Kassiri Z. Vahsen N. Benoit L. Patel MM. Nowikovsky K. Vassault A. Backx PH. Wada T. Kroemer G. Rustin P. Penninger JM. Muscle-specific loss of apoptosis-inducing factor leads to mitochondrial dysfunction, skeletal muscle atrophy, and dilated cardiomyopathy. Mol Cell Biol. 2005;25:10261–10272. doi: 10.1128/MCB.25.23.10261-10272.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Joza N. Pospisilik JA. Hangen E. Hanada T. Modjtahedi N. Penninger JM. Kroemer G. AIF: not just an apoptosis-inducing factor. Ann N Y Acad Sci. 2009;1171:2–11. doi: 10.1111/j.1749-6632.2009.04681.x. [DOI] [PubMed] [Google Scholar]
- 105.Joza N. Susin SA. Daugas E. Stanford WL. Cho SK. Li CY. Sasaki T. Elia AJ. Cheng HY. Ravagnan L. Ferri KF. Zamzami N. Wakeham A. Hakem R. Yoshida H. Kong YY. Mak TW. Zuniga-Pflucker JC. Kroemer G. Penninger JM. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 106.Kaneko T. Li L. Li SS. The SH3 domain: a family of versatile peptide- and protein-recognition module. Front Biosci. 2008;13:4938–4952. doi: 10.2741/3053. [DOI] [PubMed] [Google Scholar]
- 107.Kang YH. Yi MJ. Kim MJ. Park MT. Bae S. Kang CM. Cho CK. Park IC. Park MJ. Rhee CH. Hong SI. Chung HY. Lee YS. Lee SJ. Caspase-independent cell death by arsenic trioxide in human cervical cancer cells: reactive oxygen species-mediated poly(ADP-ribose) polymerase-1 activation signals apoptosis-inducing factor release from mitochondria. Cancer Res. 2004;64:8960–8967. doi: 10.1158/0008-5472.CAN-04-1830. [DOI] [PubMed] [Google Scholar]
- 108.Kar P. Chakraborti T. Samanta K. Chakraborti S. Submitochondrial localization of associated μ-calpain and calpastatin. Arch Biochem Biophys. 2008;470:176–186. doi: 10.1016/j.abb.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 109.Kar P. Chakraborti T. Roy S. Choudhury R. Chakraborti S. Identification of calpastatin and μ-calpain and studies of their association in pulmonary smooth muscle mitochondria. Arch Biochem Biophys. 2007;466:290–299. doi: 10.1016/j.abb.2007.07.022. [DOI] [PubMed] [Google Scholar]
- 110.Karbowski M. Lee YJ. Gaume B. Jeong SY. Frank S. Nechushtan A. Santel A. Fuller M. Smith CL. Youle RJ. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol. 2002;159:931–938. doi: 10.1083/jcb.200209124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Karbowski M. Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. doi: 10.1038/sj.cdd.4401260. [DOI] [PubMed] [Google Scholar]
- 112.Karkkainen S. Hiipakka M. Wang JH. Kleino I. Vaha-Jaakkola M. Renkema GH. Liss M. Wagner R. Saksela K. Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 2006;7:186–191. doi: 10.1038/sj.embor.7400596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karras GI. Kustatscher G. Buhecha HR. Allen MD. Pugieux C. Sait F. Bycroft M. Ladurner AG. The macro domain is an ADP-ribose binding module. EMBO J. 2005;24:1911–1920. doi: 10.1038/sj.emboj.7600664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kedersha N. Anderson P. Stress granules: sites of mRNA triage that regulate mRNA stability and translatability. Biochem Soc Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- 115.Kiang JG. Tsokos GC. Heat shock protein 70 kDa: molecular biology, biochemistry, and physiology. Pharmacol Ther. 1998;80:183–201. doi: 10.1016/s0163-7258(98)00028-x. [DOI] [PubMed] [Google Scholar]
- 116.Kim JT. Kim KD. Song EY. Lee HG. Kim JW. Chae SK. Kim E. Lee MS. Yang Y. Lim JS. Apoptosis-inducing factor (AIF) inhibits protein synthesis by interacting with the eukaryotic translation initiation factor 3 subunit p44 (eIF3g) FEBS Lett. 2006;580:6375–6383. doi: 10.1016/j.febslet.2006.10.049. [DOI] [PubMed] [Google Scholar]
- 117.Kim KM. Yi EC. Baker D. Zhang KY. Post-translational modification of the N-terminal His tag interferes with the crystallization of the wild-type and mutant SH3 domains from chicken src tyrosine kinase. Acta Crystallogr D Biol Crystallogr. 2001;57:759–762. doi: 10.1107/s0907444901002918. [DOI] [PubMed] [Google Scholar]
- 118.Kim TH. Zhao Y. Ding WX. Shin JN. He X. Seo YW. Chen J. Rabinowich H. Amoscato AA. Yin XM. Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome c release. Mol Biol Cell. 2004;15:3061–3072. doi: 10.1091/mbc.E03-12-0864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kleiger G. Eisenberg D. GXXXG and GXXXA motifs stabilize FAD and NAD(P)-binding Rossmann folds through Cα-H…O hydrogen bonds and van der waals interactions. J Mol Biol. 2002;323:69–76. doi: 10.1016/s0022-2836(02)00885-9. [DOI] [PubMed] [Google Scholar]
