

Abstract
Purpose:
Increased actomyosin contraction of the dense band of actin cytoskeleton at the apical junctional complex (perijunctional actomyosin ring, PAMR) breaks down the barrier integrity of corneal endothelium. This study has investigated the efficacy of statins, which inhibit activation of RhoA, in opposing the thrombin-induced loss of barrier integrity of monolayers of cultured bovine corneal endothelium.
Methods:
Myosin light chain (MLC) phosphorylation, a biochemical measure of actomyosin contraction, was assayed by urea–glycerol gel electrophoresis, followed by western blot analysis. The locus of MLC phosphorylation and changes in the organization of the PAMR were visualized by immunostaining. Phosphorylation of MYPT1, a regulatory subunit of myosin light-chain phosphatase (MLCP), was assessed by Western blot analysis to determine down-regulation of RhoA. The barrier integrity was assessed in terms of trans-endothelial electrical resistance (TER), and further confirmed by determining permeability to FITC dextran (10 kDa) and distribution of ZO-1, a marker of tight junctional assembly.
Results:
Lovastatin, a prototype of lipophilic statins, induced MLC dephosphorylation under basal conditions. It opposed increase in phosphorylation of MLC and MYPT1 in response to thrombin and nocodazole, agents known to activate RhoA in the endothelium. Pretreatment with the statin opposed the thrombin- and nocodazole-induced disruption of the PAMR and the thrombin-induced decline in TER. Lovastatin also opposed the thrombin- and nocodazole-induced increase in permeability to FITC dextran and redistribution of ZO-1. However, upon supplementation with GGPP (geranylgeranyl pyrophosphate), lovastatin failed to oppose the effects of thrombin and nocodazole on the PAMR, ppMLC, and ZO-1 distribution.
Conclusions:
Lovastatin attenuates RhoA activation in the corneal endothelium presumably by reducing its isoprenylation. This underlies the suppression of the thrombin-induced loss in barrier integrity of the corneal endothelium.
Introduction
The corneal endothelium is a leaky monolayer at the posterior surface of the cornea.1–3 It is responsible for hydration control of the corneal stroma, which is essential for corneal transparency. The hydration control is dependent on the barrier and fluid pump functions of the endothelium. While the latter is brought about by active ion transport mechanisms at the apical and basolateral membranes of the endothelium,4 the former restrains rate of fluid leak into the stroma. This leak is driven by an imbibition pressure because of the hydrophilic glycosaminoglycans in the stroma. Under healthy conditions, the fluid leak into the stroma is counterbalanced by the active fluid transport by the endothelium from stroma to the anterior chamber.4–6 In a variety of inflammatory situations, corneal edema is induced because of loss in the endothelial barrier integrity.4 The aim of this study is to determine if statins, which are known to suppress barrier dysfunction in certain vascular endothelium,7–9 are also similarly useful in the corneal endothelium.
The barrier integrity of the epithelial/endothelial monolayers is conferred by the tight junctions (TJs) at the apical junctional complex. These possess transmembrane proteins (ie, occludins, claudins, and junctional adhesion molecules), which interact with their counterparts in the neighboring cells. Such interactions, which are responsible for occlusion of the paracellular space, are stabilized by intercellular tethering forces generated by the adherens junctions (AJs), which are proximal to TJs. The cytoplasmic domains of the transmembrane molecules of TJs and of the AJs are structurally and functionally linked to a thick band of cortical actin cytoskeleton at the apical junctional complex (called the perijunctional actomyosin ring, or PAMR) via adapter molecules, such as ZO-1 (zonula occludens-1).10 Our recent studies with corneal endothelium have demonstrated that an increased actomyosin contraction of the PAMR breaks down the barrier integrity, presumably through a reduction of the intercellular tethering forces at the TJs and AJs. This finding is similar to those reported in the vascular endothelium.3,11–14 The actomyosin contraction of the actin cytoskeleton, including that of the PAMR, is increased by phosphorylation of the regulatory light chain of myosin II (also called myosin light chain, or MLC).15,16 Activation of RhoA, small GTPase of Rho family, is known to increase MLC phosphorylation through its downstream effector, Rho kinase.17,18 Accordingly, a variety of inflammatory mediators that activate RhoA–Rho kinase axis are known to break down the barrier integrity.19
Statins, which are employed in the treatment of hyperlipidemia, are inhibitors of HMG-CoA (3-hydroxy-3-methylglutaryl-coenzyme) reductase. This results in a decrease in the biosynthesis of mevalonate so that cholesterol formation is reduced.20,21 Along this cholesterol biosynthetic pathway, geranylgeranyl pyrophosphate (GGPP) and farnesyl pyrophosphate (FPP) are formed downstream of mevalonate.20,22 These isoprenoids serve as lipid attachments for the post-translational modifications of Rho and Ras family GTPases.20 Specifically, GGPP is transferred onto RhoA, by the activity of geranylgeranyltransferase. This modification, referred to as isoprenylation, is essential for activation of RhoA.23,24 Accordingly, many studies8,24 have shown that statins inhibit RhoA activation induced by thrombin. This effect of statins is known to limit thrombin-induced breakdown of the barrier integrity in vascular endothelia.
In our previous studies,12,13 we demonstrated that thrombin breaks down the barrier integrity of corneal endothelium through RhoA-mediated MLC phophorylation and consequent disruption of the PAMR. In this study, we have investigated whether lovastatin, a lipophilic prototype of statins, can prevent thrombin-induced loss of the barrier integrity in cultured bovine corneal endothelial monolayers. Our principal findings indicate that lovastatin opposes the thrombin-induced MLC phosphorylation and also limits the loss of barrier integrity induced by the protease.
Methods
Drugs and chemicals
Nocodazole, lovastatin, GGPP, and α-tubulin antibody were procured from Sigma Aldrich Co. (St. Louis, MO). Bovine α-thrombin was obtained from Enzyme Research (South Bend, IN). Polyclonal MLC antibody (E201) was generously provided by Dr. Patricia Gallagher (IUPUI, School of Medicine, Indianapolis, IN). Antibodies against ppMLC (Thr18/Ser19) and phospho-MYPT1 (Thr853) were obtained from Cell Signaling Technology (Danvers, MA). The antibody against ZO-1 was procured from Zymed labs (San Francisco, CA). Texas red conjugated phalloidin, goat anti-mouse Alexa-488, rabbit anti-mouse Alexa-488, and anti-fade agent were obtained from Molecular Probes (Eugene, OR). The enhanced chemiluminescence kit was obtained from Amersham-Pharmacia Biotech (Piscataway, NJ).
Cell culture
Primary cultures of bovine corneal endothelial cells were established, as described previously, from fresh eyes obtained from a local slaughterhouse.3,13 The growth medium contained Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% bovine calf serum and an antibiotic–antimycotic mixture (penicillin, 100 U/mL; streptomycin, 100 μg/mL; and amphotericin-B, 0.25 μg/mL). Cells were cultured at 37°C under 5% CO2. The medium was changed every 2 days. When the cells in the culture reached confluence, they were subcultured and cells from the first and second passages were used for experiments. Cell culture supplies were from Invitrogen (Grand Island, NY).
MLC phosphorylation assay
MLC phosphorylation was assayed by urea–glycerol gel electrophoresis, followed by immunoblotting, as described previously.12,13 Cells grown on 60-mm Petri dishes were treated with desired agents, which were followed by an addition of 1 mL of ice-cold lysis buffer containing trichloroacetic acid and dithiothreitol. Protein was extracted using a urea sample buffer containing 8 M urea, 10 mM dithiothreitol, 20 mM Tris base, 23 mM glycine, and 0.04% bromophenol blue. They were subjected to electrophoresis on a polyacrylamide gel, and then transferred onto a nitrocellulose membrane. The phosphorylated and non-phosphorylated MLC were detected by western blotting, using a polyclonal anti-MLC antibody (E201; 1:3,000 dilution). Blots were washed with Tris buffer saline Tween (TBST) and then visualized using the enhanced chemiluminescence kit. The migration rate of MLC was in the following order: diphosphorylated form > monophosphorylated form > non-phosphorylated form, identified as “PP,” “P,” and “NP,” respectively, on gel pictures. A custom-made software program was used to quantify the intensities of MLC bands on the blots. The fraction of phosphorylated MLC (denoted as pMLC and expressed in %) was calculated by dividing the total moles of phosphorylated MLC (PP + P) by the total moles of MLC (NP + P + PP).
