

Abstract
Certain cancers may be auxotrophic for a particular amino acid and amino acid deprivation is one method to treat these tumors. Arginine deprivation is a novel approach to target tumors which lack argininosuccinate synthetase (ASS) expression. ASS is a key enzyme which converts citrulline to arginine. Tumors which usually do not express ASS include melanoma, hepatocellular carcinoma, some mesotheliomas and some renal cell cancers. Arginine can be degraded by several enzymes including arginine deiminase (ADI). Although ADI is a microbial enzyme from mycoplasma, it has high affinity to arginine and catalyzes arginine to citrulline and ammonia. Citrulline can be recycled back to arginine in normal cells which express ASS, whereas ASS(−) tumor cells cannot. A pegylated form of ADI (ADI-PEG20) has been formulated and has shown in vitro and in vivo activity against melanoma and hepatocellular carcinoma. ADI-PEG20 induces apoptosis in melanoma cell lines. However, arginine deprivation can also induce ASS expression in certain melanoma cell lines which can lead to in-vitro drug resistance. Phase I and II clinical trials with ADI-PEG20 have been conducted in patients with melanoma and hepatocellular carcinoma and antitumor activity has been demonstrated in both cancers. This article reviews our laboratory and clinical experience as well as others with ADI-PEG20 as an antineoplastic agent. Future direction in utilizing this agent is also discussed.
Keywords: Arginine, melanoma, arginine deiminase, hepatocellular carcinoma
INTRODUCTION
One type of targeted therapy for human cancers involves the depletion of key amino acids needed by tumors to survive. The best example of this is asparaginase which is used to treat acute lymphoblastic leukemia (ALL), a common type of leukemia in children and young adults [1, 2]. This anticancer enzyme depletes or lowers the asparagine blood level. While asparagine is a nonessential amino acid in humans, ALL cells require it to survive and proliferate. Thus, depletion of this amino acid by asparaginase therapy is generally well tolerated since normal human cells do not require asparagine. Thus, eliminating a certain amino acid which is nonessential in humans but for which a particular cancer is auxotrophic may be a selective method of targeting malignancies.
Arginine is another nonessential amino acid in adult humans but is essential for rapidly proliferating cells. Thus, arginine deprivation can be exploited as a potential targeted therapy for treatment of various cancers. In this paper we review the rationale for this form of therapy and our own as well as other laboratory and clinical data.
Why Arginine Deprivation Leads to Cell Death in Certain Tumor Cell Lines ?
Arginine is involved in multiple pathways which are involved in major cellular functions such as nitric oxide production, creatine production and polyamine synthesis [3, 4]. In humans, arginine is derived primarily from diet, turnover of body proteins and de-novo synthesis via intestinal –renal axis [4]. Although arginine can be synthesized, it is considered a semi-essential amino acid in adult humans since endogenous production is insufficient when cells are under stress or need to proliferate [5, 6]. In tumor cells, arginine influences their growth/proliferation [5, 7–9] and diet restriction has been shown to inhibit metastatic tumor growth [8].
De-novo synthesis of arginine occurs primarily in the proximal renal tubule via urea cycle. However, this capability is also found in many other cells [6]. Arginine is synthesized from citrulline in two steps via the urea cycle. The enzyme argininosuccinate synthetase catalyzes the conversion of L-citrulline and aspartic acid to argininosuccinate which is then further converted to L-arginine and fumaric acid by argininosuccinate lyase, L-arginine can be degraded to L-ornithine by the urea cycle enzyme arginase. L-ornithine is then converted back to L-citrulline by ornithine transcarbamyl transferase (OCT) and then recycle back to arginine by ASS/ASL (see Fig. 1). Thus, the ability to regenerate arginine from citrulline depends on the amount/activity of ASS and ASL [10, 11]. In fact, Wheatley has suggested that these two enzymes are tightly coupled [12–14] and that the sensitivity of cells to arginine deprivation depends on their ability to regenerate arginine from the alternative intermediate (ornithine, citrulline and argininosuccinate) in the urea cycle [9, 12–14].
Fig. (1).

Enzymes involved in urea cycle. The dot line on ADI designate its action on extracellular conversion of arginine to citrulline and ammonia.
Several laboratories have reported that certain tumor cell lines such as melanoma, hepatocellular carcinoma, renal cell carcinoma and some mesotheliomas do not express ASS, and hence become auxotrophic for arginine [9, 11, 15–18]. Consequently, arginine degrading enzyme has been shown by several laboratories to have antitumor effect in these tumors [9, 13, 19–25]. To further prove that the lack of ASS expression in melanoma cell lines is a key to render them susceptible to cell death upon arginine deprivation, we and others have transfected melanoma cells with ASS. These transfectants are resistant to arginine deprivation [15, 18].
Arginine can be catabolized by three enzymes: arginase, arginine decarboxylase and arginine deiminase.(ADI). Arginase has been tested in experimental animals since 1950, however, the therapy has failed to produce major responses. The reason for failure could be due to the fact that this enzyme has low affinity for arginine and thus a large amount of the enzyme will be required. The optimal pH for arginase is also high (9.5) [26] and cannot be achieved. However, recently, Wheatly et al. have shown that arginase is effective from pH 7.2–9 [27]. Importantly, arginase catabolizes arginine to ornithine. It is uncertain whether other normal tissues except liver and small bowel can synthesize ornithine to citrulline due to the fact that OCT, a key enzyme required for this reaction is expressed primarily in liver. In other tissues, OCT gene is hypermethylated and hence not expressed. Thus, one may encounter normal tissue toxicity. These drawbacks may explain why the development of arginase as an antitumor agent has failed despite its activity seen in vitro [28, 29]. Nevertheless, recently pegylated arginase has been shown to have activity in hepatocellular carcinoma both in vitro and in vivo due to lack of OCT expression in certain hepatocellular carcinoma cell lines [30]. On the other hand, arginine deiminase, a microbial enzyme from mycoplasma which catabolizes arginine to citrulline and ammonia, is more attractive due to the fact that tumor cells which lack ASS will not be able to synthesize intracellular arginine from citrulline while normal cells are able to regenerate arginine. This approach will avoid normal tissue toxicity. Furthermore, this enzyme is active in physiologic pH and has a high affinity for arginine. In fact, ADI has been reported to have antitumor activity both in vitro and in vivo in melanoma and hepatocellular carcinoma cell lines and in other cell lines [15–18, 31–34]. However, one of the drawbacks of ADI is that it is not produced by humans and, consequently, it is highly antigenic. In addition, ADI also has a short half life. To overcome these drawbacks, a pegylated form of ADI (ADI-PEG20) has been developed by Polaris, Inc (formerly Phoenix Pharmacologics) [35]and after extensive in vitro testing and in-vivo study [15, 34], the drug has entered into clinical trial for both hepatocellular carcinoma and melanoma.