- 120.Klein JA. Longo-Guess CM. Rossmann MP. Seburn KL. Hurd RE. Frankel WN. Bronson RT. Ackerman SL. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 121.Kluck RM. Esposti MD. Perkins G. Renken C. Kuwana T. Bossy-Wetzel E. Goldberg M. Allen T. Barber MJ. Green DR. Newmeyer DD. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kokoszka JE. Waymire KG. Levy SE. Sligh JE. Cai J. Jones DP. MacGregor GR. Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kondo K. Obitsu S. Ohta S. Matsunami K. Otsuka H. Teshima R. Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and MEK inhibition. J Biol Chem. 2010;285:13079–13091. doi: 10.1074/jbc.M109.044206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Koonin EV. Aravind L. Origin and evolution of eukaryotic apoptosis: the bacterial connection. Cell Death Differ. 2002;9:394–404. doi: 10.1038/sj.cdd.4400991. [DOI] [PubMed] [Google Scholar]
- 125.Kuwana T. Mackey MR. Perkins G. Ellisman MH. Latterich M. Schneiter R. Green DR. Newmeyer DD. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 126.Kwon CH. Park JY. Kim TH. Woo JS. Kim YK. Ciglitazone induces apoptosis via activation of p38 MAPK and AIF nuclear translocation mediated by reactive oxygen species and Ca2+ in opossum kidney cells. Toxicology. 2009;257:1–9. doi: 10.1016/j.tox.2008.11.019. [DOI] [PubMed] [Google Scholar]
- 127.Landshamer S. Hoehn M. Barth N. Duvezin-Caubet S. Schwake G. Tobaben S. Kazhdan I. Becattini B. Zahler S. Vollmar A. Pellecchia M. Reichert A. Plesnila N. Wagner E. Culmsee C. Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 2008;15:1553–1563. doi: 10.1038/cdd.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Larkin MA. Blackshields G. Brown NP. Chenna R. McGettigan PA. McWilliam H. Valentin F. Wallace IM. Wilm A. Lopez R. Thompson JD. Gibson TJ. Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 129.Larochette N. Decaudin D. Jacotot E. Brenner C. Marzo I. Susin SA. Zamzami N. Xie Z. Reed J. Kroemer G. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp Cell Res. 1999;249:413–421. doi: 10.1006/excr.1999.4519. [DOI] [PubMed] [Google Scholar]
- 130.Lee YJ. Jeong SY. Karbowski M. Smith CL. Youle RJ. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell. 2004;15:5001–5011. doi: 10.1091/mbc.E04-04-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lemasters JJ. Qian T. Bradham CA. Brenner DA. Cascio WE. Trost LC. Nishimura Y. Nieminen AL. Herman B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- 132.Li F. Mao HP. Ruchalski KL. Wang YH. Choy W. Schwartz JH. Borkan SC. Heat stress prevents mitochondrial injury in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol. 2002;283:C917–C926. doi: 10.1152/ajpcell.00517.2001. [DOI] [PubMed] [Google Scholar]
- 133.Li X. Klaus JA. Zhang J. Xu Z. Kibler KK. Andrabi SA. Rao K. Yang ZJ. Dawson TM. Dawson VL. Koehler RC. Contributions of poly(ADP-ribose) polymerase-1 and −2 to nuclear translocation of apoptosis-inducing factor and injury from focal cerebral ischemia. J Neurochem. 2010;113:1013–1022. doi: 10.1111/j.1471-4159.2010.06667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lindsten T. Ross AJ. King A. Zong WX. Rathmell JC. Shiels HA. Ulrich E. Waymire KG. Mahar P. Frauwirth K. Chen Y. Wei M. Eng VM. Adelman DM. Simon MC. Ma A. Golden JA. Evan G. Korsmeyer SJ. MacGregor GR. Thompson CB. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liou AK. Zhou Z. Pei W. Lim TM. Yin XM. Chen J. BimEL up-regulation potentiates AIF translocation and cell death in response to MPTP. FASEB J. 2005;19:1350–1352. doi: 10.1096/fj.04-3258fje. [DOI] [PubMed] [Google Scholar]
- 136.Loeffler M. Daugas E. Susin SA. Zamzami N. Metivier D. Nieminen AL. Brothers G. Penninger JM. Kroemer G. Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J. 2001;15:758–767. doi: 10.1096/fj.00-0388com. [DOI] [PubMed] [Google Scholar]
- 137.Lorenzo HK. Susin SA. Penninger J. Kroemer G. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- 138.Lu C. Zhu F. Cho YY. Tang F. Zykova T. Ma WY. Bode AM. Dong Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol Cell. 2006;23:121–132. doi: 10.1016/j.molcel.2006.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lui JC. Kong SK. Heat shock protein 70 inhibits the nuclear import of apoptosis-inducing factor to avoid DNA fragmentation in TF-1 cells during erythropoiesis. FEBS Lett. 2007;581:109–117. doi: 10.1016/j.febslet.2006.11.082. [DOI] [PubMed] [Google Scholar]
- 140.Maeda H. Sahara H. Mori Y. Torigo T. Kamiguchi K. Tamura Y. Hirata K. Sato N. Biological heterogeneity of the peptide-binding motif of the 70-kDa heat shock protein by surface plasmon resonance analysis. J Biol Chem. 2007;282:26956–26962. doi: 10.1074/jbc.M703436200. [DOI] [PubMed] [Google Scholar]
