

Abstract
Long-term treatment of mouse cancer cells with interferon-α (IFN-α) converts parental B16 melanoma cells to B16α vaccine cells. Inoculation of syngeneic mice with UV-irradiated B16α vaccine cells triggers immunity to the parental B16 tumor that is mediated by host macrophages, T cells, and NK cells. Lymph node cells from mice inoculated with irradiated B16α vaccine cells, but not with irradiated parental cells, proliferate when cultured in vitro, suggesting long-term in vivo activation of lymphoid cells. Both IL-15 mRNA and IL-15 protein are highly induced in B16α vaccine cells. The bulk of the induced IL-15 is shown to be cell-associated, either cytoplasmic or membranous. The current study investigated the feasibility of applying the B16α vaccination protocol to generate a cancer vaccine against murine RM-1 prostate carcinoma. In comparison to B16α vaccine cells, long-term IFN-α–treated RM-1 cells (RM-1α vaccine cells) showed significant IL-15 mRNA induction but relatively low IL-15 protein up-regulation. When UV-irradiated, a 3-fold increase in intracellular IL-15 was observed in RM-1α vaccine cells, suggesting UV damage may have negated a possible control mechanism for IL-15 synthesis. Efficacy of in vivo vaccination of syngeneic mice with UV-irradiated RM-1α and B16α vaccine cells showed correlation between high IL-15 level and high vaccine efficacy in B16α cells compared to low IL-15 level and low vaccine efficacy in RM-1α cells. This supports the concept that the induction of IL-15 in tumor cells can be useful for creating whole-cell cancer vaccines.
Introduction
Cancer is a major cause of morbidity and mortality around the world. Despite the best treatment regimens of surgery, radiation therapy, chemotherapy, and biological therapy, ∼50% of all cancer patients will die of their disease. Clearly, new therapeutic approaches are needed. One promising new area involves inoculation with killed, whole tumor cells as vaccines. Working with murine B16 melanoma as a tumor model for human cancers, we have shown that parental cancer cells induced to express intracellular interleukin-15 (IL-15) by long-term (2 weeks) in vitro IFN-α treatment can become potent vaccine cells (B16α cells) (Wu and Fleischmann 1998; Wu and Fleischmann 2001; Fleischmann and Wu 2004; Fleischmann and Wu 2005; Wu and others 2007). When lethally irradiated and inoculated into mice, IL-15-containing vaccine cells activate the host immune system to develop tumor immunity. The tumor immunity protects mice from challenge with parental tumor, protects mice from metastases, and even cures mice with preexisting parental tumors. The development of tumor immunity in response to B16α vaccination is dependent upon the function of CD8+ T cells, NK cells, IL-12, and, particularly, CD4+ T cells (Wu and Fleischmann 2001). Lymph node cells harvested from mice inoculated with the vaccine cells spontaneously proliferate when established in tissue culture as long as 5 weeks after the last vaccine inoculation (Wu and others 2007), suggesting that vaccinations with the vaccine cells trigger a potent and long-lasting stimulation of T cells.
While sharing some characteristics with IL-2 (Grabstein and others 1994), IL-15 is thought to be essential for NK and T-cell development (Dubois and others 2008) and is known to increase the antitumor activity of tumor-specific T cells (Klebanoff and others 2004). IL-15 is unique in its ability to promote proliferation, long-term survival, and activation of CD8+ memory T cells (Berand and others 1994). In addition, IL-15 is also a T-cell chemoattractant (Wilkinson and Liew 1995), so it plays multiple roles in T-cell activation and survival. Unlike IL-2, whose production is restricted to T lymphocytes, IL-15 mRNA is constitutively expressed in a variety of tissues including macrophages, monocytes, epithelial cells, fibroblasts, heart, lung, liver, kidney, placenta, and skeletal muscle (Grabstein and others 1994; Bamford and others 1996). However, it has been difficult to demonstrate IL-15 in the lysates or supernatants of many cells that express such mRNA (Grabstein and others 1994; Bamford and others 1996). As previously reported, IL-15 transcription and translation are significantly enhanced in IFN-α–treated B16α vaccine cells when compared to the untreated parental cells (Wu and others 2007). Induced IL-15 protein accumulates mostly inside the cytoplasm and, to some extent, on the cell membrane, but no secretory IL-15 is detectable in the tissue culture fluid of B16α cells (Wu and others 2007).
In this study, the relative IL-15 induction levels and vaccine efficacies for IFN-α–treated murine B16α melanoma and RM-1α prostate cancer vaccines are evaluated and compared.
Materials and Methods
Tumor cells
Murine B16-F1 melanoma cells (B16 cells, C57Bl/6 background) (Fidler 1973) were grown in plastic tissue culture dishes (Corning, Corning, NY) in a growth medium consisting of Eagle’s Minimal Essential Medium (MEM, Earle’s base; Gibco, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT), 100 units/mL penicillin (Gibco), 100 µg/mL streptomycin (Gibco), and 11 µg/mL gentamicin (Sigma-Aldrich, St. Louis, MO). Murine RM-1 prostate carcinoma cells (C57Bl/6 background) (Baley and others 1995) were maintained in DMEM (Gibco) supplemented with 10% FBS, 10 mM HEPE (Gibco), and the antibiotics mentioned earlier. The cells were grown in an incubator (National Appliances Company, Winchester, VA) at 37°C in a humidified atmosphere containing 5% CO2 and were routinely passaged every 3–4 days.
For passaging cells, parental cells and IFN-α–treated vaccine cells were released from plastic culture dishes with trypsin (0.25%)–EDTA (1 mM) solution (Gibco) for 5 min. The cells were pelleted in a clinical centrifuge (Fisher Scientific, Hampton, NH), washed with 10 mL growth medium, and resuspended to the desired concentration in growth medium with or without IFN-α.
Interferon
The IFN-α employed in this study was recombinant DNA-derived HuIFN-αA/D and was obtained from PBL Biomedical Laboratories (rHuIFN-αA/D; Piscataway, NJ). rHuIFN-αA/D is called “universal interferon” because of its similar level of biological activity on cells from a variety of different animal species (Kramer and others 1983). Stock IFN-α preparations (106 units/mL) contained no detectable endotoxin by Limulus amebocyte lysate test (<0.5 ng/mL of endotoxin; Sigma Diagnostics, St. Louis, MO).
IFN-α titers were determined by plaque reduction assay in microtiter plates (Campbell and others 1975) in comparison with the mouse IFN-α/β international reference standard Gu02-901-511 and were expressed as International Reference Units/mL (IU/mL).