Antitumor Activity of ADI-PEG20 in Melanoma Cells
We and others have shown that degrading arginine in the culture media using ADI or exposure of melanoma cell lines to arginine free media with citrulline and NH4Cl supplement can inhibit growth as well as have cytotoxic effects in melanoma cell lines [15, 18]. The IC50 after 7 days of exposure ranged from 0.01–0.3 μg/ml among 16 melanoma cell lines tested [15]. In our laboratory, we have studied the growth inhibitory effect of ADI-PEG20 in 4 melanoma cell lines, (A-375, SK-Mel2, A-2058 and Mel-1220 all ASS (−) one normal human fibroblast (BJ-1) and one ASS (+) NSCLC line. The growth inhibitory effect ranged from 0.05–0.09 μg/ml after 3 days exposure in 4 melanoma cell lines tested (see Table 1) and greater than 1 in BJ-1 and NSCLCS cell lines [18]. Interestingly, at 72 hr. there was still arginine remaining in the media (12.8 μM for Sk-Mel-2 and 23.5 μM for A2058 and no detectable arginine for A375 and MEL-1220). However, if one incubates EMEM media with 0.05 μg/ml of ADI-PEG20, there are no detectable arginine levels at 48 hr. Thus, it appears that the capability of intra-cellular machinery to maintain arginine levels either by degradation of other nonessential proteins or decreased proliferation or turning on ASS through translation or transcription is different among melanoma cell lines. These differences may explain why some melanoma cell lines can become resistant to this form of treatment.
Table 1.
Growth Inhibitory Effect of ADI-PEG20
| Cell Lines | ID50 (ug/ml) | Arginine (uM) at 72 hr | Citrulline (uM) at 72 hr |
|---|---|---|---|
| A2058 | 0.088 ± 0.008 | 23.5 | 367.5 |
| A375 | 0.055 ± 0.001 | ND | 317.5 |
| Sk-Mel-2 | 0.070 ± 0.008 | 12.8 | 329.2 |
| Mel-1220 | 0.090 ± 0.008 | 0 | 3 |
| BJ-1 | > 1 | 1.6 | 404 |
| NSCLCS* | > 1 | 20.27 | 603 |
RPMI media (base line Arginine: 971 uM).
EMEM media (base line arginine: 400 uM).
Antitumor Activity of ADI-PEG20 in Hepatocellular Carcinoma
ADI-PEG20 has been shown to have antitumor activity in some hepatocellular carcinoma cell lines which lack ASS expression [15]. However, recently Cheng et al. have studied 5 HCC cell lines (HepG2, Hep3B, PLC/PRF/5, Huh7 and Sk-HEP-1) and reported that these cell lines express ASS and are not sensitive to ADI [30]. On the other hand, these cell lines lack OCT expression and are sensitive to arginase [30]. Antitumor activity was also seen in vivo using PEG-arginase, but the toxicity profile was not discussed. It is of interest why hepatocellular carcinoma cells lack OCT or ASS expression since normal hepatocytes possess these enzymes. Thus, it is not yet known how frequent hepatocellular carcinoma cells do not express ASS or OCT or both. Nevertheless, Dillion et al. have reported that all 51 hepatoma tumor samples are deficient in ASS by immunohistochemical staining [36]
Antitumor Activity of ADI-PEG20 in other Cell Lines
Certain mesothelioma cell lines (2591 and MSTO) do not express ASS and are sensitive to ADI-PEG20 while two other cell lines (226,2461) express high levels [17]. Immunohistochemical study showed 63% of mesothelioma express low levels of ASS [17]. Based on this study, clinical trials in mesothelioma have been initiated [Szlosarck, personal communication]. ADI-PEG20 is also active in certain renal cell cancer cell lines which do not express ASS [16]. Interestingly, some renal cell cancers also are negative for ASS by immunohistochemical staining despite high levels of these enzymes found in the kidney. Other cell lines which are sensitive to ADI-PEG20 include neuroblastoma cell lines [37] and human T and B lymphoblastic cell lines [31–33], but not a myeloid leukemic cell line (HL-60). In this report, ADI-PEG20 has better antitumor activity than L-asparaginase [31]. Tumor cell lines which express low levels of ASS may also be sensitive to arginine deprivation [11]. In contrast, Kang et al. [38] have shown that depletion of arginine with ADI isolated from sertoli cells did not induce apoptosis in DU145 prostate cancer cell lines, however, it protected them from paclitaxel-induced apoptosis. This is not surprising since DU145 possesses ASS, and arginine depletion will result in retardation of cell growth which renders them resistant to chemotherapy.