- 141.Maestre I. Jordan J. Calvo S. Reig JA. Cena V. Soria B. Prentki M. Roche E. Mitochondrial dysfunction is involved in apoptosis induced by serum withdrawal and fatty acids in the beta-cell line INS-1. Endocrinology. 2003;144:335–345. doi: 10.1210/en.2001-211282. [DOI] [PubMed] [Google Scholar]
- 142.Maiuri MC. Zalckvar E. Kimchi A. Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 143.Manchen ST. Hubberstey AV. Human Scythe contains a functional nuclear localization sequence and remains in the nucleus during staurosporine-induced apoptosis. Biochem Biophys Res Commun. 2001;287:1075–1082. doi: 10.1006/bbrc.2001.5701. [DOI] [PubMed] [Google Scholar]
- 144.Mannella CA. Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta. 2006;1763:542–548. doi: 10.1016/j.bbamcr.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 145.Manteca A. Sanchez J. Recombinant cyclophilins lack nuclease activity. J Bacteriol. 2004;186:6325–6326. doi: 10.1128/JB.186.18.6325-6326.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martin SJ. Reutelingsperger CP. McGahon AJ. Rader JA. van Schie RC. LaFace DM. Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Martin SJ. Reutelingsperger CP. McGahon AJ. Rader JA. van Schie RC. LaFace DM. Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Martinez LO. Agerholm-Larsen B. Wang N. Chen W. Tall AR. Phosphorylation of a pest sequence in ABCA1 promotes calpain degradation and is reversed by ApoA-I. J Biol Chem. 2003;278:37368–37374. doi: 10.1074/jbc.M307161200. [DOI] [PubMed] [Google Scholar]
- 149.Masutani M. Sonenberg N. Yokoyama S. Imataka H. Reconstitution reveals the functional core of mammalian eIF3. EMBO J. 2007;26:3373–3383. doi: 10.1038/sj.emboj.7601765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mate MJ. Ortiz-Lombardia M. Boitel B. Haouz A. Tello D. Susin SA. Penninger J. Kroemer G. Alzari PM. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat Struct Biol. 2002;9:442–446. doi: 10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- 151.Matsumori Y. Hong SM. Aoyama K. Fan Y. Kayama T. Sheldon RA. Vexler ZS. Ferriero DM. Weinstein PR. Liu J. Hsp70 overexpression sequesters AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- 152.Mazzalupo S. Cooley L. Illuminating the role of caspases during Drosophila oogenesis. Cell Death Differ. 2006;13:1950–1959. doi: 10.1038/sj.cdd.4401892. [DOI] [PubMed] [Google Scholar]
- 153.Milarski KL. Morimoto RI. Mutational analysis of the human HSP70 protein: distinct domains for nucleolar localization and adenosine triphosphate binding. J Cell Biol. 1989;109:1947–1962. doi: 10.1083/jcb.109.5.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Min L. Fulton DB. Andreotti AH. A case study of proline isomerization in cell signaling. Front Biosci. 2005;10:385–397. doi: 10.2741/1536. [DOI] [PubMed] [Google Scholar]
- 155.Miramar MD. Costantini P. Ravagnan L. Saraiva LM. Haouzi D. Brothers G. Penninger JM. Peleato ML. Kroemer G. Susin SA. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J Biol Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 156.Mizukoshi S. Nakazawa M. Sato K. Ozaki T. Metoki T. Ishiguro SI. Activation of mitochondrial calpain and release of apoptosis-indusing factor from mitochondria in RCS rat retinal degeneration. Exp Eye Res. 2010;91:353–361. doi: 10.1016/j.exer.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 157.Modjtahedi N. Giordanetto F. Madeo F. Kroemer G. Apoptosis-inducing factor: vital and lethal. Trends Cell Biol. 2006;16:264–272. doi: 10.1016/j.tcb.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 158.Montague JW. Hughes FM., Jr. Cidlowski JA. Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis-trans-isomerase activity. Potential roles of cyclophilins in apoptosis. J Biol Chem. 1997;272:6677–6684. doi: 10.1074/jbc.272.10.6677. [DOI] [PubMed] [Google Scholar]
- 159.Moubarak RS. Yuste VJ. Artus C. Bouharrour A. Greer PA. Menissier-de Murcia J. Susin SA. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol. 2007;27:4844–4862. doi: 10.1128/MCB.02141-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mufti AR. Burstein E. Duckett CS. XIAP: cell death regulation meets copper homeostasis. Arch Biochem Biophys. 2007;463:168–174. doi: 10.1016/j.abb.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Munoz-Pinedo C. Guio-Carrion A. Goldstein JC. Fitzgerald P. Newmeyer DD. Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci U S A. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Norberg E. Gogvadze V. Ott M. Horn M. Uhlen P. Orrenius S. Zhivotovsky B. An increase in intracellular Ca2+ is required for the activation of mitochondrial calpain to release AIF during cell death. Cell Death Differ. 2008;15:1857–1864. doi: 10.1038/cdd.2008.123. [DOI] [PubMed] [Google Scholar]
