

Abstract
The cytoplasmic dynein complex is fundamentally important to all eukaryotic cells for transporting a variety of essential cargoes along microtubules within the cell. This complex also plays more specialized roles in neurons. The complex consists of 11 types of protein that interact with each other and with external adaptors, regulators and cargoes. Despite the importance of the cytoplasmic dynein complex, we know comparatively little of the roles of each component protein, and in mammals few mutants exist that allow us to explore the effects of defects in dynein-controlled processes in the context of the whole organism. Here we have taken a genotype-driven approach in mouse (Mus musculus) to analyze the role of one subunit, the dynein light intermediate chain 1 (Dync1li1). We find that, surprisingly, an N235Y point mutation in this protein results in altered neuronal development, as shown from in vivo studies in the developing cortex, and analyses of electrophysiological function. Moreover, mutant mice display increased anxiety, thus linking dynein functions to a behavioral phenotype in mammals for the first time. These results demonstrate the important role that dynein-controlled processes play in the correct development and function of the mammalian nervous system.
Introduction
In mammals cytoplasmic dynein 1 is a large complex of proteins whose constituent members are defined by their molecular weights: the heavy chain (encoded by a single gene Dync1h1); the intermediate chains (Dync1i1, Dync1i2); the light-intermediate chains (Dync1li1, Dync1li2); the light chains (Dynlt1, Dynlt3, Dynlrb1, Dynlrb2, Dynll1, Dynll2) (Pfister et al., 2005, 2006; Höök and Vallee, 2006; Levy and Holzbaur, 2006). The stoichiometry of the intact complex is not known exactly, but at its core lies a homodimer of heavy chains, which binds to microtubules enabling cytoplasmic dynein to move in an ATP-dependent manner (Gennerich et al., 2007). The other subunits are thought to maintain the stability of the complex, to modulate its activity, and to interact with accessory and cargo proteins. However, while it has been established that the individual dynein subunits can have complex cell-specific splicing patterns (Pfister et al., 1996a,b; Salata et al., 2001; Ha et al., 2008; Kuta et al., 2010), we know comparatively little regarding the specific functions of the separate subunits.
One way to study dynein function is to investigate individual genes/protein mutations, however, in mammals the only DYNC1 (cytoplasmic dynein) mutations so far described are four alleles of mouse Dync1h1, one of which is a knock-out (Harada et al., 1998; Hafezparast et al., 2003; Chen et al., 2007). The point mutants (all detected in phenotype-driven screens) give highly informative phenotypes compared with those seen in knock-out animals and have uncovered roles that are not detected in null animals (Harada et al., 1998; Hafezparast et al., 2003; Kieran et al., 2005; Chen et al., 2007; Banks and Fisher, 2008; Ilieva et al., 2008; Dupuis et al., 2009).
To further investigate the role of individual dynein subunits, we have used a genetic technique, random chemical mutagenesis, and then taken a genotype-driven approach using a mouse with a point mutation in the dynein light intermediate chain 1, DYNC1LI1. We chose this subunit because it binds important cargos including pericentrin (Tynan et al., 2000) and Na+ channels (Zheng et al., 2008), and in Aspergillus DYNC1LI1 stabilizes the interaction of heavy and intermediate chains (Zhang et al., 2009). Thus it is likely that dynein light intermediate chains play critical roles within the cell and therefore in the organism as a whole. However, at the time of writing no mammalian model of light intermediate chain dysfunction has been published.
We identified and characterized a mouse carrying a point mutation in Dync1li1. We find mutant mice have increased anxiety-like behavior, changes in cortical and peripheral neuronal outgrowth and branching, hypotrophy of sensory neurons and electrophysiological defects and at the cellular level defects in Golgi reassembly and endosome sorting and trafficking. Thus this genotype-driven approach, characterizing a mouse with a relatively subtle point mutation, has revealed a range of unsuspected essential functions dependent on the cytoplasmic dynein complex.
Materials and Methods
Animal studies
Wherever possible, all methods were undertaken blind to mouse genotype, which was decoded after the results were collected. The animal studies were performed under guidance issued by the UK Medical Research Council in Responsibility in the Use of Animals for Medical Research (1993) and under license from the UK Home Office.
Heteroduplex screening the MRC Mammalian Genetics Unit ENU DNA archive
The Dync1li1N235Y mutation arose in an ENU mutagenesis experiment at the MRC Mammalian Genetics Unit, MRC Harwell, UK (Nolan et al., 2000). Briefly, BALB/cAnN male mice are treated with ENU which results in random point mutations in the sperm of these animals. These males are crossed with C3H/HeH females to produce F1(C3H/HeH × BALB/cAnN) progeny carrying heterozygous random point mutations. Genomic DNAs are archived from F1(C3H/HeH × BALB/cAnN) males for subsequent screening; sperm samples are archived in parallel for rederivation of mouse lines. Genomic DNAs from archive were analyzed by heteroduplex analysis following the protocols outlined in (Quwailid et al., 2004). Please see supplemental material (available at www.jneurosci.org) for primer sequences and complete methodology.
Rederivation and genotyping of Dync1li1N235Y mouse strain
Frozen sperm from the Dync1li1N235Y mutant F1 male mouse was used for in vitro fertilization (IVF) with C57BL/6J females; we used 50 two-cell stage embryos for implantation into two pseudo-pregnant C57BL/6J females, resulting in 12 animals, of which 3 females and 1 male were heterozygotes and were used as founders for our Dync1li1N235Y colony. Subsequently the sex of the Dync1li1N235Y heterozygote animal used in backcrosses was alternated for each generation so that nuclear, mitochondrial and Y chromosome DNA was all of C57BL/6J origin.
DNA was extracted from mouse tail biopsies using a genomic DNA isolation kit (Promega UK Ltd). Genotyping was performed by allele-specific PCR. Two amplifications were performed per genomic DNA sample: one PCR amplified the wild-type Dync1li1 allele only (DLIC1WT forward: ATCTCTGGCCGCCAAGGTCC; DLIC1WT reverse: AGCACTGGTAGGCCCAGATT) generating a fragment of 427 bp. The second PCR amplified the Dync1li1N235Y allele only (DLIC1N235Y forward: TTACCACTGAGCCATCTCTG; DLIC1N235Y reverse: AGCACTGGTAGGCCCAGGTA) generating a fragment of 440 bp. Both reactions also contained primers to amplify an unrelated DNA fragment of 590 bp from Dync1i2 (control forward: GTTGACAGGATTTAATTGGCC; control reverse: TGTGACAATGGCAACGTCAG). PCRs were performed for 35 cycles, annealing temperature 66°C, using MegaMix Gold (Microzone) according to the manufacturer's instructions. Products were visualized on a 2% agarose gel. If the control fragment was absent, the reaction was “failed.” If the control fragment was present samples in which the wild-type Dync1li1 allele and not the Dync1li1N235Y allele amplified were genotyped as Dync1li1+/+; those with the Dync1li1N235Y allele and not the wild-type allele were scored as Dync1li1N235Y/N235Y; samples in which both the wild-type allele and the Dync1li1N235Y allele amplified were genotyped as Dync1li1N235Y/+.
Behavioral tests
Please see supplemental material (available at www.jneurosci.org) for methods for open field behavior, food and water intake, glucose preference, wheel running, accelerating rotarod, modified SHIRPA, spontaneous alternation and water maze.
Spontaneous locomotor activity.
Spontaneous locomotor activity was assessed in transparent plastic cages with two horizontal photocell beams located along the long axis of each cage, during a single 2 h test session (without habituation to the test cage). Female mice were used only and were placed individually into the activity cages, and the total number of beam breaks made by each mouse was recorded.
Plus maze.
The plus maze had four arms (each 27.5 × 5.5 cm) arranged in a cross formation: two “closed” arms bordered by 30-cm-high walls and two “open” arms without walls. The maze was elevated 50 cm above the ground and illuminated by bright white light. Female mice were used only and were placed at the distal end of a closed arm and their movements were recorded for 5 min. Behavioral analysis was performed using an automated tracking system (Ethovision XT) which measured the time spent in each region, the number of entries to each region, the latency to enter an open arm and the distance traveled.
Successive alleys.
The successive alleys apparatus consisted of four increasingly anxiogenic, linearly connected wooden alleys. Each alley was 45 cm in length. The alleys had the following dimensions: alley 1 = 9 cm wide, 29-cm-high walls, painted black; alley 2 = 9 cm wide, 2.5-cm-high walls, painted gray; alley 3 = 6.7 cm wide, 0.5-cm-high walls, painted white; alley 4 = 5 cm wide, 0.3-cm-high walls, painted white. A 2 cm step down led from alley 2 to alley 3, with a further 0.5 cm step down between alleys 3 and 4. The apparatus was elevated 1 m above the floor in a well lit laboratory. Female mice used only and were placed individually at the closed end of Alley 1, facing the wall. Each trial lasted for 300 s, during which time the latency to first enter, the total time spent in, and the number of entries into, each section were recorded.
Weight lifting.
Strength testing was performed using a set of seven weights of linearly increasing mass. Each mouse was held by the tail, and its forepaws were allowed to grasp a ball of wire mesh to which a weight was attached; female mice were used only. If the mouse lifted this weight for 3 s it was tested on progressively heavier ones. The weight lifting score was calculated from the heaviest weight lifted and the number of seconds for which the weight was held.
CatWalk.
Female mice were used only and were allowed to walk spontaneously up and down the glass CatWalk walkway while images were collected by a video camera placed below. Three crosses of the walkway were analyzed for each animal and the data averaged. A number of parameters were examined, including footstep sequence, front and hind base of support, regularity, and paw print size, angle and intensity.
Cortical neuron dendrite analysis
Uterine horns were removed from time-mated heterozygous Dync1li1N235Y/+ mice at embryonic day 15 (E15) under anesthesia (isoflurane, 2.5 L/min). Approximately 0.5 μl of solution containing enhanced green fluorescent protein (GFP)-pCIG2 expression plasmid (2 μg/μl) and Fast Green dye was injected into the lateral ventricle using a micropipette under pressure (FemtoJet, Eppendorf). Electroporation paddles were oriented to target GFP incorporation to neurons eventually populating the PFC. Electroporated embryos were placed back into the mother and allowed to develop normally, until either E17 or postnatal day 15 (P15). Embryos harvested at E17 were used to prepare dissociated cortical neuron cultures, according to established methods (Banker and Goslin, 1998): briefly, dissected cortices were trypsinized (0.125%, Invitrogen), dissociated and plated into the wells of poly-l-lysine (0.01%, Sigma)-coated Labtek chamber slides at a density of 1 × 105 cells per chamber. Cultures were maintained in Neurobasal medium with supplements (B-27, Glutamax and penicillin/streptomycin, Invitrogen), until they were fixed at 10 days in vitro (DIV) with 4% paraformaldehyde (PFA) and immunolabeled with an antibody to GFP (1:100, MorphoSys). Up to 20 GFP-labeled neurons per embryo were imaged using a DeltaVision microscope and Softworks software. Neurolucida software was used to trace photographed neurons and analyze dendritic characteristics in wild-type and Dync1li1N235Y/N235Y neurons. In all cases data were checked for normal distribution using the D'Agostino and Pearson test, then compared for statistical differences using the t test or nonparametric Mann–Whitney U test.