Real-time reverse transcriptase-polymerase chain reaction
Induction of IL-15 mRNA was quantified by two-step real-time reverse transcriptase-polymerase chain reaction (real-time RT-PCR). Cells were grown for various durations on 100-mm tissue culture dishes in growth media with or without 3,000 IU/mL IFN-α treatment. Three days after the last passage, the cells were given fresh media with or without IFN-α treatment. At 24 h after the last fluid change, total RNA was isolated from sets of cells with the RNeasy kit (Qiagen, Valencia, CA) according to the manufacturer’s protocol and quantified by spectrophotometry using the BioMate 3 spectrophotometer (Thermo Electron, Waltham, MA). Equal quantities of mRNA (2 µg) were added to each RT-PCR. Random hexamer-primed reverse transcription with TaqMan® Reverse Transcription Reagents (Applied Biosystems, Foster City, CA) was performed at 25°C for 10 min followed by 48°C for 30 min and 95°C for 5 min. Quantitative PCR was performed with 1/10th of the RT product for prostate cancer samples. For melanoma samples, quantitative PCR was performed with 1/5th of the RT product in order to obtain consistent amplifications in the parental samples that carried very low background levels of IL-15 mRNA (Wu and others 2007). The PCR primer set used included the sequences 5′-GAAACAGTAAGAAACGTGCTCTACCTT-3′ for forward priming and 5′-TCACATTCCTTGCAGCCAGAT-3′ for reverse priming and was purchased from BioSynthesis (Lewisville, TX) (Wu and others 2007). The primers were designed to span exon–exon junctions in order to avoid detecting genomic DNA. The TaqMan® MGB probe had the sequence 5′-CACTCTGTCTTCTAACAAG-3′ and was labeled with 6FAM™ (Applied Biosystems) (Wu and others 2007). All primers and probe sequences were searched against the Celera database to confirm specificity. The PCR reactions were performed with Ampli Taq Gold (Applied Biosystems) under the following cycling conditions on an iCycler with Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA): 1 cycle at 50°C for 2 min and 95°C for 10 min; 45 cycles at 95°C for 15 sec and 60°C for 1 min (Wu and others 2007).
For quantification, the comparative cycle threshold (Ct) method was used (Livak and Schmittgen 2001). The amount of target was obtained by normalizing to an endogenous reference, murine glyceraldehyde-3-phosphate dehydrogenase (Applied Biosystems), and calibrated against uninduced parental tumor cells as fold increase in expression. When visualized on 2% agarose gel with ethidium bromide staining, the relative intensities of PCR products amplified by real-time RT-PCR matched the relative levels of mRNA expression calculated by the comparative Ct method (Wu and others 2007).
IL-15 enzyme-linked immunosorbent assays (ELISAs)
IL-15 protein levels in melanoma and prostate cancer cells were determined as released IL-15, as cytoplasmic IL-15, and as cell membrane-bound IL-15. Cells were grown for various durations in growth media with or without 3,000 IU/mL IFN-α treatment. Three days after the last passage, the cells were given fresh media with or without 3,000 IU/mL IFN-α treatment. IL-15 determinations were made at 48 h after the last fluid change.
To determine secretory IL-15, supernatant fluids from cells grown on 100-mm tissue culture dishes were harvested, centrifuged to remove any floating cells, and concentrated 30-fold from 15 mL to 0.5 mL (Amicon Ultra Centrifugal Filter Devices; Millipore, Bedford, MA) (Wu and others 2007). The number of cells in each Petri dish was determined by enumeration. The levels of IL-15 in the samples were determined using a DuoSet Mouse IL-15 ELISA Development System (R & D Systems, Minneapolis, MN). ELISA plates (Falcon, Lincoln Park, NJ) were pre-absorbed with IL-15 capturing antibodies (4.0 µg/mL) overnight at room temperature. The plates were washed 3 times with PBS containing 0.05% Tween 20 and blocked with PBS containing 1% bovine serum albumin for 1 h at room temperature. Samples and serial dilutions of the recombinant DNA-derived IL-15, included in the ELISA kit for generation of a standard curve, were applied to the plates in duplicate (100 µL/well). Following a 2-h incubation at room temperature, the plates were washed 3 times followed by a 2-h incubation at room temperature with biotin-conjugated anti-IL-15 detection antibody (400 ng/mL). The plates were washed 3 times again and incubated in the dark at room temperature for 20 min with streptavidin–horseradish peroxidase. The plates were washed 3 more times, followed by a 20-min incubation in the dark at room temperature with tetramethylbenzidine substrate mixture. The reaction was stopped by adding 2 N H2SO4. The absorbance was measured at 450 nm with a reference wavelength of 595 nm using an ELISA plate reader (Bio-Rad Laboratories, Hercules, CA). According to the linear range of the standard curve constructed, the sensitivity of the assay was estimated to be 6.25 pg. Results were expressed as picograms of IL-15 per 106 cells by comparison of the data from samples with the standard curve (Wu and others 2007).
For determining cytoplasmic IL-15, cell monolayers grown on 100-mm tissue culture dishes were washed 3 times with Hanks basal salt solution (HBSS; Gibco). The cells were removed from the Petri dishes with trypsin–EDTA solution, spun down in a clinical centrifuge, and resuspended in 10 mL of HBSS. The number of cells in each tissue culture dish was determined by enumeration. The cells were pelleted again and resuspended in 0.5 mL of lysis buffer containing 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.1% NP-40, 1 mM PMSF (Sigma), and 0.2 U/mL aprotinin (Sigma) (Sugiura and others 2002; Wu and others 2007). The cell suspensions were sonicated at 2 W for 30 s in an icy water bath using a sonic dismembrator (Fisher Scientific). The lysates were centrifuged at 5,000g to remove cellular fragments before use in ELISA (Sugiura and others 2002; Wu and others 2007). Results were expressed as picograms of IL-15 per 106 cells by comparison of the data from samples with the standard curve.
To determine cell membrane-associated IL-15, a modification of the Duoset Mouse IL-15 ELISA Development System was used. Equal numbers of untreated parental cells and IFN-treated α-cells were seeded in duplicate into 96-well microtiter plates suitable for cell adherence (Falcon). Since RM-1 cells detached easily from the microtiter plates, the plates were washed 2 times with PBS after removal of culture media followed by a 30-min heat fixing at 80°C. The plates were then incubated with biotin-conjugated anti-IL-15 detection antibody (400 ng/mL) at room temperature for 2 h. Serial dilutions of recombinant IL-15 were assayed simultaneously on plates with capturing antibodies. The whole-cell ELISA plates were washed 3 times again with PBS followed by a 20-min incubation in the dark at room temperature with streptavidin–horseradish peroxidase. The plates were washed 3 more times followed by a 20-min incubation in the dark at room temperature with the substrate solution. The reactions were stopped and analyzed as described earlier. Since the whole-cell assay did not involve the use of capturing antibodies as the sandwich assay did for the detection of IL-15 standards and harvested IL-15 from cell lysates and culture supernatants, relative comparisons of IL-15 surface expression in fold enhancement instead of absolute titers were inferred from the standard curve (Wu and others 2007).
Confocal fluorescence microscopy
Cells were set (105.0 to 105.7 cells/well) in Lab-tek CCG 8-well cell culture plates (Fisher) in growth medium plus or minus IFN-α. After 24 h of incubation, the cells were fixed by adding 0.3 mL/well of 2% paraformaldehyde (Nichols and others 2001). Following a 30-min incubation on ice, the formaldehyde was decanted. The cells were permeabilized by adding 0.3 mL/well of 0.1% Triton X-100. Following a 15-min incubation on ice, the Triton X-100 was decanted. The cells were washed once with room temperature PBS and once with PBS at 4°C. To block nonspecific binding of antibodies, the cells were incubated for 10 min on ice with 0.4 mL/well of 5% bovine serum albumin in PBS (BSA/PBS). After decanting the BSA/PBS, the cells were overlaid with 0.3 mL/well PBS/BSA and 0.3 mL/well of 2.5 µg/mL polyclonal rabbit anti-mouse IL-15 antibody (primary antibody; eBioscience, San Diego, CA) and incubated for 30 min on ice. The fluid was decanted and the cells were washed 3 times with PBS at 4°C. After decanting the PBS, the cells were overlaid with 0.3 mL/well BSA/PBS and 0.3 mL/well of 10 µg/mL goat [F(ab′)2] anti-rabbit IgG secondary antibody conjugated with FITC (fluorescein isothiocyanate; Pierce, Rockford, IL) and incubated for 30 min on ice. After decanting the fluid, the cells were washed 3 times with PBS at 4°C. The cell nuclei were stained on ice with 0.3 mL/well of 0.1 µg/mL DAPI (Molecular Probes, Eugene, OR) for 5 min. The cells were washed 3 times with PBS at 4°C. The cells were observed using a confocal microscope (Zeiss LSM 510 UV META laser scanning confocal microscope; Zeiss, Thornwood, NY).
