

Abstract
Purpose:
The objectives of this work were (i) to screen ocular hypotensive prostaglandin (PGF2α) analogs—bimatoprost, latanoprost, and travoprost as well as their free acid forms—for interaction with efflux pumps on the cornea and (ii) to assess the modulation of efflux upon co-administration of these prostaglandin analogs.
Methods:
Cultured rabbit primary corneal epithelial cells (rPCEC) were employed as an in vitro model for rabbit cornea. Transporter-specific interaction studies were carried out using Madin-Darby canine kidney (MDCK) cells overexpressing MDR1, MRP1, MRP2, MRP5, and BCRP. Freshly excised rabbit cornea was used as an ex vivo model to determine transcorneal permeability.
Results:
Cellular accumulation studies clearly showed that all prostaglandin analogs and their free acid forms are substrates of MRP1, MRP2, and MRP5. Bimatoprost was the only prostaglandin analog in this study to interact with P-gp. In addition, none of these molecules showed any affinity for BCRP. Ki values of these prostaglandin analogs obtained from dose-dependent inhibition of erythromycin efflux in rPCEC showed bimatoprost (82.54 μM) and travoprost (94.77 μM) to have similar but higher affinity to efflux pumps than latanoprost (163.20 μM). Ex vivo studies showed that the permeation of these molecules across cornea was significantly elevated in the presence of specific efflux modulators. Finally, both in vitro and ex vivo experiments demonstrated that the efflux of these prostaglandin analogs could be modulated by co-administering them together.
Conclusion:
Bimatoprost, latanoprost, travoprost, and their free acid forms are substrates of multiple drug efflux pumps on the cornea. Co-administration of these molecules together is a viable strategy to overcome efflux, which could simultaneously elicit a synergistic pharmacological effect, since these molecules have been shown to activate different receptor population for the reduction of intraocular pressure (IOP).
Introduction
Topical drug administration is the most preferred and convenient route to treat diseases affecting the anterior segment of the eye. The ability of a drug molecule to achieve therapeutic levels in the anterior segment predominantly depends on the rate and extent of corneal permeation. However, ocular bioavailability following topical administration is extremely limited (<5%) due to a variety of precorneal factors.1 Low ocular bioavailability has also been attributed to the lipoidal nature of the corneal epithelium and the water-laden stroma that act as rate-limiting barriers for hydrophilic and lipophilic molecules, respectively.
In addition to the tight junctions expressed by the corneal epithelium, more recently, efflux pumps such as P-glycoprotein (P-gp) and multidrug resistance-associated proteins (MRP) have also shown to play a role in restricting ocular bioavailability.2 , 3 A number of efflux pumps has been identified on the human and rabbit corneal epithelial cells. P-gp (MDR1) was amongst the first found to be functionally active on the rabbit cornea.4 Very recently, a half transporter of MDR1, that is, breast cancer resistance protein (BCRP) has been identified on the human corneal epithelial cells and appears to play a role in drug efflux.5 Earlier reports from our laboratory have revealed the molecular evidence of MRP2 and MRP5 on the human as well as rabbit primary corneal epithelial cells.6–8 In addition, mRNA expression levels of other isoforms of MRP, MRP1 and MRP3, have been reported in the human cornea, though functional activity and localization still remain to be assessed.9 , 10 With an array of efflux transporters being identified on the corneal epithelium and along with our earlier report that clearly demonstrates the role of efflux pumps in restricting ocular bioavailability, it is imperative to screen potential ocular therapeutic agents for interaction with efflux pumps.
Ocular hypotensive drugs (anti-glaucoma) represent an important class of therapeutic agents that are primarily absorbed through the cornea, following topical administration. Among the various classes of glaucoma medications such as β-blockers, carbonic anhydrase inhibitors, and α2-adrenergic agonists, prostaglandin analogs (PGF2α analogs) have emerged as first-line treatment option due to its superior efficacy. Naturally occurring prostaglandins, especially the F-series are relatively polar due to the presence of a carboxylic acid moiety and several hydroxyl groups, resulting in poor permeation across biological membranes. Hence prodrugs of PGF2α were synthesized that resulted in the currently marketed drugs such as bimatoprost, latanoprost, and travoprost. Latanoprost and travoprost are isopropyl ester prodrugs of PGF2α, whereas bimatoprost is an ethyl amide prodrug of 17-phenyl-PGF2α (Fig. 1).
FIG. 1. .
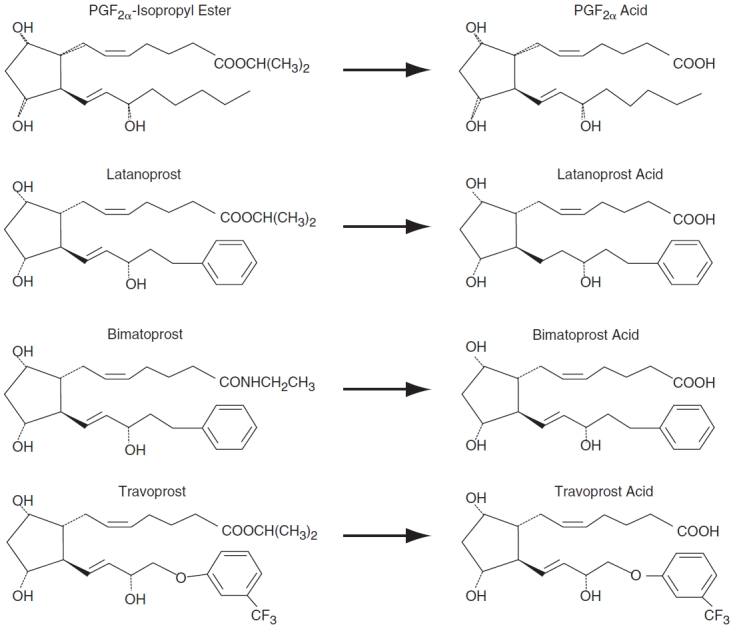
Chemical structures of prostaglandin analogs—bimatoprost, latanoprost, travoprost, and their respective free acid forms.
