

Abstract
Assays for ATPases have been enabled for high-throughput screening (HTS) by employing firefly luciferase to detect the remaining ATP in the assay. However, for any enzyme assay, measurement of product formation is a more sensitive assay design. Recently, technologies that allow detection of the ADP product from ATPase reactions have been described using fluorescent methods of detection. We describe here the characterization of a bioluminescent assay that employs firefly luciferase in a coupled-enzyme assay format to enable detection of ADP levels from ATPase assays (ADP-Glo®, Promega Corp.). We determined the performance of the ADP-Glo assay in 1,536-well microtiter plates using the protein kinase Clk4 and a 1,352 member kinase focused combinatorial library. The ADP-Glo assay was compared to the Clk4 assay performed using a bioluminescence ATP-depletion format (Kinase-Glo™, Promega Corp). We performed this analysis using quantitative HTS (qHTS) where we determined potency values for all library members and identified ∼300 compounds with potencies ranging from as low as 50 nM to >10 µM, yielding a robust dataset for the comparison. Both assay formats showed high performance (Z′-factors ∼0.9) and showed a similar potency distribution for the actives. We conclude that the bioluminescence ADP detection assay system is a viable generic alternative to the widely used ATP-depletion assay for ATPases and discuss the advantages and disadvantages of both approaches.
INTRODUCTION
Accurate determination of enzymological parameters requires the measurement of the initial rate of reaction that in principle can be determined by following either the formation of product or depletion of substrate provided that <5% of the substrate is converted.1 In high-throughput screening (HTS), a signal:background (S:B) ratio of ≥2-fold is generally targeted. In some cases, such as the cleavage of a pro-fluorescent peptide substrate by a protease to form a fluorescent product, this S:B can be achieved or exceeded with a %conversion as low as ∼10%. However, HTS assays for classes of enzymes such as protein kinases have been performed using either product formation or substrate depletion, oftentimes at much higher conversion levels to achieve sufficient S:B ratios.
Protein kinases are one of the largest and most widely explored classes of ATPases. For protein kinases, a number of HTS assays have been developed aimed at measuring the products of the kinase reaction.2 For example, phosphorylated peptide has been measured with Molecular Devices IMAP technology3–7 using metal-chelated particles to immobilize fluorescently labeled phosphorylated peptides with fluorescent-polarization-based detection, Invitrogen’s Z-lyte™ technology8 that is based on FRET-labeled peptides, which are differentially protected from proteolysis upon phosphorylation, and scintillation proximity assays (SPA)9 to capture the radiolabeled phosphorylated peptide. Another antibody-free fluorescent assay system has been developed that employs a series of coupling enzymes to detect ADP (ADP Hunter™).10 More recently, generic systems for ATPases have been developed that detect the ADP product using an ADP-specific antibody labeled with a red-shifted fluorophore suitable for detection fluorescent-polarization (BellBrooks Lab, Transcreener™; Alexa633 is used λex 612 nm λem 670 nm). The red-shifted fluorescence limits spectroscopic interference by compounds and the ratiometric nature of fluorescent-polarization measurements aides in minimizing artifacts due to liquid handling.11,12 Further, this system shows good assay performance at low (∼10%) conversion levels.
To date, the application of bioluminescence to ATPase assays has relied on a substrate depletion format. In these assays, the ATP dependence of firefly luciferase is used to measure the remaining ATP concentration where the luminescence signal is inversely proportional to kinase activity.13–15 To provide a S:B of ∼2-fold, the substrate must be depleted by at least 50%. While operating enzyme assays under these high conversion conditions is not at all optimal for classical enzymological studies, this is acceptable for HTS as shifts in potency are typically <2-fold with %conversion <80%.16,17 Further, given that a typical HTS assay will show variability in potency determinations of ∼2- to 3-fold, shifts due to high conversions in the range of 50%–80% will not be easily discernable from the assay noise. Therefore, although ATP depletion involves performing the assay at high %conversions, the reduced apparent potency values obtained are not as much of a liability as one would expect and substrate depletion has thus become a popular choice to perform generic HTS assays for ATPases, particularly protein kinases.
In this manuscript, we focus on the comparison of 2 bioluminescence-based assays for protein kinases where one format used substrate depletion and the other used product formation. Both the substrate depletion assay (Kinase-Glo™) and the product formation assay (ADP-Glo®) employed the same variant of firefly luciferase to generate the assay signal (Ultra-Glo® luciferase).18 For the evaluation, we used the kinase Clk4, a kinase thought to modulate pre-mRNA splicing.19 To measure the performance and compare apparent potency values between the 2 assays, we screened a 1,352 member combinatorial library at 7 concentrations using a quantitative HTS approach20 (qHTS) to generate IC50 values for every library member from concentration–response curves (CRCs). The library was found to be highly active against Clk4 and hundreds of IC50 values were obtained that covered >3 orders of magnitude in potency. This provided a robust pharmacological dataset to compare the 2 assays. We found that both assays performed extremely well in a 1,536-well microtiter plate format and showed comparable potency values for active compounds. We conclude that the miniaturized bioluminescence assay for quantification of ADP levels is a practical HTS assay for ATPases.