UV inactivation of long-term IFN-treated RM-1α and B16α vaccine cells
RM-1 prostate cancer cells and B16 melanoma cells were grown in growth media with or without 3,000 IU/mL IFN-α treatment for 16 days. Three days after the last passage, cells were given fresh media with or without 3,000 IU/mL IFN-α treatment. One day after the last fluid change, parental cell and vaccine cell monolayers in the culture media were lethally UV-irradiated or left alone without UV treatment and harvested as described below. UV-irradiated cells were exposed to a 15-W ultraviolet-light (UV-light) source delivering 4 erg × s−1 × m−2 for 25 min at a distance of 26 cm. Another 24 h later, the remaining cell monolayers were washed 3 times, detached from tissue culture plates by trypsin–EDTA solution, pelleted by centrifugation, resuspended, enumerated, pelleted by centrifugation, resuspended (final volume = 0.5 mL), and sonicated. The sonicates were assayed for the presence of IL-15 by ELISA.
In vivo RM-1α prostate cancer vaccine model
Parental RM-1 cells were treated for 2 weeks or more with 300 units/mL or 3,000 units/mL of IFN-α to create RM-1α cancer vaccine cells. Three days after the last passage, the cells were given a final fluid change. One day later, RM-1α vaccine cell or RM-1 parental cell monolayers were exposed to a 15-W ultraviolet-light (UV-light) source as described earlier. The UV-light-irradiated cells were washed 3 times with HBSS and removed from the plastic culture dishes with a 5-min, 37°C treatment with trypsin–EDTA solution (Gibco). The cells were washed 1 more time in HBSS by pelleting and resuspension. The final cell suspension was counted and adjusted to 107 cells/mL with HBSS. A volume of 0.1 mL was inoculated intraperitoneally (total inoculum = 106 cells) once a week for a total of 6 weeks.
For live tumor challenge, RM-1 parental cell monolayers were detached by incubation with trypsin–EDTA solution, washed once with fresh culture medium, centrifuged, and resuspended in fresh culture medium. Immediately after the last vaccination, mice were inoculated i.p. into the right mid-abdominal region with a RM-1 inoculum of 105 live cells in 0.1 mL.
Statistics
Data were evaluated by Student’s t-test and ANOVA using StatView (Abacus Concepts, Berkeley, CA) and by Fisher’s exact test.
Results
In vitro IL-15 mRNA expression in B16α and RM-1α cells
The method for the creation of efficacious tumor vaccines involves the treatment of tumor cells for ≥2 weeks with rHuIFN-αA/D, a recombinant DNA-derived universal interferon. The long-term in vitro interferon treatment causes B16 tumor cells to become B16α vaccine cells (Wu and Fleischmann 1998; Wu and Fleischmann 2001; Wu and others 2007). As previously reported, 3,000 U/mL IFN-αA/D is the optimal dosage for generating B16α vaccine cells (Wu and Fleischmann 1998). Since long-term in vitro IFN-α treatment induces substantial levels of IL-15 mRNA and cell-associated IL-15 protein in B16α cells, IFN-induced IL-15 was hypothesized to play a major role in antitumor immunity (Wu and others 2007). Thus, vaccine cells created for other tumor types such as RM-1 murine prostate cancer cells would be expected to express cell-associated IL-15. To test IL-15 induction in RM-1α cells, RM-1 parental cells were treated in vitro with 3,000 U/mL IFN-αA/D for 24 h and 15 days to generate RM-1α 24 h cells and RM-1α vaccine cells, respectively. Simultaneously, B16α 24 h cells and B16α vaccine cells were generated the same way for comparison. RM-1 and B16 parental cells that had been identically treated (except for not being exposed to IFN-α) were employed as controls.
Figure 1 shows that 24 h after in vitro IFN treatment, IL-15 mRNA increased about 76-fold in B16α 24 h cells but only 19-fold in RM-1α 24 h cells in comparison to parental controls. When a 15-day treatment schedule was employed, IL-15 mRNA increased about 750-fold in B16α vaccine cells but only 35-fold in RM-1α vaccine cells. These data suggest that IFN-α up-regulated IL-15 gene transcription in both cell lines as early as 24 h after the introduction of IFN, with a significantly lower level of activation in RM-1α 24 h cells. IL-15 mRNA induction in long-term IFN-treated B16α vaccine cells was about 10-fold stronger than in short-term treated B16α 24 h cells. However, a marginal increase (1.8-fold) was seen for long-term IFN-treated RM-1α vaccine cells over short-term treated RM-1α 24 h cells.
FIG. 1. .
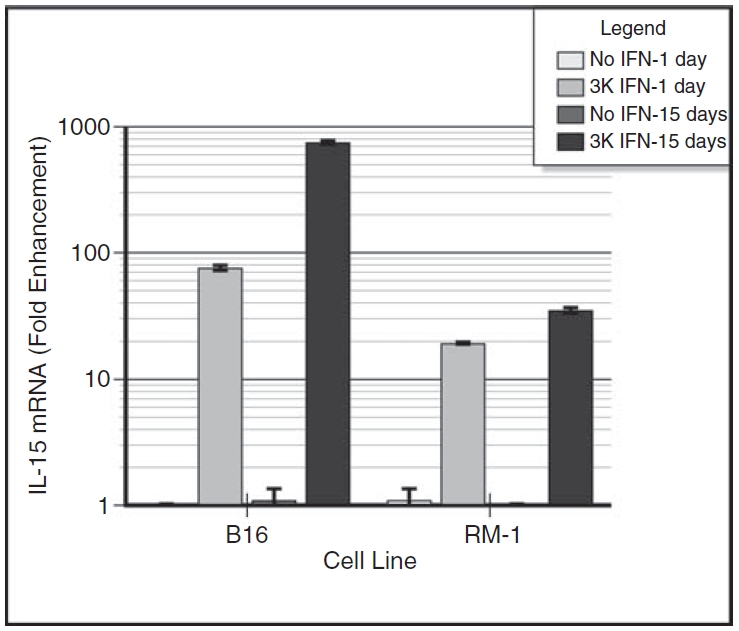
Induction of interleukin-15 (IL-15) mRNA by interferon-α (IFN-α) treatment of B16α cells and RM-1α cells. Monolayers of B16α and RM-1α cells were grown in medium containing 3,000 units/mL of IFN-α for 1 day or for 15 days. Control monolayers of B16 parental cells were given medium alone without IFN treatment. At 1 day after providing fresh medium with or without IFN-α, the cells were lysed and the RNA harvested was subjected to real-time RT-PCR. The levels of IL-15 mRNA and GAPDH mRNA were determined. After adjusting for the levels of GAPDH mRNA in each of the preparations, the level of IL-15 mRNA in parental cells grown in medium alone was set at 1 and the level of IL-15 mRNA in IFN-treated cells was expressed as fold enhancement. The data are expressed as fold enhancement (mean ± SE) in IL-15 mRNA level versus days of IFN treatment. Student’s t-test analysis: day 1 B16 parental cells versus day 1 B16α cells: P < 0.0001; day 1 RM-1 parental cells versus day 1 RM-1α cells: P < 0.0001; day 15 B16 parental versus day 15 B16α cells: P < 0.0001; day 15 RM-1 parental cells versus day 15 RM-1α cells: P < 0.0001; day 1 B16α cells versus day 15 B16α cells: P < 0.0001; day 1 RM-1α cells versus day 15 RM-1α cells: P < 0.0018.