The rationale to screen these potent PGF2α analogs was triggered by a recent article by Reid and colleagues, who reported that PGF1α, PGF2α, and PGA1 are high-affinity inhibitors and therefore presumably are substrates of MRP4.11 Other prostaglandins such as PGA1 and PGE1 inhibited MRP4 and MRP5 transport of cAMP, suggesting that these prostaglandins are transported by MRP4/5.12 Moreover, MRP1, a transporter closely related to MRP4/5, has been shown to transport the glutathione conjugate of PGA1.13 MRP1 and MRP2 have also been shown to translocate another prostanoid, the proinflammatory leukotriene, LTC4.14–16 These reports clearly demonstrate that prostaglandins are transported by various isoforms of MRP. Since corneal epithelium expresses various isoforms of MRP such as MRP1, MRP2, MRP3, and MRP5, it was very important to examine if the corneal permeation of prostaglandin analogs is limited by these pumps. These molecules may very well be substrates for more than one isoform of MRP, since overlapping substrate specificities for MRP1–4 have been reported.17
As described earlier, one of the viable strategies to overcome efflux is to co-administer the drug along with another substrate/inhibitor drug that can modulate efflux as well as impart a synergistic pharmacological effect in the treatment regimen.3 A dual advantage is realized since the efflux is overcome by competitive inhibition and the co-administered molecule is also indicated in the treatment regimen. If the prostaglandin analogs were found to interact with efflux pumps on the corneal epithelium, a similar strategy can be adopted by administering these agents in combination. Various reports indicate that bimatoprost, which is an ethyl amide prodrug of 17-phenyl-PGF2α, is classified as a prostamide and exerts its action in lowering intraocular pressure (IOP) through a novel prostamide receptor.18 − 21 On the other hand, its metabolite, bimatoprost-free acid (17-phenyl-PGF2α) is also shown to lower the IOP by interacting with prostanoid FP receptors, thus being classified as a potent FP receptor agonist.22–24 On the contrary, both latanoprost and travoprost are isopropyl ester prodrugs, which readily get converted to its free acid form in the aqueous humor and iris–ciliary body.25 , 26 It is clear that the free acid form of latanoprost and travoprost are involved in lowering the IOP by prostanoid FP receptor signaling.25 , 26 Assuming that the free acid form is the predominant species of bimatoprost, latanoprost, and travoprost, it could still be important to co-administer them together, since these molecules exhibit binding affinities to prostaglandin receptor subtypes, such as EP1 [K i = 119 nM (latanoprost acid); 95 nM (bimatoprost acid)] and EP3 [K i = 387 nM (bimatoprost acid)] receptors.27 Knockout of EP3 receptors caused a significant increase in the IOP reduction levels following prostaglandin analog administration relative to wild-type mice.28 Moreover, clinical reports also reveal that IOP effects of the labeled concentrations of bimatoprost or travoprost were additives to that of latanoprost, with bimatoprost showing a greater additive response than travoprost.29 In addition, patients unresponsive to latanoprost have been shown to respond to bimatoprost therapy, which clearly suggests that these molecules including the free acid forms might stimulate different receptor populations.30 Even if the free acid form is the major species in the aqueous humor, since they differ in their affinities to prostaglandin receptor subtypes, a possibility exists that there is an additive/synergistic pharmacological effect resulting from co-administration of these agents. The effective doses of these prostaglandin analogs may be reduced that can substantially reduce potential ocular side effects arising from repeated administration such as conjunctival hyperemia, iris cysts, cystoid macular edema, anterior uveitis, and reactivation of herpes simplex keratitis.31
Therefore, the objectives of this work are (i) to screen bimatoprost, latanoprost, and travoprost as well as their free acid forms for interaction with efflux pumps: MDR1, MRP1, MRP2, MRP5, and BCRP and (ii) to assess the modulation of efflux upon co-administration of these prostaglandin analogs.
Methods
Materials
Bimatoprost, latanoprost, and travoprost, their respective free acids, and PGF2α diethyl amide were purchased from Cayman Chemicals (Ann Arbor, MI). MK571, a specific inhibitor of MRP was procured from Biomol International (Plymouth Meeting, PA). GF120918 was a generous gift from GlaxoSmithKline Ltd. [14C] Erythromycin (specific activity 51.3 mCi/mmol) was obtained from PerkinElmer Life and Analytical Sciences (Boston, MA). Stock solutions of the prostaglandin analogs were prepared in ethanol at a concentration of 10 mg/mL. Stock solutions of GF120918 (1 mg/mL) and MK571 (25 mg/mL) were prepared in dimethyl sulfoxide (DMSO) and aliquots were diluted in Dulbecco’s phosphate-buffered saline (DPBS) to achieve the desired concentration.
MDCK-WT (wild-type) cells and MDCK cells transfected with the human MDR1 gene (MDCK-MDR1), human MRP1 gene (MDCK-MRP1), human MRP2 gene (MDCK-MRP2), human MRP5 gene (MDCK-MRP5), and human BCRP gene (MDCK-BCRP) were generously provided by Drs. A. Schinkel and P. Borst (The Netherlands Cancer Institute, Amsterdam, Netherlands). Cell culture supplies that included minimum essential medium (MEM, for rPCEC), Dulbecco’s modified Eagle’s medium (DMEM, for MDCK-WT, -MDR1, -MRP1, -MRP2, -MRP5, and -BCRP cells), trypsin–EDTA solution, nonessential amino acids, and fetal bovine serum were procured from Invitrogen (Carlsbad, CA). Penicillin, streptomycin, sodium bicarbonate, lactalbumin, HEPES, amphotericin B, and polymyxin B sulfate were obtained from Sigma-Aldrich. Culture flasks (75-cm2 growth area) were procured from MidSci (St. Louis, MO). Twelve-well culture plates (3.8-cm2 growth area per well) were obtained from Corning Costar Corp (Cambridge, MA).
New Zealand albino male rabbits weighing between 2.0 and 2.5 kg were purchased from Myrtle’s Rabbitry (Thompson Station, TN). Studies were performed according to the animal protocol stated in ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Ketamine HCl and Rompun (Xylazine®) were purchased from Fort Dodge Animal Health (Fort Dodge, IA) and Bayer Animal Health (Shawnee Mission, KS), respectively. All other chemicals were obtained from Sigma-Aldrich (St. Louis, MO). The solvents were of HPLC grade and obtained from Fisher Scientific Company (St. Louis, MO).
Methods
Cell culture. Rabbit corneal epithelial cells were cultured according to a previously published method from our laboratory.4 Cells were nourished with culture medium comprising MEM, 10% FBS, HEPES, sodium bicarbonate, penicillin, streptomycin sulfate, and 1% (v/v) nonessential amino acids, adjusted to pH 7.4. Cells were grown in 75-cm2 culture flasks and maintained at 37°C, in a humidified atmosphere of 5% CO2 and 90% relative humidity. Culture medium was replaced every alternate day. Cells were subcultured after 7 days (subculture ratio, 1:5) with 0.25% trypsin containing 0.53 mM EDTA and plated at a density of 250,000 cells/well on 12-well culture plates.
MDCK-WT and MDCK-transfected cells overexpressing efflux proteins were grown in culture medium comprising DMEM, 10% FBS, HEPES, sodium bicarbonate, penicillin, streptomycin sulfate, and 1% (v/v) nonessential amino acid, adjusted to pH 7.4. Cells were grown under similar conditions as mentioned above and plated at a density of 250,000 cells/well on 12-well culture plates.
Cellular accumulation studies
Preparation of drug solutions. Cellular accumulation studies were conducted on rPCEC (8–10 days post-seeding, passages 5–10) and MDCK-WT, -MDR1, -MRP1, -MRP2, -MRP5, and -BCRP cells (4–6 days post-seeding, passages 5–15). The medium was aspirated and cells were washed twice (once in 10 min) with Dulbecco’s phosphate-buffered saline (DPBS; pH 7.4). Stock solutions of GF120918, MK571, and the prostaglandin analogs were prepared as mentioned earlier. Test solutions were then prepared by adding aliquots from the respective stock solutions and diluting with DPBS, pH 7.4, to achieve required concentration. The organic solvent concentration in the final solution did not exceed 1% (v/v) and appropriate amount was added to control.
Radioactive studies. Cellular accumulation was initiated by adding 0.5 mL of [14C] erythromycin (0.2 μCi/mL) prepared in DPBS, pH 7.4, in the absence and presence of test compounds. Incubation with drug solution was carried out for 15 min at 37°C. Following incubation, the drug solution was aspirated and cell monolayers were washed twice with 1 mL of ice-cold stop solution (210 mM KCl, 2 mM HEPES) to arrest cellular accumulation. Finally, cells were lysed with 1 mL of 0.3% NaOH containing 0.1% Triton-X solution and stored overnight. Cell-associated radioactivity was quantified with a scintillation counter and the rate of cellular accumulation of [14C] erythromycin was normalized to the protein content in each well.
Non-radioactive studies. Cellular accumulation was initiated by adding 0.5 mL of drug solution (in the presence or absence of competing substrates) to the wells. Incubation was carried out over a period of 15 min at 37°C. At the end of incubation period, drug solution was removed and the cell monolayers were washed twice with 1 mL of ice-cold stop solution to arrest cellular accumulation. Finally, 0.5 mL of DPBS, pH 7.4, was added to each well and the culture plates were stored at −80°C overnight to allow the cells to lyse. The following day, intracellular drug concentration was quantified by liquid chromatography tandem mass spectrometry (LC/MS/MS). The rate of cellular accumulation of the drug was normalized to the protein content in each well.