MATERIALS AND METHODS
Reagents
Magnesium chloride and Tris (pH 7.5) were purchased from Quality Biological. Bovine serum albumin fraction V (BSA), ADP, ATP, Triton X-100, and positive control TG003 (cat # 300801-52-9) were obtained from Sigma-Aldrich. Certified A.C.S. grade dimethylsulfoxide (DMSO) was acquired from Fisher. Enzyme Clk4 (E.C. 2.7.12.1) was purchased from Invitrogen (cat # PV3839), while the RS repeat peptide substrate was acquired from AnaSpec (cat # 61722). Both the Kinase-Glo and the beta test kit ADP-Glo detection reagents were from Promega Corp., Madison, WI.
Compound library
The combinatorial library was obtained from Pharmacopeia (now owned by Ligand Pharmaceuticals). This library is a collection of 1,352 analogs containing scaffolds known to target protein kinases. For qHTS, the library was prepared as 7 inter-plate dilutions21 to establish a concentration–response series starting from 10 mM stock concentration that was diluted to 0.64 µM using serial dilutions of 1:5.
Control plates preparation
Sixteen point titrations (2-fold dilutions in 50/50 DMSO/ water) of 525 µM ATP and 450 µM ADP stock solutions were prepared. In a column-wise orientation, the ATP titrations were transferred to one 384-well plate (for Kinase-Glo assay) and the ADP titrations to another (for ADP-Glo assay). Seven microliters of the dilution series was then transferred to column 1 of the respective 1,536-well control plates using a CyBi-Well 384 liquid handling robot (CyBio, Jena, Germany). The positive control inhibitor, TG003,19 was dissolved in DMSO to a stock solution of 10 mM. A 16-point, 2-fold serial dilution series in DMSO was prepared and transferred to column 2 of each of the 1,536-well control plates using the same procedure. Seven microliters of DMSO and 10 mM of TG003 were also added to both control plates in columns 3 and 4, respectively.
qHTS Clk4 kinase-Glo assay
Two microliters/well of substrate–buffer solution (100 µM RS peptide, 1 µM ATP, 25 mM Tris pH7.5, 10 mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100 final concentration) was dispensed into 1,536-well assay plates (Greiner, solid white medium-binding plates) with Aurora Discovery BioRAPTR Flying Reagent Dispenser (FRD; Beckton-Dickenson, Franklin Lakes, NJ).22 Using a Kalypsys pin tool equipped with a 1,536-pin tool,23 23 nL of compound solution was transferred to the assay plate. One microliter/well Clk4-buffer solution (25 nM Clk4, 25 mM Tris pH 7.5, 10 mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100, final concentrations) was then added using the FRD yielding a total kinase reaction volume of 3 µL/well. After 4.5 h of room-temperature incubation, 3 uL Kinase-Glo reagent was added for a final assay volume of 6 uL/well. Luminescence was detected with the ViewLux plate reader (Perkin Elmer, Waltham, MA) after 5-min incubation using a 5-s exposure time and 2 times binning.
ADP-Glo assay optimization and reagent preparation
Prior to the kinetic reaction, detection reagents were prepared according to manufacturer’s protocol. In brief, 15 µL of ADP-Glo Enzyme I was added per milliliter of ADP-Glo Buffer I to form ADP-Glo Reagent I, while 8 uL of ADP-Glo Enzyme II was added per milliliter of Buffer II (made by adding 16 µL Substrate II to each 10 mL of Kinase-Glo MAX reagent) to form ADP-Glo Reagent II (we note that the current formulation of ADP-Glo does not require these assembly steps).
Linearity of the kit was also assessed using various concentrations of ADP to ATP mixture in the ADP-Glo kinase reaction buffer (100 µM RS peptide, 1× supplied ADP-Glo Buffer A, 2 mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100 final concentrations) as described in the manufacturer’s protocol. Twelve mixtures representing different %conversions were generated as follows: 100% conversion (1 µM ADP:0 µM ATP); 80% conversion (0.8 µM ADP:0.2 µM ATP); 60% conversion (0.6 µM ADP:0.4 µM ATP); 40% conversion (0.4 µM ADP:0.6 µM ATP); 20% conversion (0.2 µM ADP:0.8 µM ATP); 10% conversion (0.1 µM ADP:0.9 µM ATP); 5% conversion (0.05 µM ADP:0.95 µM ATP); 4% conversion (0.04 µM ADP:0.96 µM ATP); 3% conversion (0.03 µM ADP:0.97 µM ATP); 2% conversion (0.02 µM ADP:0.98 µM ATP); 1% conversion (0.01 µM ADP:0.99 µM ATP); and 0% conversion (0 µM ADP:1 µM ATP). The solutions were arranged in a column-wise order and 2.5 µL of each ADP/ATP mixture was transferred to a 1,536-well assay plate (Greiner, solid white medium-binding plates) using CyBi-Well 384 pipetter. Next, 2.5 µL of ADP-Glo Reagent I was added to each well using the FRD and incubated at room temperature. After 45 min, 5 µL ADP-Glo Reagent II was added to each well and luminescence signal was detected with the ViewLux plate reader (Perkin Elmer) after 30 min at ambient temperature incubation and using a 5-s exposure time with 2 times binning. Obtained results were plotted using GraphPad Prism to determine kit performance.