- 163.Norberg E. Gogvadze V. Vakifahmetoglu H. Orrenius S. Zhivotovsky B. Oxidative modification sensitizes mitochondrial apoptosis-inducing factor to calpain-mediated processing. Free Radic Biol Med. 2010;48:791–797. doi: 10.1016/j.freeradbiomed.2009.12.020. [DOI] [PubMed] [Google Scholar]
- 164.Norberg E. Orrenius S. Zhivotovsky B. Mitochondrial regulation of cell death: processing of apoptosis-inducing factor (AIF) Biochem Biophys Res Commun. 2010;396:395–100. doi: 10.1016/j.bbrc.2010.02.163. [DOI] [PubMed] [Google Scholar]
- 165.Obchoei S. Wongkhan S. Wongkham C. Li M. Yao Q. Chen C. Cyclophilin A: potential functions and therapeutic target for human cancer. Med Sci Monit. 2009;15:RA221–RA232. [PubMed] [Google Scholar]
- 166.Olichon A. Guillou E. Delettre C. Landes T. Arnaune-Pelloquin L. Emorine LJ. Mils V. Daloyau M. Hamel C. Amati-Bonneau P. Bonneau D. Reynier P. Lenaers G. Belenguer P. Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta. 2006;1763:500–509. doi: 10.1016/j.bbamcr.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 167.Oppenheim RW. Blomgren K. Ethell DW. Koike M. Komatsu M. Prevette D. Roth KA. Uchiyama Y. Vinsant S. Zhu C. Developing postmitotic mammalian neurons in vivo lacking Apaf-1 undergo programmed cell death by a caspase-independent, nonapoptotic pathway involving autophagy. J Neurosci. 2008;28:1490–1497. doi: 10.1523/JNEUROSCI.4575-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Otera H. Ohsakaya S. Nagaura Z. Ishihara N. Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ozaki T. Tomita H. Tamai M. Ishiguro S. Characteristics of mitochondrial calpains. J Biochem. 2007;142:365–376. doi: 10.1093/jb/mvm143. [DOI] [PubMed] [Google Scholar]
- 170.Ozaki T. Yamashita T. Ishiguro S. ERp57-associated mitochondrial micro-calpain truncates apoptosis-inducing factor. Biochim Biophys Acta. 2008;1783:1955–1963. doi: 10.1016/j.bbamcr.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 171.Pardo J. Perez-Galan P. Gamen S. Marzo I. Monleon I. Kaspar AA. Susin SA. Kroemer G. Krensky AM. Naval J. Anel A. A role of the mitochondrial apoptosis-inducing factor in granulysin-induced apoptosis. J Immunol. 2001;167:1222–1229. doi: 10.4049/jimmunol.167.3.1222. [DOI] [PubMed] [Google Scholar]
- 172.Park SY. Kim HY. Lee JH. Yoon KH. Chang MS. Park SK. The age-dependent induction of apoptosis-inducing factor (AIF) in the human semitendinosus skeletal muscle. Cell Mol Biol Lett. 2009;15:1–12. doi: 10.2478/s11658-009-0030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Park YC. Jeong JH. Park KJ. Choi HJ. Park YM. Jeong BK. Higuchi Y. Yoo YH. Sulindac activates nuclear translocation of AIF, DFF40 and endonuclease G but not induces oligonucleosomal DNA fragmentation in HT-29 cells. Life Sci. 2005;77:2059–2070. doi: 10.1016/j.lfs.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 174.Paumard P. Vaillier J. Coulary B. Schaeffer J. Soubannier V. Mueller DM. Brethes D. di Rago JP. Velours J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002;21:221–230. doi: 10.1093/emboj/21.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Petit PX. Goubern M. Diolez P. Susin SA. Zamzami N. Kroemer G. Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 1998;426:111–116. doi: 10.1016/s0014-5793(98)00318-4. [DOI] [PubMed] [Google Scholar]
- 176.Pinto DM. Flaus A. Structure and Function of Histone H2AX. Subcell Biochem. 2010;50:55–78. doi: 10.1007/978-90-481-3471-7_4. [DOI] [PubMed] [Google Scholar]
- 177.Pleschke JM. Kleczkowska HE. Strohm M. Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275:40974–40980. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 178.Polster BM. Basanez G. Etxebarria A. Hardwick JM. Nicholls DG. Calpain I induces cleavage and release of apoptosis-inducing factor from isolated mitochondria. J Biol Chem. 2005;280:6447–6454. doi: 10.1074/jbc.M413269200. [DOI] [PubMed] [Google Scholar]
- 179.Porter AG. Urbano AG. Does apoptosis-inducing factor (AIF) have both life and death functions in cells? Bioessays. 2006;28:834–843. doi: 10.1002/bies.20444. [DOI] [PubMed] [Google Scholar]
- 180.Pospisilik JA. Knauf C. Joza N. Benit P. Orthofer M. Cani PD. Ebersberger I. Nakashima T. Sarao R. Neely G. Esterbauer H. Kozlov A. Kahn CR. Kroemer G. Rustin P. Burcelin R. Penninger JM. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell. 2007;131:476–491. doi: 10.1016/j.cell.2007.08.047. [DOI] [PubMed] [Google Scholar]