MYPT1 phosphorylation assay
Phosphorylated MYPT1 was determined by immunoblot analysis using a phospho-specific antibody. The assay was carried out using cultured confluent bovine corneal endothelial cells (BCEC). Cells were maintained in regular media with 0.1% serum overnight before treatment with desired agents. Following treatment, cells were washed 2 times with 1× PBS and lysed with lysis buffer. Then the lysates were centrifuged at 14,000 rpm for 10 min at 4°C. Cell lysate proteins were resolved by SDS-PAGE and electrophoretically transferred on to nitrocellulose membrane. The membrane was incubated overnight at 4°C with primary antibody for MYPT1 (Thr853) (1:1,000/α-tubulin (1:5,000) and then incubated for 1 h with appropriate HRP (horseradish peroxidase)-conjugated secondary antibody. The bands were identified by enhanced chemiluminescence kit.
Immunocytochemistry for F-actin, ZO-1, and diphosphorylated MLC
Cells grown on coverslips were washed with PBS after desired drug treatment, fixed with 3.7% paraformaldehyde, and permeabilized using 0.2% Triton X-100 for 5 min. This was followed by staining for F-actin by phalloidin conjugated to Texas Red (1:1,000) for 45–60 min at room temperature. Cells were stained for ZO-1 by fixing the cells with fixation buffer after drug treatment. This was followed by permeabilization with 0.01% saponin in PBS. The cells were exposed to blocking buffer for 45 min, then incubated with the antibody for ZO-1 (1:25) overnight at 4°C. This was followed by washing and incubation with secondary antibody (goat anti-mouse Alexa flour 488; 1:1,000). Similarly, cells were stained for diphosphorylated MLC (ppMLC) using the antibody at 1:25 dilution. Stained cells were mounted with an anti-fade agent and visualized using an epifluorescence microscope equipped with a 60× oil immersion objective and 1.2 NA (Nikon, Tokyo, Japan).
Measurement of trans-endothelial electrical resistance
The trans-endothelial electrical resistance (TER) was measured by ECIS device (ECIS 1600R; Applied Biophysics, Inc., Troy, NY). Cells were seeded at a density of 5 × 105 cells on gold electrodes (8W10E+; Applied Biophysics, Inc.) and grown to confluence. The confluency was monitored through spreading data acquired overnight. After the cellular monolayers reached steady resistance values, they were taken for experiments. The wells were washed twice with serum-free media and allowed to equilibrate for an hour before drug treatment. The resistive portion of impedance (ie, TER) normalized to its initial value at time zero was monitored continuously and taken as a measure of barrier integrity.
Measurement of permeability to FITC dextran
Cells were grown on 0.2 μm pore-size collagen IV (1 mg/mL)-coated tissue culture inserts (Nunc, Fisher Scientific, Pittsburgh, PA) to confluence. The monolayers were then serum-starved for an hour and either left untreated or exposed in triplicate with desired agents. Following treatment, FITC dextran (10 kDa) dissolved in the Ringer solution was placed in the apical compartment at 0.4 μg/mL and put back in the incubator for 2 h. Samples were then taken from the basolateral chamber for fluorescence measurements. Cells were pretreated with lovastatin for 20 h before exposure to thrombin. Fluorescence was measured by excitation at 492 nm and the emission was collected at 520 nm.
Statistical analysis
Statistical comparisons were made by analysis of variance (ANOVA) with Bonferroni’s post-test analysis using GraphPad Prism™ software (GraphPad Software Inc., San Diego, CA). A value of P < 0.05 was considered statistically significant. Data are expressed as mean ± SE. “N” represents number of independent experiments.
Results
Effect of lovastatin on the thrombin-induced MLC phosphorylation
Figure 1 shows the influence of thrombin on MLC phosphorylation with and without pretreatment with 10 μM lovastatin for 24 h. Exposure to 2 U/mL of thrombin for 5 min led to increased MLC phosphorylation (Fig. 1A) as shown previously.12,13 Treatment with lovastatin alone caused MLC dephosphorylation, and also opposed the thrombin-induced MLC phosphorylation. Densitometric analysis of blots similar to that shown in Figure 1A is summarized in Figure 1B. These data show that thrombin increases MLC phosphorylation by ∼50% when compared to untreated cells only in the absence of pretreatment with lovastatin. This influence of lovastatin was further confirmed by immunostaining diphosphorylated MLC (ppMLC). In untreated cells, ppMLC along the cell periphery was moderate, suggestive of basal level of MLC phosphorylation (Fig. 2A). Treatment with lovastatin led to a decrease in ppMLC (Fig. 2B) when compared to untreated cells in consistence with Figure 1. Exposure to thrombin led to increased ppMLC along the cell periphery (Fig. 2C) in consistence with Figure 1. We observe that ppMLC staining is in a punctate pattern, as we have noted previously in response to histamine.14 Pretreatment with lovastatin opposed the thrombin response (Fig. 2D) in consistence with MLC phosphorylation shown in Figure 1.
FIG. 1. .
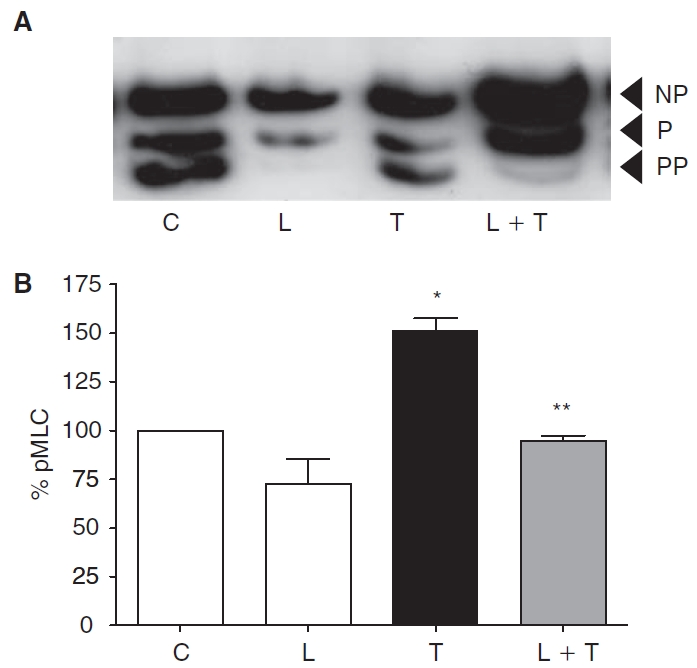
Effect of lovastatin on myosin light chain (MLC) phosphorylation. MLC phosphorylation was assayed by urea–glycerol gel electrophoresis, followed by western blotting. Typical blot depicting changes in status of MLC phosphorylation (A) (NP, non-phosphorylated MLC; P, monophosphorylated MLC; PP, diphosphorylated MLC). Untreated cells (C) show a basal level of MLC phosphorylation. Treatment with 10 μM lovastatin (L) for 24 h induces MLC dephosphorylation. Exposure to 2 U/mL thrombin (T) for 2 min induces increased MLC phosphorylation. Pretreatment with lovastatin opposes MLC phosphorylation induced by thrombin. Bar graph of densitometric analysis (B) of the representative experiments (N = 3) shown in panel A expressed as %pMLC. Lovastatin significantly opposes thrombin-induced MLC phosphorylation. *Significantly greater than control (P < 0.05). **Significantly less than thrombin treatment (P < 0.05).