Corneal transport studies
A typical side-by-side diffusion apparatus was used. New Zealand albino rabbits weighing 2.0 to 2.5 kg were euthanized by an overdose of pentobarbital through the marginal ear vein. The eyes were carefully enucleated and washed with ice-cold DPBS (pH 7.4) to remove any traces of blood. Subsequently, a small incision was made in the sclera and the cornea was excised carefully. The cornea was mounted in such a way that the epithelium faced the donor chamber (tear to stromal-directed sampling). The side-by-side diffusion apparatus was maintained at a temperature of 34°C (corneal temperature in vivo) by circulating water through the jacketed chambers of diffusion apparatus. Drug solution (3 mL) was added to the half-chamber facing the epithelial side of the cornea (donor chamber). In the other half-chamber (receiver chamber), 3.2 mL of DPBS (maintained at 34°C) was added and both the chambers were stirred continuously with magnetic stir bars. The buffer volume in the receiver chamber was maintained slightly higher in order to generate higher hydrostatic pressure to maintain the shape of cornea throughout an experiment. Sink conditions were maintained throughout the experiment. Two hundred microliter aliquots were removed from the receiver chamber at appropriate time intervals and replaced with an equal volume of DPBS (pH 7.4, maintained at 34°C). Storing aqueous solutions of prostaglandin analogs for more than a day is usually not recommended, and hence all samples were processed immediately (refer to sample preparation section) at the end of an experiment. Further analysis of the samples was carried out by LC/MS/MS.
Analytical procedure
Scintillation counter. Radioactivity in cellular accumulation experiments with [14C] erythromycin was measured by a scintillation counter (Model LS 6500; Beckman Instruments Inc., Fullerton, CA).
LC/MS/MS
Sample preparation. All prostaglandin analogs were analyzed by LC/MS/MS. Sample preparation was carried out with liquid–liquid extraction technique. PGF2α diethyl amide was used as an internal standard for the analysis of all prostaglandin analogs in the positive mode of multiple reaction monitoring (MRM). Their corresponding free acids were analyzed in the negative mode using appropriate free acid form of other prostaglandin analog depending upon the analyte being measured. The organic solvent, ethyl acetate, was utilized to extract the drug from the aqueous phase.
(i) Cellular accumulation studies: Culture plates stored at −80°C were first thawed at room temperature. Subsequently, the samples were centrifuged at 12,500g for 10 min to separate cellular debris. Following centrifugation, 450 μL of the sample was removed and 25 μL of the corresponding internal standard was added. When both prostaglandin analog and their respective free acid were analyzed, 25 μL of the internal standard for both the positive and negative ion analytes was added accordingly. The samples were vortexed for 10–15 s following the addition of internal standard(s). One and a half milliliter of the organic solvent, ethyl acetate was then added to the sample–internal standard mixture and vortexed for 3 min. For efficient separation of the aqueous and organic layers, samples were centrifuged at 10,000g for 5 min. After centrifugation, the organic layer was collected and dried in vacuum. The residue was reconstituted in 75 μL of distilled deionized water (DDW) with 0.1% formic acid and 50 μL was injected onto the LC/MS/MS for analysis. When both the prostaglandins and their free acid forms were analyzed, the injection volume was reduced to 25 μL for each analyte detection in the positive and negative modes. Standard solutions in DPBS were also extracted and quantitated by following an identical procedure.
(ii) Corneal transport studies: In these experiments, 25 μL of the corresponding internal standard(s) was added to the sample. The samples were vortexed for 10–15 s following the addition of internal standard(s). After that, 500 μL of ethyl acetate was added to the sample–internal standard mixture and vortexed for 3 min. Subsequent steps were similar to those mentioned in the previous section.
LC/MS/MS. QTrap® LC/MS/MS mass spectrometer (Applied Biosystems, Foster City, CA) equipped with Agilent 1100 Series quaternary pump (Agilent G1311A), vacuum degasser (Agilent G1379A), and autosampler (Agilent G1367A; Agilent Technology Inc., Palo Alto, CA) was employed to analyze samples from cellular accumulation, transport, and ocular tissue distribution studies. HPLC separation was performed on a XTerra® MS C18 column 50 × 2.1 mm, 3.5 μm (Waters, Milford, MA). The mobile phase consisted of 65% acetonitrile and 35% water with 0.1% formic acid, pumped at a flow rate of 0.3 mL/min. Analysis time was 4 min per run and all analytes eluted within 1–2 min. Multiple reaction monitoring (MRM) mode was utilized to detect the compound of interest. The mass spectrometer was operated in the positive ion detection mode for the quantification of all prostaglandin analogs (bimatoprost, latanoprost, and travoprost). Negative ion detection mode was used for the quantification of free acid forms. The precursor and the secondary ions generated were: bimatoprost +416.1/362.1; latanoprost +433.0/105.0; travoprost +500.9/207.0; PGF2α diethyl amide (internal standard) +444.3/426.3; bimatoprost-free acid –387.0/342.7; latanoprost-free acid –389.0/344.9; and travoprost-free acid −456.9/160.9. The turbo ion spray setting and collision gas pressure were optimized (IS voltage: ±4500 V, temperature: 300°C, nebulizer gas: 40 psi, curtain gas: 30 psi). MS/MS was performed using nitrogen as collision gas. Peak areas for all components were automatically integrated with Analyst™ software and peak-area ratios (analyte peak area/IS peak area) were plotted against concentration by weighted linear regression. The analytical data with the current MRM method showed excellent linearity extending to nanomolar range. The limits of quantification were found to be 15 ng/mL for all prostaglandin analogs and their respective free acid forms. The method generated rapid and reproducible results.
Data treatment
Permeability measurements across rabbit cornea. Cumulative amounts transported across cell monolayers and rabbit cornea were plotted as a function of time. Linear regression of the amounts transported as a function of time yielded the rate of transport across the cell monolayer (dM/dt). Rate divided by the cross-sectional area available for transport (A) generated steady-state flux as shown in Equation 1.
![]()
Slopes were then obtained from the linear portion of the curve to calculate apparent permeability (P app) through normalization of the steady-state flux to the donor concentration (C d) according to Equation 2.
![]()
Affinity measurements. For dose response studies in which [14C] erythromycin uptake was inhibited, the inhibitory effect of unlabeled bimatoprost, latanoprost, and travoprost was described by Equation 3.

where x denotes the logarithm of the concentration of the prostaglandin analog, Y is the cellular accumulation of [14C] erythromycin, IC50 represents the inhibitor concentration where the efflux of [14C] erythromycin is inhibited by 50%, and H is the Hill constant. Y starts at a minimum (min) value (at low inhibitor concentration) and then plateaus at a maximum (max) value (at high inhibitor concentration) resulting in a sigmoidal shape. IC50, estimated from Equation 3, was used to calculate the K i of the prostaglandin analogs by the method of Cheng and Prusoff in which K i is equivalent to IC50/(1 + C/K m), where C indicates the concentration of the ligand.32 Data were fitted to Equation 3 with a transformed nonlinear regression curve analysis program (GraphPad Prism version 4.0, GraphPad Software Inc., San Diego, CA).
Statistical analysis
Cellular accumulation studies were conducted at least in quadruplicate and corneal transport experiments were conducted at least in triplicate. All results are expressed as mean ± standard deviation (SD). Student’s t-test was applied to determine statistical significance between 2 groups, with P < 0.05 being considered to be statistically significant.
Results
Cellular accumulation of [14C] erythromycin in rPCEC in the presence of specific inhibitors, prostaglandin analogs, and their corresponding free acids
Cellular accumulation of [14C] erythromycin was significantly elevated (160% and 370% relative to control) in the presence of GF120918 (P-gp inhibitor) and MK571 (MRP inhibitor), respectively (Fig. 2A). In addition, cellular accumulation of [14C] erythromycin increased significantly in the presence of all prostaglandin analogs—bimatoprost, latanoprost, and travoprost (Fig. 2A). In contrast, cellular accumulation of [14C] erythromycin did not alter significantly when the corresponding free acid forms of prostaglandin analogs, with the exception of travoprost-free acid, were co-incubated with [14C] erythromycin (Fig. 2B).