During optimization, 4 different enzyme concentrations (0, 10, 25, and 50 nM Clk4), using 3 different incubation times (30 min, 1 h, and 3 h) were tested to determine the appropriate enzyme concentration and incubation period.
qHTS Clk4 ADP-Glo assay
For the ADP-Glo kinase assay, 2 uL/well of substrate–buffer solution (100 µM RS peptide, 1 µM ATP, 1× ADP-Glo Buffer A, mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100, final concentrations) was dispensed into 1,536-well plates (Greiner, solid white, medium-binding assay plates) with the FRD. Twenty-three nanoliters of compound and control solutions were transferred to the assay plate (as described above) followed by 0.5 uL/well Clk4-buffer solution (25 nM Clk4, 1× ADP-Glo Buffer A, 10 mM MgCl2, 0.5 mM EGTA, 2.5 mM DTT, 0.01% Triton X-100 final concentration) dispensing for a total kinase reaction volume of 2.5 µL/well. After 1 h of room-temperature incubation, 2.5 uL ADP-Glo reagent was added and the reaction was incubated at room temperature for 45 min to stop the kinetic reaction and degrade residual ATP. The ADP product was then converted to ATP by adding 5 uL per well of ADP-Glo Reagent II to yield a total assay volume of 10 µL/well. Luminescence was detected after 30-min room-temperature incubation with the Perkin Elmer Viewlux.
Data analysis
The qHTS concentration–response data for each compound were fit using software developed in-house (http://www.ncgc.nih.gov/pub/openhts/curvefit/ for sample application) to the standard 4-parameter Hill equation. Our method for classification of CRC fitting has been described elsewhere.20,24 In brief, in qHTS CRC classes are categorized based on efficacy and quality of fit (r2). Complete CRCs that contain 2 asymptotes are designated as class 1, incomplete CRCs that have only one asymptote are class 2, poorly fit CRCs with activity at the highest tested concentrations are class 3, and inactive data that does not show any concentration–response relationship are class 4. High-quality fit curves (r2 > 0.9) with high (>80%) or lower (30%–80%) efficacies are subdivided into subclasses a or b, respectively.
Minimum significant ratio (MSR) values for individual runs were calculated using normalized, background-corrected data from the control titrations of each assay plate.24 The minimum significant ratio represents the smallest potency ratio between 2 determinations that is significant and poor reproducibility of potency values will be reflected by an increase in MSR values.25,26
Retrospective analysis to calculate false positive (FP), false negative (FN), and true positives (TP) was performed as previously described.24 In brief, compounds yielding CRCs assigned to the highest confidence CRC classes (classes 1 and 2) are considered active. CRCs with activities exceeding the threshold at the test concentration are counted toward the TP set, while those with activities within the statistical cutoff are considered FN, as these are associated with high-confidence CRCs but are not found as positive in the single concentration assay. Compounds showing no concentration–response relationship are classified as inactive (class 4). The majority of these compounds are counted as true negatives (TN) as their activities fall within the activity threshold limit. However, if an inactive compound’s activity is found to be beyond the threshold at a single concentration, it is counted as a FP. In the case when a compound’s data is successfully fit by the Hill equation but assigned a lower confidence CRC class (eg, class 3), it is counted as TP if the measured activity is beyond the activity threshold at the test concentration, but not counted toward the FN count if its activity falls below the threshold.
Identification of active scaffold clusters
Active compounds were defined by being associated with high-quality (classes 1 and 2) CRCs. These were examined for the presence of one of the 6 scaffolds that formed the basis of the combinatorial library. Compounds of lower confidence CRC classes (termed inconclusive) as well as inactive compounds were also examined for structural similarity to the active series and placed in 1 of the 6 scaffold clusters.
RESULTS
Principle of the Bioluminescent assays
The 2 bioluminescence assay systems evaluated here are shown in Figure 1. Measurement of ATP depletion employed the Kinase-Glo assay system where a firefly luciferase detection reagent containing d-luciferin and buffer components are added to detect the remaining ATP following the Clk4 kinase assay (Fig. 1A). The second system, ADP-Glo measures kinase activity by quantifying the amount of ADP formed after kinase reaction. Bioluminescent detection of ADP levels is achieved through the addition of 2 different detection reagents (Fig. 1B). First, a reagent that stops the protein kinase reaction and depletes the remaining ATP is added. Then a second reagent is added to stop ATP degradation. In addition, the second reagent also contains an enzyme that efficiently converts the ADP to ATP and the same firefly luciferase/d-luciferin components present in Kinase-Glo that generates a luminescent signal that is proportional to the concentration of ADP produced in the original kinase reaction. The 2 assay formats, therefore, show opposite luminescence signal changes in response to protein kinase inhibitors (Fig. 1C).
Fig. 1. .

Principle of bioluminescent substrate depletion and product formation assays. Illustrated are the assay principles for protein kinases (Clk4 was used here). (A) ATP-depletion format where a detection reagent containing firefly luciferase is added after the kinase reaction to detect the remaining ATP. (B) ADP-formation assay where following the kinase reaction a reagent that depletes the remaining ATP is added (Reagent I) followed by a reagent that converts the ADP formed in the protein kinase reaction to ATP as well as firefly luciferase that detects the ATP levels (Reagent II). (C) The luminescent response (presented as RLUs, or relative light units) is inversely proportional to the kinase activity in the ATP-depletion format (Kinase-Glo assay) while the luminescent response is directly proportional to kinase activity in the ADP-formation format (ADP-Glo assay).