- 181.Praefcke GJ. McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- 182.Rashmi R. Kumar S. Karunagaran D. Human colon cancer cells lacking Bax resist curcumin-induced apoptosis and Bax requirement is dispensable with ectopic expression of Smac or downregulation of Bcl-XL. Carcinogenesis. 2005;26:713–723. doi: 10.1093/carcin/bgi025. [DOI] [PubMed] [Google Scholar]
- 183.Ravagnan L. Gurbuxani S. Susin SA. Maisse C. Daugas E. Zamzami N. Mak T. Jaattela M. Penninger JM. Garrido C. Kroemer G. Heat-shock protein 70 antagonizes apoptosis-inducing factor. Nat Cell Biol. 2001;3:839–843. doi: 10.1038/ncb0901-839. [DOI] [PubMed] [Google Scholar]
- 184.Ravagnan L. Roumier T. Kroemer G. Mitochondria, the killer organelles and their weapons. J Cell Physiol. 2002;192:131–137. doi: 10.1002/jcp.10111. [DOI] [PubMed] [Google Scholar]
- 185.Rechsteiner M. Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1996;21:267–271. [PubMed] [Google Scholar]
- 186.Rossi MN. Carbone M. Mostocotto C. Mancone C. Tripodi M. Maione R. Amati P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J Biol Chem. 2009;284:31616–31624. doi: 10.1074/jbc.M109.025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Roue G. Pichereau V. Lincet H. Colomer D. Sola B. Cyclin D1 mediates resistance to apoptosis through upregulation of molecular chaperones and consequent redistribution of cell death regulators. Oncogene. 2008;27:4909–4920. doi: 10.1038/onc.2008.126. [DOI] [PubMed] [Google Scholar]
- 188.Ruchalski K. Mao H. Li Z. Wang Z. Gillers S. Wang Y. Mosser DD. Gabai V. Schwartz JH. Borkan SC. Distinct hsp70 domains mediate apoptosis-inducing factor release and nuclear accumulation. J Biol Chem. 2006;281:7873–7880. doi: 10.1074/jbc.M513728200. [DOI] [PubMed] [Google Scholar]
- 189.Ruchalski K. Mao H. Singh SK. Wang Y. Mosser DD. Li F. Schwartz JH. Borkan SC. HSP72 inhibits apoptosis-inducing factor release in ATP-depleted renal epithelial cells. Am J Physiol Cell Physiol. 2003;285:C1483–1493. doi: 10.1152/ajpcell.00049.2003. [DOI] [PubMed] [Google Scholar]
- 190.Sanna MG. da Silva Correia J. Ducrey O. Lee J. Nomoto K. Schrantz N. Deveraux QL. Ulevitch RJ. IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol Cell Biol. 2002;22:1754–1766. doi: 10.1128/MCB.22.6.1754-1766.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Scharstuhl A. Mutsaers HA. Pennings SW. Russel FG. Wagener FA. Involvement of VDAC, Bax and ceramides in the efflux of AIF from mitochondria during curcumin-induced apoptosis. PLoS One. 2009;4:e6688. doi: 10.1371/journal.pone.0006688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–7190. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- 193.Schimmer AD. Dalili S. Batey RA. Riedl SJ. Targeting XIAP for the treatment of malignancy. Cell Death Differ. 2006;13:179–188. doi: 10.1038/sj.cdd.4401826. [DOI] [PubMed] [Google Scholar]
- 194.Schinzel AC. Takeuchi O. Huang Z. Fisher JK. Zhou Z. Rubens J. Hetz C. Danial NN. Moskowitz MA. Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Schmitt E. Maingret L. Puig PE. Rerole AL. Ghiringhelli F. Hammann A. Solary E. Kroemer G. Garrido C. Heat shock protein 70 neutralization exerts potent antitumor effects in animal models of colon cancer and melanoma. Cancer Res. 2006;66:4191–4197. doi: 10.1158/0008-5472.CAN-05-3778. [DOI] [PubMed] [Google Scholar]
- 196.Schmitt E. Parcellier A. Gurbuxani S. Cande C. Hammann A. Morales MC. Hunt CR. Dix DJ. Kroemer RT. Giordanetto F. Jaattela M. Penninger JM. Pance A. Kroemer G. Garrido C. Chemosensitization by a non-apoptogenic heat shock protein 70-binding apoptosis-inducing factor mutant. Cancer Res. 2003;63:8233–8240. [PubMed] [Google Scholar]
- 197.Schulthess FT. Katz S. Ardestani A. Kawahira H. Georgia S. Bosco D. Bhushan A. Maedler K. Deletion of the mitochondrial flavoprotein apoptosis inducing factor (AIF) induces β-cell apoptosis and impairs β-cell mass. PLoS One. 2009;4:e4394. doi: 10.1371/journal.pone.0004394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Scorrano L. Ashiya M. Buttle K. Weiler S. Oakes SA. Mannella CA. Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 199.Scovassi AI. Soldani C. Veneroni P. Bottone MG. Pellicciari C. Changes of mitochondria and relocation of the apoptosis-inducing factor during apoptosis. Ann N Y Acad Sci. 2009;1171:12–17. doi: 10.1111/j.1749-6632.2009.04707.x. [DOI] [PubMed] [Google Scholar]
- 200.Senda T. Yamada T. Sakurai N. Kubota M. Nishizaki T. Masai E. Fukuda M. Mitsuidagger Y. Crystal structure of NADH-dependent ferredoxin reductase component in biphenyl dioxygenase. J Mol Biol. 2000;304:397–410. doi: 10.1006/jmbi.2000.4200. [DOI] [PubMed] [Google Scholar]