FIG. 2. .
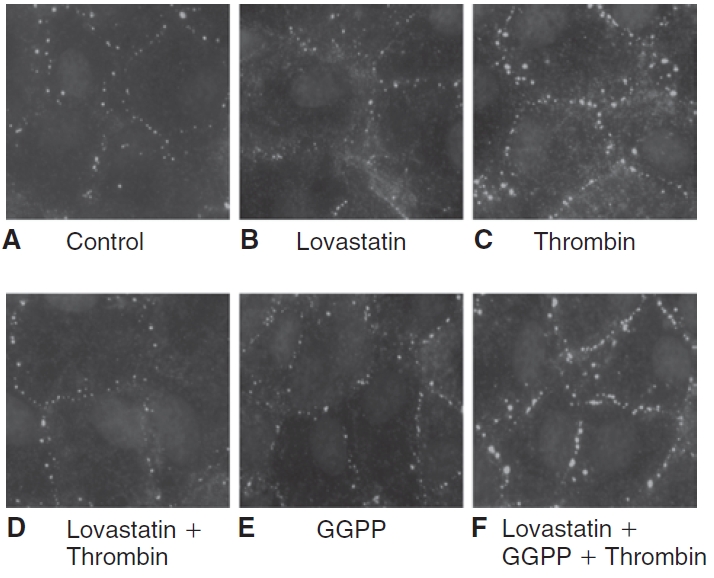
Influence of lovastatin on the localization of diphosphorylated myosin light chain (ppMLC). The localization of ppMLC was ascertained by immunostaining, followed by imaging with an epifluorescence microscope. The images are representative of 3 independent experiments. In untreated cells (A), ppMLC staining in the cell periphery is moderate, indicating a basal level of MLC phosphorylation. Treatment with 10 μM lovastatin for 24 h (B) reduces ppMLC when compared to untreated cells, indicating MLC dephosphorylation. Exposure to 2 U/mL of thrombin for 2 min (C) increases the punctate localization of ppMLC in the cell periphery, indicating increased MLC phosphorylation. Pretreatment with lovastatin (D) opposes the thrombin response. Upon treatment with 10 μM geranylgeranyl pyrophosphate (GGPP) for 24 h (E), the localization of ppMLC is similar to that of untreated cells. On co-treatment of 10 μM GGPP with 10 μM lovastatin for 24 h followed by exposure to thrombin (F), lovastatin fails to oppose thrombin-induced increase in ppMLC.
As an alternative to thrombin, we employed nocodazole to induce MLC phosphorylation. This agent, which induces microtubule disassembly,25–28 is known to activate RhoA. Figure 3 shows the influence of nocodazole on MLC phosphorylation with and without lovastatin pretreatment. As expected, treatment with 2 μM nocodazole for 30 min led to an increased MLC phosphorylation similar to the thrombin response (Fig. 3A). However, upon pretreatment with 10 μM lovastatin for 24 h, the nocodazole effect on MLC phosphorylation was reduced. Densitometric analysis of all the experiments, similar to that shown in Figure 3A, is summarized in Figure 3B. It is evident that nocodazole-induced MLC phosphorylation is opposed by pretreatment with lovastatin. As shown in Figure 4C, nocodazole also increased ppMLC at the cell periphery similar to thrombin. Pretreatment with lovastatin opposed the nocodazole response (Fig. 4D).
FIG. 3. .
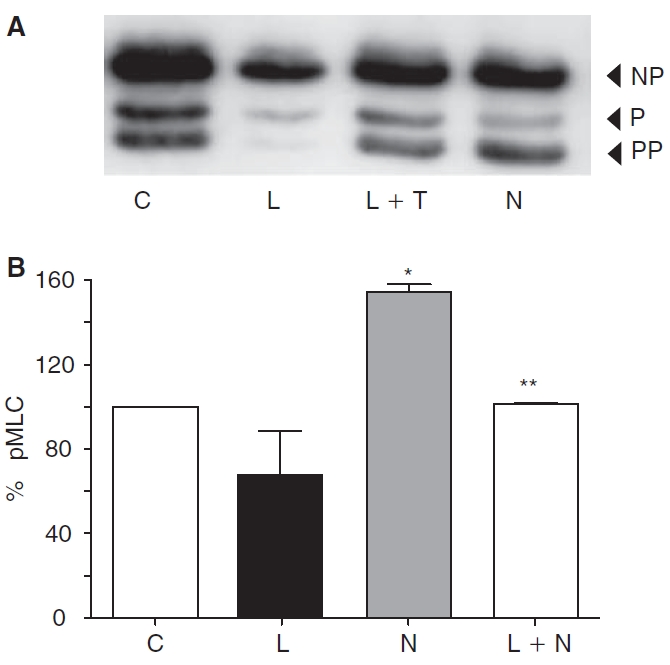
Effect of lovastatin on nocodazole-induced myosin light chain (MLC) phosphorylation. Western blot depicting changes in MLC phosphorylation in response to nocodazole with or without the presence of lovastatin (A). Exposure to 2 μM nocodazole (N) for 30 min induces increased MLC phosphorylation when compared to untreated cells (C). Pretreatment with lovastatin (L) opposes nocodazole-induced MLC phosphorylation. Bar graph of densitometric analysis (B) of the representative experiments (N = 3) shown in panel A. Lovastatin significantly opposes nocodazole-induced MLC phosphorylation. *Significantly greater than control (P < 0.05). **Significantly less than nocodazole treatment (P < 0.05).
FIG. 4. .
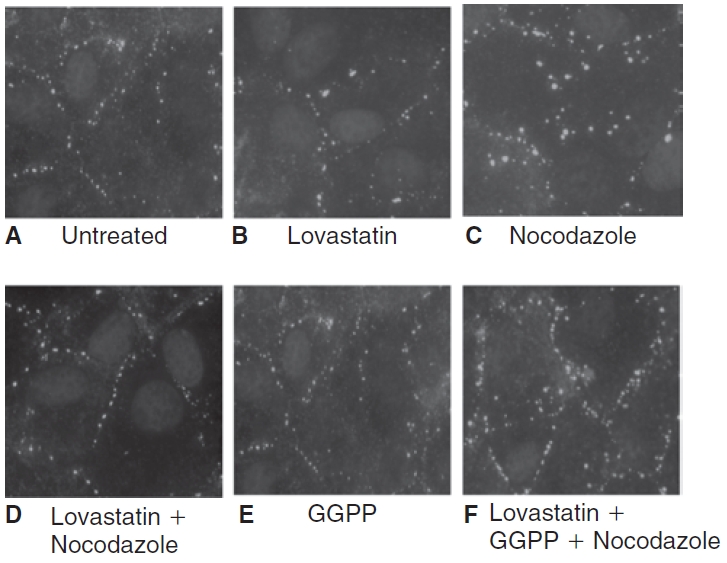
Influence of lovastatin on nocodazole-induced changes in localization of diphosphorylated myosin light chain (ppMLC). The images are representative of 3 independent experiments. In untreated cells (A), the localization of ppMLC in the cortical region of the cell is less dense, indicative of a basal level of MLC phosphorylation. Treatment with 10 μM lovastatin for 24 h (B) reduces ppMLC, correlating with MLC dephosphorylation. Exposure to 2 μM nocodazole for 30 min (C) increases ppMLC in the cortical areas, which is suggestive of increased MLC phosphorylation. Pretreatment with lovastatin (D) opposes the nocodazole response. Upon treatment with 10 μM geranylgeranyl pyrophosphate (GGPP) for 24 h (E), the staining for ppMLC is similar to that of untreated cells. On co-treatment of 10 μM GGPP with 10 μM lovastatin for 24 h (F), lovastatin fails to oppose nocodazole-induced increase in ppMLC.