Cytoplasmic IL-15 in B16α and RM-1α cells
Since up-regulation of IL-15 mRNA does not necessarily correspond to successful translation of the protein, cellular IL-15 was analyzed in 3 different ways by ELISA. It was possible that IL-15 protein was induced and could be detected in the cytoplasm of the vaccine cells. To test this possibility, RM-1α and B16α cells that had been maintained for 48 h or 15 days in 3,000 units/mL of IFN-α were harvested and counted 48 h after addition of fresh medium containing IFN-α. B16 and RM-1 cells that had been identically treated (except for not being exposed to IFN-α) were employed as a control. Cell lysates were tested by ELISA for the presence of IL-15.
Figure 2 shows the summated results of 2 experiments with essentially identical results. The data are expressed as IL-15 in picograms per 106 cells. Lysates from B16α cells treated with IFN for 48 h contained measurable amounts of IL-15, about 23 pg/million cells on average, representing no significant increase in IL-15 expression over the parental controls (20 pg/million cells). Lysates from B16α cells treated with IFN for 2 weeks contained about 157 pg/million cells at 48 h after addition of fresh media with IFN-α, corresponding to a substantial 6.8-fold increase in IL-15 expression over the parental controls (23 pg/million cells). Contrarily, lysates from either parental RM-1 or RM-1α cells treated with IFN for only 48 h contained baseline levels of IL-15, also resulting in no significant increase over the parental controls. RM-1α vaccine cells treated with IFN for 15 days contained a low but measurable amount of 6.8 pg/million cells, corresponding to a 3.2-fold increase in IL-15 induction from the RM-1 parental cells, which contained a background level of 2.1 pg/million cells.
FIG. 2. .
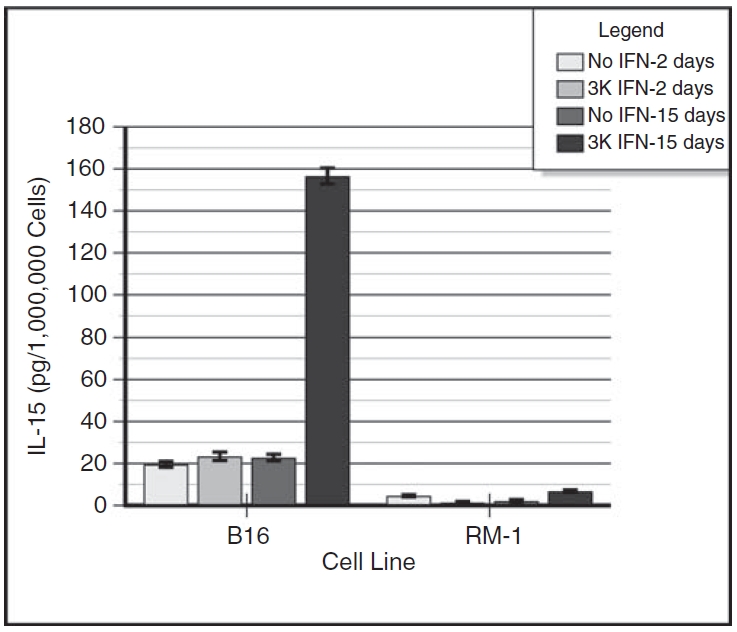
Induction of intracellular interleukin-15 (IL-15) in interferon-α (IFN-α)–treated B16α cells and RM-1α cells. Monolayers of B16α and RM-1α cells were grown in medium containing 3,000 units/mL of IFN-α for 2 days or for 15 days. Control monolayers of parental cells were given medium alone without IFN treatment. At 2 days after providing fresh medium with or without IFN-α, the cells were harvested. The supernatant fluids were decanted, spun to remove floating cells, concentrated 30-fold (final volume = 0.5 mL), and assayed for the presence of IL-15 by ELISA. The cells were harvested for sonication, and the sonicates were assayed for the presence of IL-15 by ELISA. Standard curves of IL-15 were run concomitantly with the samples. The ELISA had a sensitivity of 6.25 pg. The data are expressed as IL-15 in picograms per 106 cells (mean ± SE) versus days of IFN treatment. ANOVA analysis for intracellular IL-15: day 2 B16 parental cells versus day 2 B16α cells: P = NS; day 2 RM-1 parental cells versus day 2 RM-1α cells: P = NS; day 16 B16 parental cells versus day 16 B16α cells: P < 0.0001; day 16 RM-1 parental cells versus day 16 RM-1α cells: P < 0.0001; day 2 B16α cells versus day 16 B16α cells: P < 0.0001; day 2 RM-1α cells versus day 16 RM-1α cells: P < 0.0001.
Secretory IL-15 in B16α and RM-1α cells
To determine the amount of secretory IL-15, supernatant fluids from the cell monolayers described earlier were harvested 48 h after the last medium change, concentrated 30-folds, and assayed for the presence of IL-15 by ELISA. IL-15 levels for both IFN-treated cells and parental cells were below the detection limit of the ELISA (6.25 pg) (data not shown). Thus, neither supernatant fluids from short-term and long-term treated RM-1α and B16α cells nor supernatant fluids from RM-1 and B16 parental cells contained a detectable level of IL-15.
Membrane-associated IL-15 in B16α and RM-1α vaccine cells
IL-15 naturally produced (Musso and others 1999) or synthesized in cells transfected with the gene for IL-15 (Budagian and others 2004) has been reported to be membrane-associated. Thus, it was important to determine whether our vaccine cells expressed IL-15 on their membrane surface. To test this possibility, RM-1α and B16α cells that had been maintained for 15 days in 3,000 units/mL of IFN-α were assayed for the presence of membrane-bound IL-15 at 48 h after addition of fresh media containing IFN-α. B16 and RM-1 cells that had been identically treated (except for not being exposed to IFN-α) were employed as a control.
There were detectable amounts of IL-15 on the cell surfaces of B16α vaccine cells and B16 parental cells. Figure 3 shows the summated results of 4 experiments with essentially identical results. To obtain a relative quantification, the data are expressed as percent of parental cell control. At 48 h after fresh IFN-α treatment, B16α vaccine cells expressed 1.6-fold more membrane-bound IL-15 than B16 parental cells. However, there was no significant increase in membrane-associated IL-15 on the cell surfaces of RM-1α vaccine cells.
FIG. 3. .