Optimization of the ADP-Glo assay
Assay performance was evaluated at different time points (yielding different %conversions) to determine the optimum incubation time. Results of the ADP-Glo showed a leftward shift in IC50 value as the percent conversion is decreased (10% conversion) bringing the observed IC50 value closer to the published TG003 IC50 of 15 nM.19 Performance of the ADP assay was examined by calculating the Z′-factor and the ratio of the observed IC50 to the reported value. We observed that the assay run at 10% conversion, 30-min reaction showed a Z′-factor that ranged from 0.37 to 0.39. More optimal performance for the ADP-Glo assay was found for a 1 h kinase reaction, giving a Z′-factor >0.6, S:B = 11, and IC50 within 2-fold of the reported value. Under these conditions, the ADP/ATP titration curve showed an r2 = 0.96 ± 0.03. The Kinase-Glo assay, on the other hand, showed equivalent assay performance at ∼55% conversion after 4.5 h kinase reaction.
qHTS assay protocols and chemical library
The 2 final 1,536-well protocols used to compare the assay performance are shown in Table 1. The Kinase-Glo protocol contains fewer steps but generally requires a longer kinase reaction incubation time (step 5, Table 1; PubChem AID: 1770)27,28 to achieve higher substrate conversion for adequate S:B if equivalent enzyme levels are used. However, the ADP-Glo protocol (PubChem AID: 1771) requires 2 additional incubation steps to deplete the remaining ATP (step 6, Table 1) and generate the bioluminescent ATP response from the ADP product (step 8, Table 1). Therefore, at least for the Clk4 assay, there was a 2-fold overall savings in time for the ADP-Glo assay although 2 additional liquid handling and incubation steps are required for this protocol.
Table 1. .
Assay Protocols for 1536-Well Microtiter Plates Used for qHTS
| Step |
Kinase-Glo |
ADP-Glo |
||||
|---|---|---|---|---|---|---|
| Parameter | Value | Description | Parameter | Value | Description | |
| 1 | Reagent | 2 µL | ATP/peptide | Reagent | 2 µL | ATP/peptide |
| 2 | Library | 23 nL | 0.5 nM–46 µM | Library | 23 nL | 0.6 nM–55.2 µM |
| 3 | Controls | 23 nL | TG003 | Controls | 23 nL | TG003 |
| 4 | Reagent | 1 µL | Clk4 | Reagent | 0.5 µL | Clk4 |
| 5 | Time | 4.5 h | r.t. incubation | Time | 1 h | r.t. incubation |
| 6 | Reagent | 3 µL | Kinase-Glo | Reagent | 2.5 µL | Deplete ATP |
| 7 | Read | 2 s | ViewLux | Time | 45 min | r.t. incubation |
| 8 | Reagent | 5 µL | ADP ◊ ATP/Luc | |||
| 9 | Time | 30 min | r.t. incubation | |||
| 10 | Read | 2 s | ViewLux | |||
| Step | Notes | Notes |
|---|---|---|
| 1 | 100 µM RS peptide 1 µM ATP (final) concentration in buffer: FRD dispense | 100 µM RS peptide 1 µM ATP (final) concentration in buffer: FRD dispense |
| 2 | Pin-tool transfer compound library for a (final) range of 46 µM–0.5 nM | Pin-tool transfer compound library for a (final) range of 55.2 µM–0.6 nM |
| 3 | Pin-tool transfer of controls | Pin-tool transfer of controls |
| 4 | Clk4 at 25 nM final, FRD dispense | Clk4 at 25 nM final, FRD dispense |
| 5 | Kinase reaction, room-temperature incubation | Kinase reaction, room-temperature incubation |
| 6 | Detection Kinase-Glo | ADP-Glo Reagent I |
| 7 | Perkin Elmer Viewlux, clear filter | ATP depletion, room-temperature incubation |
| 8 | ADP-Glo Reagent II | |
| 9 | Room-temperature incubation | |
| 10 | Perkin Elmer Viewlux, clear filter |
The relative efficiencies of these protocols will vary with the type of enzyme. For example, the differences in time savings noted above could be lessened by using higher enzyme in the Kinase-Glo reaction provided that linear conditions are maintained and the cost of enzyme preparation is not prohibitive. In general, the differences in time savings between the 2 protocols will be less when faster turnover enzymes are used at equivalent enzyme concentrations because the enzyme incubation time step is minimized relative to the rest of the assay procedure. The K m for ATP affects the Kinase-Glo optimization more so than ADP-Glo. This is because the ADP-Glo assay can be used over a wide range of ATP concentrations (the assay is linear at up to 1 mM ATP)29 while the choice of ATP-depletion detection reagent depends on the ATP concentration in the assay (Kinase-Glo, Kinase-Glo Plus, and Kinase-Glo Max are available for low, intermediate, and high ATP ranges, respectively).30 Therefore, variation of ATP concentration for the purpose of optimization or mechanism of action studies will be more readily performed in the ADP-Glo assay format.
To evaluate the performance of each assay in HTS, we used the 1,536-well protocols to screen a combinatorial library focused against protein kinases (Fig. 2). This library contains substituted 4-methyl-6-phenylquinazoline (1), 4-methyl-2-phenylquinazoline (2), phenylpyrimidines (3–5), and pteridinone (6) scaffolds known to target protein kinases.31,32 The 1,352 member library contains several hundred analogs around each scaffold to provide dense structure–activity relationships (SAR) for active scaffolds.27,33 In addition, we had previously determined that a quinazoline inhibitor was active at Clk434 and therefore we suspected that this library would provide a large set of active compounds to compare potency values between the 2 assay formats.
Fig. 2. .