Alternatively, GFP-expressing mice at P15 were transcardially perfused with 0.9% saline, followed by 4% paraformaldehyde. Brains were removed, postfixed for 1 h in 4% PFA, then vibratome cut coronal sections (100 μm) were imaged using an SP5 Multiphoton, Leica confocal system to observe the architecture of the cortex, in particular the organization of layer II/III projection neurons and their dendritic arbors. Confocal images were collected using Leica software and were processed using Volocity, NIH ImageJ, and Photoshop (Adobe) software. Fluorescence intensity was plotted using NIH ImageJ, as a measure of GFP-positive neuron position within the neocortex.
Conditioning lesion and sensory neuron cultures
Mice of either sex were subjected to unilateral sciatic nerve crush at mid-thigh level. Three days after injury, dissociated cultures were prepared from the L4–5 dorsal root ganglia (DRGs) ipsilateral and contralateral to the nerve crush as described previously (Hanz et al., 2003). The dissociated DRGs were plated in DMEM/F12 medium supplemented with N1 and 10% horse serum on laminin-coated glass coverslips at a density of 2–5 neurons/mm2. After 20 h cultures were fixed in 4% paraformaldehyde, stained with anti-NFH (neurofilament heavy chain) and number of branches per neuron was measured using MetaMorph software on digital images captured by ImageXpressMicro (Molecular Devices) fluorescent automatic microscope at 10× magnification.
Conduction studies and dorsal root ganglion histology
Skin-nerve preparation.
Animals of either sex, 3–4 months of age, 7 wild-type and 8 Dync1li1N235Y/N235Y littermates were killed by cervical dislocation. There were no obvious gross morphological differences of the nerves on dissection. The skin nerve preparation was performed as described previously (Koltzenburg et al., 1997). The saphenous nerve with the skin of the hind leg attached was dissected free and placed “inside-up” in an organ bath. The preparation was superfused (15 ml/min) with an oxygen-saturated modified synthetic interstitial fluid solution containing the following (in mm): 123 NaCl, 3.5 KCl, 0.7 MgSO4, 1.7 NaH2PO4, 2.0 CaCl2, 9.5 sodium gluconate, 5.5 glucose, 7.5 sucrose, and 10 HEPES at a (mean ± SD) pH of 7.4 ± 0.05 and a temperature of 32.0 ± 0.5°C.
Recordings of A- and C-fiber sensory nerve compound action potential (SNAP) were made using a low noise custom-made amplifier and HP 1 Hz, LP 1 kHz filter setting without a notch filter. The nerve was stimulated with a fine monopolar steel needle as cathode and the anode placed close by in the organ bath. Values given are mean ± SEM. Statistical comparison was made with a nonpaired t test.
Motor nerve conduction velocity studies.
Animals of either sex, 4–5 months of age, 5 wild-type and 5 Dync1li1N235Y/N235Y littermates were studied in vivo. Mice were anesthetized with isoflurane and studied using a Viking Quest EMG machine (generously donated by CareFusion). Compound action potentials were recorded from small foot muscles with needle electrodes and 10 Hz HP and 10 kHz LP filters. The recording electrode was inserted perpendicularly through the plantar skin midway between the metatarsal-phalangeal joints and the ankle. The reference electrode was inserted into the hallux. The tibial nerve was stimulated at the ankle and the sciatic nerve at the sciatic notch using a pair of monopolar needle electrodes. Needle EMG was obtained with a thin (30 ga, “facial”) concentric needle electrode with a recording area of 0.03 mm2 from intrinsic foot muscles or the medial gastrocnemius muscle using 10 Hz HP and 10 kHz LP filters. Values given are mean ± SEM. Statistical comparison with made with a nonpaired t test.
Histological analyses of DRGs.
Animals of either sex, 3–4 months of age, 4 wild-type and 3 Dync1li1N235Y/N235Y littermates were used in this analysis. Cervical and lumbar DRGs were fixed in a 4% PFA and subsequently soaked in a sterile 10% sucrose solution (in PBS) for 2 h and then successively transferred to increasing concentrations of 20% sucrose for a further 2 h and finally a 30% solution overnight at 4°C. Cryoprotected lumbar or cervical DRGs were embedded in OCT and sectioned on a cryostat at 10 μm.
Fixed or unfixed skin.
Analysis of epidermal nerve fiber density using PGP9.5 (a marker for unmyelinated fibers) (Achilli et al., 2009), Merkel cells (using cytokeratin 18), and large myelinated fibers stained for neurofilament heavy chain (N52) was undertaken. Sections were double immunostained at room temperature following permeabilization with Triton-X and blockade with donkey serum either using a mouse IgG1 monoclonal anti-neurofilament 200 (N52, Sigma) antibody and a rabbit polyclonal anti-peripherin antibody (Millipore Bioscience Research Reagents, AB1530) or using the mouse IgG1 monoclonal anti-parvalbumin (235 Swant) or a rabbit polyclonal antiserum to calcitonin gene-related peptide (CGRP) (1134, Biomol International). Primary antibodies were applied overnight, secondary antibodies were an Alexa Fluor 488 donkey anti-rabbit IgG (Invitrogen, A21206) and a Cy3 donkey anti-mouse IgG (Jackson ImmunoResearch) and applied for 2 h. Slides were mounted using a Vectashield mounting media with a nuclear DAPI stain and viewed on a Zeiss Axiophot 2. Images were taken at a 20× fluor objective and captured using appropriate filters.
Data analysis
Cell counts for NFH/peripherin immunostaining.
Lumbar and cervical DRGs from each animal were analyzed separately. Captured images were viewed in Adobe Photoshop CS4. A minimum of 200 cell profiles, constituting approximately three dorsal root ganglion (DRG) sections, were analyzed for each animal and the number of cell profiles positive for each marker was recorded. Of those profiles showing nuclei, the areas and perimeters of at least 50 cells positive for each marker were recorded using a 21-inch LCD (liquid crystal display) digitalizing tablet with Adobe Photoshop software. The mean data from individual animals was then combined so that group data from mutant animals could be compared with the data from the wild-type animals.
Cell counts for parvalbumin/CGRP immunostaining.
As with the NFH/peripherin immunostaining, the lumbar and cervical DRGs were analyzed separately. A minimum of 400 cell profiles were counted for each animal and areas were recorded for those profiles which showed immunoreactivity for each marker using the same software as for the NFH/peripherin counts. The nuclei of parvalbumin and CGRP-immunoreactive cells were not always visible so the areas of all the immunoreactive cell profiles were measured as opposed to just those profiles that had been sectioned in the nuclear plane.
Mouse embryonic fibroblast protocols
Creation of mouse embryonic fibroblasts.
A pregnant female was sacrificed at day 13.5 postcoitum by cervical dislocation, embryos were separated and kept in L-15 medium on ice (Invitrogen) before processing. Head and visceral organs were removed and tissues were minced using razorblades and suspended in 1.5 ml of 0.05% trypsin (Invitrogen) in DPBS supplemented with 10 mm MgCl2 and DNase I (200 U, Invitrogen). Tissue was incubated at 37°C, 20 min, transferred into 4 ml of warm medium [DMEM supplemented with 10% FBS and 1:100 (v/v) penicillin/streptomycin]. Remaining pieces of tissue were allowed to settle to the bottom of the tube and cell suspension was transferred into cell culture flasks (Nunc).
Golgi reassembly after disruption with nocodazole.
E13 mouse embryonic fibroblasts (MEFs) were grown to 70% confluence on round coverslips in 6 well plates in standard DMEM at 37°C, 3% O2. Standard DMEM was replaced by ice-cold standard DMEM containing 15 mm HEPES, cells were left on ice for 30 min. This was replaced with DMEM containing 10 μg/ml nocodazole, and cells were incubated at 37°C, 3 h, 3% O2. Nocodazole was washed from the cells 5 times in DMEM and cells returned to 3% O2, 37°C incubator to recover for 0, 30 and 50 min before being fixed in −20°C methanol, 8 min. The cells were then washed 3 times in PBS containing 0.2% gelatin from coldwater fish skin (PBSG). Samples were stained for Golgi using rabbit polyclonal anti-giantin (1:1000) (Covance) and cytoskeleton using mouse monoclonal anti-α-tubulin (1:200) (Millipore); secondary antibodies were Alexa Fluor 546-conjugated goat anti-rabbit IgG (1:200) (Invitrogen) and Alexa Fluor 488-conjugated goat anti-mouse IgG (1:200) (Invitrogen). Antibodies were diluted in either PBSG (anti-giantin) or PBS− (PBS without magnesium or calcium) for 30 min, room temperature. Finally, cells were mounted on glass slides using ProLong Gold antifade mounting medium, containing 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Invitrogen), and allowed to cure for 24 h at room temperature, then stored at 4°C.
Endosomal trafficking chase of Alexa Fluor-conjugated epidermal growth factor
E13 MEFs were grown to 80% confluence on round coverslips in 6 well plates in standard DMEM at 37°C, 3% O2 and then washed twice with PBS, without calcium or magnesium (PBS−). Cells were starved for 2 h in starving medium (DMEM, penicillin/streptomycin, and l-glutamine) at 37°C, 3% O2, before stimulation for 10 min with warmed starving medium containing 8 ng/ml Alexa Fluor 555-conjugated epidermal growth factor (EGF) (Invitrogen) and 0.1% BSA. Cells were washed twice with ice-cold PBS− and the internalized EGF was chased for 0, 20, and 40 min. Samples were then fixed at the given time points with 4% PFA for 15 min, and permeabilized for 5 min in 0.1% Triton X-100 and blocked in 2% BSA, 1 h. Samples were then mounted on glass slides using ProLong Gold antifade mounting medium containing DAPI (Invitrogen) and allowed to cure for 24 h, room temperature, then stored at 4°C.