Since MLC phosphorylation is downstream of RhoA activation, the findings in Figures 1 and 3 indicate that lovastatin opposes the thrombin- and nocodazole-induced MLC phosphorylation by opposing activation of RhoA.
When mevalonate synthesis is inhibited by statins, the availability of GGPP is reduced for isoprenylation of RhoA.23,24,29 This has been demonstrated to limit translocation of RhoA to the plasma membrane,7,24 which is involved in the activation of RhoA. To confirm that a similar effect is responsible for lovastatin-induced MLC dephosphorylation, we co-treated cells with 10 μM GGPP for 24 h and then assessed the status of MLC phosphorylation. The statin-induced MLC dephosphorylation was effectively abolished by co-treatment with GGPP (data not shown), indicating that lovastatin interferes with geranylgeranylation of RhoA, leading to its inactivation and thereby causing MLC dephosphorylation. We also determined whether supplementation with GGPP, an isoprenoid, opposed the influence of lovastatin on the thrombin-induced increase in ppMLC. On treatment with 10 μM GGPP for 24 h, ppMLC is similar to untreated cells (Fig. 2E). However, upon co-treatment of GGPP with lovastatin, thrombin increased ppMLC (Fig. 2F). Similarly, lovastatin failed to oppose increased ppMLC by nocodazole in presence of GGPP (Fig. 4F). These results suggest that the statin opposes the thrombin and nocodazole response through reduced geranylgeranylation of RhoA.
Effect of lovastatin on the thrombin-induced disruption of actin cytoskeleton
We have previously shown that thrombin-induced disruption of PAMR and formation of inter-endothelial gaps is opposed by pretreatment with Y-27632 (Rho kinase inhibitor).13 We examined whether lovastatin could mimic the effect of Y-27632. Upon treatment with 10 μM lovastatin for 24 h, the organization of the PAMR is largely unaltered (Fig. 5A vs. 5B). Exposure to 2 U/mL of thrombin for 5 min caused disruption of PAMR and formation of inter-endothelial gaps (Fig. 5C). Nocodazole had a similar effect (Fig. 5F). Pretreatment with 10 μM lovastatin for 24 h opposed the disruption of PAMR by thrombin (Fig. 5D) and nocodazole (Fig. 5G). Upon co-treatment of 10 μM lovastatin with 10 μM GGPP for 24 h followed by thrombin/nocodazole, lovastatin failed to oppose the disruption of PAMR induced by thrombin/nocodazole (Fig. 5E and 5H, respectively). These results suggest that lovastatin opposes thrombin- and nocodazole-induced disruption of the PAMR through reduced geranylgeranylation of RhoA.
FIG. 5. .
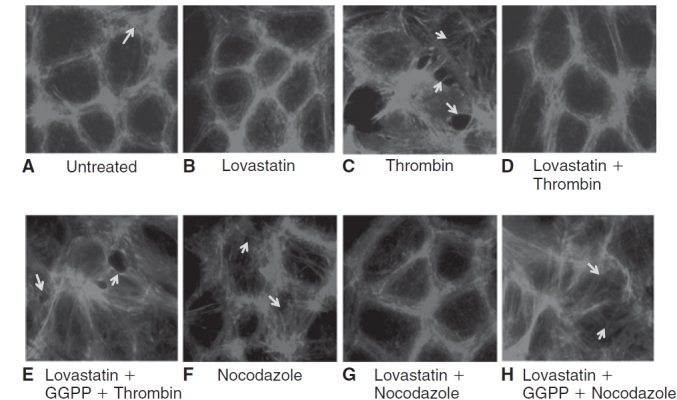
Effect of lovastatin on organization of cortical actin. The changes in organization of cortical actin were ascertained by staining for F-actin, followed by imaging with fluorescence microscopy. The images are representative of 3 independent experiments. In untreated cells (A), the characteristic organization of cortical actin in the form of perijunctional actomyosin ring (PAMR) is evident (shown by arrow). Upon treatment with 10 μM lovastatin alone for 24 h (B), the organization of actin cytoskeleton remains largely unaltered. Exposure to 2 U/mL thrombin for 5 min (C) induces disruption of the cortical actin and formation of inter-endothelial gaps (shown by arrows). Pretreatment with lovastatin (D) opposes thrombin-induced disruption of PAMR. Upon co-treatment of lovastatin with 10 μM geranylgeranyl pyrophosphate (GGPP) for 24 h (E), lovastatin fails to oppose thrombin-induced disruption of PAMR. Treatment with 2 μM nocodazole for 30 min (F) also induces disruption of PAMR (shown by arrows). Pretreatment with lovastatin opposes the nocodazole (G) response on PAMR. However, upon co-treatment of lovastatin with 10 μM GGPP for 24 h (H), lovastatin fails to oppose the influence of nocodazole on PAMR.
Effect of lovastatin on status of MYPT1 phosphorylation
MYPT1 (130 kDa) is the regulatory subunit of myosin light-chain phosphatase (MLCP). It is phosphorylated by Rho kinase at Thr853, which is a downstream effector of RhoA.30,31 Thus, MYPT1 phosphorylation status is an indicator of RhoA modulation. As shown in Figure 6, treatment with 2 U/mL thrombin for 2 min increased phosphorylation of MYPT1 at Thr853 compared to that in untreated cells. Similarly, exposure to 2 μM nocodazole for 30 min increased phosphorylation of MYPT1. However, pretreatment with 10 μM lovastatin for 24 h opposed thrombin- and nocodazole-induced increase in MYPT1 phosphorylation. Upon treatment with lovastatin, MYPT1 phosphorylation is reduced when compared to untreated cells. These results suggest that lovastatin opposes the thrombin- and nocodazole-induced RhoA activation.
FIG. 6. .
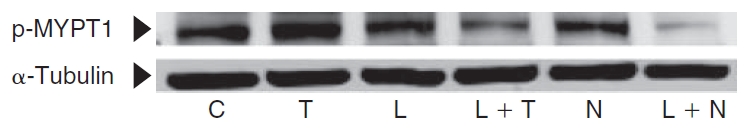
Influence of lovastatin on phosphorylation status of MYPT1, a regulatory subunit of myosin light-chain phosphatase (MLCP). Phosphorylation of MYPT1 at Thr853 was determined using a phospho-specific antibody. Treatment with 2 U/mL thrombin for 2 min or 2 μM nocodazole for 30 min increased phosphorylation of MYPT1 (Lanes 2 and 5, respectively). Upon treatment with 10 μM lovastatin for 24 h, the MYPT1 phosphorylation is suppressed when compared to untreated cells (Lane 3 vs. Lane 1). Pretreatment with lovastatin opposes the increase in MYPT1 phosphorylation induced by thrombin and nocodazole (Lanes 4 and 6, respectively).
Effect of lovastatin on the thrombin-induced loss of barrier integrity
The influence of lovastatin on the barrier integrity of endothelial monolayers was next monitored in terms of TER (see Methods). Cells incubated for 20 h with 10 μM lovastatin showed a slight increase in TER when compared to control (Fig. 7A). Treatment with 3.5 U/mL thrombin induced an immediate decline in TER that failed to recover to baseline, even after nearly 3 h. However, upon pretreatment with 10 μM lovastatin for 20 h, the thrombin-induced decline in TER was opposed and moreover, TER recovered to baseline rapidly unlike the thrombin response. Data from 6 independent experiments is summarized in terms of % reduction in TER, corresponding to the peak response of thrombin (Fig. 7B).
FIG. 7. .