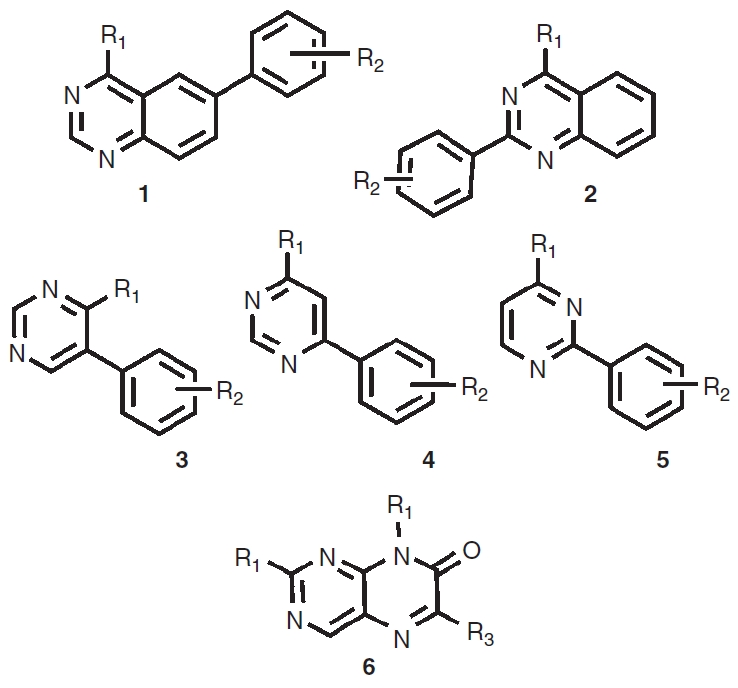
Scaffolds in the kinase-targeted combinatorial library.
Performance of the kinase-Glo and ATP-Glo
The performance of the 2 assays is summarized in Figures 3 and 4. A total of 9,436 samples were assayed in the Kinase-Glo qHTS, while the same samples were assayed in duplicate using ADP-Glo. In the qHTS, both assays showed excellent performance and Z′-factors of 0.86 were observed for both assays. The S:B was slightly higher for ADP-Glo (5-fold) than Kinase-Glo (3-fold) but the ADP-Glo assay was performed at an estimated 30% ± 5% conversion vs. 50% for Kinase-Glo. The reproducibility of potency values was estimated from the intra-plate titrations of the TG003 control where we found MSR values of 2.3 and 3.0 for ADP-Glo and Kinase-Glo assays, respectively.
Fig. 3. .
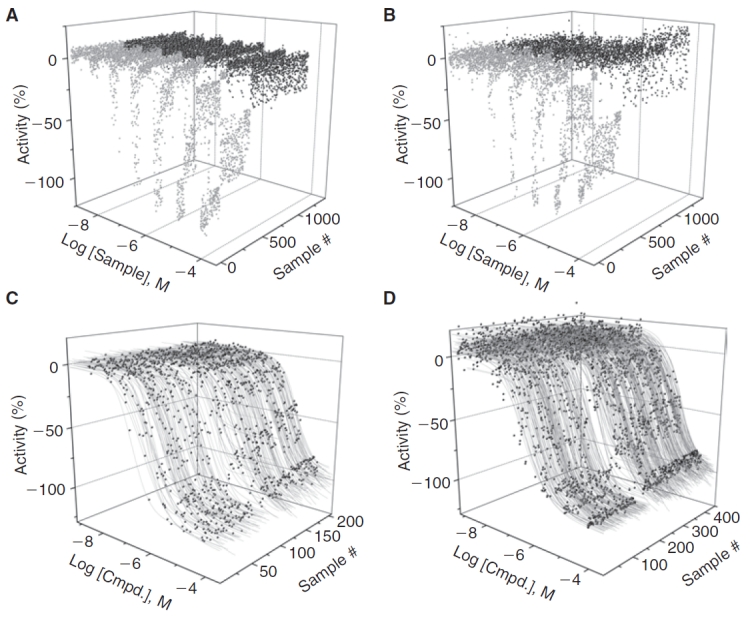
Performance of the two 1,536-well Clk4 kinase assays. Kinase-Glo (A) and ADP-Glo (B) quantitative high-throughput screening (qHTS) experiments. Light gray represents the compounds that fit to active concentration–response curve (CRC) classes and dark gray is data classified as inactive. Data for one of the ADP-Glo qHTS experiments is shown. (C). Data and curve-fits for the highest confidence CRCs classes obtained in the Kinase-Glo qHTS. (D). Data and curve-fits for the highest confidence CRCs classes obtained in the ADP-Glo qHTS, both replicates are shown.
Fig. 4. .

ADP-Glo and Kinase-Glo show excellent agreement in potency values. Comparison of potency values for run 1 (A) and run 2 (B) of the ADP-Glo quantitative high-throughput screening (qHTS) against the Kinase-Glo qHTS. Black solid circles represent potencies derived from high-quality concentration–response curve (CRC) and open squares represent potencies derived from lower confidence CRCs.
Both assays identified essentially the same inhibitors and we found good reproducibility for the potency values obtained (Fig. 4). We found nearly equal numbers of these inhibitors having high-quality CRCs (classes 1a, 1b, 2a, and 2b, showing >60% inhibition) in both assays with 332, 334, and 353 for Kinase-Glo, ADP-Glo run1, and ADP-Glo run2, respectively. The potency distribution of these actives was also similar (Table 2). Both assays were able to identify compounds with potencies <0.1 µM.
Table 2. .