Results
Origin and inheritance of Dync1li1N235Y mutation
An archive of genomic DNAs from novel mice with random point mutations has been created at the MRC Mammalian Genetics Unit, UK in an N-ethyl-N-urea (ENU) driven mutagenesis program (Nolan et al., 2000). To take a genotype driven approach to understanding dynein subunit function, we screened this archive for Dync1li1 exon 5 since it is one of the largest protein coding exons in the gene and the larger the stretch of DNA screened, the higher the probability that a mutation will be found. We amplified a 248 bp fragment (including complete 170 bp of exon 5), multiplexed at four DNA samples per well. A heteroduplex screen of 1248 amplicons (i.e., 4992 pools) showed a possible mutation in one DNA pool (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material); sequencing revealed the single mutant DNA sample (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Thus in our amplicon we detected 1 mutation per 1.24 × 106 bp, of which 0.85 × 106 bp were protein coding.
The Dync1li1 mutation was an A to T change at base pair 795 (cDNA numbering; Mouse Genome Informatics accession number MGI:2135610) causing an asparagine to tyrosine change at the highly conserved residue 235: Dync1li1N235Y (Fig. 1). This mutation was not found in either parental strain, BALB/cAnN or C3H/HeH, confirming it is not a single-nucleotide polymorphism (nor is it found in C57BL/6).
Figure 1.

Partial sequence and domains of wild-type and mutant DYNC1LI1. A, Multiple sequence alignment of cytoplasmic dynein light intermediate chain 1. Asparagine 235 is mutated in the Dync1li1N235Y mouse but is highly conserved throughout the animal kingdom and also in amoebae and fungi. B, Protein domain map of DYNC1LI1, showing the location of the DYNC1LI11N235Y mutation as a red bar. The map was created using the domains identified by Bielli et al. (2001), Hughes et al. (1995), and Tynan et al. (2000).
The Dync1li1N235Y mouse line was rederived by IVF and 4 animals (3 female, 1 male) were used as the founders for our Dync1li1N235Y colony. These mice were crossed to C57BL/6J mice and subsequent generations were backcrossed to produce a congenic strain or intercrossed to produce homozygous animals. At N5, 8 Dync1li1N235Y/+ heterozygous males were crossed with 14 Dync1li1N235Y/+ heterozygous females, producing 37 litters with an average size of 8 pups per litter. The sex ratio did not vary from normal (143 males, 147 females; χ2 = 0.09). Homozygous animals were viable and the genotype ratios were not significantly altered from Mendelian norms (79 wild type, 140 heterozygote, 71 homozygote; χ2 = 2.86). Homozygotes were fertile—two intercrosses of homozygous males and females produced litters of 11 and 7 homozygous pups.
We went on to test the mutant mice for phenotypic differences: all phenotyping was performed blind to genotype on wild-type and homozygous age-, sex-matched littermates. Note that although the backcross generation of mice used varied between experiments, all mice within each experiment were of the same generation on C57BL/6J.
Behavioral analysis and histology of brain and spinal cord
To determine whether the Dync1li1 mutation plays a role in behavior, female mice (12 wild type, 11 Dync1li1N235Y/N235Y homozygotes, N4) were assessed. We found no difference between genotypes in open field behavior, food and water intake, glucose preference or wheel running (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). However, when placed in a novel environment Dync1li1N235Y/N235Y mice displayed lower levels of spontaneous locomotor activity than wild-type littermates (F(1,21) = 4.35; p < 0.05) (Fig. 2A).
Figure 2.

Behavior of Dync1li1N235Y/N235Y mice. A, Dync1li1N235Y/N235 mice displayed significantly lower levels of spontaneous locomotor activity compared with wild-type littermates. B, On the elevated plus maze (EPM), the Dync1li1N235Y/N235Y mice spent significantly less time in the open arms and more time in the closed arms. C, They also made significantly fewer entries to both the open and closed arms. D, On the successive alleys task Dync1li1N235Y/N235Y mice spent less time in the more anxiogenic alleys compared with wild types. E, They also made fewer entries to these alleys (the Dync1li1N235Y/N235Y mice did not make any entries to alleys 3 or 4). F, Footprint analysis using the CatWalk system found that compared with wild types, Dync1li1N235Y/N235Y mice displayed a significantly greater difference between the BOS of their front legs and hind legs due to a relative narrowing of the front BOS and a widening of the hind BOS; a representative sample from one animal is shown. Green, Right front foot; dark green, right hind foot; red, left front foot; dark red, left hind foot. Animals walked from right to left.
Next, anxiety was examined using the elevated plus maze. Compared with wild types, Dync1li1N235Y/N235Y mice had a longer latency to first enter an open arm (U(11,12) = 29.0; p < 0.05), spent less time in the anxiogenic open arms (U(11,12) = 100.5; p < 0.05) and more in the closed arms (U(11,12) = 13.0; p < 0.01) (Fig. 2B) and made fewer entries to the open arms (U(11,12) = 103.0; p < 0.05) and the closed arms (U(11,12) = 112.5; p < 0.005) (Fig. 2C). However, the distance moved by the Dync1li1N235Y/N235Y mice did not differ from that of wild types (t(21) = −1.50; p = 0.15), suggesting that the reduced exploration of the open arms could not be explained by hypoactivity. In addition, Dync1li1N235Y/N235Y mice displayed significantly increased defecation during the task compared with wild types (U(11,12) = 28.0; p < 0.05).
Anxiety was further examined using the successive alleys task (Deacon et al., 2003). Again, Dync1li1N235Y/N235Y mice were slower to move into the more anxiogenic second alley (U(11,12) = 26.0; p < 0.05), spent less time in the second alley (U(11,12) = 106.0; p < 0.05) (Fig. 2D) and made fewer entries into the second alley (U(11,12) = 110.0; p < 0.01) (Fig. 2E). The small number of animals (4 wild type, 0 Dync1li1N235Y/N235Y) that ventured beyond the second alley meant that valid analyses could not be performed on alleys 3 and 4. Thus the results of the plus maze and successive alleys tasks suggest that the Dync1li1N235Y/N235Y mice display increased anxiety compared with their wild-type littermates.
At 6 months of age the performance of Dync1li1N235Y/N235Y mice on the accelerating rotarod did not differ from that of wild types (t(21) = 0.47; p = 0.64). However, Dync1li1N235Y/N235Y mice tended to be slightly stronger than wild types on a weightlifting test of grip strength (U(11,12) = 35.5; p = 0.06) (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). In addition, examination of gait using the CatWalk analysis system found that the ratio of forepaw base of support (BOS) to hindpaw BOS was significantly reduced in the homozygous mutant animals (t(21) = 3.21; p < 0.005) compared with wild-type littermates. This was due to a relative narrowing of the forepaw BOS coupled with an increase in the hindpaw BOS (Fig. 2F). We performed the modified SHIRPA test (Rogers et al., 1997, 2001), which included further rotarod and grip strength measurements, upon male and female mice up to 52 weeks of age but found no difference between homozygotes and wild-type controls (supplemental material, supplemental Fig. S2I,J, available at www.jneurosci.org as supplemental material) and no differences in tests of hippocampal spatial working memory (spontaneous alternation) (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material), or reference memory acquisition and reversal (supplemental material, supplemental Fig. S2D–H, available at www.jneurosci.org as supplemental material).
Given the behavioral and gait abnormalities, brain and cervical and lumbar spinal cord were assessed histologically. Attention was paid to prefrontal cortex (PFC), hippocampus, striatum and cerebellum, but we detected no differences between wild-type and mutant littermates in brain or spinal cord morphology or motor neuron counts in the sciatic motor pool (supplemental material, supplemental Fig. S3A–D, available at www.jneurosci.org as supplemental material).
Cortical neuron dendrite outgrowth and branching
As axonal and dendritic development is known to be mediated by cytoplasmic dynein-dependent processes, we investigated the morphology of cortical neurons, including those in the PFC. A GFP expression plasmid was transfected into the cortex of embryos at E15 by in utero electroporation, selectively labeling neurons that would in the developed brain become layer II/III cortical projection neurons. Two days after electroporation dissociated cortical cultures were prepared from fluorescing hemispheres. Ten days later GFP-expressing cells were analyzed to determine patterns of dendritic outgrowth and branching (Fig. 3A). We found the mean dendritic tree length was significantly increased in Dync1li1N235Y/N235Y neurons (Fig. 3Bi; wild type, 370.3 ± 33.46 μm; Dync1li1N235Y/N235Y, 452.6 ± 34.52 μm; p = 0.0029). Furthermore, individual dendrites terminated significantly further from the soma in Dync1li1N235Y/N235Y neurons than wild-type controls (Fig. 3Bii; wild type, 170.9 ± 4.637 μm; Dync1li1N235Y/N235Y, 182.6 ± 4.411 μm; p = 0.0281). Thus homozygous mutant dendrites grew longer and extended further from the soma than those of wild-type animals, which may have implications for correct connectivity between neurons in vivo. Despite these differences in dendritic tree length, wild-type and Dync1li1N235Y/N235Y neurons had the same average number of dendritic trees (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material; wild type, 5.405 ± 0.3388; Dync1li1N235Y/N235Y, 4.923 ± 0.2807; p = 0.4697) and the trees displayed similar branching patterns: the average number of branch points, or nodes, per dendrite was not statistically different (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material; wild type, 3.985 ± 0.3221; Dync1li1N235Y/N235Y, 4.236 ± 0.2391; p = 0.4697). In addition, the frequency of nodes per dendritic tree was statistically indistinguishable between wild-type and Dync1li1N235Y/N235Y neurons. This indicates that the variability in branching patterns observed between neurons within each genotype, was consistent between the two genotypes (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material). Finally, a Sholl analysis showed virtually identical number of intersections within given radii of the cell body for wild-type and Dync1li1N235Y/N235Y neurons (supplemental Fig. S4D, available at www.jneurosci.org as supplemental material). Therefore, increased dendrite outgrowth, but not increased branching, characterizes cortical neurons in Dync1li1N235Y/N235Y neuron cultures.
Figure 3.