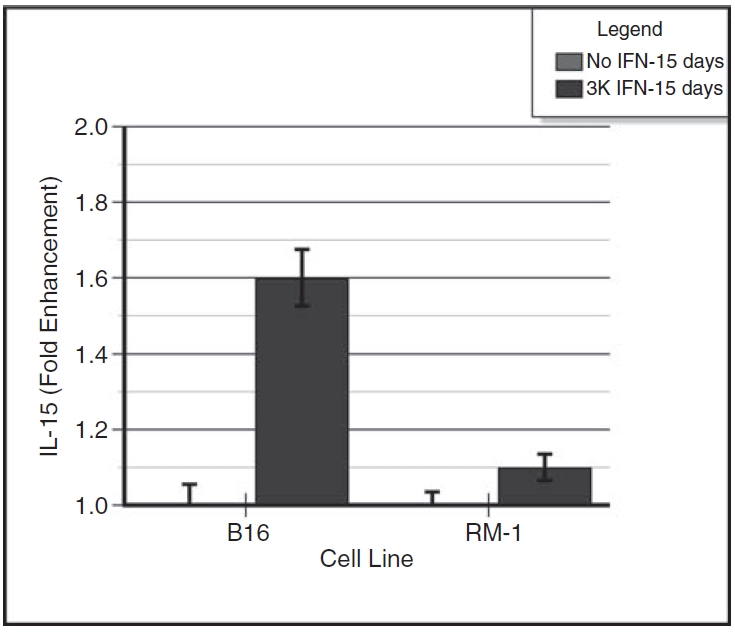
Induction of membrane-associated interleukin-15 (IL-15) in interferon-α (IFN-α)–treated B16α cells and RM-1α cells. Monolayers of B16α and RM-1α cells were grown in medium containing 3,000 units/mL of IFN-α for 15 days. Control monolayers of parental cells were given medium alone without IFN treatment. At 2 days after providing fresh medium with or without IFN-α, the cell monolayers were assayed for membrane-bound IL-15 using a whole-cell ELISA. Standard curves of IL-15 were run concomitantly with the samples. The data are expressed as IL-15 fold enhancement (mean ± SE) versus days of IFN treatment. ANOVA analysis for membrane-associated IL-15: day 16 B16 parental versus day 16 B16α cells: P < 0.0001; day 16 RM-1 parental cells versus day 16 RM-1α cells: P = NS.
Intracellular distribution of IL-15 in B16α and RM-1α vaccine cells
The intracellular distribution of IL-15 was monitored in long-term IFN-treated vaccine cells and in parental cells by confocal fluorescence microscopy. Slides containing the vaccine cells and the parental cells were fixed, permeabilized, reacted or not reacted with primary antibody to IL-15, and stained with secondary antibody conjugated to fluorescein.
Figure 4 shows representative areas of slides of the vaccine cells and the parental cells from 1 of 2 essentially identical experiments. As previously reported, IL-15-specific fluorescence in B16 parental cells (Fig. 4A) was very low and very similar to background fluorescence (negative control without primary antibody, data not shown), indicating that most of the fluorescence observed with the B16 parental cells was nonspecific. IL-15-specific fluorescence in B16α vaccine cells (Fig. 4B) was substantially higher than background fluorescence and IL-15-specific fluorescence in B16 parental cells (Fig. 4A). In B16α vaccine cells, punctate fluorescence was observed in both the nucleus (as light blue color) and cytoplasm. Within the nucleus, the fluorescence intensity was quite variable from cell to cell.
FIG. 4. .
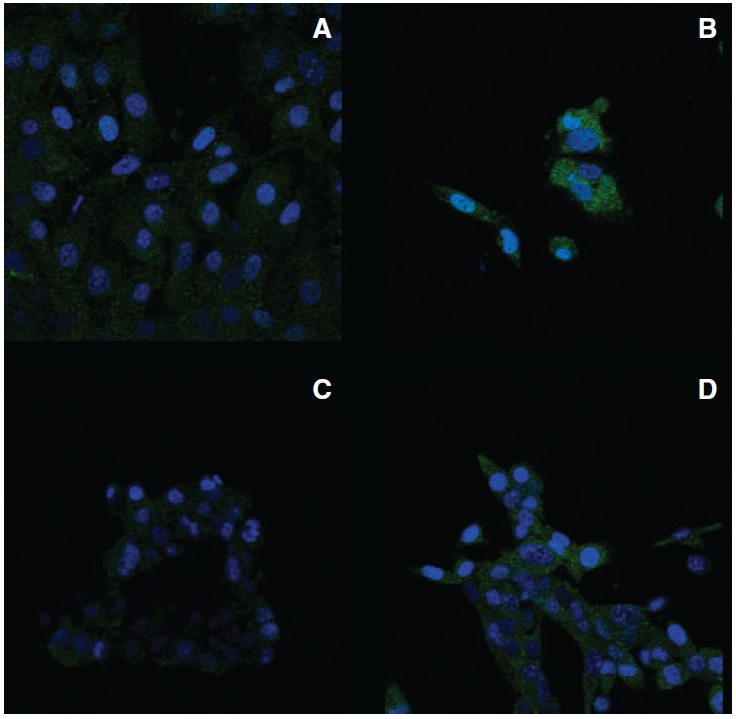
Subcellular distribution of interleukin-15 (IL-15) protein in B16α and RM-1α cells. Parental cells and interferon (IFN)-treated α-cells were cultured in the absence or presence of 3,000 IU/mL of IFN-α for 2 weeks, respectively. The cells were fixed, permeabilized, and incubated with or without (data not shown) primary antibody to IL-15 and subsequently incubated with secondary antibody to IgG conjugated with fluorescein isothiocyanate (green fluorescence). Nuclei were stained with DAPI (blue fluorescence). Fluorescence was visualized with a Zeiss LSM510 UV META confocal microscope. The illustrations represent overlays of the blue and green fluorescence images. All images have the same magnification with the scale. (A) B16 parental cells incubated with primary antibody to IL-15. (B) B16α cells incubated with primary antibody to IL-15. (C) RM-1 parental cells incubated with primary antibody to IL-15. (D) RM-1α vaccine cells incubated with primary antibody to IL-15.
Results obtained from RM-1α vaccine cells were considerably different from those obtained from B16α vaccine cells. IL-15-specific fluorescence in RM-1 parental cells (Fig. 4C) was, in parallel to B16 parental cells, very low and similar to background fluorescence. IL-15-specific fluorescence in RM-1α cells (Fig. 4D), however, showed only a modest increase relative to background fluorescence and IL-15-specific fluorescence in RM-1 parental cells. Again, IL-15-specific fluorescence in RM-1α cells was distributed in the nucleus as well as in the cytoplasm. When present, nuclear fluorescence consisted of blue punctate bodies. Overall, IL-15 fluorescence in RM-1α cells was considerably less intense than in B16α cells.
Thus, consistent with the ELISA data, IL-15 protein was present at higher levels in B16α vaccine cells than in RM-1α vaccine cells and was evenly distributed throughout the cytoplasm of both vaccine cell types.
The effect of lethal UV irradiation on cytoplasmic expression of IL-15 in RM-1α vaccine cells
While there was a significant induction of IL-15 mRNA in RM-1α vaccine cells, relatively little IL-15 protein was detected by ELISA or by confocal microscopy. It was hypothesized that IL-15 synthesis in RM-1α vaccine cells was more vigorously regulated post-transcriptionally than in B16α vaccine cells. It should be noted that these RM-1α vaccine cells used in the in vitro assays described earlier were not lethally irradiated as was the vaccine inocula given to mice. It might be anticipated that lethal irradiation of RM-1α vaccine cells would prevent the activation of the putative translational regulatory mechanism and would enhance the level of translation of IL-15. To test this possibility, RM-1α vaccine cells and B16α vaccine cells were UV-irradiated 24 h after addition of fresh media containing IFN-α (as prepared for in vivo vaccine studies) and harvested 24 h after UV irradiation for ELISA. Parental cells were included as controls and assayed in the same manner as the RM-1α vaccine cells. Non-irradiated cells of both types were also harvested for comparisons.