ADI-PEG20 Induces Apoptosis in Melanoma Cell Lines
It has been shown that ADI-PEG20 inhibits protein synthesis using 35S incorporation, but how inhibition of protein synthesis leads to cell death is not clear. Gong et al. have reported apoptotic cell death in human lymphoblastic cell lines [31, 32]. We have also seen apoptotic cell death in melanoma cells [18] (Figs. 2 and 3). Interestingly although apoptosis is seen in melanoma cells treated with ADI-PEG20 at ID50 dose for three days, these cells can survive by replenishing new media with arginine and removal of ADI-PEG20, but if one continues to add ADI-PEG20 in the new media, there is no viable cells in all three cells lines (A-375, Sk-mel2 and Mel1220) in 3 weeks. We speculate that all these cells undergo autophagy with no arginine prior to undergoing apoptosis. In fact, our preliminary data showed increased Beclin1 in these cell lines with Mel1220 having the highest Beclin1 levels. In mesothelioma, Szlosarek has shown that arginine depletion induces BAX conformation changes and mitochondrial inner membrane depolarization in ASS(−) cells which leads to apoptosis [17, 39]. Thus, the mechanism(s) which lead to apoptosis is complex and may be different among all the cell lines which lack ASS expression.
Fig. (2).

In situ end labeling apoptosis assay in A375 cell line. A: control. B: after exposure to 0.08 ug/ml for 72 hr. Treated cells undergo apopotosis shown brown staining in the nuclei
Fig. (3).
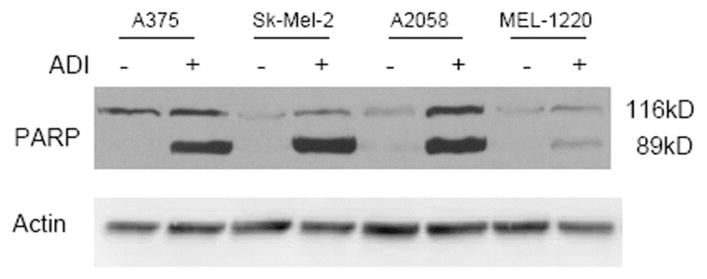
Apoptosis as detected by PARP cleavage in 4 melanoma cell lines (A375, Sk-mel-2, A2058 and MEL-1220). Untreated cell showed uncleaved PARP at 116 kD whereas treated cells showed cleaved PARP seen at 89 kD.
ADI-PEG20 Inhibits NO Production
Since arginine is the only substrate for nitric oxide synthetase [3], it is not surprising that ADI-PEG20 inhibits NO production in lipopolysaccharides and interferon challenged macrophages [40]. This observation was also seen in vivo when mice were treated with TNF and endotoxin. [40]. However, ADI-PEG20 did not inhibit NO which produces eNOS [40]. Thus, it appears that ADI-PEG20 can be used as selective modulator for NO which is produced by iNOS. The implication of these findings to tumor cell survival/carcinogenesis has not been studied. On the other hand NO can reversibly inactivate ASS via S-nitrosulation both in vitro and in vivo [41]. Thus the complex relationship of these parameters will need further investigation.
ADI-PEG20 Inhibits Angiogenesis
The effect of ADI on angiogenesis has been investigated both in vitro and in vivo. Beloussow et al. first [42] demonstrated the anti-angiogenic effect of recombinant ADI with cultured human umbilical vein endothelial cells (HUVEC). Park et al. also used HUVECs and recombinant ADI to show that ADI could inhibit angiogenesis in a dose dependent manner, and this inhibitory effect can be reversed by supplying arginine back to the culture medium [43]. Park et al. also observed that recombinant ADI exerted inhibition of angiogenesis in their in vivo studies using chick embryo and mouse model [43]. This antiangiogenetic property of ADI suggested an effective way for treating tumors. They showed that ADI was capable of inhibiting tumor cell growth using CHO and HeLa cell lines. Furthermore, Gong et al. [37] also showed that ADI was effective in inhibiting the growth of unfavorable neuroblastoma similar to other angiogenesis blocking reagents. Interestingly, ADI or these reagents was able to potentiate the effect of radiation therapy for this tumor using a mouse model.
ADI-PEG20 Lowers Hepatitis C Viral Titer
Izzo et al. have reported that ADI-PEG20 can reduce HCV titer up to 99% in 5/10 HCV-sero type 1b patient with subsequent improvement of liver function. However, it has no effect in HCV type 2c patients [44]. The exact mechanism of this is not known and may be related to decrease NO production.
ADI-PEG20 Affects mTOR Signaling
It is known that nutrient deprivation decreases mTOR signaling [45–47]. This is most likely due to decreased ATP which leads to activation of AMPK and hence negatively influences mTOR activity [48]. It is uncertain that TSC1/2 complex is involved [49]. Nutritional deprivation also positively influences the association of mTOR with raptor, the tight association of mTOR and Raptor will decrease mTOR activity [50]. We have shown that after exposing 4 melanoma cell lines to ADI-PEG20 for 72 hr, the phosphorylation of 4E-BP is substantially reduced in 3 melanoma cell lines and to a much lesser extent in A-2058 cell line (Fig. 4A). Furthermore, the amount of phosphorylation p-70S6 kinase is also reduced at 72 hr (Fig. 4B). In contrast, pAMPK which negatively influences mTOR activity is highly elevated in A-375 and Mel1220 at 72 hr., but is moderately elevated in A-2058 (Fig. 4C). There is no change in Sk-Mel 2; this cell line may have alteration in LKB. The changes in 4E-BP phosphorylation and P-70S6 kinase phosphorylation are not seen in NSCLC cell line and BJ-1 cell lines which possess ASS. Thus, arginine deprivation in the ASS (−) melanoma cells negatively impacts mTOR signaling. Inhibition of mTOR has been shown to decrease cellular proliferation and contribute to autophagy [51] which may explain antitumor activity of ADI-PEG20 in melanoma cells
Fig. (4).