FIG. 2. .
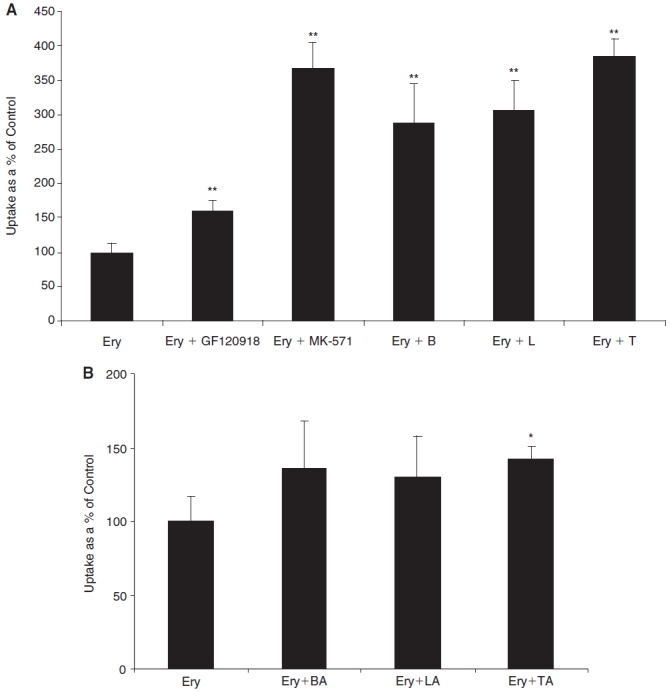
(A) Cellular accumulation of [14C] erythromycin (0.2 μCi/mL) by primary corneal epithelial cells (rPCEC) in the presence of a P-gp inhibitor (GF120918, 2 μM), MRP inhibitor (MK571, 50 μM), bimatoprost (B, 50 μM), latanoprost (L, 50 μM), and travoprost (T, 50 μM). Each data point represents mean ± SD (n = 4). ** Significant difference from control (P < 0.01). (B) Cellular accumulation of [14C] erythromycin (0.2 μCi/mL) by rPCEC in the presence of free acid forms of bimatoprost (BA, 100 μM), latanoprost (LA, 100 μM), and travoprost (TA, 100 μM). Each data point represents mean ± SD (n = 4). *Significant difference from control (P < 0.05).
Cellular accumulation of prostaglandin analogs and their corresponding free acids in MDCK-MDR1, -MRP1, -MRP2, -MRP5, -BCRP, and rPCEC in the presence of specific inhibitors
These studies were conducted with unlabeled prostaglandin analogs and their corresponding free acid forms according to the protocol described in the Methods section. Cellular accumulation studies were carried out at a concentration of 10 μM for prostaglandin analogs and at 100 μM for their free acid forms. Ten times higher concentration of the free acid form was used in order to achieve the detection levels, since the membrane permeability of the free acid forms are reported to be very low. GF120918 (2 μM) was used as the specific inhibitor in both MDCK-MDR1 as well as -BCRP cells, since it was reported to inhibit both P-gp and BCRP efflux pumps. On the other hand, MK571 (50 μM), an inhibitor known to inhibit almost all isoforms of MRP, was used for interaction studies with MDCK-MRP1, -MRP2, and -MRP5 cells. No conversion of bimatoprost to its free acid form was detected during the course of the experiment (15 min), but the free acid forms of latanoprost and travoprost were detectable. Hence, for latanoprost and travoprost, the total concentrations of both the parent as well as the free acid forms were taken into account.
Table 1 provides a comprehensive overview of all possible interactions for prostaglandin analogs and their corresponding free acid forms with various efflux pumps studied. It was observed that the cellular accumulation of bimatoprost significantly elevated in the presence of GF120918 in MDCK-MDR1 but not in MDCK-BCRP cells. Interestingly, latanoprost and travoprost did not exhibit any statistically significant increase in their cellular accumulation with GF120918 in both MDCK-MDR1 and -BCRP cells. All three prostaglandin analogs, bimatoprost, latanoprost, and travoprost, showed significant interactions with all the isoforms of MRP studied, that is, MRP1, MRP2, and MRP5. On the other hand, free acid forms of bimatoprost, latanoprost, and travoprost did not produce statistically significant interactions in MDCK-MDR1 and -BCRP cells. However, these compounds interacted significantly with all isoforms of MRP (MRP1, MRP2, and MRP5). Table 2 summarizes the substrate specificities of all the prostaglandin analogs and their free acid forms with respect to P-gp, MRP1, MRP2, MRP5, and BCRP efflux pumps.
Table 1. .
Cellular Accumulation of Prostaglandin Analogs and Their Corresponding Free Acids in the Absence and Presence of Specific Efflux Inhibitors—GF120918 2 μM (P-gp and BCRP) and MK571 50 μM (All Isoforms of MRP)
| Drug ± inhibitor |
Cellular accumulation (% control) |
|||||
|---|---|---|---|---|---|---|
| MDCK-MDR1 | MDCK-MRP1 | MDCK-MRP2 | MDCK-MRP5 | MDCK-BCRP | rPCEC | |
| Bimatoprosta | ||||||
| B | 100.0 ± 12.9 | 100.0 ± 9.6 | 100.0 ± 18.3 | 100.0 ± 12.9 | 100.0 ± 15.4 | 100.0 ± 17.6 |
| B + GF120918 | 259.3 ± 14.1** | — | — | — | 97.7 ± 17.0 | 205.7 ± 3.9** |
| B + MK571 | — | 237.8 ± 33.1** | 474.9 ± 31.7** | 209.0 ± 21.1** | — | 297.3 ± 14.4** |
| BA | 100.0 ± 4.0 | 100.0 ± 7.4 | 100.0 ± 18.1 | 100.0 ± 19.2 | 100.0 ± 5.5 | 100.0 ± 17.0 |
| BA + GF120918 | 92.3 ± 5.4 | — | — | — | 107.3 ± 23.4 | 107.6 ± 4.5 |
| BA + MK571 | — | 298.5 ± 40.1** | 414.1 ± 24.1** | 278.7 ± 26.8** | — | 276.8 ± 35.9** |
| Latanoprostb | ||||||
| L | 100 ± 11.5 | 100.0 ± 7.3 | 100.0 ± 12.9 | 100.0 ± 8.8 | 100.0 ± 13.1 | 100 ± 7.0 |
| L + GF120918 | 111.0 ± 8.2 | — | — | — | 95.9 ± 4.7 | 102.5 ± 18.4 |
| L + MK571 | — | 194.9 ± 11.2** | 355.38 ± 6.05** | 212.3 ± 26.4** | — | 267.9 ± 56.8** |
| LA | 100.0 ± 14.2 | 100.0 ± 3.0 | 100.0 ± 5.9 | 100.0 ± 7.8 | 100.0 ± 16.4 | 100.0 ± 23.7 |
| LA + GF120918 | 87.8 ± 7.6 | — | — | — | 98.3 ± 12.6 | 96.7 ± 10.0 |
| LA + MK571 | — | 267.4 ± 33.0** | 382.0 ± 91.2** | 287.9 ± 18.7** | — | 301.3 ± 40.6** |
| Travoprostc | ||||||
| T | 100 ± 7.6 | 100.0 ± 10.1 | 100.0 ± 5.7 | 100.0 ± 9.7 | 100.0 ± 18.2 | 100.0 ± 12.6 |
| T + GF120918 | 99.0 ± 5.6 | — | — | — | 89.1 ± 24.7 | 99.1 ± 11.7 |
| T + MK571 | — | 189.3 ± 19.5** | 245.9 ± 27.0** | 205.3 ± 19.7** | — | 153.1 ± 16.5** |
| TA | 100.0 ± 10.8 | 100.0 ± 17.5 | 100.0 ± 29.8 | 100.0 ± 15.3 | 100.0 ± 4.8 | 100.0 ± 7.5 |
| TA + GF120918 | 92.8 ± 13.1 | — | — | — | 104.6 ± 19.0 | 89.0 ± 12.6 |
| TA + MK571 | — | 278.0 ± 26.9** | 675.4 ± 59.2** | 313.1 ± 37.6** | — | 434.9 ± 22.8** |
aBimatoprost (B, 10 μM), bimatoprost-free acid (BA, 100 μM).
bLatanoprost (L, 10 μM), latanoprost-free acid (LA, 100 μM).
cTravoprost (T, 10 μM), travoprost-free acid (TA, 100 μM).