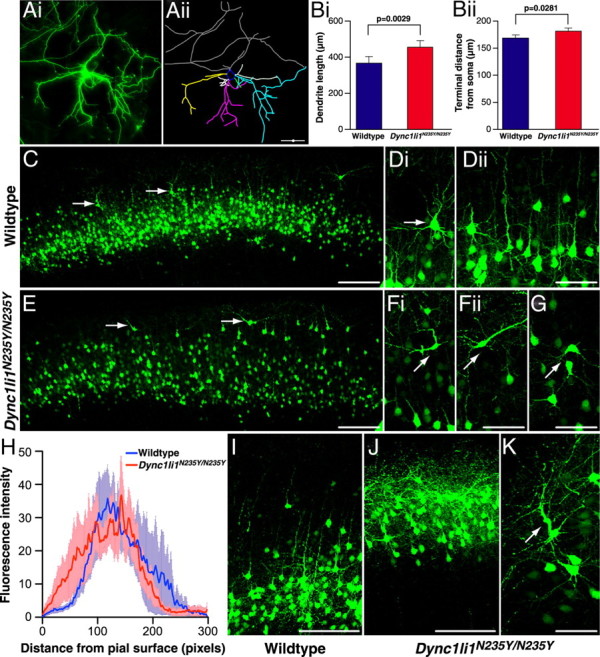
Morphology of wild-type and mutant Dync1li1N235Y/N235Y cortical neurons. A, Cortical neurons were electroporated in vivo at E15, then cultured in vitro from E17 embryos. At 10 DIV they were analyzed to compare morphology of wild-type and Dync1li1N235Y/N235Y cortical neurons. GFP-positive neurons (i) were hand-traced using Neurolucida software (ii). B, The length of dendritic trees for wild-type and Dync1li1N235Y/N235Y neurons, in vitro. i, Dync1li1N235Y/N235Y dendrites were significantly longer than in wild-type neurons (p = 0.0029). ii, Dync1li1N235Y/N235Y dendrites terminated significantly further from the soma than wild-type dendrites (p = 0.00281). C, Low magnification of the cortex from a wild-type brain electroporated with GFP at E15 and harvested at P15. GFP-positive neurons occupy a discreet band corresponding to cortical layers II/III. D, i, Higher-magnification image showing the orientation of a pyramidal neuron in layer II. ii, Neighboring neurons align in the same orientation with apical dendrites projecting toward the pial surface. E, The cortex from a Dync1li1N235Y/N235Y brain electroporated with GFP at E15 and harvested at P15 (rostrocaudally matched with C). Compared with C, GFP-positive cells were more distributed, with more neurons residing in the uppermost part of the cortex (arrows). F, The cell body and apical dendrite of neurons located closer to the pial surface in Dync1li1N235Y/N235Y mutants were mis-oriented. G, Neurons in layer II of Dync1li1N235Y/N235Y cortex also often displayed incorrect orientation. H, Comparison of the proportion of electroporated neurons located in upper layers of the neocortex, as measured by fluorescence intensity. I, Higher-magnification imaging from wild-type cortex in more caudal sections showed most GFP-positive neurons within layer II/II. J, In contrast to I, GFP-expressing cells in Dync1li1N235Y/N235Y cortex occupied a location much closer to the pial surface. K, The apical dendrite of neurons Dync1li1N235Y/N235Y cortex were often thickened and projected in an incorrect orientation. Scale bars: A, C, E, I, J, 100 μm; D, F, G, K, 50 μm.
To determine whether dendritic branching and organization defects occur in Dync1li1N235Y/N235Y neurons in vivo, embryos subjected to electroporation with GFP at E15 were left to develop normally and brains were harvested at P15. In these experiments electroporation was targeted toward the PFC, an area that has been associated with anxiety studies in lesion studies (Deacon et al., 2003). We found mutant phenotypes that could not be observed in in vitro cultures: in the PFC of Dync1li1N235Y/N235Y animals more neurons that had migrated past their appropriate layer II/III position and into the uppermost part of the cortex (Fig. 3C,E; quantitation shown in H). More caudally (Fig. 3I,J) there was a marked increase in GFP-labeled neurons in the uppermost layer of the cortex in Dync1li1N235Y/N235Y mice. Many of these neurons displaced from layer II adopted abnormal orientation in Dync1li1N235Y/N235Y animals compared with wild types (Fig. 3D,F) and also a proportion within layer II/III showed abnormal orientation (Fig. 3G). Figure 3 shows that this mis-orientation was characterized by the apical dendrite not projecting radially, toward the pial surface, but instead to varying degrees tangentially. In addition we observed neurons with abnormal apical dendrite morphology including thickening of the dendrite and often irregular trajectories (Fig. 3K, arrow).
Adult sensory neuron branching
To establish whether the Dync1li1N235Y mutation affected neuronal branching in the periphery we examined neuronal cultures from wild-type and Dync1li1N235Y/N235Y DRGs. We also examined the possibility that the Dync1li1 mutation might affect dynein-based retrograde injury signaling by performing conditioning lesion experiments as described previously (Hanz et al., 2003).
Two wild-type and two Dync1li1N235Y/N235Y male mice of 2–4-months old were used for each experiment, and three independent replicates were carried out. Mice were subjected to unilateral sciatic nerve crush at mid-thigh level; 3 d later, dissociated cultures were prepared from L4–5 DRGs ipsilateral and contralateral to the nerve crush. Neurons were fixed after 20 h in culture, stained with anti-NFH, and branching was measured. As shown in Figure 4A,B, in naive conditions NFH-positive sensory neurons from Dync1li1N235Y/N235Y mice had a greater number of branches than wild-type controls. We found similar results for neurons under injury conditions, although differences were less significant than for naive conditions, apparently due to a more marked injury response in wild-type neurons (47.6% increase in branching after injury, compared with 33.5% increase in the mutants).
Figure 4.

Dync1li1N235Y point mutation increases branching in cultured sensory neurons. A, Fluorescent images of DRG neurons from wild-type and Dync1li1N235Y homozygous mice under naive and injury conditions. Right leg sciatic nerves of wild-type and mutant mice were crush-lesioned 3 d before dissection of L4–5 DRGs for primary culture of sensory neurons; corresponding contralateral DRGs were used for naive controls. Cultures were imaged by automated fluorescent microscopy after 20 h in vitro, to obtain 150 images per slide, at magnification 10×. B, Quantification shows a significant increase in branching of naive sensory neurons from Dync1li1N235Y/N235Y homozygotes (29.4% increase over wild type). Increased branching was also observed in homozygous Dync1li1N235Y/N235 sensory neurons after injury, although the difference is less pronounced than in naive neurons, apparently due to a more robust injury response in the wild-type neurons. ***p < 10−6; *p < 0.05 (Student's t test). Scale bar, 100 μm.
To assess nerve branching in developing animals we performed whole mount neurofilament immunohistochemistry in the limbs of E13.5 animals; we found no significant difference between wild-type and Dync1li1N235Y/N235Y animals in the number of branch points (supplemental material, supplemental Figure S4, available at www.jneurosci.org as supplemental material) and whole-mount neurofilament staining did not show any obvious abnormalities in the peripheral nervous system of homozygotes at P12.5 (Christiana Ruhrberg, personal communication).
Neurophysiology of sensory neurons and dorsal root ganglia histology
Mice with mutations in the dynein heavy chain 1 subunit have a loss of cutaneous and proprioceptive sensory neurons, and so for comparison we undertook sensory nerve conduction studies of the saphenous nerve on 8 wild-type and 7 Dync1li1N235Y/N235Y homozygous male littermates (3 months of age, N3). Recordings were taken of the A (myelinated)- or C (unmyelinated)-fiber SNAPs; we found a small but significant (p < 0.05) reduction of the peak-to-peak amplitude of the A-fiber SNAP from 3.2 ± 0.3 mV in wild types to 2.5 ± 0.2 mV in homozygous mutants (Fig. 5A) but no difference in the conduction velocity (wild type, 28.8 ± 0.8 m/s; Dync1li1N235Y/N235Y, 27.1 ± 1.2 m/s; p > 0.2). With respect to the C-fibers, we found no significant reduction of the peak-to-peak amplitude of the C-fiber SNAP (wild type, 391 ± 48 μV; Dync1li1N235Y/N235Y, 385 ± 61 μV, p > 0.4) and no significant reduction of the conduction velocity (wild type, 0.86 ± 0.06 m/s; Dync1li1N235Y/N235Y, 0.92 ± 0.04 m/s; p > 0.4).
Figure 5.
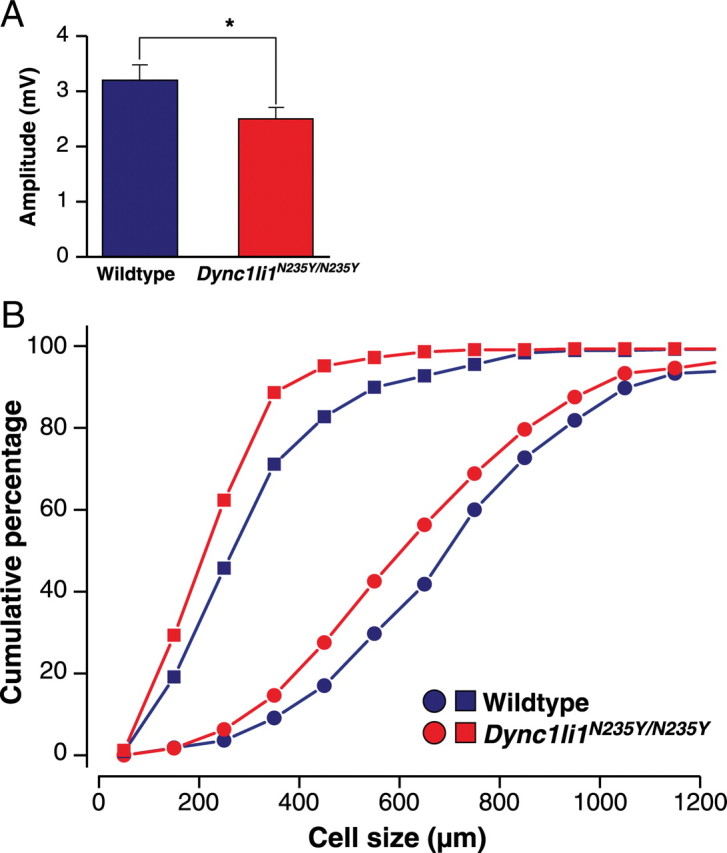
Neurophysiological and histological analysis of Dync1li1N235Y/N235Y mice. A, Dync1li1N235Y/N235 have mice a significant reduction of the peak-to-peak amplitude of A-fiber compound action potential in the saphenous nerve compared with wild-type littermates. *p < 0.05. B, QSum plots of the neuronal profile area distribution of neurofilament-immunoreactive neurons (giving rise to large myelinated A-fibers; round symbols) and of peripherin-immunoreactive cells (giving rise to C-fibers; square symbols) in lumbar dorsal root ganglia. The cell profile size in both populations is reduced in Dync1li1N235Y/N235Y mice compared with wild-type littermates.