A). The activity of mTOR was measured by its ability to phosphorylated 4E-BP. The bottom band is the unphosphorylated form. 4E-BP. 4E-BP has multiple phosphrylation which depict multiple bands seen. After treatment with ADI-PEG at 0.6 ug/ml (81 IU/ml=11.3 mg/ml) for 72 hr., the major form of 4E-BP are unphosphorylated. B). phosphoryltion of p-70S6 kinase also reflect mTOR activity. After exposure to ADI-PEG20 at 0.6 mg/ml, the phospho-p-70S6 kinase decresaed in A-375, Sk-Mel2, but increase in A-2058, which indicative of phosphorylation from other upstream protein. Mel-1220 has minimal phosphorylation of p-70S6 kinase and hence changes is not discernable. C). phospho-AMPK in 4 melanoma cell lines after exposure to ADI-PEG20 for 72 ht. Three cell lines has increased phospho-AMPK, but no changes in Sk-Mel 2.
Other Biological Effects of ADI
Since arginine serves as a precursor for polyamine (ornithine, putrescine), arginine deprivation may effect polyamine biosynthesis. In this regard, Shen et al. have shown that the addition of putrescine to ADI did not affect the antitumor activity of ADI, however it abolished the antitumor effect of DFMO, an ornithine decarboxylase inhibitor [10]. Furthermore, in CHO cells which lack ASS expression, treatment with ADI did not affect intracellular polyamine concentration. The authors conclude that low Km of ODC as well as compensation through protein degradation, efflux and uptake could be responsible for arginine supply and arginine concentration is not critical for maintaining intracellular polyamine concentration. Other effects of arginine deprivation including other protein changes such as involving heat shock protein and proteins which enhance cell susceptibility to apoptosis [52]. Recently Graboa et al. [53] has shown that arginine may play a crucial role in carcinogenesis.
Why ASS is not Expressed in Certain Tumor Types ?
ASS gene is located on chromosome 9q34.1 base pair 132, 310,092 to 132,366,481. There are many pseudogenes (14 reported) located among other chromosomes [54]. It is possible that deletion of chromosome at this region in tumor cells can account for lack of ASS expression. However, in 4 melanoma cell lines tested by southernblot analysis, the ASS DNA was present (Fig. 5). However, it is still possible that the promoter could be altered and cannot be detected by southernblot analysis. Our data [18] and other published papers [15, 17] indicate that the lack of ASS protein corresponded with low levels of mRNA expression (not detected by northernblot analysis). Recently Szlosarek et al. have found that mesothelioma cell lines which express low levels of ASS have aberrant promoter CpG methylation [17]. The authors conclude that epigentic regulation of ASS transcription plays a role in controlling ASS expression seen in mesothelioma cell lines. Our initial laboratory study did not support this concept in melanoma cell lines, and 5-Azacytidine, a known demethylating agent, does not affect ASS expression in melanoma cell lines (Savaraj, unpublished data). Thus, the key question which remains unanswered is why different tumor cell lines possess different levels of ASS expression. ASS cDNA has been cloned in 1981 [55] and is highly conserved between species. The promoter region in human and mice has been partially characterized. In human, the 5′ flanking sequence shows multiple SP-1 binding sites, AP2, GCN4, NF-KappaB, Max1, Egr and others [56, 57]. These bindng sites also are predicted using AliBaba 2.1 web search tool. We have currently cloned the promoter and constructed a series of the deleted mutant to study this question. Although the kinetic properties of ASS enzyme has been extensively studied and the crystal structure in bacteria has been identified [4], the transcriptional and translational control of ASS gene are not well understood. Two species of ASS mRNA which differ in their 5′UTR has been reported (Genbank Database). Previous published studies indicate that ASS regulation occurs at pre-translational levels [4]. Multiple factors have been shown to positively and negatively influence ASS gene and may be tissue specific. Several investigators have demonstrated that glucocorticoid, glucagon and cyclin AMP increase ASS expression whereas fatty acids repress ASS expression [58–65]. Insulin and growth hormone have negative influence on ASS in liver tissue but no effect in other tissue [66–68]. Cytokines, IL-Ib, interferon and TGF-b also can influence ASS expression [69, 70]. Recently, it has been showed that glutamine stimulates ASS expression in rat hepatocytes and Caco-2 cells through O-glycosylation of Sp1. sites [71]. The expression of ASS also has been showed to be regulated by arginine [72]. In this regard, we have demonstrated that in melanoma cell lines the expression of ASS is also influenced by arginine in media [18], but not influenced by either glutamine glucocorticoid or fatty acids (unpublished data). However, ASS gene regulation is most likely different in different tumor types. We are currently investigating the regulation of ASS in melanoma cells. It is noteworthy that normal human melanocytes (cell line HEM-l and HEM-m obtained from Sciencell) express very low levels of ASS protein which is different than normal hepatocytes.
Fig. (5).
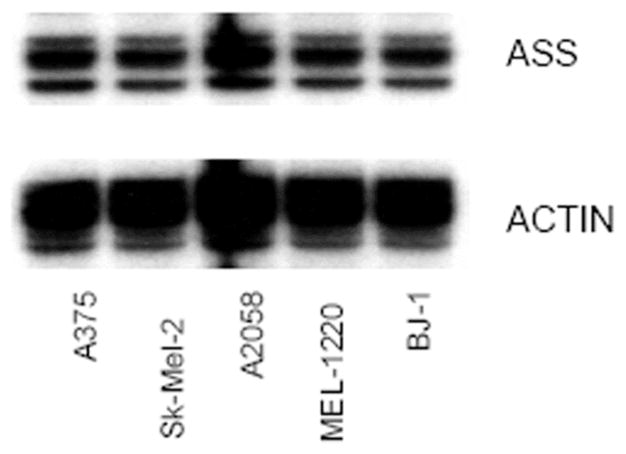
Southernblot Analysis of ASS in 4 melanoma cell lines and BJ-1 cell lines. All five cell lines show similar levels of ASS DNA.