Each data point represents mean ± SD (n = 4). *Represents significant difference from control P < 0.01.
Table 2. .
Substrate Specificities of all the Prostaglandin Analogs and Their Free Acid Forms to P-gp, MRP1, MRP2, MRP5, and BCRP Efflux Pumps
| Drug |
Interaction |
||||
|---|---|---|---|---|---|
| MDR1 | MRP1 | MRP2 | MRP5 | BCRP | |
| B | Y | Y | Y | Y | N |
| BA | N | Y | Y | Y | N |
| L | N | Y | Y | Y | N |
| LA | N | Y | Y | Y | N |
| T | N | Y | Y | Y | N |
| TA | N | Y | Y | Y | N |
Abbreviations: Y, yes; N, no.
Dose-dependent inhibition of [14C] erythromycin efflux in rPCEC in the presence of prostaglandin analogs
To determine the inhibitory potency, dose-dependent inhibition of [14C] erythromycin efflux, in the presence of varying concentrations of bimatoprost, latanoprost, and travoprost, was carried out. As shown in Fig. 3A–3C, cellular accumulation of [14C] erythromycin increased in a dose-dependent manner in the presence of all three prostaglandin analogs. A modified log [dose]–response curve was applied to fit the data in order to obtain the IC50 values. Inhibition constant, K i, was calculated by the method of Cheng and Prusoff using the equation: IC50/[1 + (C/K m)]. Michaelis constant, K m, for erythromycin was determined to be 473.74 ± 86.7 μM (data not shown). The resulting IC50 and K i values were close (up to 2 decimal points) and found to be 82.54, 163.20, and 94.77 μM for bimatoprost, latanoprost, and travoprost, respectively.
FIG. 3. .
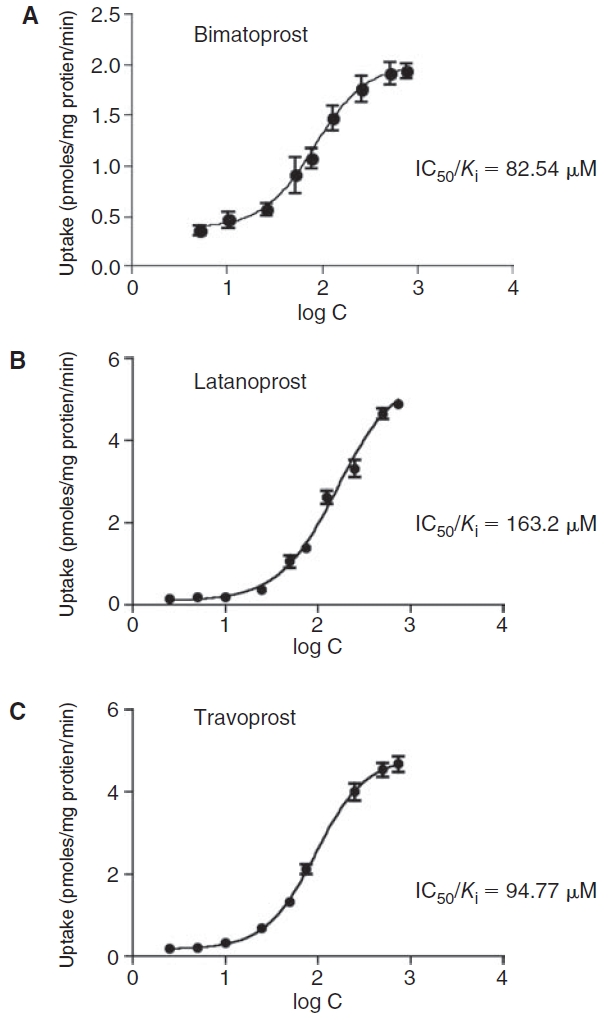
Dose-dependent inhibition of [14C] erythromycin (0.2 μCi/mL) efflux by (A) bimatoprost, (B) latanoprost, and (C) travoprost in primary corneal epithelial cells (rPCEC). Each data point represents mean ± SD (n = 4).
Corneal transport of prostaglandin analogs in the presence of specific inhibitors
Corneal transport of bimatoprost was performed alone and in the presence of both GF120918 and MK571, since bimatoprost was found to be a substrate for P-gp as well as MRP. On the other hand, corneal transport of latanoprost and travoprost were studied only in the presence of MK571. In case of bimatoprost, both the parent ester and the free acid forms were equally predominant and hence concentrations of both species were taken into account while calculating flux. But, latanoprost and travoprost hydrolyzed completely into their free acid forms and the parent ester species were below the detection limit. The corneal permeability values of bimatoprost, latanoprost, and travoprost increased by 1.67, 1.79, and 2.11 times, respectively, relative to appropriate control values (Table 3).
Table 3. .
Transcorneal Permeability Values of Bimatoprost (B, 50 μM), Latanoprost (L, 50 μM), and Travoprost (T, 50 μM) Alone and in the Presence of Specific Efflux Inhibitors (GF120918 2 μM, MK571 50 μM)
| Drug ± inhibitor | Permeability (× 105 cm/s) |
|---|---|
| B | 0.74 ± 0.05 |
| B + GF120918 + MK571 | 1.23 ± 0.016* |
| L | 2.02 ± 0.02 |
| L + MK571 | 3.61 ± 0.26** |
| T | 1.11 ± 0.14 |
| T + MK571 | 2.35 ± 0.26* |
Each data point represents mean ± SD (n = 3). (**) and (*) represent significant difference from control (P < 0.01) and (P < 0.05), respectively.
Abbreviations: B, bimatoprost; L, latanoprost; T, travoprost.
Cellular accumulation of bimatoprost in rPCEC in the presence of other prostaglandin analogs and corticosteroids
In order to examine the modulation of efflux due to co-administration, cellular accumulation of bimatoprost was carried out in the presence of other prostaglandin analogs (latanoprost, travoprost) and steroids (6α-methyl prednisolone, prednisolone). Cellular accumulation of bimatoprost was significantly elevated in the presence of all the compounds tested (Fig. 4). Travoprost (∼230%) inhibited the efflux of bimatoprost to a larger extent than latanoprost (∼180%). On the other hand, 6α-methyl prednisolone (∼200%) exhibited a higher inhibitory potential than prednisolone (∼150%).
FIG. 4. .
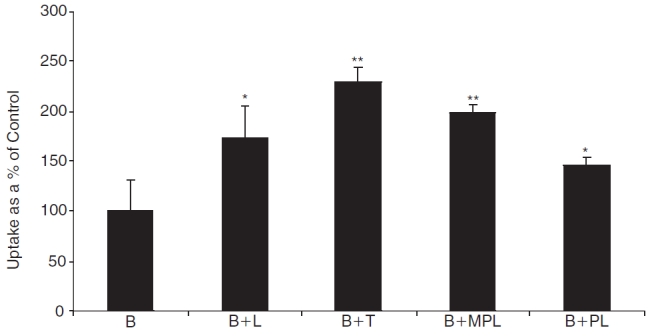
Cellular accumulation of bimatoprost (B, 10 μM) in the presence of latanoprost (B+L, 50 μM), travoprost (B+T, 50 μM), 6α-methyl prednisolone (B+MPL, 500 μM), and prednisolone (B+PL, 500 μM). Each data point represents mean ± SD (n = 4). Significant differences from control: **P < 0.01; *P < 0.05.