- 201.Seth R. Yang C. Kaushal V. Shah SV. Kaushal GP. p53-dependent caspase-2 activation in mitochondrial release of apoptosis-inducing factor and its role in renal tubular epithelial cell injury. J Biol Chem. 2005;280:31230–31239. doi: 10.1074/jbc.M503305200. [DOI] [PubMed] [Google Scholar]
- 202.Sevrioukova IF. Redox-linked conformational dynamics in apoptosis-inducing factor. J Mol Biol. 2009;390:924–938. doi: 10.1016/j.jmb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shoshan-Barmatz V. Keinan N. Abu-Hamad S. Toymkin D. Aram L. Apoptosis is regulated by the VDAC1 N-terminal region and by VDAC oligomerization: release of cytochrome c, AIF and Smac/Diablo. Biochim Biophys Acta. 2010;1797:1281–1291. doi: 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 204.Shumway SD. Maki M. Miyamoto S. The PEST domain of IκBα is necessary and sufficient for in vitro degradation by μ-calpain. J Biol Chem. 1999;274:30874–30881. doi: 10.1074/jbc.274.43.30874. [DOI] [PubMed] [Google Scholar]
- 205.Singh MH. Brooke SM. Zemlyak I. Sapolsky RM. Evidence for caspase effects on release of cytochrome c and AIF in a model of ischemia in cortical neurons. Neurosci Lett. 2010;469:179–183. doi: 10.1016/j.neulet.2009.11.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Smulson ME. Simbulan-Rosenthal CM. Boulares AH. Yakovlev A. Stoica B. Iyer S. Luo R. Haddad B. Wang ZQ. Pang T. Jung M. Dritschilo A. Rosenthal DS. Roles of poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability and functions of p53 and E2F-1. Adv Enzyme Regul. 2000;40:183–215. doi: 10.1016/s0065-2571(99)00024-2. [DOI] [PubMed] [Google Scholar]
- 207.Sokol RJ. Dahl R. Devereaux MW. Yerushalmi B. Kobak GE. Gumpricht E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr. 2005;41:235–243. doi: 10.1097/01.mpg.0000170600.80640.88. [DOI] [PubMed] [Google Scholar]
- 208.St-Denis NA. Litchfield DW. Protein kinase CK2 in health and disease: From birth to death: the role of protein kinase CK2 in the regulation of cell proliferation and survival. Cell Mol Life Sci. 2009;66:1817–1829. doi: 10.1007/s00018-009-9150-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Stambolsky P. Weisz L. Shats I. Klein Y. Goldfinger N. Oren M. Rotter V. Regulation of AIF expression by p53. Cell Death Differ. 2006;13:2140–2149. doi: 10.1038/sj.cdd.4401965. [DOI] [PubMed] [Google Scholar]
- 210.Susin SA. Daugas E. Ravagnan L. Samejima K. Zamzami N. Loeffler M. Costantini P. Ferri KF. Irinopoulou T. Prevost MC. Brothers G. Mak TW. Penninger J. Earnshaw WC. Kroemer G. Two distinct pathways leading to nuclear apoptosis. J Exp Med. 2000;192:571–580. doi: 10.1084/jem.192.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Susin SA. Lorenzo HK. Zamzami N. Marzo I. Brenner C. Larochette N. Prevost MC. Alzari PM. Kroemer G. Mitochondrial release of caspase-2 and −9 during the apoptotic process. J Exp Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Susin SA. Lorenzo HK. Zamzami N. Marzo I. Snow BE. Brothers GM. Mangion J. Jacotot E. Costantini P. Loeffler M. Larochette N. Goodlett DR. Aebersold R. Siderovski DP. Penninger JM. Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 213.Susin SA. Zamzami N. Castedo M. Daugas E. Wang HG. Geley S. Fassy F. Reed JC. Kroemer G. The central executioner of apoptosis: multiple connections between protease activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced apoptosis. J Exp Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Susin SA. Zamzami N. Castedo M. Hirsch T. Marchetti P. Macho A. Daugas E. Geuskens M. Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Susin SA. Zamzami N. Larochette N. Dallaporta B. Marzo I. Brenner C. Hirsch T. Petit PX. Geuskens M. Kroemer G. A cytofluorometric assay of nuclear apoptosis induced in a cell-free system: application to ceramide-induced apoptosis. Exp Cell Res. 1997;236:397–403. doi: 10.1006/excr.1997.3733. [DOI] [PubMed] [Google Scholar]
- 216.Tanaka H. Shimazaki H. Kimura M. Izuta H. Tsuruma K. Shimazawa M. Hara H. Apoptosis-inducing factor and cyclophilin A cotranslocate to the motor neuronal nuclei in amyotrophic lateral sclerosis model mice. CNS Neurosci Ther. 2010 doi: 10.1111/j.1755-5949.2010.00180.x. [Epub ahead of print]; Pubmed ID: 20553309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 218.Thress K. Henzel W. Shillinglaw W. Kornbluth S. Scythe: a novel reaper-binding apoptotic regulator. EMBO J. 1998;17:6135–6143. doi: 10.1093/emboj/17.21.6135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Thress K. Song J. Morimoto RI. Kornbluth S. Reversible inhibition of Hsp70 chaperone function by Scythe and Reaper. EMBO J. 2001;20:1033–1041. doi: 10.1093/emboj/20.5.