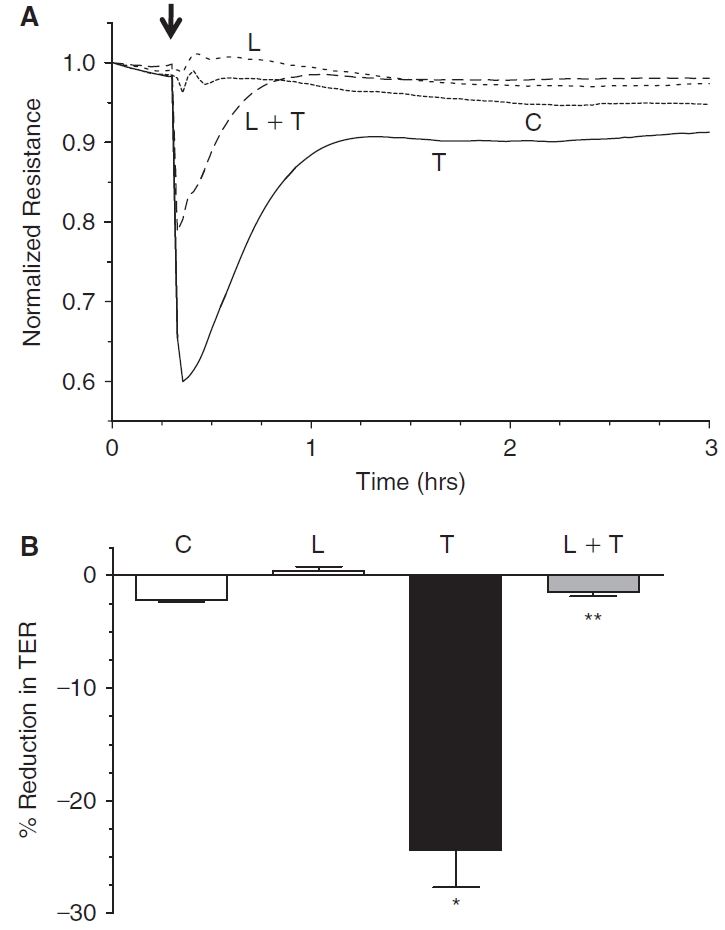
Effect of lovastatin on trans-endothelial electrical resistance (TER). The changes in TER were monitored by ECIS. In untreated cells (control), the TER remains relatively constant (A). Exposure to 3.5 U/mL thrombin induces an immediate decline in TER that fails to recover to baseline, even after nearly 3 h. Treatment with 10 μM lovastatin for 20 h slightly increases TER when compared to control. Pretreatment with 10 μM lovastatin for 20 h attenuates the extent of reduction in TER by thrombin and also facilitates faster recovery to baseline. Bar graph (B) of the peak response (<10 min) of representative experiments (N = 6) shown in Panel A. Lovastatin significantly opposes thrombin-induced reduction in TER. *Significantly greater than control (P < 0.001). **Significantly less than thrombin treatment (P < 0.001).
The loss of barrier integrity in response to thrombin is also reflected by dispersion of TJ-associated proteins, such as ZO-1, as shown, for example, in human pulmonary artery endothelial cells.32 To further establish our findings in Figure 7, we examined the influence of lovastatin on the distribution of ZO-1, with and without thrombin. In untreated cells, the ZO-1 is distributed continuously along the cell periphery (Fig. 8A). Upon treatment with lovastatin, ZO-1 distribution remained undisturbed (Fig. 8B). Treatment with 3.5 U/mL thrombin for 30 min led to dispersion of ZO-1 at the cell border (Fig. 8C). However, pretreatment with 10 μM lovastatin for 20 h opposed the thrombin response (Fig. 8D). Treatment with 10 μM GGPP alone for 20 h did not alter the localization of ZO-1 (Fig. 8E). Upon co-treatment of lovastatin with GGPP, followed by thrombin, lovastatin failed to oppose the dispersion of ZO-1 induced by thrombin (Fig. 8F). We further ascertained the influence of lovastatin on barrier integrity in the presence of thrombin by quantifying the flux of FITC dextran (10 kDa) across cultured cells. Treatment with 3.5 U/mL thrombin significantly increased the permeability to FITC dextran when compared to untreated cells (Fig. 9). Pretreatment with 10 μM lovastatin for 20 h significantly attenuated the thrombin response.
FIG. 8. .
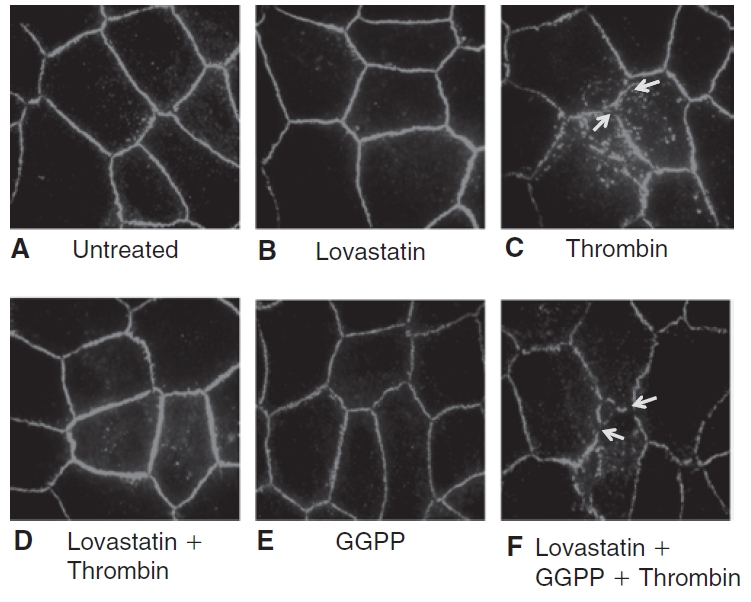
Influence of lovastatin on the localization of ZO-1, a marker of tight junctional assembly, in presence of thrombin. The localization of ZO-1 was ascertained by immunostaining, followed by imaging with fluorescence microscopy. The images are representative of 3 independent experiments. In untreated cells (A), ZO-1 localization is continuous and uniform throughout the cell periphery. Upon treatment with lovastatin alone (B), the localization of ZO-1 is unaltered. Treatment with 3.5 U/mL thrombin for 30 min (C) induces dispersion of ZO-1, indicating disorganization at the level of TJ (shown by arrows). Pretreatment with 10 μM lovastatin for 20 h (D) opposes thrombin response. Treatment with 10 μM geranylgeranyl pyrophosphate (GGPP) for 20 h (E) does not disturb the localization of ZO-1 and is similar to that of untreated cells. However, upon co-treatment of lovastatin with 10 μM GGPP for 24 h (F), lovastatin fails to oppose thrombin-induced dispersion of ZO-1.
FIG. 9. .
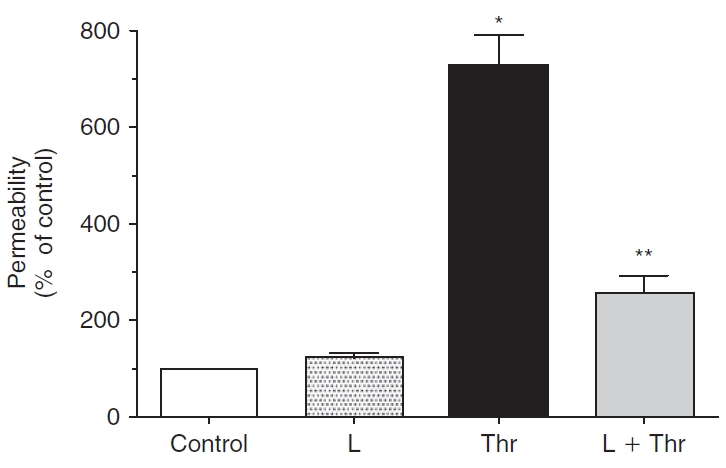
Effect of lovastatin on permeability in presence of thrombin. The changes in permeability were ascertained by quantifying the flux of FITC dextran across cells grown on porous culture inserts. Treatment with 3.5 U/mL thrombin significantly increases permeability when compared to untreated cells. Pretreatment with 10 μM lovastatin for 20 h significantly attenuates thrombin response. The increase in permeability by lovastatin itself is insignificant compared to control. The data shown are representative of 8 independent experiments. *Significantly greater than control (P < 0.001). **Significantly less than thrombin treatment (P < 0.001).