Distribution of Potency Values in the Two Bioluminescent Assays
| Assay | <0.1 µM | 0.1–1 µM | 1–10 µM | >10 µM |
|---|---|---|---|---|
| Kinase-Glo | 3 | 34 | 109 | 186 |
| ADP-Glo (1) | 2 | 35 | 99 | 199 |
| ADP-Glo (2) | 1 | 37 | 114 | 201 |
Retrospective analysis of the qHTS data was performed to determine how each of the bioluminescence assays would perform at a single tested compound concentration. For this purpose, we used the CRC information to define the true positive (TP, confirmed) set as described in Shukla et al. 24 and then asked how many of these were found or not found (FN) at a 15 µM testing concentration. Both assays found all the TPs at this concentration (100% sensitivity). Of the hits identified at only the 15 µM concentration (defined as showing a %activity >3 SD of the assay noise), 97% were confirmed as TP in the Kinase-Glo assay and 99% were confirmed as TP in the ADP-Glo assay (FP rates of 3% and 1%, respectively). The higher confirmation rate for the ADP-Glo assay is likely due to the positive luminescence response associated with the bioluminescent detection of ADP in this assay.
Bioluminescence from both assays arises due to the presence of an optimized variant of firefly luciferase from Photuris pennsylvanica (Ultra-Glo™) in the assay detection reagent. We have previously determined the SAR of firefly luciferase inhibitors and determined that formulations of Ultra-Glo are more resistant to inhibition than formulations containing Photinus Pyralis firefly luciferase.18,35 Consistent with the robust nature of the Ultra-Glo formulations and the SAR of firefly luciferase inhibitors, we did not observe any interference with Kinase-Glo in the presence of ATP alone for any of the compounds in the combinatorial library demonstrating that the Ultra-Glo luciferase used in both assays is not affected by these classes of compounds.
The ADP-Glo assay utilizes 2 additional reagents to enable detection of the ADP product. The first contains an ATP-depleting reagent and the second reagent, used for final detection, contains an enzyme capable of converting ADP ◊ ATP in addition to Ultra-Glo luciferase. Although the composition of these reagents is propriety information, we can speculate that the ADP ◊ ATP converting enzyme is pyruvate kinase (PyK), as this has been used for other coupled kinase assays.36,37 Through screening multiple species orthologs of PyK at our center, we have observed that this enzyme does not readily yield inhibitors from typical small molecule screening collections (unpublished observation), although specific activation is observed due to the allosteric properties of this enzyme. In the case of activation of PyK in the detection reagent, the ADP will only be more efficiently converted to ATP that should not affect the final results. The composition of the ATP-depleting reagent is not known, but full inhibition of this step by a compound that is also active at the targeted kinase would yield a false negative result in the ADP-Glo assay. At least for the library screened here, the near equivalence of inhibitors identified in both assays suggests that these types of library compounds do not interfere with either the ATP-depletion step or ADP ◊ ATP converting enzyme present in ADP-Glo.
SAR observed for the Clk4 inhibitors
The distribution of activity and potency for the inhibitors identified here in the bioluminescent assays is summarized in Figure 5. The quinazoline-containing scaffolds 1 and 2 were represented most prominently among the inhibitors (Fig. 5A) while the phenylpyrimidine scaffolds 3–5 showed a reduced number of actives with the 5-phenylpyrimidine (3) showing the most actives. As well, the potency distribution of inhibitors (Fig. 5B) showed that more than half the compounds associated with scaffold 1 showed IC50s between 0.1 and 10 µM (with a few at <0.1 µM) while the cluster associated scaffold 3, which contained a few compounds associated with potencies <0.1 µM, had mainly compounds with IC50s >1 µM. The 2-phenylpyrimidines (5) showed the fewest number of actives for the phenylpyrimidine class and the observed potencies were mainly in the 1–10 µM range while 6-phenylpyrimidines (4) showed an intermediate values in both quantity and potency of inhibitors. Overall, the pteridinone scaffold (6) showed the lowest percentage of inhibitors among any of the 6 scaffolds and most compounds containing this scaffold showed weak potency values or were inactive. A representative compound (PubChem CID: 3232621) was resynthesized from the scaffold 1 cluster series and we confirmed an IC50 of ∼80 nM in both bioluminescent assays (Fig. 5C). The opposite nature of the luminescent response observed for each assay (Fig. 5C) provided a good assay pair to confirm genuine Clk4 inhibition.
Fig. 5. .

Structure–activity relationships (SAR) of Clk4 inhibitors. Data from the duplicate quantitative high-throughput screening (qHTS) experiments in the ADP-Glo assay is shown. (A) The percentage of compounds in active, inconclusive, or inactive categories for each scaffold cluster. (B) Potency distribution for each scaffold cluster. (C) Raw relative light units (RLUs) for the confirmation data of a representative 4-phenyl quinazoline (scaffold 1; CID: 3232621) is shown for the Kinase-Glo assay (solid circles) and the ADP-Glo assay (open circles).
SUMMARY
Both the ATP-depletion and ADP-formation bioluminescence assays showed excellent and near-equivalent performance in terms of Z′-factor, potency values, sensitivity, and selectivity. The ATP-depletion assay requires fewer steps than the ADP-formation assay but these additional steps did not preclude miniaturization to 1,536-well assay volumes. The ability to use low conversion levels in ADP-Glo assays should be useful, particularly for ATPases where low concentrations of enzyme are needed to mitigate cost or where the turnover number is low. The directly proportional nature of the luminescence response to ADP concentration in the ADP-Glo assay should make this a good choice as either a primary or confirmatory assay for ATPases. Finally, the opposite bioluminescent responses for ATP depletion and ADP formation provide an excellent assay pair for orthogonal validation of ATPase inhibitors.