To determine whether the sensory electrophysiological defect in A-fiber SNAP was reflected histologically, DRGs from the cervical and lumbar spinal cord were stained for NFH (a marker for myelinated fibers) and peripherin (a marker for thin myelinated and unmyelinated fibers) (supplemental Figure S5A, available at www.jneurosci.org as supplemental material). We found no differences in the NFH- and peripherin-immunoreactive cell profiles in cervical or lumbar DRGs of wild-type or mutant animals (supplemental Table S1, available at www.jneurosci.org as supplemental material). However, analysis of the size distribution of labeled neuronal profiles revealed a significant shift to the left (smaller neurons) in homozygous mutants compared with wild-type littermates in lumbar DRG. Changes in cervical DRGs were smaller which is compatible with a length-dependent effect as seen in many neuropathies (Fig. 5B; Table 1).
Table 1.
Cell profile size distribution in cervical and lumbar DRGs for NFH- and peripherin-positive neurons in wild-type and Dync1li1N235Y/N235Y mice
| Cervical |
Lumbar |
|||
|---|---|---|---|---|
| Wild type (n = 3) | Dync1li1N235Y/N235Y (n = 4) | Wild type (n = 3) | Dync1li1N235Y/N235Y (n = 4) | |
| Total number of NFH-positive profile areas measured | 201 | 286 | 165 | 240 |
| Mean NFH cell profile area | 739 ± 80 (579–833) | 676 ± 37 (568–739) | 773 ± 46 (714–864) | 680 ± 33 (595–734) |
| Total number of peripherin-positive profile areas measured | 338 | 496 | 319 | 430 |
| Mean peripherin cell profile area | 339 ± 32 (279–387) | 307 ± 6 (279–323) | 351 ± 31 (294–299) | 285 ± 18 (245–319) |
For mean ± SEM values, the values in parentheses are ranges.
Neurofilament: There was no significant difference (p > 0.1, Kolmogorov–Smirnov test) in the distribution of the profile areas of cervical NFH-immunoreactive DRG neurons between wild-type mice (719 ± 324 μm2, mean ± SD; ncells = 201, nanimals = 3) and Dync1li1N235Y/N235Y littermates (664 ± 275 μm2, mean ± SD; ncells = 286, nanimals = 4). Data not shown. The distribution of the profile areas of lumbar NFH-immunoreactive DRG neurons is significantly different (p < 0.025, Kolmogorov–Smirnov test) between wild-type mice (765 ± 296 μm2, mean ± SD; ncells = 165, nanimals = 3) and Dync1li1N235Y/N235Y littermates (686 ± 280 μm2, mean ± SD; ncells = 240, nanimals = 4) (Fig. 5B).
Peripherin: The distribution of the profile areas of cervical peripherin-immunoreactive DRG neurons is significantly different (p < 0.025, Kolmogorov–Smirnov test) between wild-type mice (335 ± 152 μm2, mean ± SD; ncells = 338, nanimals = 3) and Dync1li1N235Y/N235Y littermates (307 ± 165 μm2, mean ± SD; ncells = 496, nanimals = 4). Data not shown. The distribution of the profile areas of lumbar peripherin-immunoreactive DRG neurons is significantly different (p < 0.001, Kolmogorov–Smirnov test) between wild-type mice (353 ± 199 μm2, mean ± SD; ncells = 319, nanimals = 3) and Dync1li1N235Y/N235Y littermates (286 ± 151 μm2, mean ± SD; ncells = 430, nanimals = 4) (Fig. 4C).
We also stained cervical and lumbar DRGs for calcitonin gene-related peptide (CGRP, a marker of peptidergic nociceptors) and parvalbumin (stains prioprioceptors and mechanoreceptors) (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material). Similarly, we found no differences in the percentage of CGRP- and parvalbumin-immunoreactive cells in the cervical and lumbar DRGs of wild-type and mutant animals (supplemental Table S2, available at www.jneurosci.org as supplemental material), but there was a significant shift to smaller neurons in homozygous mutants compared with wild-type littermates (Table 2).
Table 2.
Cell profile size distribution in cervical and lumbar DRG for CGRP- and parvalbumin-positive neurons
| Cervical |
Lumbar |
|||
|---|---|---|---|---|
| Wild type (n = 3) | Dync1li1N235Y/N235Y (n = 4) | Wild type (n = 3) | Dync1li1N235Y/N235Y (n = 3) | |
| Total number of CGRP-positive profile areas measured | 239 | 315 | 255 | 389 |
| Mean CGRP cell profile area | 459 ± 69 (378–595) | 420 ± 27 (383–498) | 485 ± 26 (469–502) | 435 ± 26 (395–484) |
| Total number of parvalbumin-positive profile areas measured | 264 | 285 | 233 | 339 |
| Mean parvalbumin cell profile area | 508 ± 26 (477–559) | 477 ± 21 (416–509) | 474 ± 7 (418–578) | 378 ± 7 (364–388) |
For mean ± SEM values, the values in parentheses are ranges.
CGRP: The distribution of the profile areas of cervical CGRP-immunoreactive DRG neurons is significantly different (p < 0.005, Kolmogorov–Smirnov test) between wild-type mice (478 ± 276 μm2, mean ± SD; ncells = 239, nanimals = 3) and Dync1li1N235Y/N235Y littermates (421 ± 229 μm2, mean ± SD; ncells = 315, nanimals = 4). Data not shown. The distribution of the profile areas of lumbar CGRP-immunoreactive DRG neurons is significantly different (p < 0.005, Kolmogorov–Smirnov test) between wild-type mice (484 ± 280 μm2, mean ± SD; ncells = 255, nanimals = 3) and Dync1li1N235Y/N235Y littermates (432 ± 254 μm2, mean ± SD; ncells = 389, nanimals = 4). Data not shown.
Parvalbumin: The distribution of the profile areas of cervical parvalbumin-immunoreactive DRG neurons is significantly different (p < 0.001, Kolmogorov–Smirnov test) between wild-type mice (503 ± 260 μm2, mean ± SD; ncells = 264, nanimals = 3) and Dync1li1N235Y/N235Y littermates (468 ± 282 μm2, mean ± SD; ncells = 285, nanimals = 4). Data not shown. The distribution of the profile areas of lumbar parvalbumin-immunoreactive DRG neurons is significantly different (p < 0.001, Kolmogorov–Smirnov test) between wild-type mice (465 ± 276 μm2, mean ± SD; ncells = 233, nanimals = 3) and Dync1li1N235Y/N235Y littermates (376 ± 196 μm2, mean ± SD; ncells = 393, nanimals = 4) (Fig. 5D).
We found no loss of epidermal nerve fiber density between wild-type and mutant animals, and no reduction in the number of Merkel cells, whose survival depends on innervation by mechanoreceptive sensory neurons. Together these findings indicate that there is no distal degeneration of large myelinated or unmyelinated sensory neurons. To establish whether the Dync1li1N235Y mutation had any effect upon the saphenous nerve itself, a histological analysis was performed upon the saphenous nerves of 3 wild types and 3 homozygotes and we found no difference in average fiber size, fiber size distribution, g-ratio, or average axonal diameter (supplemental material, supplemental Fig. S5C–F, supplemental Table S3, available at www.jneurosci.org as supplemental material).
As Dync1li1N235Y/N235Y animals have a different gait from wild-type littermates, motor nerve conduction velocity was assessed wild-type and Dync1li1N235Y/N235Y littermates and we found no differences between genotypes (supplemental material, available at www.jneurosci.org). In aggregate these findings indicate that there is no loss of subpopulations of neurons, but a hypotrophy of all cell types and no dying back neuropathy of motor, or large and small diameter sensory neurons.
Dynein-controlled processes in Dync1li1N235Y/N235Y mouse embryonic fibroblasts
We studied the effect of the Dync1li1N235Y mutation on two important cellular processes known to involve cytoplasmic dynein: maintenance of Golgi integrity and endosomal movements. Golgi abnormalities have been reported in both Dync1h1Loa (Hafezparast et al., 2003) and in Drosophila with a mutation in the dynein light intermediate chain (Zheng et al., 2008; Palmer et al., 2009). We analyzed the effect of the Dync1li1N235Y mutation on reassembly of Golgi complex in MEFs, following exposure to nocodazole. We found Golgi reassembly progressed throughout the recovery time and was almost complete in wild-type cells within 50 min, thus there was no significant difference in the ratio of Golgi spots per unit area in wild-type cells compared with the corresponding untreated wild-type cells at the 50 min time point (p = 0.31) (Fig. 6A,B; supplemental Fig. S6, available at www.jneurosci.org as supplemental material). However, there was a defect in Golgi complex reassembly in Dync1li1N235Y/N235Y cells, when untreated and treated cells at the 50 min recovery time point were compared (p = 0.008). No statistically significant differences (p = 0.06) were observed in the ratio of Golgi spots per unit area in heterozygous Dync1li1N235Y/+ cells under these two conditions (supplemental Fig. S6, available at www.jneurosci.org as supplemental material).
Figure 6.

Golgi reassembly in wild-type and Dync1li1N235Y/N235Y MEFs. A, MEFs were treated with cold (4°C) for 20 min and nocodazole for 3 h, washed and then allowed to recover at 37°C for the times indicated. Golgi are shown in red, cytoskeleton in green (α-tubulin) and nuclei in blue (DAPI). The scale bar represents 30 μm. B, The total number of discrete spots and the total area of Golgi complex were measured to calculate the ratio of spots/total area per cell; 5 cells per genotype were assessed. In wild-type and heterozygous cells, after 50 min of recovery, the Golgi complex has reformed and is not significantly different from that in untreated cells. However, in the Dync1li1N235Y/N235Y homozygous mutant cells, the Golgi complex has a significant defect in recovery after 50 min compared with untreated cells and treated wild-type and heterozygous cells at the same time point.
As dynein is responsible for the trafficking and sorting of endosomes within the cell (Driskell et al., 2007) we investigated the effect of the Dync1li1N235Y mutation on endosomal dynamics. Wild-type and homozgygous MEFs were pulsed with Alexa Fluor 555-conjugated EGF, then fixed at set time points (Fig. 7A,B), and the number of EGF-positive vesicles in each cell was counted. When we compared the number of EGF-positive vesicles, as a measure of their trafficking to the lysosome for degradation, at 20 and 40 min time points, we observed significant differences between wild-type and homozygous MEFs (p = 0.0032 and p = 0.0007 respectively) which may indicate a slower rate of degradation.
Figure 7.