Figure 5 shows the relative effects of UV irradiation on RM-1α cells as compared to B16α cells. For direct comparative purposes, the data are plotted as fold increase relative to the time 0 parental cells. The data show that UV-irradiated RM-1α cells that would otherwise have limited IL-15 translation without UV irradiation (7 pg/million cells) exhibited a further 2.7-fold enhancement in IL-15 translation with UV irradiation (19 pg/million cells). In contrast, the UV-irradiated B16α cells that would otherwise have robust IL-15 translation without UV irradiation failed to show a significant enhancement in IL-15 translation with UV irradiation.
FIG. 5. .
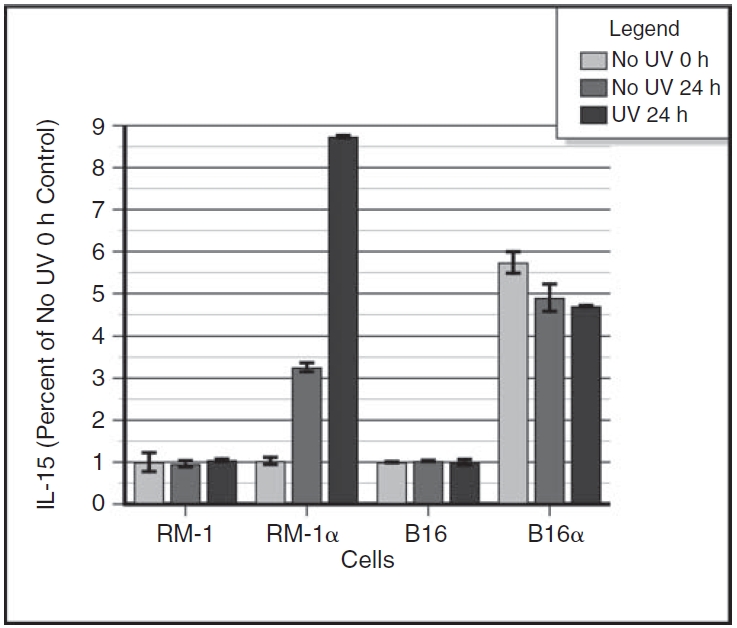
Relative levels of enhancement of interleukin-15 (IL-15) translation in UV-irradiated RM-1α and B16α vaccine cells. Monolayers of RM-1α cells and B16α cells were grown in medium containing 3,000 units/mL of interferon-α (IFN-α) for 15 days. Control monolayers of parental cells were given medium alone without IFN treatment. At 24 h after providing fresh medium with or without IFN-α, cell monolayers were subjected to lethal UV irradiation, left alone without UV treatment, or harvested for sonication. Another 24 h later, the remaining cell monolayers were harvested for sonication. The sonicates were assayed for the presence of IL-15 by ELISA. Standard curves of IL-15 were run concomitantly with the samples. The data are plotted as percent of untreated control cells. Data for non-irradiated and UV-irradiated B16 and B16α melanoma cells are also plotted. The data are expressed as percent of no UV 0 h control (RM-1 cells or B16 cells) divided by 100. ANOVA analysis for intracellular IL-15 in the RM-1 and RM-1α prostate cancer cell groups: no UV 0 h RM-1 parental cells versus no UV 24 h RM-1 parental cells versus UV 24 h RM-1 parental cells: P = NS; no UV 0 h RM-1 parental cells versus no UV 0 h RM-1α cells: P = NS; no UV 24 h RM-1 parental cells versus no UV 24 h RM-1α cells: P < 0.0001; UV 24 h RM-1 parental cells versus UV 24 h RM-1α cells: P < 0.0001; no UV 0 h RM-1α cells versus no UV 24 h RM-1α cells: P < 0.0001; no UV 24 h RM-1α cells versus UV 24 h RM-1α cells: P < 0.0001. ANOVA analysis for intracellular IL-15 in the B16 and B16α melanoma cell groups: no UV 0 h B16 parental cells versus no UV 24 h B16 parental cells versus UV 24 h B16 parental cells: P = NS; no UV 0 h B16 parental cells versus no UV 0 h B16α cells: P < 0.0001; no UV 24 h B16 parental cells versus no UV 24 h B16α cells: P < 0.0001; UV 24 h B16 parental cells versus UV 24 h B16α cells: P < 0.0001; no UV 0 h B16α cells versus no UV 24 h B16α cells: P = 0.021; no UV 24 h B16α cells versus UV 24 h B16α cells: P = NS.
Vaccination efficacy of RM-1α vaccine cells
It was possible that the levels of cell-associated IL-15 induced in our vaccine cells might not correlate to vaccine efficacies. Therefore, the comparative vaccine efficacies of B16α vaccine cells and RM-1α vaccine cells were determined. A 60% survival rate has been previously reported for mice vaccinated 4 times at weekly intervals with UV-irradiated B16α vaccine cells followed by a live tumor challenge of a million B16 parental cells (Wu and Fleischmann 1998; Wu and Fleischmann 2001). Since the initial range finding experiments (data not shown) indicated that 4 vaccinations with inactivated RM-1α vaccine cells did not induce significant tumor immunity, 6 vaccinations were employed for the vaccination experiments depicted in Figure 6. To test the IFN-α dosage effect in generating efficacious RM-1α vaccines, RM-1 parental cells were untreated, treated in vitro for about 2 weeks with 300 IU/mL IFN-α, or treated for the same duration with 3,000 IU/mL IFN-α. The data show that 6 vaccinations gave no protection to mice vaccinated with inactivated RM-1 parental cells. A modest, but significant, 30% protection was observed with inactivated RM-1α vaccine cells generated by the 300 IU/mL IFN-α treatment. A significantly greater 60% protection was observed with RM-1α vaccine cells generated by the 3,000 IU/mL IFN-α treatment. These results were similar to our previous observations with 4 vaccinations with B16α vaccine cells (Wu and Fleischmann 1998; Wu and Fleischmann 2001; Fleischmann and Wu 2004; Fleischmann and Wu 2005), in which the 300 IU/mL IFN-α treatment (21%) was less effective than the 3,000 IU/mL IFN-α treatment (60%) in generating an efficacious vaccine. Overall, the more limited vaccination efficacy of RM-1α vaccine cells (6 vaccinations) correlated with the low level of IL-15 expression. This was in contrast to the higher vaccination efficacy of B16α vaccine cells (4 vaccinations) that correlated with a higher IL-15 expression.
FIG. 6. .