The inhibition of the thrombin-induced dispersion of ZO-1 and increase in permeability to FITC dextran by lovastatin was further tested with nocodazole. Treatment with 2 μM nocodazole for 30 min led to discontinuities in ZO-1 distribution, indicating disturbance in the structural integrity of TJs (Fig. 10C). Pretreatment with lovastatin opposed the nocodazole response (Fig. 10D). Upon co-treatment of lovastatin with GGPP, lovastatin failed to oppose the dispersion of ZO-1 induced by nocodazole (Fig. 10F). Similarly, the nocodazole-induced increase in permeability to FITC dextran was attenuated by pretreatment with lovastatin (Fig. 11). These results confirm that lovastatin opposes thrombin- and nocodazole-induced loss of barrier integrity through down-regulation of RhoA activation.
FIG. 10. .
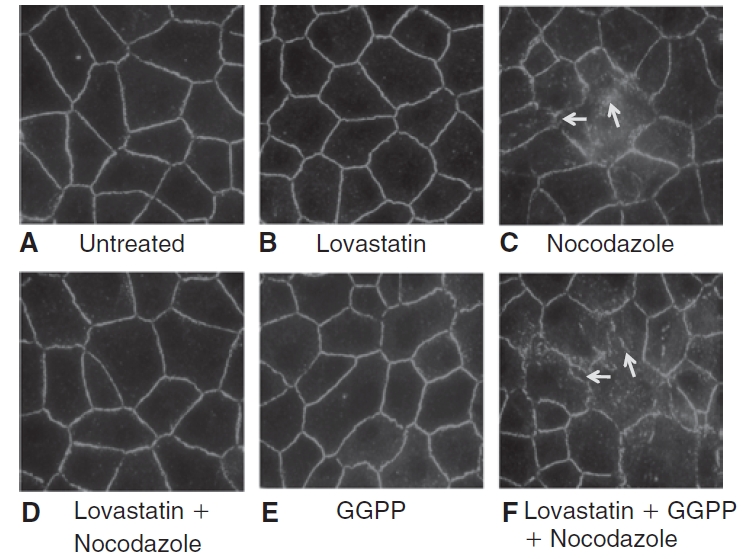
Influence of lovastatin on ZO-1, a marker of tight junctional assembly, localization in presence of nocodazole. The images shown are representative of 3 independent experiments. In untreated cells (A), continuous peripheral pattern of ZO-1 localization is evident. Treatment with 10 μM lovastatin for 20 h (B) does not alter the pattern of ZO-1 localization and is similar to those of untreated cells. Treatment with 2 μM nocodazole for 30 min (C) induces discontinuities in ZO-1 localization (shown by arrows). Pretreatment with lovastatin (D) opposes nocodazole response. On treatment with 10 μM geranylgeranyl pyrophosphate (GGPP) for 20 h (E), the localization of ZO-1 is similar to that of untreated cells. However, upon co-treatment of lovastatin with 10 μM GGPP for 24 h (F), lovastatin fails to oppose nocodazole-induced dispersion of ZO-1.
FIG. 11. .
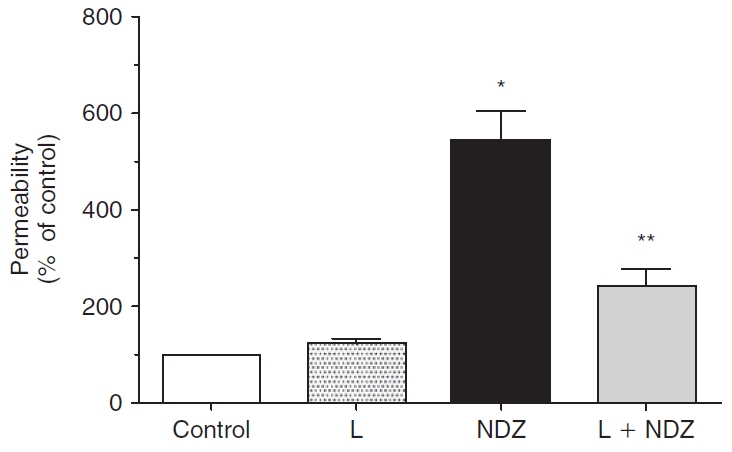
Effect of lovastatin on permeability in presence of nocodazole. Treatment with 2 μM nocodazole significantly increases permeability when compared to control. Pretreatment with lovastatin significantly attenuates nocodazole response. The data shown are representative of 8 independent experiments. *Significantly greater than control (P < 0.001). **Significantly less than nocodazole treatment (P < 0.001).
Discussion
Our recent studies3,12–14 have demonstrated that the barrier integrity of corneal endothelium is regulated by actomyosin contraction of the PAMR. In the case of thrombin,13 we have shown that the protease activates PAR-1 receptors leading to enhanced MLC phosphorylation, disruption of the PAMR, and loss of barrier integrity. Such a loss was opposed by Y-27632,13 a Rho kinase inhibitor, implying that the thrombin effects are mediated by RhoA–Rho kinase axis. This study examined the efficacy of lovastatin to oppose the thrombin effect by down-regulation of RhoA activation. Our results show that pretreatment with lovastatin opposes the thrombin-induced loss in the barrier integrity by inhibition of MLC phosphorylation induced by the protease.
Our rationale for the use of lovastatin to rescue the barrier integrity is based on its ability to reduce isoprenylation of RhoA by blocking the formation of GGPP.20–22 It is well known that isoprenylation of RhoA is a prerequisite for translocation of the small GTPase to the plasma membrane and subsequent activation of its effector, Rho kinase.20,33,34 With reduced Rho kinase activity, phosphorylation of its downstream substrate MYPT1, a myosin-binding subunit of the myosin light-chain phosphatase (MLCP), would be reduced.30,31 Specifically, it is known that Rho kinase phosphorylates MYPT1 at Thr853 and Thr696. These phosphorylations result in inhibition of the MLCP activity, and consequently, result in enhanced MLC phosphorylation.35,36 Therefore, we expected lovastatin to reduce loss of barrier integrity in response to thrombin and nocodazole, which are able to induce MLC phosphorylation.
Consistent with suppression of activation of RhoA and consequent reduction in Rho kinase activity, we found that lovastatin induced MLC dephosphorylation and also opposed MLC phosphorylation by thrombin and nocodazole (Figs. 1–4). Several studies have shown that nocodazole, which causes disassembly of microtubules,37 induces actomyosin contraction25–28,38 through RhoA activation.25,28 The efficacy of lovastatin to oppose the nocodazole response on MLC phosphorylation further confirms our findings with thrombin. Similar findings of the statin-induced MLC dephosphorylation have been reported in other cell types, including vascular endothelium.9,39,40 In our recent study, we have also observed that microtubule disassembly induced by thrombin can be suppressed by pretreatment with paclitaxel, a microtubule stabilizing agent.41 Similar barrier dysfunction involving microtubule disassembly is well known in vascular endothelium as well.25,27 Hence, our current findings indicate that the effect of lovastatin is only downstream of microtubule disassembly induced by thrombin and nocodazole. Given the pleiotropic effects of statins, further work would be needed to rule out if lovastatin can also stabilize the microtubule and thereby oppose the effects of thrombin and nocodazole similar to paclitaxel. However, in agreement with its effects on MLC phosphorylation as claimed above, we have shown that lovastatin opposes the thrombin- and nocodazole-induced disruption of the PAMR (Fig. 5D and 5G, respectively) and MYPT1 phosphorylation (Fig. 6). These results reaffirm that the effect of the statin is likely to be inhibition of activation of RhoA. In accordance with these findings, lovastatin also opposed the thrombin-induced decline in TER (Fig. 7). The effect on thrombin-induced loss in TER with and without pretreatment with lovastatin is further demonstrated at the level of ZO-1 (Fig. 8D) and permeability to FITC dextran (Fig. 9). In a similar fashion, we also found that lovastatin is able to oppose the nocodazole-induced ZO-1 dislocation (Fig. 10D) and increase in the permeability to FITC dextran (Fig. 11). Finally, upon co-treatment of GGPP with lovastatin, the latter failed to oppose the influence of thrombin/nocodazole on ppMLC (Figs. 2F and 4F, respectively), PAMR (Fig. 5E and 5H, respectively), and ZO-1 (Figs. 8F and 10F, respectively). These results, taken together, indicate that lovastatin prevents the loss in endothelial barrier integrity in response to thrombin and nocodazole through inhibition of geranylgeranylation of RhoA.