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Tovchigrechko A. Vakser IA. GRAMM-X public web server for protein-protein docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Urbano A. Lakshmanan U. Choo PH. Kwan JC. Ng PY. Guo K. Dhakshinamoorthy S. Porter A. AIF suppresses chemical stress-induced apoptosis and maintains the transformed state of tumor cells. EMBO J. 2005;24:2815–2826. doi: 10.1038/sj.emboj.7600746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Uren RT. Dewson G. Bonzon C. Lithgow T. Newmeyer DD. Kluck RM. Mitochondrial release of pro-apoptotic proteins: electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J Biol Chem. 2005;280:2266–2274. doi: 10.1074/jbc.M411106200. [DOI] [PubMed] [Google Scholar]
- 223.Vahsen N. Cande C. Briere JJ. Benit P. Joza N. Larochette N. Mastroberardino PG. Pequignot MO. Casares N. Lazar V. Feraud O. Debili N. Wissing S. Engelhardt S. Madeo F. Piacentini M. Penninger JM. Schagger H. Rustin P. Kroemer G. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Vahsen N. Cande C. Dupaigne P. Giordanetto F. Kroemer RT. Herker E. Scholz S. Modjtahedi N. Madeo F. Le Cam E. Kroemer G. Physical interaction of apoptosis-inducing factor with DNA and RNA. Oncogene. 2006;25:1763–1774. doi: 10.1038/sj.onc.1209206. [DOI] [PubMed] [Google Scholar]
- 225.van Empel VP. Bertrand AT. van der Nagel R. Kostin S. Doevendans PA. Crijns HJ. de Wit E. Sluiter W. Ackerman SL. De Windt LJ. Downregulation of apoptosis-inducing factor in harlequin mutant mice sensitizes the myocardium to oxidative stress-related cell death and pressure overload-induced decompensation. Circ Res. 2005;96:e92–e101. doi: 10.1161/01.RES.0000172081.30327.28. [DOI] [PubMed] [Google Scholar]
- 226.van Empel VP. Bertrand AT. van Oort RJ. van der Nagel R. Engelen M. van Rijen HV. Doevendans PA. Crijns HJ. Ackerman SL. Sluiter W. De Windt LJ. EUK-8, a superoxide dismutase and catalase mimetic, reduces cardiac oxidative stress and ameliorates pressure overload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol. 2006;48:824–832. doi: 10.1016/j.jacc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 227.Vosler PS. Sun D. Wang S. Gao Y. Kintner DB. Signore AP. Cao G. Chen J. Calcium dysregulation induces apoptosis-inducing factor release: cross-talk between PARP-1- and calpain-signaling pathways. Exp Neurol. 2009;218:213–220. doi: 10.1016/j.expneurol.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wang H. Yu SW. Koh DW. Lew J. Coombs C. Bowers W. Federoff HJ. Poirier GG. Dawson TM. Dawson VL. Apoptosis-inducing factor substitutes for caspase executioners in NMDA-triggered excitotoxic neuronal death. J Neurosci. 2004;24:10963–10973. doi: 10.1523/JNEUROSCI.3461-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wang N. Chen W. Linsel-Nitschke P. Martinez LO. Agerholm-Larsen B. Silver DL. Tall AR. A PEST sequence in ABCA1 regulates degradation by calpain protease and stabilization of ABCA1 by apoA-I. J Clin Invest. 2003;111:99–107. doi: 10.1172/JCI16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Wang P. Heitman J. The cyclophilins. Genome Biol. 2005;6:226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Wang X. Wang J. Gengyo-Ando K. Gu L. Sun CL. Yang C. Shi Y. Kobayashi T. Mitani S. Xie XS. Xue D. C. elegans mitochondrial factor WAH-1 promotes phosphatidylserine externalization in apoptotic cells through phospholipid scramblase SCRM-1. Nat Cell Biol. 2007;9:541–549. doi: 10.1038/ncb1574. [DOI] [PubMed] [Google Scholar]
- 232.Wang X. Yang C. Chai J. Shi Y. Xue D. Mechanisms of AIF-mediated apoptotic DNA degradation in Caenorhabditis elegans. Science. 2002;298:1587–1592. doi: 10.1126/science.1076194. [DOI] [PubMed] [Google Scholar]
- 233.Wang Y. Dawson VL. Dawson TM. Poly(ADP-ribose) signals to mitochondrial AIF: a key event in parthanatos. Exp Neurol. 2009;218:193–202. doi: 10.1016/j.expneurol.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wang Y. Kim NS. Li X. Greer PA. Koehler RC. Dawson VL. Dawson TM. Calpain activation is not required for AIF translocation in PARP-1-dependent cell death (parthanatos) J Neurochem. 2009;110:687–696. doi: 10.1111/j.1471-4159.2009.06167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Watanabe M. Adachi S. Matsubara H. Imai T. Yui Y. Mizushima Y. Hiraumi Y. Watanabe K. Kamitsuji Y. Toyokuni SY. Hosoi H. Sugimoto T. Toguchida J. Nakahata T. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int J Cancer. 2009;124:55–67. doi: 10.1002/ijc.23897. [DOI] [PubMed] [Google Scholar]