Arginine Deprivation Induced Melanoma ASS Protein Expression
It has been shown that arginine levels can regulate ASS expression in lymphoblastoma cells and epithelial cells. Similarly, we have found that exposing melanoma cell lines to ADI-PEG20 or arginine free media with citrulline and NH4Cl2 supplement for 3 days, ASS protein as detected by westernblot was found in three cell lines. However, the levels of ASS expression are different: A2058 has more ASS expression (2.76 fold) followed by Sk-Mel-2 (2.03) and A375 (1.52) while MEL-1220 is unable to produce ASS protein. However, upon replenishment with normal media, the ASS protein expression is again repressed [18]. Fig. (6) summarized these findings. Interestingly, in cell lines (BJ-1 and NSCLCS) which constitutively express ASS the levels of ASS did not change upon exposure to arginine free media. We have further investigated whether the increase in ASS protein occurs at the transcription level. However, we were unable to detect ASS mRNA by northern-blot analysis (Fig. 7). We have developed a real time RT-PCR method to quantify ASS mRNA expression, only A-2058 showed an increase in mRNA expression at 48 and 72 hr (Fig. 8). The other three cell lines including BJ-1 (normal human fibroblast) did not show an increase in ASS mRNA upon arginine deprivation. The ASS mRNA also returned to normal upon placing the cells back in normal media. However, upon continued exposure to arginine free media, A-2058 is able to turn on a substantial amount of mRNA which could be detected by northernblot analysis (Fig. 7).
Fig (6).
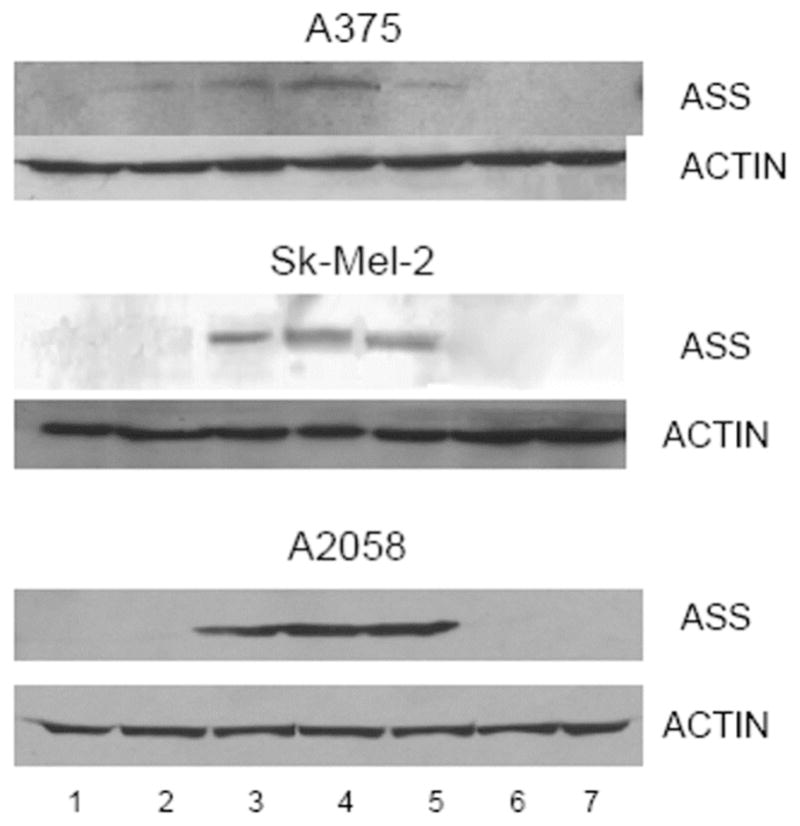
Immunoblot of ASS protein in 4 melanoma cell lines (A375, Sk-mel-2, A2058 and MEL-1220) before and after exposure arginine free media supplemented with citrulline and NH4Cl for 24, 48 and 72 hr. Afterward, cells were washed, and replenished with normal EMEM media for 24, 48 and 72 hr. Lane 1: control. Lane 2: 24 hrs. on arginine free media Lane 3: 48 hrs on arginine free media Lane 4: 72 hrs on arginine free media Lane 5: Removal of arginine free media and changed to normal EMEM media for 24 hrs. Lane 6: 48 hrs. on normal media. Lane 7: 72 hrs. on normal media. Similar results were obtained with arginine free media with no citrulline supplement and ADI-PEG20 treated media. with citrulline and NH4Cl supplement.
Fig. (7).
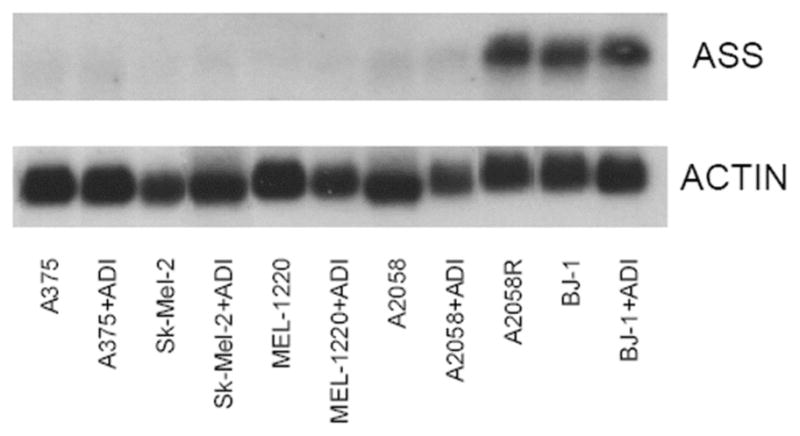
Northernblot Analysis of ASS in a panel of melanoma cell line and BJ-1 cells. Lane 1: A375. Lane 2: A375 after exposure to ADI-PEG20 for 72 hrs. Lane 3: SK-Mel-2 Lane 4: Sk-Mel-2 after exposure to ADI-PEG20 for 72 hrs. Lane 5: MEL-1220. Lane 6: MEL-1220 after exposure to ADI-PEG20 for 72 hrs. Lane 7: A2058. Lane 8: A2058 after exposure to ADI-PEG20 for 72 hrs. Lane 9: A2058R. Lane 10: BJ-1. Lane 11: BJ-1 after exposure to ADI-PEG20 for 72 hrs. Note: only A2058R and BJ-1 possess 1.9 kb ASS mRNA and there is no differences in ASS mRNA in BJ-1 cells after exposure to ADI-PEG20.