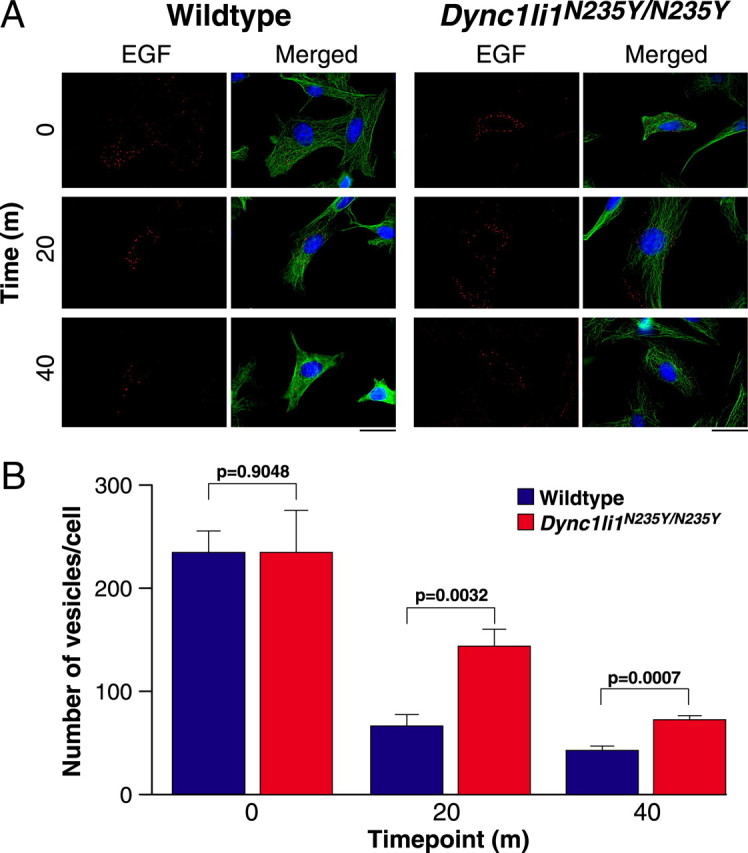
Endosomal trafficking chase of Alexa Fluor 555-conjugated EGF in wild-type and Dync1li1N235Y/N235Y MEFs. A, MEFs were pulsed with EGF for 10 min and chased at 37°C for the times indicated. EGF is shown in red, cytoskeleton (α-tubulin) in green and nuclei in blue (DAPI). Ten cells per genotype per time point were assessed. Scale bar, 30 μm. B, The number of EGF-positive vesicles per cell in wild-type and homozygous MEFs at 0, 20, and 40 min time points. At 0 min wild-type and Dync1li1N235Y/N235Y homozygous mutant cells have similar numbers of vesicles, however, after 20 and 40 min Dync1liN235Y/N235 homozygous mutant cells have significantly more vesicles remaining than wild-type cells.
Dynein subunit levels and interactions
We generated antibodies against wild-type DYNC1LI1, and the dynein intermediate chain protein 1, DYNC1I1 (supplemental material, supplemental Fig. S7, available at www.jneurosci.org as supplemental material). To investigate whether the DYNC1LI1 mutation affected its stability or expression, or its interactions with other components of the dynein complex, we assessed various cytoplasmic dynein subunit levels in brain from 12-month old wild-type and Dync1li1N235Y/N235Y mice and found no differences in individual protein levels, except for a significant increase in the levels of DYNLT3 in Dync1li1N235Y/N235Y mice compared with wild-type controls (p = 0.001; supplemental Fig. S8A,B, supplemental Table S4, available at www.jneurosci.org as supplemental material). Coimmunoprecipitation studies indicated DYNC1LI1N235Y was still bound within the complex, and also that there was a significant decrease in the amount of coprecipitating DYNLL in mutant samples when compared with wild type (p = 0.000005; supplemental material, supplemental Fig. S8C–E, supplemental Table S5, available at www.jneurosci.org as supplemental material). We found no differences in the autophagy marker LC3 II or in the cellular localization of DYNC1LI1 or its cargo RAB4 between wild-type and mutant animals in cortical neurons (supplemental material, supplemental Fig. S8F–K, available at www.jneurosci.org as supplemental material).
Discussion
We have identified and characterized a novel mouse line carrying an ENU-derived mutation causing an asparagine to tyrosine change in DYNC1LI1 at highly conserved residue 235. This is the first published mammalian mutant of a dynein light intermediate chain and allows us to assess aspects of dynein function that have not been highlighted thus far in the available mouse models of dynein dysfunction. Dync1li1N235Y/N235Y homozygote mice show increased anxiety-like behavior and altered gait in behavioral tests and have neurons with altered morphologies and electrophysiological properties. The Dync1li1N235Y mutation changes Golgi formation and endosomal trafficking, and the level expression and association of two other subunits of the dynein complex.
Our behavioral studies reveal that Dync1li1N235Y/N235Y homozygotes display greater levels of anxiety-like behavior than wild-type littermates on both the elevated plus maze and successive alleys task. Although these tasks can be influenced by changes in baseline levels of locomotor activity, the two groups did not differ in the distance traveled on the elevated plus maze, suggesting that the reduced exploration of the open arms by the Dync1li1N235Y/N235Y mice could not be explained by hypoactivity, and no hypoactivity was found in wheel running tests. Despite this, it is difficult to conclusively disassociate anxiety and locomotor activity as these two factors are closely linked (reduced locomotor activity potentially may result from increased anxiety as well as potentially acting as a confound in the measurement of anxiety) (Milner and Crabbe, 2008). Because of this difficulty, defecation was used as a non-locomotor measure of anxiety (DeFries et al., 1978). The increased defecation observed in Dync1li1N235Y/N235Y mice during behavioral testing supports the hypothesis that these animals display increased levels of anxiety.
Dync1li1N235Y/N235Y homozygotes have defects in cortical projection neuron morphology and the laminar positioning of this neuronal population, which indicates a migration defect affecting the cortex and prefrontal cortex. Alterations in neuronal morphology in the medial PFC have been found to be induced by chronic stress (Goldwater et al., 2009), as well as anxiety (Pascual and Zamora-León, 2007). Stress linked neuronal migration defects have also been observed in the hippocampus (Keays et al., 2007; Scobie et al., 2009) and basolateral amygdala (Kudo et al., 2007; Carim-Todd et al., 2009). A number of other mouse models with PFC-related behavioral deficits show defects in neuronal morphology or migration. However, linking behavior and migration defects is complex, for example migration defects observed in this study are similar to those in the Lmx1a (dreher) mutant mouse (overmigration of layer II neurons into layer I), but the two mouse lines show markedly different behavioral phenotypes—dreher mice show circling behavior, balance abnormalities, hyperactivity and deafness, none of which we see in Dync1li1N235Y/N235Y homozygotes) (Costa et al., 2001).
Dynein is involved in neuronal migration through its interactions with LIS1 (Tsai et al., 2007). Lis1 mutant mice have severe defects in neuronal migration in the brain, including in the same cortical regions disrupted in Dync1li1N235Y/N235Y homozygotes (Hirotsune et al., 1998). However, Lis1 mutant mice do not show changes in anxiety (Paylor et al., 1999) suggesting the interesting possibility that neuronal defects observed in Dync1li1N235Y/N235Y mice may be due to interactions with pathways other than those regulated by LIS1.
We note that importins, transported by cytoplasmic dynein (Perry and Fainzilber, 2009), are involved in synapse-to-nucleus signaling downstream of NMDA receptors (Thompson et al., 2004; Dieterich et al., 2008; Jeffrey et al., 2009), and NMDA receptors are important mediators of anxiety (Barkus et al., 2010). It is possible the Dync1li1N235Y mutation affects importin transport in response to NMDA receptor activation, thus altering anxiety in these mice.
Previous mouse studies have shown DYNC1H1 plays an important role in prioprioception and sensory neuronal function (Chen et al., 2007; Ilieva et al., 2008). Similarly the Dync1li1N235Y mutation alters the firing properties, neuron size and morphology of sensory nerves. Thus as sensory nerve deficits have been found now in mouse models with mutations in two different cytoplasmic dynein subunits, possibly DRG neurons are more reliant on dynein-mediated processes than other neuron populations. Cell type specificity may be due to different cargos transported by dynein within the different cells—studies in Drosophila show the dynein light intermediate chain interacts with a class of degenerin/epithelial sodium channels (Zheng et al., 2008) that in mammals appear to be found exclusively in DRG (Benson et al., 2002).
Mutation of the light intermediate chain in Drosophila cause dendritic and axonal defects in neurons, such as branching defects and a reduction of the dendritic arbor (Satoh et al., 2008; Zheng et al., 2008). However, these neuronal phenotypes differ from those found in the present study: in Drosophila mutations in the light intermediate chain cause a reduction in the length and number of dendrite branches (Satoh et al., 2008; Zheng et al., 2008), whereas the Dync1li1N235Y/N235Y mice have an increase in dendrite length in cortical neurons (with no changes in branching) and an increase in the number of dendrite branches in DRG neurons. These differences may arise because the reported Drosophila mutations cause loss of function, while the Dync1li1N235Y may be a novel gain of function.
The light intermediate chains play an essential role in endosomal dynamics in cells through their interactions with the RAB protein family (Bielli et al., 2001; Satoh et al., 2008; Zheng et al., 2008). Studies of the cytoplasmic dynein light intermediate chain in Drosophila suggest the cellular machinery controlling dendrite branching is transported in endosomes and Golgi outposts (Satoh et al., 2008; Zheng et al., 2008). Our data from the Dync1li1N235Y mutation support this view to some extent: we found the Dync1li1N235Y mutation reduces the trafficking of Golgi fragments and endosomes. However, while branching defects were observed in DRG neurons in Dync1li1N235Y/N235Y mice, we found no such defects in cortical neurons or in the peripheral nerves in the developing limb. In contrast, the cortical neurons of Dync1li1N235Y/N235Y mice showed other morphological abnormalities such as increased dendrite outgrowth. These results imply that dynein-mediated processes may mediate different aspects of neuronal morphology in different neuronal populations.
We also note that the Dync1li1N235Y mutation lies at the periphery of a known binding site for pericentrin. The primary function of pericentrin is thought to be the control of the mitotic spindle and the assembly of the centriole. Interestingly, a recent study of Cep120 (a centriole localized protein) hypothesized that defects in centriole formation may lead to alterations in neuronal migration (Mahjoub et al., 2010). Furthermore, there is also evidence to suggest that pericentrin may play a role in ciliogenesis (Miyoshi et al., 2006) and that ependymal cilia are responsible for directing CSF flow (Lang et al., 2006). Because abnormal CSF flow is linked to gait abnormalities [for example, in the human condition, normal pressure hydrocephalus (Gideon et al., 1994)], it is possible that the Dync1li1N235Y mutation causes abnormalities in pericentrin localization/function that affect cilia and CSF flow, and thus locomotion in this mouse. Future studies of the Dync1li1N235Y mutation should look at the interaction between the mutated light-intermediate chain 1 and pericentrin.