The putative pleiotropic effects of statins (ie, effects beyond cholesterol lowering) are attributed to the reduction in isoprenylation of not only RhoA, but also Rac1, the other prominent member of the Rho family GTPases.42 The reduction in the Rac1 activity in response to statins has been attributed to the reduced NADPH oxidase activity and a consequent decline in the levels of reactive oxygen species.43 Since neither the expression of the NADPH oxidase complex nor the relevance of Rac1 signaling in the regulation of the barrier integrity in the corneal endothelium is known, the scope of our experiments were limited to the effects on MLC phosphorylation, which is downstream of RhoA–Rho kinase signaling. This focus is also consistent with the fact that RhoA is considered to be the major target of statins in comparison to other members of the Rho family of GTPases.44 Statins through down-regulation of RhoA–Rho kinase axis is also known to up-regulate NO signaling in vascular endothelium through enhanced activity of eNOS.42 Although NO signaling can induce MLC dephosphorylation, expression of eNOS-like activity is not known in the corneal endothelium. Thus, our observations can be taken to suggest that lovastatin largely down-regulated Rho kinase leading to the observed MLC dephosphorylation and suppression of the thrombin effects on the barrier integrity.
Although our study focused on thrombin, we note that a variety of proinflammatory molecules are known to break down the barrier integrity through enhanced actomyosin interaction.19 It is also well known that the corneal endothelium is subjected to inflammatory stress during corneal allograft rejection, anterior uveitis, viral infections, and iatrogenic trauma (eg, cataract surgery).4,45,46 Moreover, microtubule disassembly by nocodazole in this study also mimics a variety of situations including oxidative stress,47,48 exposure to TNF-α,49 and cold temperature.50 Therefore, the observed efficacy of lovastatin against the effects of nocodazole and thrombin warrants further studies to determine if statins are useful in other situations involving loss of barrier integrity secondary to microtubule disassembly.
Conclusions
This study demonstrates that in the corneal endothelium, lovastatin opposes the thrombin-induced increased MLC phosphorylation by down-regulation of RhoA. This pharmacological effect underlies the observed suppression of the thrombin-induced loss in the barrier integrity of the corneal endothelium.
Acknowledgments
This work was supported by NIH grant R21-EY019119 and Faculty Research Grant, VP of Research, IU Bloomington, IN (S.P.S.).
Contributor Information
Mahesh Shivanna, School of Optometry, Indiana University, Bloomington, Indiana..
Supriya S. Jalimarada, School of Optometry, Indiana University, Bloomington, Indiana.
Sangly P. Srinivas, School of Optometry, Indiana University, Bloomington, Indiana.
References
- 1.Lim J.J., Fischbarg J. Electrical properties of rabbit corneal endothelium as determined from impedance measurements. Biophys. J. 1981;36:677–695. doi: 10.1016/S0006-3495(81)84758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noske W., Fromm M., Levarlet B., et al. Tight junctions of the human corneal endothelium: morphological and electrophysiological features. Ger. J. Ophthalmol. 1994;3:253–257. [PubMed] [Google Scholar]
- 3.Srinivas S.P., Satpathy M., Gallagher P., et al. Adenosine induces dephosphorylation of myosin II regulatory light chain in cultured bovine corneal endothelial cells. Exp. Eye Res. 2004;79:543–551. doi: 10.1016/j.exer.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 4.Edelhauser H.F. The balance between corneal transparency and edema: the Proctor Lecture. Invest. Ophthalmol. Vis. Sci. 2006;47:1754–1767. doi: 10.1167/iovs.05-1139. [DOI] [PubMed] [Google Scholar]
- 5.Fischbarg J., Lim J.J. Fluid and electrolyte transports across corneal endothelium. Curr. Top. Eye Res. 1984;4:201–223. [PubMed] [Google Scholar]
- 6.Riley M. Pump and leak in regulation of fluid transport in rabbit cornea. Curr. Eye Res. 1985;4:371–376. doi: 10.3109/02713688509025150. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson J.R., Dudek S.M., Birukov K.G., et al. Cytoskeletal activation and altered gene expression in endothelial barrier regulation by simvastatin. Am. J. Respir. Cell Mol. Biol. 2004;30:662–670. doi: 10.1165/rcmb.2003-0267OC. [DOI] [PubMed] [Google Scholar]
- 8.van Nieuw Amerongen G.P., Vermeer M.A., Nègre-Aminou P., et al. Simvastatin improves disturbed endothelial barrier function. Circulation. 2000;102:2803–2809. doi: 10.1161/01.cir.102.23.2803. [DOI] [PubMed] [Google Scholar]
- 9.Zeng L., Xu H., Chew T.L., et al. HMG CoA reductase inhibition modulates VEGF-induced endothelial cell hyperpermeability by preventing RhoA activation and myosin regulatory light chain phosphorylation. FASEB J. 2005;19:1845–1847. doi: 10.1096/fj.05-4240fje. [DOI] [PubMed] [Google Scholar]
- 10.Turner J.R., Angle J.M., Black E.D., et al. PKC-dependent regulation of transepithelial resistance: roles of MLC and MLC kinase. Am. J. Physiol. 1999;277:C554–C562. doi: 10.1152/ajpcell.1999.277.3.C554. [DOI] [PubMed] [Google Scholar]
- 11.Somlyo A.P., Somlyo A.V. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol. Rev. 2003;83:1325–1358. doi: 10.1152/physrev.00023.2003. [DOI] [PubMed] [Google Scholar]
- 12.Satpathy M., Gallagher P., Jin Y., et al. Extracellular ATP opposes thrombin-induced myosin light chain phosphorylation and loss of barrier integrity in corneal endothelial cells. Exp. Eye Res. 2005;81:183–192. doi: 10.1016/j.exer.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 13.Satpathy M., Gallagher P., Lizotte-Waniewski M., et al. Thrombin-induced phosphorylation of the regulatory light chain of myosin II in cultured bovine corneal endothelial cells. Exp. Eye Res. 2004;79:477–486. doi: 10.1016/j.exer.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 14.Srinivas S.P., Satpathy M., Guo Y., et al. Histamine-induced phosphorylation of the regulatory light chain of myosin II disrupts the barrier integrity of corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 2006;47:4011–4018. doi: 10.1167/iovs.05-1127. [DOI] [PubMed] [Google Scholar]
- 15.Kamm K.E., Stull J.T. Dedicated myosin light chain kinases with diverse cellular functions. J. Biol. Chem. 2001;276:4527–4530. doi: 10.1074/jbc.R000028200. [DOI] [PubMed] [Google Scholar]
- 16.Somlyo A.P., Somlyo A.V. Signal transduction by G-proteins, rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J. Physiol. (Lond.) 2000;522(Pt 2):177–185. doi: 10.1111/j.1469-7793.2000.t01-2-00177.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Nieuw Amerongen G.P., Draijer R., Vermeer M.A., et al. Transient and prolonged increase in endothelial permeability induced by histamine and thrombin: role of protein kinases, calcium, and RhoA. Circ. Res. 1998;83:1115–1123. doi: 10.1161/01.res.83.11.1115. [DOI] [PubMed] [Google Scholar]
- 18.van Nieuw Amerongen G.P., van Delft S., Vermeer M.A., et al. Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ. Res. 2000;87:335–340. doi: 10.1161/01.res.87.4.335. [DOI] [PubMed] [Google Scholar]