Fig (8).
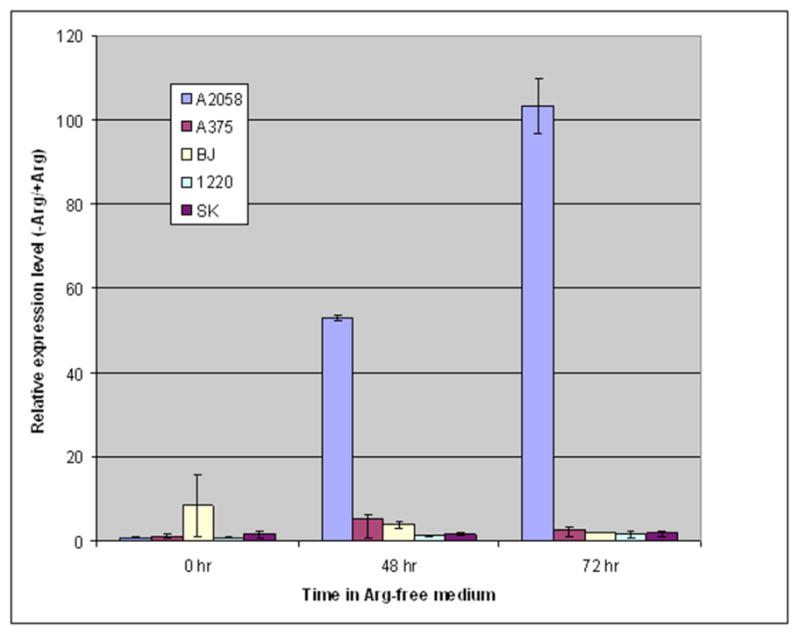
The relative expression level of ASS mRNA detected with real-time PCR in different cell lines in arginine-free (− Arg) media as compared with that in normal media (+Arg). The quantification method used was proposed by Pfaffl in Nucleic Acid Research, 29:2002–2007. GAPDH was used as the reference gene. The hours on the graph indicates the time for which the cells had been cultured in −Arg or +Arg media.
Resistance to ADI-PEG20 in Melanoma
From our melanoma cell line data as well as primary culture obtained from patients, it appears that cell lines or primary culture which can turn on ASS mRNA upon arginine deprivation are most likely to develop resistance to ADI. We are able to generate ADI-PEG20 resistant cell line in A-2058, but not in A-375, Skmel-2 or Mel 1220. These three cell lines are unable to turn on ASS mRNA upon arginine deprivation. The transient increase in ASS protein seen in A-375 and Sk-mel2 is mostly mediated by transient increase in translation. Once ASS mRNA is induced to certain extent, the ASS expression become stable and cannot be reversed (Fig. 9). Why certain cell lines can rapidly turn on ASS mRNA is not known and we are currently investigating the transcription control of ASS mRNA.
Fig. (9).
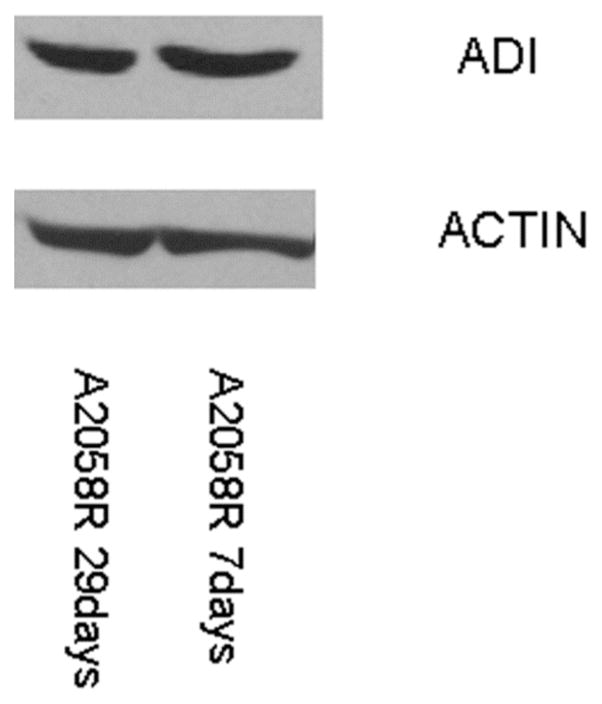
Immunoblot of ASS protein in A-2058R (resistance to ADI) after culture in normal media for 7 and 29 days. The expression is stable with no changes.
FUTURE LABORATORY DIRECTIONS
The work in our laboratory as well as others support the antitumor activity of ADI-PEG20 in ASS(−) tumor types. Thus far, melanoma and hepatoma appear to be the suitable tumors for this approach due to the lack of ASS expression. Certain mesothelioma and renal cell cancers may also be considered for this approach. However, development of drug resistance secondary to ASS expression may be problematic but could be potentially overcome. Currently, we are developing a method to identify those tumors which are ASS negative but can be induced after repeated treatment. These tumors will most likely require combination treatment which will kill cells rapidly and hence the tumor cells are not able to turn on ASS. Another possibility is to inhibit ASS expression using a pharmacological approach. We are currently investigating these different approaches in melanoma.
CLINICAL STUDIES WITH ADI-PEG20
We reported on a Phase I/II trial of ADI-PEG20 in patients with metastatic melanoma [73]. Two cohort dose-escalation studies were performed: a phase I study in the U.S. and a phase I/II study in Italy. The phase I study in the U.S. enrolled 15 patients in 4 cohorts. ADI-PEG20 treatment consisted of weekly intramuscular injections on days 1, 15, and 22. The first 3 cohorts consisted of 3 patients each and each cohort received ADI-PEG20 at 20, 40 or 80 U/m2. Subsequently, 6 patients were treated at 160 U/m2 which was considered the optimum biological dose (OBD).