- 236.Wei MC. Zong WX. Cheng EH. Lindsten T. Panoutsakopoulou V. Ross AJ. Roth KA. MacGregor GR. Thompson CB. Korsmeyer SJ. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292:727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Widlak P. Garrard WT. Discovery, regulation, and action of the major apoptotic nucleases DFF40/CAD and endonuclease G. J Cell Biochem. 2005;94:1078–1087. doi: 10.1002/jcb.20409. [DOI] [PubMed] [Google Scholar]
- 238.Wilkinson JC. Wilkinson AS. Galban S. Csomos RA. Duckett CS. Apoptosis-inducing factor is a target for ubiquitination through interaction with XIAP. Mol Cell Biol. 2008;28:237–247. doi: 10.1128/MCB.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wissing S. Ludovico P. Herker E. Buttner S. Engelhardt SM. Decker T. Link A. Proksch A. Rodrigues F. Corte-Real M. Frohlich KU. Manns J. Cande C. Sigrist SJ. Kroemer G. Madeo F. An AIF orthologue regulates apoptosis in yeast. J Cell Biol. 2004;166:969–974. doi: 10.1083/jcb.200404138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Yamashita D. Miller JM. Jiang HY. Minami SB. Schacht J. AIF and EndoG in noise-induced hearing loss. Neuroreport. 2004;15:2719–2722. [PubMed] [Google Scholar]
- 241.Yan CH. Yang YP. Qin ZH. Gu ZL. Reid P. Liang ZQ. Autophagy is involved in cytotoxic effects of crotoxin in human breast cancer cell line MCF-7 cells. Acta Pharmacol Sin. 2007;28:540–548. doi: 10.1111/j.1745-7254.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 242.Ye H. Cande C. Stephanou NC. Jiang S. Gurbuxani S. Larochette N. Daugas E. Garrido C. Kroemer G. Wu H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol. 2002;9:680–684. doi: 10.1038/nsb836. [DOI] [PubMed] [Google Scholar]
- 243.Yu SW. Andrabi SA. Wang H. Kim NS. Poirier GG. Dawson TM. Dawson VL. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Yu SW. Wang H. Poitras MF. Coombs C. Bowers WJ. Federoff HJ. Poirier GG. Dawson TM. Dawson VL. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 245.Yu SW. Wang Y. Frydenlund DS. Ottersen OP. Dawson VL. Dawson TM. Outer mitochondrial membrane localization of apoptosis-inducing factor: mechanistic implications for release. ASN Neuro. 2009;1:e00021. doi: 10.1042/AN20090046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Yuste VJ. Moubarak RS. Delettre C. Bras M. Sancho P. Robert N. d'Alayer J. Susin SA. Cysteine protease inhibition prevents mitochondrial apoptosis-inducing factor (AIF) release. Cell Death Differ. 2005;12:1445–1448. doi: 10.1038/sj.cdd.4401687. [DOI] [PubMed] [Google Scholar]
- 247.Yuste VJ. Sanchez-Lopez I. Sole C. Moubarak RS. Bayascas JR. Dolcet X. Encinas M. Susin SA. Comella JX. The contribution of apoptosis-inducing factor, caspase-activated DNase, and inhibitor of caspase-activated DNase to the nuclear phenotype and DNA degradation during apoptosis. J Biol Chem. 2005;280:35670–35683. doi: 10.1074/jbc.M504015200. [DOI] [PubMed] [Google Scholar]
- 248.Zamzami N. Susin SA. Marchetti P. Hirsch T. Gomez-Monterrey I. Castedo M. Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Zanna C. Ghelli A. Porcelli AM. Karbowski M. Youle RJ. Schimpf S. Wissinger B. Pinti M. Cossarizza A. Vidoni S. Valentino ML. Rugolo M. Carelli V. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain. 2008;131:352–367. doi: 10.1093/brain/awm335. [DOI] [PubMed] [Google Scholar]
- 250.Zemlyak I. Brooke SM. Singh MH. Sapolsky RM. Effects of overexpression of antioxidants on the release of cytochrome c and apoptosis-inducing factor in the model of ischemia. Neurosci Lett. 2009;453:182–185. doi: 10.1016/j.neulet.2009.02.020. [DOI] [PubMed] [Google Scholar]
- 251.Zhang J. Xu M. Apoptotic DNA fragmentation and tissue homeostasis. Trends Cell Biol. 2002;12:84–89. doi: 10.1016/s0962-8924(01)02206-1. [DOI] [PubMed] [Google Scholar]
- 252.Zhang Y. Han T. Zhu Q. Zhang W. Bao W. Fu HJ. Yang J. Huang XJ. Wei JX. Meng YL. Zhao J. Cao YX. Jia LT. Yangi AG. The proapoptotic activity of C-terminal domain of apoptosis-inducing factor (AIF) is separated from its N-terminal. Biol Res. 2009;42:249–260. [PubMed] [Google Scholar]
- 253.Zhu C. Wang X. Deinum J. Huang Z. Gao J. Modjtahedi N. Neagu MR. Nilsson M. Eriksson PS. Hagberg H. Luban J. Kroemer G. Blomgren K. Cyclophilin A participates in the nuclear translocation of apoptosis-inducing factor in neurons after cerebral hypoxia-ischemia. J Exp Med. 2007;204:1741–1748. doi: 10.1084/jem.20070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Zhu Q. Xu YM. Wang LF. Zhang Y. Wang F. Zhao J. Jia LT. Zhang WG. Yang AG. Heat shock protein 70 silencing enhances apoptosis inducing factor-mediated cell death in hepatocellular carcinoma HepG2 cells. Cancer Biol Ther. 2009;8:792–798. doi: 10.4161/cbt.8.9.8127. [DOI] [PubMed] [Google Scholar]