ACKNOWLEDGMENTS
This research was supported by the Molecular Libraries Initiative of the NIH Roadmap for Medical Research and the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. The authors wish to acknowledge Paul Shinn for help with compound management.
ABBREVIATIONS:
- ADP
adenosine diphosphate
- AID
assay identifier used in PubChem
- ATP
adenosine triphosphate
- CID
compound identifier used in PubChem
- Clk4
CDC-like kinase 4
- CRC
concentration-response curve
- DMSO
dimethyl sulfoxide
- FN
false negative
- FP
false positive
- FRD
Flying Reagent Dispenser
- HTS
high-throughput screening
- IC50
the half maximal inhibitory concentration
- IMAP
immobilized metal assay for phosphochemicals
- MSR
minimum significance ration
- Pyk
pyruvate kinase
- qHTS
quantitative HTS
- SAR
structure-activity relationships
- S:B
signal to background ratio
- TP
true positive.
Contributor Information
Cordelle Tanega, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland..
Min Shen, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland..
Bryan T. Mott, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland.
Craig J. Thomas, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland.
Ryan MacArthur, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland..
James Inglese, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland..
Douglas S. Auld, NIH Chemical Genomics Center, National Institutes of Health, Bethesda, Maryland.
AUTHOR DISCLOSURE STATEMENT
C.T., M.S., B.T.M., C.J.T., R.M., J.I., and D.S.A. are employees of the National Institutes of Health.
REFERENCES
- 1.Segel HI. Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady State Enzyme Systems. John Wiley and Sons; New York: 1975. [Google Scholar]
- 2.Eglen RM, Reisine T. The current status of drug discovery against the human kinome. Assay Drug Dev Technol. 2009;7:22–43. doi: 10.1089/adt.2008.164. [DOI] [PubMed] [Google Scholar]
- 3.Beasley JR, Dunn DA, Walker TL, Parlato SM, Lehrach JM, Auld DS. Evaluation of compound interference in immobilized metal ion affinity-based fluorescence polarization detection with a four million member compound collection. Assay Drug Dev Technol. 2003;1:455–459. doi: 10.1089/154065803322163768. [DOI] [PubMed] [Google Scholar]
- 4.Gaudet EA, Huang KS, Zhang Y, Huang W, Mark D, Sportsman JR. A homogeneous fluorescence polarization assay adaptable for a range of protein serine/threonine and tyrosine kinases. J Biomol Screen. 2003;8:164–175. doi: 10.1177/1087057103252309. [DOI] [PubMed] [Google Scholar]
- 5.Loomans EE, van Doornmalen AM, Wat JW, Zaman GJ. High-throughput screening with immobilized metal ion affinity-based fluorescence polarization detection, a homogeneous assay for protein kinases. Assay Drug Dev Technol. 2003;1:445–453. doi: 10.1089/154065803322163759. [DOI] [PubMed] [Google Scholar]
- 6.Sportsman JR, Gaudet EA, Boge A. Immobilized metal ion affinity-based fluorescence polarization (IMAP): advances in kinase screening. Assay Drug Dev Technol. 2004;2:205–214. doi: 10.1089/154065804323056549. [DOI] [PubMed] [Google Scholar]
- 7.Turek-Etienne TC, Kober TP, Stafford JM, Bryant RW. Development of a fluorescence polarization AKT serine/threonine kinase assay using an immobilized metal ion affinity-based technology. Assay Drug Dev Technol. 2003;1:545–553. doi: 10.1089/154065803322302808. [DOI] [PubMed] [Google Scholar]
- 8.Rodems SM, Hamman BD, Lin C, Zhao J, Shah S, Heidary D, et al. A FRET-based assay platform for ultra-high density drug screening of protein kinases and phosphatases. Assay Drug Dev Technol. 2002;1:9–19. doi: 10.1089/154065802761001266. [DOI] [PubMed] [Google Scholar]
- 9.Glickman JF, Schmid A, Ferrand S. Scintillation proximity assays in high-throughput screening. Assay Drug Dev Technol. 2008;6:433–455. doi: 10.1089/adt.2008.135. [DOI] [PubMed] [Google Scholar]
- 10.Charter NW, Kauffman L, Singh R, Eglen RM. A generic, homogenous method for measuring kinase and inhibitor activity via adenosine 5′-diphosphate accumulation. J Biomol Screen. 2006;11:390–399. doi: 10.1177/1087057106286829. [DOI] [PubMed] [Google Scholar]
- 11.Huss KL, Blonigen PE, Campbell RM. Development of a Transcreener kinase assay for protein kinase A and demonstration of concordance of data with a filter-binding assay format. J Biomol Screen. 2007;12:578–584. doi: 10.1177/1087057107300221. [DOI] [PubMed] [Google Scholar]
- 12.Kleman-Leyer KM, Klink TA, Kopp AL, Westermeyer TA, Koeff MD, Larson BR, et al. Characterization and optimization of a red-shifted fluorescence polarization ADP detection assay. Assay Drug Dev Technol. 2009;7:56–67. doi: 10.1089/adt.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan F, Wood KV. Bioluminescent assays for high-throughput screening. Assay Drug Dev Technol. 2007;5:127–136. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- 14.Schröter T, Minond D, Weiser A, Dao C, Habel J, Spicer T, et al. Comparison of miniaturized time-resolved fluorescence resonance energy transfer and enzyme-coupled luciferase high-throughput screening assays to discover inhibitors of Rho-kinase II (ROCK-II) J Biomol Screen. 2008;13:17–28. doi: 10.1177/1087057107310806. [DOI] [PubMed] [Google Scholar]