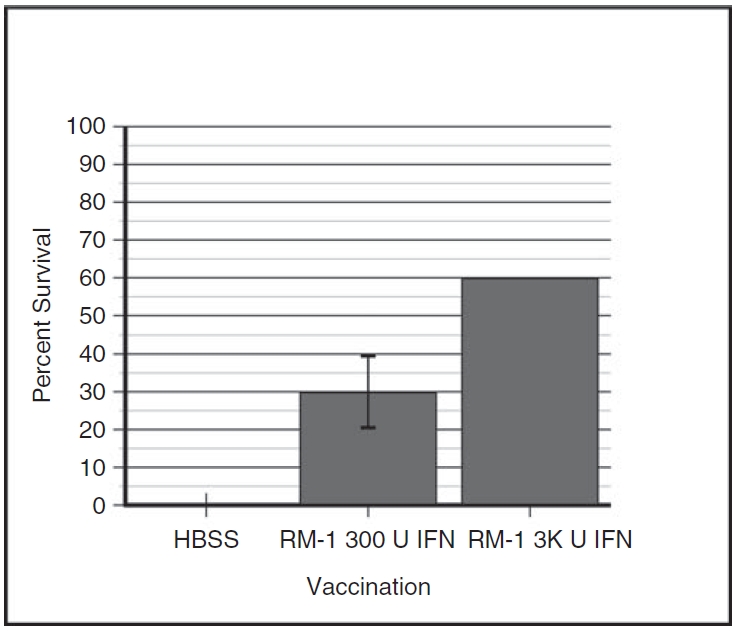
Efficacy of lethally inactivated RM-1α vaccine cells. Mice were inoculated intraperitoneally (i.p.) on days –35, –28, –21, –14, –7, and 0 with 106 inactivated RM-1α vaccine cells. The inactivated RM-1α vaccine cells were pretreated for >2 weeks with 300 or 3,000 IU/mL of interferon-α (IFN-α). Control mice were mock inoculated with the carrier (HBSS). On day 0, all mice were challenged i.p. with 105 live RM-1 parental cells. The graph plots the combined data of 2 experiments (10 mice per group in each of 2 experiments) as percent survival (mean ± SE) versus types of vaccine. Fisher’s exact test for HBSS-treated versus vaccinated vaccine cells treated with 300 IU/mL of IFN-α: P = 0.01; HBSS-treated versus vaccinated with vaccine cells treated with 3,000 IU/mL of IFN-α: P < 0.0001; vaccinated with vaccine cells treated with 300 IU/mL of IFN-α versus vaccinated with vaccine cells treated with 3,000 IU/mL of IFN-α: P = 0.043.
Discussion
In our attempts to generate general principles for creating effective, IL-15-based whole-cell vaccines against a garden variety of cancers, it was imperative to know whether mouse tumor types other than B16 melanoma were IL-15 inducible and whether the amount of IL-15 induced would correlate to the vaccine’s antitumor efficacy. To address this question, RM-1 prostate cancer cells were evaluated in comparison to B16 melanoma cells.
Interestingly, there are fundamental differences between B16 melanoma and RM-1 prostate cancer in IL-15 synthesis and regulation. At the mRNA level, both B16 and RM-1 parental cells treated with IFN-α for 24 h (B16α 24 h and RM-1α 24 h cells) had a significant up-regulation in IL-15 mRNA although the degree of up-regulation in B16α 24 h cells was 3.5-fold greater than in RM-1α 24 h cells (67-fold vs. 19-fold). The data suggest that RM-1 cells have a considerably higher threshold for IL-15 mRNA induction by IFN-α treatment. This is further evident in the long-term IFN-α–treated cells. While IL-15 mRNA induction in long-term treated B16α vaccine cells was about 10-fold stronger than in short-term treated B16α 24 h cells, only a marginal increase (1.8-folds) was seen for long-term treated RM-1α vaccine cells over short-term treated RM-1α 24 h cells, indicative of a 21-fold greater induction (750-fold vs. 35-fold).
Although IL-15 mRNA is known to be constitutively expressed in several cell types, IL-15 translation in these cells is usually tightly controlled (Grabstein and others 1994; Bamford and others 1996). IL-15 translation in RM-1α cells seems to follow that suit. While there were 19-fold and 35-fold up-regulation in IL-15 mRNA expression in short-term and long-term IFN-treated RM-1α cells, respectively, only long-term treated RM-1α vaccine cells contained a measurable amount (6.8 pg/million cells) of cytoplasmic IL-15 protein by ELISA. In addition, no up-regulation of membrane associate IL-15 was detected with long-term treated RM-1α vaccine cells. Contrarily, short-term treated B16α 48 h cells had already expressed about 23 pg/million cells cytoplasmic IL-15 protein. Long-term IFN treatment further increased that intracellular content of IL-15 protein 6.8-fold to 157 pg/million cells, as seen with 2-week treated B16α vaccine cells. On the cell surface, 2-week treated B16α vaccine cells also expressed 1.6-fold more membrane-bound IL-15 than B16 parental cells. The evidence suggests that B16α cells possess a lower threshold for IL-15 induction by IFN-α treatment than RM-1α cells at both transcriptional and translational levels. Further, the data also suggest that IL-15 transcriptional up-regulation in B16α cells, whether treated with IFN for short term or long term, correlates nicely with translational up-regulation in IL-15 expression. Confocal microscopy performed on long-term treated RM-1α vaccine cells confirmed the ELISA results that a substantially less amount of intracellular IL-15 was expressed by these cells than by long-term treated B16α vaccine cells.
It was challenging to explain why 19-fold and 35-fold levels of transcriptional up-regulation in short-term and long-term IFN-treated RM-1α cells, respectively, failed to generate more substantial IL-15 translation. One possible explanation is that cell types other than dendritic and monocytic lineage (the primary producers of IL-15) are not specialized in IL-15 synthesis and may require activation of additional genes to promote IL-15 translation. A second possible explanation is that synthesis of a potent immunostimulant like IL-15 is down-regulated by regulatory proteins when induced in non-immune cell types. There is some support for such regulation, as it has been shown that IL-15 expression is post-transcriptionally regulated by elements such as 12 upstream AUGs of the 5′ untranslated region (UTR), a 48-aa signal peptide, and the C-terminus of the mature protein (Bamford and others 1996; Bamford and others 1998).
When UV-inactivated, RM-1α vaccine cells that have a modest induction of IL-15 translation in the absence of UV irradiation showed a 2.7-fold increase in their cytoplasmic IL-15 protein content compared with non-UV-inactivated RM-1α cells. B16α cells that have a robust induction of IL-15 translation failed to show an increase in the level of IL-15 translation when UV-irradiated. It appeared that cell damage, such as that rendered by UV light, blocked the induction of the mechanism responsible for down-regulation of IL-15 translation in RM-1α cells. Thus, the data from UV-inactivated RM-1α vaccine cells support the second possibility.
It has been hypothesized that IL-15 expression induced in the long-term IFN-α–treated cells plays a major role in activating antitumor immunity (Wu and others 2007). In vitro observations with RM-1α vaccine cells, even after UV inactivation, had suggested that they would be relatively less efficacious in inducing antitumor immunity in mice when compared to B16α vaccine. The evidence from RM-1α vaccination experiments supports this original hypothesis. The vaccination efficacy of an IL-15-containing whole-cell vaccine does correlate to the amount of its IL-15 content.
As evidenced in this study, IL-15 induction in mouse tumor cell vaccines by in vitro IFN-α treatment varies greatly from one tumor type to another. Thus, extending these studies to human tumor cell vaccines will most probably require transfection of the human tumor cells with an IL-15 construct that overcomes translational and/or post-translational regulatory controls, causing the tumor cells to accumulate high levels of cell-associated IL-15 and enabling them to serve as an efficacious vaccine.
The development of human cancer vaccines would be envisioned as follows. Cancer cells harvested by biopsy would be expanded in vitro and transfected with a genetically manipulated IL-15 gene construct. Studies are currently underway to develop a genetically manipulated IL-15 construct that would permit a high level of IL-15 translation and would cause the sequestration of the IL-15 within the cytoplasm of IL-15-transfected cancer vaccine cells. The IL-15-transfected cancer vaccine cells would become, in essence, “bags” containing high concentrations of IL-15 and expressing tumor antigens. After lethally irradiation and inoculation back into the patient, the cancer vaccine cells would survive for a few days before undergoing lysis. During the few days between the inoculation of the cancer vaccine cells and the lysis of the cancer vaccine cells, tissue-infiltrating lymphocytes would be expected to gather at the site of the inoculation. These tissue-infiltrating lymphocytes would recognize tumor antigens on the surface of the cancer vaccine cells but would be initially tolerant to the tumor antigens. As the cancer vaccine cells lyse, they would release their IL-15. The released IL-15 would activate the tumor-infiltrating lymphocytes to break their tolerance to the tumor antigens expressed on the surface of the cancer vaccine cells, initiating tumor immunity. The activated tumor-infiltrating lymphocytes would then travel through the body, recognizing and killing cancer cells wherever they might be present, thus curing the patient.