The Italian cohort enrolled 24 patients in the phase I trial. Patients were enrolled onto one of six cohorts. The first 4 cohorts consisted of 3 patients each and each cohort was treated with one cycle ADI-PEG20 at 40, 80, 160 or 320 U/m2. Six patients received one cycle ADI-PEG20 at 640 U/m2. The first cycle was composed of therapy on days 1, 15 and 22. Furthermore, 6 patients were treated on a cohort of 3 cycles that consisted of 4 weekly injections at the OBD.
One objective of these studies was to determine the pharmacokinetics/pharmacodynamics, safety and antitumor activity of the drug. The pharmacokinetics/pharmacodynamics studies showed that a dose of ADI-PEG20 at 160 U/m2 lowered the plasma arginine level from a baseline level of approximately 130 μmol/L to less than 2 μmol/L for at least 7 days. Furthermore, the nitric oxide plasma levels were also decreased with no measurable effect on blood pressure or heart rate.
In this clinical trial no grade 3 or 4 toxicities were attributed to the drug. The most common side effect was mild discomfort at the injection site. Two patients had hypotension within 20–40 minutes of treatment. One of the patients had a history of hypertension and had been on antihypertensive medication. The other patient had hypotension after the fourth injection of ADI-PEG20 but received six further treatments of the drug without recurrence of the hypotension. Abnormalities in the laboratory tests that were noted during the trial include elevated serum uric acid levels, mild elevation in blood fibrinogen levels and rarely elevation of the serum lipase and amylase levels. No patient had clinical evidence of pancreatitis.
In terms of response, none of the 15 patients enrolled in the phase I trial in the U.S. had a response. In the Italian phase I/II part of the trial, 6 of 24 phase I/II patients had a response to treatment, including one complete response and 5 partial responses, for a total response rate of 25%. Fourteen patients had stable disease for ≥ 1 cycle of therapy and 6 patients had stable disease for 3 months of the trial. All patients with stable disease or response had received the OBD or higher ADI-PEG20 dose.
One objective of the trial was to investigate the immunogenicity of ADI-PEG20. In terms of immunogenicity of the drug, none of the patients developed measurable enzyme-neutralizing antibody-directed activity in the plasma samples. This is consistent with the lack of allergic reactions seen clinically. In terms of the maximum tolerated dose, 6 patients were treated at the highest dose of 640 U/m2 intramuscularly weekly. Except for discomfort at the injection site (grade 1), no major toxicities were noted. Since 640U/m2 was the highest dose that could be easily administered by intramuscular injection, it was concluded that the maximum tolerated dose exceeded the dose that could be conveniently administered by this route.
Currently, we are performing a phase II trial of ADI-PEG20 in advanced melanoma using a weekly dose of 160 U/m2 intramuscularly. Preliminary results from this trial have been presented (ASCO, 2005). Twenty patients with locally advanced or metastatic melanoma have been treated so far. In terms of response, 4 patients have had a partial response and 3 patients had a minor response (<50% reduction) for a tumor control rate of 35%. In addition, 3 patients had a mixed response suggesting evidence of antitumor activity. Interestingly, one patient who had a partial response at a dose of 160 U/m2 weekly developed progression of disease. The dose was increased and the patient again responded to treatment. This suggests that there may be a dose response or perhaps development of drug resistance due to neutralizing antibody which was overcome by increased dose of the drug. The sites of response include lung, nodal disease, soft tissue and skin. This is not unexpected since patients with disease in these organs are more likely to respond than patients with other visceral disease. We did observe a mixed response in a patient with extensive hepatic metastases using ADI-PEG20.
In terms of toxicity, one patient had grade 3 neutropenia which resolved when the drug was stopped. Another patient had grade ¾ neutropenia. Subsequently, he received filgrastim every 2 weeks which completely prevented further neutropenia and enabled him to continue to receive the ADI-PEG20 weekly. Other toxicities were mild and mainly consisted of fatigue and discomfort at the injection site. No alopecia, nausea, vomiting, diarrhea or major organ toxicities were noted. No patient stopped drug due to toxicity.
Preliminary data suggest that the presence of ASS expression in the tumor may predict response to ADI-PEG20. Two patients had melanoma tumor samples tested for ASS expression prior to treatment and were ASS (−). Both patients had a clinical response to ADI-PEG20 but later developed progression of disease. Tumor samples were obtained at the time of their disease progression and the tumor stained ASS positive. One patient whose melanoma tumor stained ASS positive prior to ADI-PEG20 did not respond to treatment. However, 7 of 11 patients whose tumor were ASS (−) prior to treatment had some evidence of antitumor activity (either partial, minor or mixed response) with ADI-PEG20. This study is continuing to accrue patients and further investigation is needed to confirm the correlation between ASS expression of the tumor and clinical response to treatment.
ADI-PEG20 has also been studied in clinical trials in patients with unresectable hepatocellular carcinoma. A patient with unresectable hepatocellular carcinoma was treated with escalating doses of ADI-PEG20 as a single patient exemption [74]. The tumor reduced in size and there was a reduction in serum alpha-fetoprotein levels. No toxicity due to the drug was noted. This encouraging response led to a phase I/II trial of ADI-PEG20 in patients with unresectable hepatocellular carcinoma [75]. Patients with hepatocellular carcinoma were accrued from the Pascale Cancer Institute in Naples, Italy. Nineteen patients were entered into the trial in 1 of 4 cohorts. The initial dose levels of ADI-PEG20 were 20, 40, or 80 U/m2. Subsequent patients were treated at a starting dose of 160 U/m2 which was determined to be the optimum biological dose that lowered the plasma arginine level to non-detectable levels (<2 μM/L) for at least 7 days. In terms of response, 2 patients had complete response and 7 patients had a partial response for a total response rate of 47%. Duration of response was defined as the time from start of treatment until progression. The median duration of response was >400 days (range: 37 to >680 days). In terms of toxicity, the side effects include pain at the injection site, elevation of serum uric acid and fibrinogen, and occasional elevation of the serum lipase and amylase levels. No clinical evidence of pancreatitis occurred. No evidence of neutralizing antibody production was found.