Corneal transport of bimatoprost in the presence of other prostaglandin analogs
Transcorneal permeation experiments were carried out to delineate the modulation of bimatoprost efflux in the presence of other prostaglandin analogs such as latanoprost and travoprost. As seen in Figure 5, the permeability of bimatoprost (0.74 ± 0.05 × 10−5 cm/s) across rabbit cornea was significantly higher in the presence of latanoprost (1.02 ± 0.09 × 10−5 cm/s) and travoprost (1.32 ± 0.05 × 10−5 cm/s). Travoprost showed a stronger inhibition of bimatoprost efflux since it increased the corneal permeability of bimatoprost by 1.8 times, relative to latanoprost that raised the permeability by 1.4 times.
FIG. 5. .
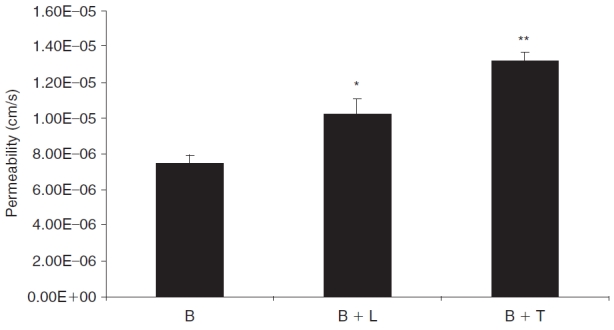
Transcorneal permeability of bimatoprost (B, 50 μM) in the presence of latanoprost (B+L, 50 μM) and travoprost (B+T, 50 μM) across freshly excised rabbit cornea. Each data point represents mean ± SD (n = 3). Significant differences from control: **P < 0.01; *P < 0.05.
Discussion
The role of efflux pumps on corneal drug absorption following topical administration is a new and emerging field. Multidrug resistance pumps such as P-gp, MRP, and BCRP can severely limit corneal drug absorption. Poor ocular bioavailability following topical administration has always been attributed to precorneal factors and highly selective corneal epithelium, but not to any efflux pump that might efflux molecules out of the corneal epithelium into the precorneal fluid. But in fact, recent reports from our laboratory clearly demonstrate the significance of efflux on corneal epithelium,2 , 3 , 5 , 7 , 8 which might be due to the combination of more than one of the following reasons: (i) precorneal factors such as drug solution drainage, continuous tear turnover, blinking, nasolacrimal drainage, and loss due to absorption by conjunctival blood vessels and lymphatics play a significant role in limiting the precorneal drug concentration. On an average there is a 10-fold decrease in the precorneal drug concentration, within 5–10 min of topical administration, due to all these factors acting simultaneously.33 Therefore, such significantly lower precorneal drug concentration makes the efflux transporters on the corneal epithelium highly relevant. (ii) A variety of efflux transporters are expressed on the human corneal epithelium that includes multidrug resistance pumps such as P-gp, BCRP, and various isoforms of MRP.5 , 8 , 10 (iii) Finally, it has been very clear from the recent literature that these multidrug resistance pumps have overlapping substrate specificities. A drug molecule is often categorized as a substrate to more than one efflux protein, thus enduring a synergistic efflux action by multiple efflux pumps. Hence, drastic reduction in precorneal drug concentration, combined with a multitude of efflux pumps with overlapping substrate specificities, renders efflux a significant factor in the ocular absorption of drugs applied topically to the eye. Previous work from this laboratory has already reported that corneal absorption of erythromycin, a substrate of both P-gp and MRP2, is restricted by the efflux pumps on the corneal epithelium.3 In this study, the objective is to screen several anti-glaucoma drugs to examine if their corneal permeation is restricted due to efflux pumps. Efflux is more clinically relevant in case of anti-glaucoma drugs, since most are delivered as topical eye drops. In addition, prostaglandin analogs, which in most cases are the first-line treatment option, are administered at a relatively low concentration (bimatoprost—0.03%, latanoprost—0.005%, and travoprost—0.004%), thus making efflux to be of high clinical relevance.
Though protein expression levels of P-gp, BCRP, and various isoforms of MRPs have been reported on the corneal epithelium, the functional activity has been demonstrated only for P-gp and MRP2. Previous study from this laboratory indicates that erythromycin can be selected as a good model substrate to study both P-gp- and MRP2-mediated efflux.3 Hence, a preliminary interaction experiment was carried out in rPCEC, by studying cellular accumulation of erythromycin in the presence of prostaglandin analogs. A significant increase in erythromycin uptake in the presence of GF120918 and MK571 corroborated with our earlier result that both P-gp and MRP are functionally active on the rabbit corneal epithelium (Fig. 2A). Moreover, the extent of enhancement in the presence of MK571 was significantly higher than that obtained with GF120918, which suggests the possibility of isoforms of MRP, other than MRP2 that may be active. Moreover, all prostaglandin analogs including bimatoprost, latanoprost, and travoprost significantly elevated cellular accumulation of erythromycin that strongly indicates that these PGF2α analogs interact with efflux pumps (Fig. 2A). Surprisingly, the free acid forms of these prostaglandin analogs did not increase the uptake of erythromycin significantly (with the exception of travoprost) (Fig. 2B). The reason for this could be attributed to inadequate concentration levels of these prostaglandins achieved intracellularly, owing to their poor membrane permeability. Hence, experiments were carried out with the free acid forms as substrates, by co-incubating with specific efflux inhibitors for further confirmation.
Functional activity of efflux transporters such as MRP1, MRP5, and BCRP have been reported recently from this laboratory.5 , 7 , 8 Hence, delineation of the affinity of these prostaglandin analogs toward MRP1, MRP5, and BCRP, in addition to P-gp and MRP2, are warranted. Since literature reports suggest rapid conversion of PGF2α analogs to their respective free acid forms in the cornea, interaction studies delineating their affinity toward efflux pumps are necessary.25 , 26 The results clearly show that bimatoprost interacts with P-gp, whereas other prostaglandin analogs do not (Table 1). The difference in chemical structures of these prostaglandin analogs might help explain their affinity toward P-gp. Carbon-1 position of bimatoprost has an ethyl amide group, whereas both latanoprost and travoprost possess an isopropyl ester group (Fig. 1). Also, there is a saturated double bond between the carbon-13 and carbon-14 positions of latanoprost, which is retained in case of bimatoprost and travoprost. In addition, travoprost has a phenoxy group at the carbon-16 position and a trifluoromethyl group at the meta position on the phenoxy ring. The phenoxy group is absent in case of bimatoprost and latanoprost, but these molecules possess a phenyl ring at the carbon-17 position (Fig. 1). Hence, interaction of bimatoprost (but not latanoprost and travoprost) with P-gp could possibly be explained by the ethyl amide group at carbon-1 position of bimatoprost. It is further strengthened by the fact that even the free acid form of bimatoprost (devoid of ethyl amide linkage at carbon-1 position) does not interact with P-gp.