In Dync1li1N235Y/N235Y mice our quantified Western blot studies indicate increased levels of DYNLT3. We cannot tell whether this light chain lies within the complex or not from our westerns, but our immunoprecipitation experiments reveal that while the interaction of the intermediate chains in the core complex is maintained, (1) the increased DYNLT3 levels were not incorporated into the complete complex, and (2) there is a reduction in binding of the DYNLL light chains. Currently there is no evidence of a direct interaction between the dynein light intermediate and light chains. Instead the light chains are thought to interact with the intermediate chains within the core complex. It is possible the Dync1li1N235Y mutation alters the confirmation of the intermediate chains in the complex and this change affects the association of the light chains.
Our knowledge of the roles of the individual cytoplasmic dynein subunits comes from studies in nonmammalian organisms and in transfected cell lines, with the exception of the heavy chain, for which four mutant alleles are known in mouse: (1) a gene targeted knock-out; the point mutants (2) Legs at odd angles (Dync1h1Loa) and (3) Cramping 1 (Dync1h1Cra1); (4) a 9 bp coding deletion Sprawling (Dync1h1Swl) (Harada et al., 1998; Hafezparast et al., 2003; Chen et al., 2007). Homozygous knock-out (null) mice die early in embryogenesis and there is no reported phenotype for heterozygous knock-outs (Harada et al., 1998). In contrast Dync1h1Loa, Dync1h1Cra1 and Dync1h1Swl heterozygotes have major defects in the proprioceptive system and locomotor abnormalities (homozygotes die in embryogenesis/at birth) (Harada et al., 1998; Hafezparast et al., 2003; Kieran et al., 2005; Chen et al., 2007; Banks and Fisher, 2008; Ilieva et al., 2008; Dupuis et al., 2009). We note that the phenotype of the Dync1li1N235Y mouse strain is different from that of the four heavy chain mutants, particularly with respect to behavior. The heavy chain and Dync1li1N235Y mutants all give rise to defects in Golgi reassembly (Hafezparast et al., 2003) and EGF trafficking (Hafezparast et al., 2003; our unpublished data) both of which are known to be dynein-dependent processes. However, heterozygous Dync1h1Loa/+ mice develop an obvious gait abnormality (“low-based, reptilian”) easily visible by eye, plus significant defects in rotarod and grip-strength, none of which we find in the Dync1li1N235Y/N235Y mutants at comparable ages (Hafezparast et al., 2003). There are also significant reductions in axon numbers in the saphenous nerve of Dync1h1Loa/+ mice and more profound neurophysiological changes in nerve conduction properties compared with the Dync1li1N235Y/N235Y mutants (AlQatari et al., 2009). We note that we did not find significant alterations in peripheral nerve branching at E13.5, whereas this was see in both heterozygous and homozygous Dync1h1Loa mutants. A deeper comparison of the different subunit mutants at the cellular level would shed more light on the role of specific subunits, versus the entire complex, and here we clearly show phenotypes that have not been described before for in any of the heavy chain mutants. We note that Ori-McKenney, Vallee and colleagues have recently shown altered processivity of dynein with the Dync1h1Loa heavy chain mutation (Ori-McKenney et al., 2010), such a change is unlikely in the Dync1li1N235Y light intermediate chain mutation, which would presumably explain the somewhat different phenotypes of mutant mice, although this needs to be investigated.
Here we have used freely available mouse genetics resources to take a focused genotype-driven approach, working with a point mutation likely to be more informative than a knock-out, to analyzing one more of the 11 cytoplasmic dynein subunit proteins and have uncovered diverse roles at the level of the whole organism, nervous system and cell. The Dync1li1N235Y mouse has opened up new avenues for investigation of the roles of cytoplasmic dynein and its individual components, and gives new insight into the functioning and architecture of the mammalian nervous system.
Footnotes
This work was funded by the Wellcome Trust, UK Medical Research Council and Biotechnology and Biological Sciences Research Council, the ENDOCYTE Research and Training Network funded by the European Union, the Brain Research Trust, Israel Science Foundation and Legacy Foundation program in neurodegeneration research, and the National Alliance for Research on Schizophrenia and Depression. We are grateful to Christiana Ruhrberg for developmental analysis of embryos and thank Ray Young for graphics. The Dync1li1N235 mouse line is freely available via the European Mutant Mouse Archive.
The authors declare no competing financial interest.
References
- Achilli F, Bros-Facer V, Williams HP, Banks GT, AlQatari M, Chia R, Tucci V, Groves M, Nickols CD, Seburn KL, Kendall R, Cader MZ, Talbot K, van Minnen J, Burgess RW, Brandner S, Martin JE, Koltzenburg M, Greensmith L, Nolan PM, et al. A novel mouse model with a point mutation in glycyl-tRNA synthetase (Gars) has sensory and motor phenotypes and profoundly reduced enzyme activity in homozygotes. Dis Mod Mech. 2009;2:359–373. doi: 10.1242/dmm.002527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlQatari M, Vastani N, Bros-Facer V, Groves M, Greensmith L, Fisher EMC, Koltzenburg M. Mutations of the cytoplasmic dynein heavy chain gene Dync1h1 cause a severe sensory neuropathy. J Peripher Nerv Syst. 2009;14(S2):6. [Google Scholar]
- Banker G, Goslin K. Cambridge, MA: Massachusetts Institute of Technology; 1998. Culturing nerve cells. [Google Scholar]
- Banks GT, Fisher EM. Cytoplasmic dynein could be key to understanding neurodegeneration. Genome Biol. 2008;9:214. doi: 10.1186/gb-2008-9-3-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkus C, McHugh SB, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM. Hippocampal NMDA receptors and anxiety: at the interface between cognition and emotion. Eur J Pharmacol. 2010;626:49–56. doi: 10.1016/j.ejphar.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson CJ, Xie J, Wemmie JA, Price MP, Henss JM, Welsh MJ, Snyder PM. Heteromultimers of DEG/ENaC subunits form H+-gated channels in mouse sensory neurons 1. Proc Natl Acad Sci U S A. 2002;99:2338–2343. doi: 10.1073/pnas.032678399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielli A, Thörnqvist PO, Hendrick AG, Finn R, Fitzgerald K, McCaffrey MW. The small GTPase Rab4A interacts with the central region of cytoplasmic dynein light intermediate chain-1. Biochem Biophys Res Commun. 2001;281:1141–1153. doi: 10.1006/bbrc.2001.4468. [DOI] [PubMed] [Google Scholar]
- Carim-Todd L, Bath KG, Fulgenzi G, Yanpallewar S, Jing D, Barrick CA, Becker J, Buckley H, Dorsey SG, Lee FS, Tessarollo L. Endogenous truncated TrkB: T1 receptor regulates neuronal complexity and TrkB kinase receptor function in vivo. J Neurosci. 2009;29:678–685. doi: 10.1523/JNEUROSCI.5060-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic dynein heavy chain 1 gene. J Neurosci. 2007;27:14515–14524. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa C, Harding B, Copp AJ. Neuronal migration defects in the Dreher (Lmx1a) mutant mouse: role of disorders of the glial limiting membrane. Cereb Cortex. 2001;11:498–505. doi: 10.1093/cercor/11.6.498. [DOI] [PubMed] [Google Scholar]
- Deacon RM, Penny C, Rawlins JN. Effects of medial prefrontal cortex cytotoxic lesions in mice. Behav Brain Res. 2003;139:139–155. doi: 10.1016/s0166-4328(02)00225-5. [DOI] [PubMed] [Google Scholar]
- DeFries JC, Gervais MC, Thomas EA. Response to 30 generations of selection for open-field activity in laboratory mice. Behav Genet. 1978;8:3–13. doi: 10.1007/BF01067700. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Karpova A, Mikhaylova M, Zdobnova I, König I, Landwehr M, Kreutz M, Smalla KH, Richter K, Landgraf P, Reissner C, Boeckers TM, Zuschratter W, Spilker C, Seidenbecher CI, Garner CC, Gundelfinger ED, Kreutz MR. Caldendrin-Jacob: a protein liaison that couples NMDA receptor signalling to the nucleus. PLoS Biol. 2008;6:e34. doi: 10.1371/journal.pbio.0060034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driskell OJ, Mironov A, Allan VJ, Woodman PG. Dynein is required for receptor sorting and the morphogenesis of early endosomes. Nat Cell Biol. 2007;9:113–120. doi: 10.1038/ncb1525. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Fergani A, Braunstein KE, Eschbach J, Holl N, Rene F, Gonzalez De Aguilar JL, Zoerner B, Schwalenstocker B, Ludolph AC, Loeffler JP. Mice with a mutation in the dynein heavy chain 1 gene display sensory neuropathy but lack motor neuron disease. Exp Neurol. 2009;215:146–152. doi: 10.1016/j.expneurol.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Gennerich A, Carter AP, Reck-Peterson SL, Vale RD. Force-induced bidirectional stepping of cytoplasmic dynein. Cell. 2007;131:952–965. doi: 10.1016/j.cell.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon P, Ståhlberg F, Thomsen C, Gjerris F, Sørensen PS, Henriksen O. Cerebrospinal fluid flow and production in patients with normal pressure hydrocephalus studied by MRI. Neuroradiology. 1994;36:210–215. doi: 10.1007/BF00588133. [DOI] [PubMed] [Google Scholar]
- Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience. 2009;164:798–808. doi: 10.1016/j.neuroscience.2009.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J, Lo KW, Myers KR, Carr TM, Humsi MK, Rasoul BA, Segal RA, Pfister KK. A neuron-specific cytoplasmic dynein isoform preferentially transports TrkB signaling endosomes. J Cell Biol. 2008;181:1027–1039. doi: 10.1083/jcb.200803150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, Priestley JV, Averill S, King VR, Ball S, Peters J, Toda T, Yamamoto A, Hiraoka Y, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Hanz S, Perlson E, Willis D, Zheng JQ, Massarwa R, Huerta JJ, Koltzenburg M, Kohler M, van-Minnen J, Twiss JL, Fainzilber M. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron. 2003;40:1095–1104. doi: 10.1016/s0896-6273(03)00770-0. [DOI] [PubMed] [Google Scholar]