Contributor Information
Tzu G. Wu, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Joana R. Perdigão, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Theresa K. Umhoefer, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Jade Cao, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota..
David A. Ansari, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Thomas B. Albrecht, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas. Infectious Disease and Toxicology Optical Imaging Center, University of Texas Medical Branch, Galveston, Texas.
Eugene P. Knutson, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas.
William A. Rose, II, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas..
Angela J. Jorgensen, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Lee M. Ryan, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Linda E. Abdalla, Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
William Robert Fleischmann, Jr., Department of Urologic Surgery, University of Minnesota Medical School, Minneapolis, Minnesota.
Acknowledgments
This work was supported, in part, by grants from the Prostate Cancer Research and Education Foundation (W.R.F.), a grant (No. DAMD 17021059) from the U.S. Department of Defense (W.R.F.), and a grant (No. ES10018) from the National Institute of Environmental Health Sciences (T.B.A.). The contents of this work are solely the responsibility of the authors and do not represent the official views of the granting agencies.
Author Disclosure Statement
No competing financial interests exist.
References
- Baley PA, Yoshida K, Qian W, Sehgal I, Thompson TC. Progression to androgen insensitivity in a novel in vitro mouse model for prostate cancer. J Steroid Biochem Mol Biol. 1995;52((5)):403–413. doi: 10.1016/0960-0760(95)00001-g. [DOI] [PubMed] [Google Scholar]
- Bamford RN, Battiata AP, Burton JD, Sharma H, Waldmann TA. Interleukin (IL) 15/IL-T production by the adult T-cell leukemia cell line HuT-102 is associated with a human T-cell lymphotrophic virus type I R region/IL-15 fusion message that lacks many upstream AUGs that normally attenuate IL-15 mRNA translation. Proc Nat Acad Sci USA. 1996;93:2897–2902. doi: 10.1073/pnas.93.7.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford RN, DeFilippis AP, Azimi N, Kurys G, Waldmann TA. The 5′ UTR, signal peptide and 3′ coding sequences of IL-15 participate in its multifaceted translational control. J Immunol. 1998;160:4418–4426. [PubMed] [Google Scholar]
- Berard M, Brandt K, Bulfone-Paus S, Trough DF. IL-15 promotes the survival of naive and memory phenotype CD8+ T cells. J Immunol. 1994;170:5018–5026. doi: 10.4049/jimmunol.170.10.5018. [DOI] [PubMed] [Google Scholar]
- Budagian V, Bulanova E, Orinska Z, Thon L, Mamat U, Bellosta P, Basilico C, Adam D, Paus R, Bulfone-Paus S. Reverse signaling through membrane-bound interleukin-15. J Biol Chem. 2004;279:42192–42201. doi: 10.1074/jbc.M403182200. [DOI] [PubMed] [Google Scholar]
- Campbell JB, Grunberger T, Kochman MA, White SL. A microplate reduction assay for human and mouse interferon. Can J Microbiol. 1975;21:1247–1253. doi: 10.1139/m75-186. [DOI] [PubMed] [Google Scholar]
- Dubois S, Patel HJ, Zhang M, Waldmann TA, Müller JR. Preassociation of IL-15 with IL-15R alpha-IgG1-Fc enhances its activity on proliferation of NK and CD8+/CD44high T cells and its antitumor action. J Immunol. 2008;180((4)):2099–2106. doi: 10.4049/jimmunol.180.4.2099. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Selection of successive tumor lines for metastasis. Nature New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- Fleischmann WR, Jr, Wu TG. Development of a vaccine against cancers. Med Razgl. 2004;43((suppl 5)):51–61. [Google Scholar]
- Fleischmann WR, Jr, Wu TG. Carr DJJ. Interferon Methods and Protocols. Totowa; Humana Press: 2005. Development of an interferon-based cancer vaccine protocol: application to several types of murine cancers; Methods in molecular medicine; pp. 151–165. [DOI] [PubMed] [Google Scholar]
- Grabstein KH, Eisenmann J, Shanebeck K, Rauch C, Srinivasan S, Fung V, Beers C, Richardson J, Schoenborn MA, Ahdieh M, Johnson L, Alderson MR, Watson JD, Anderson D, Giri JG. Cloning of a T cell growth factor that interacts with the α chain of the interleukin-2 receptor. Science. 1994;264:965–968. doi: 10.1126/science.8178155. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR, Grewal N, Spiess PJ, Antony PA, Palmer DC, Tagaya Y, Rosenberg SA, Waldmann TA, Restifo NP. IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8 T cells. Proc Natl Acad Sci USA. 2004;101:1969–1974. doi: 10.1073/pnas.0307298101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MJ, Dennin R, Kramer C, Jones G, Connell E, Rolon N, Gruarin A, Kale R, Trown PW. Cell and virus sensitivity studies with recombinant human alpha interferons. J Interferon Res. 1983;3:425–435. doi: 10.1089/jir.1983.3.425. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Musso T, Calosso L, Zucca M, Millesimo M, Ravarino D, Giovarelli M, Malavasi F, Ponzi AN, Paus R, Bulfone-Paus S. Human monocytes constitutively express membrane-bound, biologically active, and interferon-©-upregulated interleukin-15. Blood. 1999;93:3531–3539. [PubMed] [Google Scholar]
- Nichols JE, Niles JA, Roberts NJ., Jr Human lymphocyte apoptosis after exposure to influenza A virus. J Virol. 2001;73:5921–5929. doi: 10.1128/JVI.73.13.5921-5929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura T, Harigai M, Kawaguchi Y, Takagi K, Fukasawa C, Ohsako-Higami S, Ohta S, Tanaka M, Hara M, Kamatani N. Increased IL-15 production of muscle cells in polymyositis and dermatomyositis. Int Immunol. 2002;14:917–924. doi: 10.1093/intimm/dxf062. [DOI] [PubMed] [Google Scholar]
- Wilkinson PC, Liew FY. Chemoattraction of human blood T lymphocytes by interleukin-15. J Exp Med. 1995;181:1255–1259. doi: 10.1084/jem.181.3.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TY, Fleischmann WR., Jr Efficacy of B16 melanoma cells exposed in vitro to long-term IFN-α treatment (B16α cells) as a tumor vaccine in mice. J Interferon Cytokine Res. 1998;18:829–839. doi: 10.1089/jir.1998.18.829. [DOI] [PubMed] [Google Scholar]
- Wu TY, Fleischmann WR., Jr Murine B16 melanoma vaccination-induced tumor immunity: identification of specific immune cells and functions involved. J Interferon Cytokine Res. 2001;21:1117–1127. doi: 10.1089/107999001317205259. [DOI] [PubMed] [Google Scholar]
- Wu TG, Rose WA II, Albrecht TB, Knutson EP, König R, Perdigão JR, Nguyen AP, Fleischmann WR., Jr IFN-alpha-induced murine B16 melanoma cancer vaccine cells: induction and accumulation of cell-associated IL-15. J Interferon Cytokine Res. 2007;27((1)):13–22. doi: 10.1089/jir.2007.0039. [DOI] [PubMed] [Google Scholar]