- 19.Mehta D., Malik A.B. Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- 20.Liao J.K., Laufs U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stancu C., Sima A. Statins: mechanism of action and effects. J. Cell. Mol. Med. 2001;5:378–387. doi: 10.1111/j.1582-4934.2001.tb00172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takemoto M., Liao J.K. Pleiotropic effects of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibitors. Arterioscler. Thromb. Vasc. Biol. 2001;21:1712–1719. doi: 10.1161/hq1101.098486. [DOI] [PubMed] [Google Scholar]
- 23.Endres M., Laufs U. Effects of statins on endothelium and signaling mechanisms. Stroke. 2004;35:2708–2711. doi: 10.1161/01.STR.0000143319.73503.38. [DOI] [PubMed] [Google Scholar]
- 24.Ohkawara H., Ishibashi T., Sakamoto T., et al. Thrombin-induced rapid geranylgeranylation of RhoA as an essential process for RhoA activation in endothelial cells. J. Biol. Chem. 2005;280:10182–10188. doi: 10.1074/jbc.M409547200. [DOI] [PubMed] [Google Scholar]
- 25.Birukova A.A., Adyshev D., Gorshkov B., et al. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;290:L540–L548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 26.Birukova A.A., Birukov K.G., Adyshev D., et al. Involvement of microtubules and Rho pathway in TGF-beta1-induced lung vascular barrier dysfunction. J. Cell. Physiol. 2005;204:934–947. doi: 10.1002/jcp.20359. [DOI] [PubMed] [Google Scholar]
- 27.Birukova A.A., Birukov K.G., Smurova K., et al. Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 2004;18:1879–1890. doi: 10.1096/fj.04-2328com. [DOI] [PubMed] [Google Scholar]
- 28.Birukova A.A., Smurova K., Birukov K.G., et al. Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J. Cell. Physiol. 2004;201:55–70. doi: 10.1002/jcp.20055. [DOI] [PubMed] [Google Scholar]
- 29.Laufs U., Kilter H., Konkol C., et al. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc. Res. 2002;53:911–920. doi: 10.1016/s0008-6363(01)00540-5. [DOI] [PubMed] [Google Scholar]
- 30.Murthy K.S. Signaling for contraction and relaxation in smooth muscle of the gut. Annu. Rev. Physiol. 2006;68:345–374. doi: 10.1146/annurev.physiol.68.040504.094707. [DOI] [PubMed] [Google Scholar]
- 31.Sriwai W., Zhou H., Murthy K.S. G(q)-dependent signalling by the lysophosphatidic acid receptor LPA(3) in gastric smooth muscle: reciprocal regulation of MYPT1 phosphorylation by Rho kinase and cAMP-independent PKA. Biochem. J. 2008;411:543–551. doi: 10.1042/bj20071299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawkitinarong K., Linz-McGillem L., Birukov K.G., et al. Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am. J. Respir. Cell Mol. Biol. 2004;31:517–527. doi: 10.1165/rcmb.2003-0432OC. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinopoulos P.A., Karamouzis M.V., Papavassiliou A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 34.Zhang F.L., Casey P.J. Protein prenylation: molecular mechanisms and functional consequences. Annu. Rev. Biochem. 1996;65:241–269. doi: 10.1146/annurev.bi.65.070196.001325. [DOI] [PubMed] [Google Scholar]
- 35.van Nieuw Amerongen G.P., Beckers C.M., Achekar I.D., et al. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler. Thromb. Vasc. Biol. 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 36.van Nieuw Amerongen G.P., Musters R.J., Eringa E.C., et al. Thrombin-induced endothelial barrier disruption in intact microvessels: role of RhoA/Rho kinase-myosin phosphatase axis. Am. J. Physiol., Cell Physiol. 2008;294:C1234–C1241. doi: 10.1152/ajpcell.00551.2007. [DOI] [PubMed] [Google Scholar]
- 37.Eilers U., Klumperman J., Hauri H.P. Nocodazole, a microtubule-active drug, interferes with apical protein delivery in cultured intestinal epithelial cells (Caco-2) J. Cell Biol. 1989;108:13–22. doi: 10.1083/jcb.108.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kolodney M.S., Elson E.L. Contraction due to microtubule disruption is associated with increased phosphorylation of myosin regulatory light chain. Proc. Natl. Acad. Sci. USA. 1995;92:10252–10256. doi: 10.1073/pnas.92.22.10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuhlmann C.R., Lessmann V., Luhmann H.J. Fluvastatin stabilizes the blood-brain barrier in vitro by nitric oxide-dependent dephosphorylation of myosin light chains. Neuropharmacology. 2006;51:907–913. doi: 10.1016/j.neuropharm.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 40.Song J., Deng P.F., Stinnett S.S., et al. Effects of cholesterol-lowering statins on the aqueous humor outflow pathway. Invest. Ophthalmol. Vis. Sci. 2005;46:2424–2432. doi: 10.1167/iovs.04-0776. [DOI] [PubMed] [Google Scholar]
- 41.Jalimarada S.S., Shivanna M., Kini V., et al. Microtubule disassembly breaks down the barrier integrity of corneal endothelium. Exp. Eye Res. 2009;89:333–343. doi: 10.1016/j.exer.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laufs U., Liao J.K. Isoprenoid metabolism and the pleiotropic effects of statins. Curr. Atheroscler. Rep. 2003;5:372–378. doi: 10.1007/s11883-003-0008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen W., Pendyala S., Natarajan V., et al. Endothelial cell barrier protection by simvastatin: GTPase regulation and NADPH oxidase inhibition. Am. J. Physiol. Lung Cell Mol. Physiol. 2008;295:L575–L583. doi: 10.1152/ajplung.00428.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao J.K. Does it matter whether or not a lipid-lowering agent inhibits Rho kinase? Curr. Atheroscler. Rep. 2007;9:384–388. doi: 10.1007/s11883-007-0049-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.George A.J., Larkin D.F. Corneal transplantation: the forgotten graft. Am. J. Transplant. 2004;4:678–685. doi: 10.1111/j.1600-6143.2004.00417.x. [DOI] [PubMed] [Google Scholar]
- 46.Niederkorn J.Y. Immune mechanisms of corneal allograft rejection. Curr. Eye Res. 2007;32:1005–1016. doi: 10.1080/02713680701767884. [DOI] [PubMed] [Google Scholar]
- 47.Banan A., Farhadi A., Fields J.Z., et al. The delta-isoform of protein kinase C causes inducible nitric-oxide synthase and nitric oxide up-regulation: key mechanism for oxidant-induced carbonylation, nitration, and disassembly of the microtubule cytoskeleton and hyperpermeability of barrier of intestinal epithelia. J. Pharmacol. Exp. Ther. 2003;305:482–494. doi: 10.1124/jpet.102.047308. [DOI] [PubMed] [Google Scholar]
- 48.Banan A., Fields J.Z., Talmage D.A., et al. PKC-zeta is required in EGF protection of microtubules and intestinal barrier integrity against oxidant injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2002;282:G794–G808. doi: 10.1152/ajpgi.00284.2001. [DOI] [PubMed] [Google Scholar]
- 49.Petrache I., Birukova A., Ramirez S.I., et al. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am. J. Respir. Cell Mol. Biol. 2003;28:574–581. doi: 10.1165/rcmb.2002-0075OC. [DOI] [PubMed] [Google Scholar]
- 50.Suzuki S., Bing H., Sugawara T., et al. Paclitaxel prevents loss of pulmonary endothelial barrier integrity during cold preservation. Transplantation. 2004;78:524–529. doi: 10.1097/01.tp.0000131951.72851.57. [DOI] [PubMed] [Google Scholar]