- Harada A, Takei Y, Kanai Y, Tanaka Y, Nonaka S, Hirokawa N. Golgi vesiculation and lysosome dispersion in cells lacking cytoplasmic dynein. J Cell Biol. 1998;141:51–59. doi: 10.1083/jcb.141.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- Höök P, Vallee RB. The dynein family at a glance. J Cell Sci. 2006;119:4369–4371. doi: 10.1242/jcs.03176. [DOI] [PubMed] [Google Scholar]
- Hughes SM, Vaughan KT, Herskovits JS, Vallee RB. Molecular analysis of a cytoplasmic dynein light intermediate chain reveals homology to a family of ATPases. J Cell Sci. 1995;108:17–24. doi: 10.1242/jcs.108.1.17. [DOI] [PubMed] [Google Scholar]
- Ilieva HS, Yamanaka K, Malkmus S, Kakinohana O, Yaksh T, Marsala M, Cleveland DW. Mutant dynein (Loa) triggers proprioceptive axon loss that extends survival only in the SOD1 ALS model with highest motor neuron death. Proc Natl Acad Sci U S A. 2008;105:12599–12604. doi: 10.1073/pnas.0805422105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey RA, Ch'ng TH, O'Dell TJ, Martin KC. Activity-dependent anchoring of importin alpha at the synapse involves regulated binding to the cytoplasmic tail of the NR1-1a subunit of the NMDA receptor. J Neurosci. 2009;29:15613–15620. doi: 10.1523/JNEUROSCI.3314-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keays DA, Tian G, Poirier K, Huang GJ, Siebold C, Cleak J, Oliver PL, Fray M, Harvey RJ, Molnár Z, Piñon MC, Dear N, Valdar W, Brown SD, Davies KE, Rawlins JN, Cowan NJ, Nolan P, Chelly J, Flint J. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell. 2007;128:45–57. doi: 10.1016/j.cell.2006.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieran D, Hafezparast M, Bohnert S, Dick JR, Martin J, Schiavo G, Fisher EM, Greensmith L. A mutation in dynein rescues axonal transport defects and extends the lifespan of ALS mice. J Cell Biol. 2005;169:561–567. doi: 10.1083/jcb.200501085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, Stucky CL, Lewin GR. Receptive properties of mouse sensory neurons innervating hairy skin. J Neurophysiol. 1997;78:1841–1850. doi: 10.1152/jn.1997.78.4.1841. [DOI] [PubMed] [Google Scholar]
- Kudo T, Fujii T, Ikegami S, Inokuchi K, Takayama Y, Ikehara Y, Nishihara S, Togayachi A, Takahashi S, Tachibana K, Yuasa S, Narimatsu H. Mice lacking alpha1,3-fucosyltransferase IX demonstrate disappearance of Lewis x structure in brain and increased anxiety-like behaviors. Glycobiology. 2007;17:1–9. doi: 10.1093/glycob/cwl047. [DOI] [PubMed] [Google Scholar]
- Kuta A, Deng W, Morsi El-Kadi A, Banks GT, Hafezparast M, Pfister KK, Fisher EM. Mouse cytoplasmic dynein intermediate chains: identification of new isoforms, alternative splicing and tissue distribution of transcripts. PLoS One. 2010;5:e11682. doi: 10.1371/journal.pone.0011682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang B, Song B, Davidson W, MacKenzie A, Smith N, McCaig CD, Harmar AJ, Shen S. Expression of the human PAC1 receptor leads to dose-dependent hydrocephalus-related abnormalities in mice. J Clin Invest. 2006;116:1924–1934. doi: 10.1172/JCI27597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JR, Holzbaur EL. Cytoplasmic dynein/dynactin function and dysfunction in motor neurons. Int J Dev Neurosci. 2006;24:103–111. doi: 10.1016/j.ijdevneu.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Mahjoub MR, Xie Z, Stearns T. Cep120 is asymmetrically localized to the daughter centriole and is essential for centriole assembly. J Cell Biol. 2010;191:331–346. doi: 10.1083/jcb.201003009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milner LC, Crabbe JC. Three murine anxiety models: results from multiple inbred strain comparisons. Genes Brain Behav. 2008;7:496–505. doi: 10.1111/j.1601-183X.2007.00385.x. [DOI] [PubMed] [Google Scholar]
- Miyoshi K, Onishi K, Asanuma M, Miyazaki I, Diaz-Corrales FJ, Ogawa N. Embryonic expression of pericentrin suggests universal roles in ciliogenesis. Dev Genes Evol. 2006;216:537–542. doi: 10.1007/s00427-006-0065-8. [DOI] [PubMed] [Google Scholar]
- Nolan PM, Peters J, Strivens M, Rogers D, Hagan J, Spurr N, Gray IC, Vizor L, Brooker D, Whitehill E, Washbourne R, Hough T, Greenaway S, Hewitt M, Liu X, McCormack S, Pickford K, Selley R, Wells C, Tymowska-Lalanne Z, et al. A systematic, genome-wide, phenotype-driven mutagenesis programme for gene function studies in the mouse. Nat Genet. 2000;25:440–443. doi: 10.1038/78140. [DOI] [PubMed] [Google Scholar]
- Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity 1. Nat Cell Biol. 2010;12:1228–1234. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer KJ, Hughes H, Stephens DJ. Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol Biol Cell. 2009;20:2885–2899. doi: 10.1091/mbc.E08-12-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual R, Zamora-León SP. Effects of neonatal maternal deprivation and postweaning environmental complexity on dendritic morphology of prefrontal pyramidal neurons in the rat. Acta Neurobiol Exp (Wars) 2007;67:471–479. doi: 10.55782/ane-2007-1663. [DOI] [PubMed] [Google Scholar]
- Paylor R, Hirotsune S, Gambello MJ, Yuva-Paylor L, Crawley JN, Wynshaw-Boris A. Impaired learning and motor behavior in heterozygous Pafah1b1 (Lis1) mutant mice. Learn Mem. 1999;6:521–537. doi: 10.1101/lm.6.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry RB, Fainzilber M. Nuclear transport factors in neuronal function. Semin Cell Dev Biol. 2009;20:600–606. doi: 10.1016/j.semcdb.2009.04.014. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Salata MW, Dillman JF, 3rd, Torre E, Lye RJ. Identification and developmental regulation of a neuron-specific subunit of cytoplasmic dynein. Mol Biol Cell. 1996a;7:331–343. doi: 10.1091/mbc.7.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Salata MW, Dillman JF, 3rd, Vaughan KT, Vallee RB, Torre E, Lye RJ. Differential expression and phosphorylation of the 74-kDa intermediate chains of cytoplasmic dynein in cultured neurons and glia. J Biol Chem. 1996b;271:1687–1694. doi: 10.1074/jbc.271.3.1687. [DOI] [PubMed] [Google Scholar]
- Pfister KK, Fisher EM, Gibbons IR, Hays TS, Holzbaur EL, McIntosh JR, Porter ME, Schroer TA, Vaughan KT, Witman GB, King SM, Vallee RB. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171:411–413. doi: 10.1083/jcb.200508078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister KK, Shah PR, Hummerich H, Russ A, Cotton J, Annuar AA, King SM, Fisher EM. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1. doi: 10.1371/journal.pgen.0020001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quwailid MM, Hugill A, Dear N, Vizor L, Wells S, Horner E, Fuller S, Weedon J, McMath H, Woodman P, Edwards D, Campbell D, Rodger S, Carey J, Roberts A, Glenister P, Lalanne Z, Parkinson N, Coghill EL, McKeone R, et al. A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm Genome. 2004;15:585–591. doi: 10.1007/s00335-004-2379-z. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Fisher EM, Brown SD, Peters J, Hunter AJ, Martin JE. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm Genome. 1997;8:711–713. doi: 10.1007/s003359900551. [DOI] [PubMed] [Google Scholar]
- Rogers DC, Peters J, Martin JE, Ball S, Nicholson SJ, Witherden AS, Hafezparast M, Latcham J, Robinson TL, Quilter CA, Fisher EM. SHIRPA, a protocol for behavioral assessment: validation for longitudinal study of neurological dysfunction in mice. Neurosci Lett. 2001;306:89–92. doi: 10.1016/s0304-3940(01)01885-7. [DOI] [PubMed] [Google Scholar]
- Salata MW, Dillman JF, 3rd, Lye RJ, Pfister KK. Growth factor regulation of cytoplasmic dynein intermediate chain subunit expression preceding neurite extension. J Neurosci Res. 2001;65:408–416. doi: 10.1002/jnr.1168. [DOI] [PubMed] [Google Scholar]
- Satoh D, Sato D, Tsuyama T, Saito M, Ohkura H, Rolls MM, Ishikawa F, Uemura T. Spatial control of branching within dendritic arbors by dynein-dependent transport of Rab5-endosomes. Nat Cell Biol. 2008;10:1164–1171. doi: 10.1038/ncb1776. [DOI] [PubMed] [Google Scholar]
- Scobie KN, Hall BJ, Wilke SA, Klemenhagen KC, Fujii-Kuriyama Y, Ghosh A, Hen R, Sahay A. Kruppel-like factor 9 is necessary for late-phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis 1. J Neurosci. 2009;29:9875–9887. doi: 10.1523/JNEUROSCI.2260-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KR, Otis KO, Chen DY, Zhao Y, O'Dell TJ, Martin KC. Synapse to nucleus signaling during long-term synaptic plasticity; a role for the classical active nuclear import pathway. Neuron. 2004;44:997–1009. doi: 10.1016/j.neuron.2004.11.025. [DOI] [PubMed] [Google Scholar]
- Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10:970–979. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- Tynan SH, Purohit A, Doxsey SJ, Vallee RB. Light intermediate chain 1 defines a functional subfraction of cytoplasmic dynein which binds to pericentrin. J Biol Chem. 2000;275:32763–32768. doi: 10.1074/jbc.M001536200. [DOI] [PubMed] [Google Scholar]
- Zhang J, Li S, Musa S, Zhou H, Xiang X. Dynein light intermediate chain in Aspergillus nidulans is essential for the interaction between heavy and intermediate chains. J Biol Chem. 2009;284:34760–34768. doi: 10.1074/jbc.M109.026872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Wildonger J, Ye B, Zhang Y, Kita A, Younger SH, Zimmerman S, Jan LY, Jan YN. Dynein is required for polarized dendritic transport and uniform microtubule orientation in axons. Nat Cell Biol. 2008;10:1172–1180. doi: 10.1038/ncb1777. [DOI] [PMC free article] [PubMed] [Google Scholar]