On the other hand, the free acid forms of these prostaglandin analogs are excellent substrates for MRP1, MRP2, and MRP5 (Table 1). Hence, it is evident that during the interaction experiment with erythromycin, adequate inhibitory intracellular concentration levels of these molecules was not achieved due to their poor membrane permeability characteristics. It is interesting to note that all the prostaglandin analogs along with their free acid forms did not interact with BCRP. Since latanoprost, travoprost, and their free acid forms are not substrates of P-gp, it is logical that these molecules do not interact with BCRP as well, since BCRP possesses one half of the MDR1 P-gp protein structure. Due to the structural similarity between P-gp and BCRP, both efflux transporters share common substrates. Surprisingly, bimatoprost being a substrate for P-gp did not interact with BCRP (Table 1). Thus, molecular binding sites of these proteins need to be investigated to understand this anomaly. Similar results have been obtained with lopinavir in the past, which is a good substrate of P-gp but not BCRP1.34 The extent of inhibition with the free acid forms of the prostaglandin analogs in the presence of MK571 in MDCK-MRP1, -MRP2, and -MRP5 cells is higher than their parent ester moieties. A slight reduction in the affinity of the prostaglandin analogs toward efflux pumps in comparison to their free acid forms could be due to their structural modification. Our laboratory has reported that dipeptide prodrug derivatization of efflux pump substrates such as quinidine, saquinavir, and lopinavir causes significant reduction in their affinity toward efflux pumps.35–37 This hypothesis holds true even in this study, where affinity toward efflux pumps is slightly diminished due to prodrug modification. But, it is important to note that complete evasion of efflux could be realized by simultaneously targeting an influx nutrient transporter.35–37 Finally, the extent of efflux inhibition in the presence of specific inhibitors in rPCEC might be comparatively less, since it is a primary culture in comparison to MDCK-transfected cell lines employed in this study, which overexpresses efflux proteins.
Dose-dependent inhibition studies on rPCEC evaluated the affinity of the prostaglandin analogs toward efflux pumps. As illustrated in Figure 3A–3C, all prostaglandin analogs inhibited the efflux of erythromycin in a dose-dependent manner with bimatoprost and travoprost exhibiting similar IC50 and K i values (<100 μM). On the other hand, latanoprost exhibited a comparatively higher IC50 and K i value. Again, a comparatively stronger affinity of bimatoprost and travoprost could be linked to the ethyl amide linkage at carbon-1 position and the phenoxy ring at carbon-16 position with a m-trifluoromethyl group, in the respective molecules (Fig. 1). Lineweaver–Burk transformation revealed a competitive inhibition, suggesting that all the prostaglandin analogs and erythromycin probably share a common binding site on the transporter (data not shown).
Ex vivo transport studies across freshly isolated rabbit cornea showed that the absorption of these prostaglandin analogs across cornea is restricted by efflux pumps, which is consistent with in vitro results that these molecules are good substrates of drug efflux pumps (Table 3). Corneal permeability of these prostaglandin analogs are in agreement with previously reported values in the literature.9 , 21 It was noticed that both latanoprost and travoprost (ester prodrugs) rapidly hydrolyze to its free acid form thus substantiating the fact that the free acid form is responsible for IOP reduction. But in case of bimatoprost (amide prodrug), hydrolysis was partial, though the free acid form was the predominant species. Significant increase in corneal permeability in the presence of specific inhibitors suggests that these molecules are probably effluxed out of the corneal epithelium into the tear fluid.
Earlier work from this laboratory has demonstrated that co-administration of drugs is a viable strategy to overcome efflux. This was proven in the case of erythromycin, when co-administered with steroids. Since, these prostaglandin analogs were found to be substrates of drug efflux pumps, one of the practical solutions to overcome their efflux is by administering these molecules in combination. This strategy might not only result in their efflux being modulated or inhibited, but might also result in a synergistic pharmacological effect, since these molecules could activate different receptor populations to reduce IOP. Such a strategy might also present an option to reduce the dose significantly, which in turn could possibly reduce the ocular side effects involved with prostaglandin therapy. Hence, in vitro experiments were carried out to test this strategy by studying cellular accumulation and transport of bimatoprost in combination with latanoprost and travoprost. Cellular accumulation results in rPCEC showed that the efflux of bimatoprost was significantly inhibited in the presence of other prostaglandin analogs and steroids (Fig. 4). Hence, if glaucoma is also associated with inflammation, co-administration of steroids could not only play a therapeutic role in the treatment regimen but also modulate the efflux of prostaglandin analogs, thus playing a dual role. In both in vitro cellular accumulation and ex vivo corneal transport studies, it was observed that the extent of inhibition of bimatoprost efflux was higher in the presence of travoprost than latanoprost (Figs. 4 and 5). This result could again be associated with the affinity patterns as exhibited by their individual K i values (latanoprost = 163.20 μM and travoprost = 94.77 μM).
Thus, our study clearly indicates that currently marketed prostaglandin analogs such as bimatoprost, latanoprost, and travoprost are substrates for multiple drug efflux pumps. Their hydrolysis product (free acid form), which is an active metabolite, is also a substrate for drug efflux pumps on cornea. Ex vivo transport studies clearly indicate that the permeation of these molecules across cornea is restricted due to active efflux. But administration of these molecules in combination is shown to overcome efflux by competitive inhibition. Amongst the prostaglandin analogs, bimatoprost and travoprost exhibit higher efflux inhibition potential than latanoprost. Therefore, it is a viable strategy to administer these prostaglandin analogs in combination, so as to overcome efflux and simultaneously elicit a synergistic pharmacological effect, since these molecules have been shown to activate different receptor population for the reduction of IOP. If other conditions such as inflammation is associated with high IOP, then introduction of steroids in the treatment regimen might also result in modulation of efflux. Finally, efflux modulation could be seen as a strategy to reduce the effective dose, since most of the topically administered agents are associated with ocular side effects following repeated administration.
Contributor Information
Sudharshan Hariharan, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri–Kansas City, Kansas City, Missouri..
Mukul Minocha, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri–Kansas City, Kansas City, Missouri..
Gyan P. Mishra, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri–Kansas City, Kansas City, Missouri.
Dhananjay Pal, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri–Kansas City, Kansas City, Missouri..
Rohit Krishna, Vision Research Center, University of Missouri–Kansas City, Kansas City, Missouri..
Ashim K. Mitra, Division of Pharmaceutical Sciences, School of Pharmacy, University of Missouri–Kansas City, Kansas City, Missouri.; Vision Research Center, University of Missouri–Kansas City, Kansas City, Missouri.