- 15.Singh P, Harden BJ, Lillywhite BJ, Broad PM. Identification of kinase inhibitors by an ATP depletion method. Assay Drug Dev Technol. 2004;2:161–169. doi: 10.1089/154065804323056503. [DOI] [PubMed] [Google Scholar]
- 16.Wu G, Yuan Y, Hodge CN. Determining appropriate substrate conversion for enzymatic assays in high-throughput screening. J Biomol Screen. 2003;8:694–700. doi: 10.1177/1087057103260050. [DOI] [PubMed] [Google Scholar]
- 17.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, et al. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 18.Auld DS, Zhang YQ, Southall NT, Rai G, Landsman M, Maclure J, et al. A basis for reduced chemical library inhibition of firefly luciferase obtained from directed evolution. J Med Chem. 2009;52:1450–1458. doi: 10.1021/jm8014525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muraki M, Ohkawara B, Hosoya T, Onogi H, Koizumi J, Koizumi T, et al. Manipulation of alternative splicing by a newly developed inhibitor of Clks. J Biol Chem. 2004;279:24246–24254. doi: 10.1074/jbc.M314298200. [DOI] [PubMed] [Google Scholar]
- 20.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasgar A, Shinn P, Jadhav A, Auld DS, Michael S, Zheng W, et al. Compound management for quantitative high-throughput screening. J Assoc Lab Automation. 2008;13:79–89. doi: 10.1016/j.jala.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niles WD, Coassin PJ. Piezo- and solenoid valve-based liquid dispensing for miniaturized assays. Assay Drug Dev Technol. 2005;3:189–202. doi: 10.1089/adt.2005.3.189. [DOI] [PubMed] [Google Scholar]
- 23.Cleveland PH, Koutz PJ. Nanoliter dispensing for uHTS using pin tools. Assay Drug Dev Technol. 2005;3:213–225. doi: 10.1089/adt.2005.3.213. [DOI] [PubMed] [Google Scholar]
- 24.Shukla SJ, Nguyen DT, Macarthur R, Simeonov A, Frazee WJ, Hallis TM, et al. Identification of pregnane X receptor ligands using time-resolved fluorescence resonance energy transfer and quantitative high-throughput screening. Assay Drug Dev Technol. 2009;7:143–169. doi: 10.1089/adt.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eastwood BJ, Farmen MW, Iversen PW, Craft TJ, Smallwood JK, Garbison KE, et al. The minimum significant ratio: a statistical parameter to characterize the reproducibility of potency estimates from concentration-response assays and estimation by replicate-experiment studies. J Biomol Screen. 2006;11:253–261. doi: 10.1177/1087057105285611. [DOI] [PubMed] [Google Scholar]
- 26.MacArthur R, Leister W, Veith H, Shinn P, Southall N, Austin CP, et al. Monitoring compound integrity with cytochrome P450 assays and qHTS. J Biomol Screen. 2009;14:538–546. doi: 10.1177/1087057109336954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bolton EE, Wang Y, Thiessen PA, Bryant SH. PubChem: integrated platform of small molecules and biological activities. Annu Rep Comput Chem. 2008;4:217–241. [Google Scholar]
- 28. http://pubchem.ncbi.nlm.nih.gov/
- 29. http://www.promega.com/tbs/tm313/tm313.pdf
- 30. http://www.promega.com/tbs/tb372/tb372.pdf
- 31.Chao Q, Deng L, Shih H, Leoni LM, Genini D, Carson DA, et al. Substituted isoquinolines and as potential antiinflammatory agents: Synthesis and biological evaluation of inhibitors of tumor necrosis factor alpha. J Med Chem. 1999;42:3860–3873. doi: 10.1021/jm9805900. [DOI] [PubMed] [Google Scholar]
- 32.Fry DW, Kraker AJ, McMichael A, Ambroso LA, Nelson JM, Leopold WR, et al. A specific inhibitor of the epidermal growth factor receptor tyrosine kinase. Science. 1994;265:1093–1095. doi: 10.1126/science.8066447. [DOI] [PubMed] [Google Scholar]
- 33. http://www.ncbi.nlm.nih.gov/sites/entrez?db=pcsubstance&term=PCOP
- 34.Mott BT, Tanega C, Shen M, Maloney DJ, Shinn P, Leister W, et al. Evaluation of substituted 6-arylquinazolin-4-amines as potent and selective inhibitors of cdc2-like kinases (Clk) Bioorg Med Chem Lett. 2009;19:6700–6705. doi: 10.1016/j.bmcl.2009.09.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auld DS, Southall NT, Jadhav A, Johnson RL, Diller DJ, Simeonov A, et al. Characterization of chemical libraries for luciferase inhibitory activity. J Med Chem. 2008;51:2372–2386. doi: 10.1021/jm701302v. [DOI] [PubMed] [Google Scholar]
- 36.Ching TM. A sensitive and simple assay of starch synthase activity with pyruvate kinase and luciferase. Anal Biochem. 1982;122:139–143. doi: 10.1016/0003-2697(82)90262-7. [DOI] [PubMed] [Google Scholar]
- 37.Jenkins WT. The pyruvate kinase-coupled assay for ATPases: a critical analysis. Anal Biochem. 1991;194:136–139. doi: 10.1016/0003-2697(91)90160-u. [DOI] [PubMed] [Google Scholar]