Acknowledgments
This work was supported by NIH grants R01EY09171-14 and R01EY10659-12 and a grant from Saint Luke’s Hospital Foundation, Kansas City.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Macha S., Hughes P.M., Mitra A.K. Mitra A.K. Ophthalmic Drug Delivery Systems. ed. New York: Marcel Dekker; 2003. Overview of ocular drug delivery; pp. 1–12. [Google Scholar]
- 2.Dey S., Gunda S., Mitra A.K. Pharmacokinetics of erythromycin in rabbit corneas after single-dose infusion: role of P-glycoprotein as a barrier to in vivo ocular drug absorption. J. Pharmacol. Exp. Ther. 2004;311:246–255. doi: 10.1124/jpet.104.069583. [DOI] [PubMed] [Google Scholar]
- 3.Hariharan S., Gunda S., Mishra G.P., et al. Enhanced corneal absorption of erythromycin by modulating P-glycoprotein and MRP mediated efflux with corticosteroids. Pharm. Res. 2009;26:1270–1282. doi: 10.1007/s11095-008-9741-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dey S., Patel J., Anand B.S., et al. Molecular evidence and functional expression of P-glycoprotein (MDR1) in human and rabbit cornea and corneal epithelial cell lines. Invest. Ophthalmol. Vis. Sci. 2003;44:2909–2918. doi: 10.1167/iovs.02-1142. [DOI] [PubMed] [Google Scholar]
- 5.Karla P.K., Earla R., Boddu S.H., et al. Molecular expression and functional evidence of a drug efflux pump (BCRP) in human corneal epithelial cells. Curr. Eye Res. 2009;34:1–9. doi: 10.1080/02713680802518251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karla P.K., Pal D., Mitra A.K. Molecular evidence and functional expression of multidrug resistance associated protein (MRP) in rabbit corneal epithelial cells. Exp. Eye Res. 2007;84:53–60. doi: 10.1016/j.exer.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Karla P.K., Pal D., Quinn T., et al. Molecular evidence and functional expression of a novel drug efflux pump (ABCC2) in human corneal epithelium and rabbit cornea and its role in ocular drug efflux. Int. J. Pharm. 2007;336:12–21. doi: 10.1016/j.ijpharm.2006.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karla P.K., Quinn T.L., Herndon B.L., et al. Expression of multidrug resistance associated protein 5 (MRP5) on cornea and its role in drug efflux. J. Ocul. Pharmacol. Ther. 2009;25:121–132. doi: 10.1089/jop.2008.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiang C.D., Batugo M., Gale D.C., et al. Characterization of human corneal epithelial cell model as a surrogate for corneal permeability assessment: metabolism and transport. Drug Metab. Dispos. 2009;37:992–998. doi: 10.1124/dmd.108.026286. [DOI] [PubMed] [Google Scholar]
- 10.Zhang T., Xiang C.D., Gale D., et al. Drug transporter and cytochrome P450 mRNA expression in human ocular barriers: implications for ocular drug disposition. Drug Metab. Dispos. 2008;36:1300–1307. doi: 10.1124/dmd.108.021121. [DOI] [PubMed] [Google Scholar]
- 11.Reid G., Wielinga P., Zelcer N., et al. The human multidrug resistance protein MRP4 functions as a prostaglandin efflux transporter and is inhibited by nonsteroidal antiinflammatory drugs. Proc. Natl. Acad. Sci. USA. 2003;100:9244–9249. doi: 10.1073/pnas.1033060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wielinga P.R., van der Heijden I., Reid G., et al. Characterization of the MRP4- and MRP5-mediated transport of cyclic nucleotides from intact cells. J. Biol. Chem. 2003;278:17664–17671. doi: 10.1074/jbc.M212723200. [DOI] [PubMed] [Google Scholar]
- 13.Evers R., Cnubben N.H., Wijnholds J., et al. Transport of glutathione prostaglandin A conjugates by the multidrug resistance protein 1. FEBS Lett. 1997;419:112–116. doi: 10.1016/s0014-5793(97)01442-7. [DOI] [PubMed] [Google Scholar]
- 14.Cui Y., Konig J., Buchholz J.K., et al. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol. Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- 15.Jedlitschky G., Leier I., Buchholz U., et al. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance-associated protein. Cancer Res. 1994;54:4833–4836. [PubMed] [Google Scholar]
- 16.Leier I., Jedlitschky G., Buchholz U., et al. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J. Biol. Chem. 1994;269:27807–27810. [PubMed] [Google Scholar]
- 17.Borst P., Elferink R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- 18.Cantor L.B. Clinical pharmacology of bimatoprost. Expert Opin. Drug Metab. Toxicol. 2005;1:151–157. doi: 10.1517/17425255.1.1.151. [DOI] [PubMed] [Google Scholar]
- 19.Chen J., Senior J., Marshall K., et al. Studies using isolated uterine and other preparations show bimatoprost and prostanoid FP agonists have different activity profiles. Br. J. Pharmacol. 2005;144:493–501. doi: 10.1038/sj.bjp.0706044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matias I., Chen J., De Petrocellis L., et al. Prostaglandin ethanolamides (prostamides): in vitro pharmacology and metabolism. J. Pharmacol. Exp. Ther. 2004;309:745–757. doi: 10.1124/jpet.103.061705. [DOI] [PubMed] [Google Scholar]
- 21.Woodward D.F., Krauss A.H., Chen J., et al. The pharmacology of bimatoprost (Lumigan) Surv. Ophthalmol. 2001;45((Suppl 4)):S337–S345. doi: 10.1016/s0039-6257(01)00224-7. [DOI] [PubMed] [Google Scholar]
- 22.Sharif N.A., Kelly C.R., Crider J.Y. Agonist activity of bimatoprost, travoprost, latanoprost, unoprostone isopropyl ester and other prostaglandin analogs at the cloned human ciliary body FP prostaglandin receptor. J. Ocul. Pharmacol. Ther. 2002;18:313–324. doi: 10.1089/10807680260218489. [DOI] [PubMed] [Google Scholar]
- 23.Sharif N.A., Williams G.W., Kelly C.R. Bimatoprost and its free acid are prostaglandin FP receptor agonists. Eur. J. Pharmacol. 2001;432:211–213. doi: 10.1016/s0014-2999(01)01486-8. [DOI] [PubMed] [Google Scholar]
- 24.Woodward D.F., Krauss A.H., Chen J., et al. Pharmacological characterization of a novel antiglaucoma agent, Bimatoprost (AGN 192024) J. Pharmacol. Exp. Ther. 2003;305:772–785. doi: 10.1124/jpet.102.047837. [DOI] [PubMed] [Google Scholar]
- 25.Stjernschantz J.W. From PGF(2alpha)-isopropyl ester to latanoprost: a review of the development of xalatan: the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 2001;42:1134–1145. [PubMed] [Google Scholar]
- 26.Waugh J., Jarvis B. Travoprost. Drugs Aging. 2002;19:465–471. doi: 10.2165/00002512-200219060-00005. [DOI] [PubMed] [Google Scholar]
- 27.Sharif N.A., Kelly C.R., Crider J.Y., et al. Ocular hypotensive FP prostaglandin (PG) analogs: PG receptor subtype binding affinities and selectivities, and agonist potencies at FP and other PG receptors in cultured cells. J. Ocul. Pharmacol. Ther. 2003;19:501–515. doi: 10.1089/108076803322660422. [DOI] [PubMed] [Google Scholar]
- 28.Ota T., Aihara M., Saeki T., et al. The effects of prostaglandin analogues on prostanoid EP1, EP2, and EP3 receptor-deficient mice. Invest. Ophthalmol. Vis. Sci. 2006;47:3395–3399. doi: 10.1167/iovs.06-0100. [DOI] [PubMed] [Google Scholar]
- 29.Gagliuso D.J., Wang R.F., Mittag T.W., et al. Additivity of bimatoprost or travoprost to latanoprost in glaucomatous monkey eyes. Arch Ophthalmol. 2004;122:1342–1347. doi: 10.1001/archopht.122.9.1342. [DOI] [PubMed] [Google Scholar]
- 30.Gandolfi S.A., Cimino L. Effect of bimatoprost on patients with primary open-angle glaucoma or ocular hypertension who are nonresponders to latanoprost. Ophthalmology. 2003;110:609–614. doi: 10.1016/S0161-6420(02)01891-2. [DOI] [PubMed] [Google Scholar]
- 31.Alm A., Grierson I., Shields M.B. Side effects associated with prostaglandin analog therapy. Surv. Ophthalmol. 2008;53((Suppl 1)):S93–S105. doi: 10.1016/j.survophthal.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 32.Cheng Y., Prusoff W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 33.Mishima S., Gasset A., Klyce S.D., Jr., et al. Determination of tear volume and tear flow. Invest. Ophthalmol. 1966;5:264–276. [PubMed] [Google Scholar]
- 34.Agarwal S., Pal D., Mitra A.K. Both P-gp and MRP2 mediate transport of lopinavir, a protease inhibitor. Int. J. Pharm. 2007;339:139–147. doi: 10.1016/j.ijpharm.2007.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agarwal S., Boddu S.H., Jain R., et al. Peptide prodrugs: improved oral absorption of lopinavir, a HIV protease inhibitor. Int. J. Pharm. 2008;359:7–14. doi: 10.1016/j.ijpharm.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jain R., Agarwal S., Majumdar S., et al. Evasion of P-gp mediated cellular efflux and permeability enhancement of HIV-protease inhibitor saquinavir by prodrug modification. Int. J. Pharm. 2005;303:8–19. doi: 10.1016/j.ijpharm.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 37.Katragadda S., Talluri R.S., Mitra A.K. Modulation of P-glycoprotein-mediated efflux by prodrug derivatization: an approach involving peptide transporter-mediated influx across rabbit cornea. J. Ocul. Pharmacol. Ther. 2006;22:110–120. doi: 10.1089/jop.2006.22.110. [DOI] [PubMed] [Google Scholar]