A second phase I/II trial of weekly ADI-PEG20 was performed at M.D. Anderson Hospital (ASCO, 2005). The phase I part of the trial involved escalating doses of the drug up to 160 U/m2. The phase II part of the trial used a starting dose of 160 U/m2 which was considered the OBD but allowed dose escalation up to ≤240 U/m2. Thirty-five patients were enrolled onto the study. In terms of response, one patient had a response which allowed the patient to be resectable. In addition, 16 patients had stable disease. Four patients did not complete the trial due to either allergic reaction or intercurrent disease. Twenty-eight patients had disease progression. The reported mean time for progression was 3.4 months with a range of 1 to 13 months. All patients had undetectable plasma arginine levels after ADI-PEG20 treatment (< 2 μM). For the one patient who had a response and became resectable, death occurred due to portal vein thrombosis and resulting live failure. Autopsy showed minimal viable tumor left.
In terms of toxicity, the side effects were more pronounced compared to the phase I trial in Italy. Twelve patients had grade 3 toxicity consisting mainly of liver function or serum electrolyte abnormalities. In addition, 3 patients had grade 4 toxicity consisting of liver function abnormalities or elevation of serum lipase. Subsequently, we participated in as phase 2B testing of ADI-PEG20 in patients with unresectable hepatocellular carcinoma in conjunction with the Pascale Cancer Institute. Patients received ADI-PEG20 at 80 or 160 U/m2. The primary endpoint was overall survival. An interim analysis showed that the median survival from time of diagnosis was 11.7 months and the median survival from first treatment was 8.8 months. The 12 month survival rate was 36.7%. These numbers are comparable to those reported from the Pascale Cancer Institute and from a phase I/II trial reported from M.D. Anderson [75]. Interestingly, of the 9 patients treated at the University of Miami, two patients had stable disease for at least 12 months. One patient had stable disease for one year and the other patient had stable disease for >2 years with no side effects noted from the treatment except for discomfort at the injection site. This patient is continuing to receive treatment with weekly ADI-PEG20 injections.
CONCLUDING REMARKS
Arginine deprivation is a novel and promising targeted approach to the treatment of certain tumors which cannot form arginine from the urea cycle and hence are auxotrophic for arginine. In particular, melanoma and hepatocellular carcinoma and possibly certain mesotheliomas and renal cell cancers may be sensitive to this therapy. While new therapies are emerging for renal cell cancer, the other cancers are difficult to treat with drug therapy and for patients with unresectable disease, the response rates and overall survival remains poor. Thus, there remains a great need to develop new drug therapies for these malignancies.
Due to its low incidence of major side effects such as myelo-suppression and gastrointestinal toxicity or other major organ toxicity, arginine deprivation may be easily combined with other anti-cancer agents to increase the response rate. Our in vitro laboratory data suggest that ADI-PEG20 may be synergistic with DNA damaging agents such as cisplatin and temozolomide (AACR, 2007). This appears to be due to the inability of ASS (−)For MDR1, the primers were the mdr1 specific sequences GGAGTGTCCGTGGATCACAAG (residues 1909–1930) and TGTTCAGGATCATCAATTCTTGT (residues 2218–2241). These primers were selected at regions that are only 36.4 and 37.5% similar to the corresponding region of mdr-3 cDNA. Thus, they should not recognize the mdr-3 gene. The resulting PCR product from these primers was 232 bp. tumor cells to repair DNA in the absence of arginine. Combination of ADI-PEG20 with these DNA damaging agents and other drugs are currently being studied for future clinical trial.
The development of drug resistance due to induction of ASS gene expression is a potential problem in certain tumors. This form of resistance may be overcome by understanding the transcription/translation control of this gene. Pharmacologic manipulation to control ASS expression and prevent drug resistance is being investigated in our laboratory.
Acknowledgments
This work is supported by a grant (R01CA109578) from NIH and a research grant from VA. We would like to thank Polaris Inc. for providing ADI-PEG20 and analysis of arginine and citrulline.
References
- 1.Asselin BL. Adv Exp Med Biol. 1999;457:621–9. [PubMed] [Google Scholar]
- 2.Muller HJ, Boos J. Crit Rev Oncol Hematol. 1998;28:97–113. doi: 10.1016/s1040-8428(98)00015-8. [DOI] [PubMed] [Google Scholar]
- 3.Wu G, Morris SM., Jr Biochem J. 1998;336(Pt 1):1–17. doi: 10.1042/bj3360001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Husson A, Brasse-Lagnel C, Fairand A, Renouf S, Lavoinne A. Eur J Biochem. 2003;270:1887–99. doi: 10.1046/j.1432-1033.2003.03559.x. [DOI] [PubMed] [Google Scholar]
- 5.Lind DS. J Nutr. 2004;134:2837S–2841S. doi: 10.1093/jn/134.10.2837S. discussion 2853S. [DOI] [PubMed] [Google Scholar]
- 6.Morris SM., Jr Am J Clin Nutr. 2006;83:508S–512S. doi: 10.1093/ajcn/83.2.508S. [DOI] [PubMed] [Google Scholar]
- 7.Caso G, McNurlan MA, McMillan ND, Eremin O, Garlick PJ. Clin Sci (Lond) 2004;107:371–9. doi: 10.1042/CS20040096. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez GG, Byus CV. Cancer Res. 1991;51:2932–9. [PubMed] [Google Scholar]
- 9.Wheatley DN, Campbell E. Br J Cancer. 2003;89:573–6. doi: 10.1038/sj.bjc.6601134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shen LJ, Beloussow K, Shen WC. Cancer Lett. 2006;231:30–5. doi: 10.1016/j.canlet.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 11.Shen LJ, Lin WC, Beloussow K, Shen WC. Cancer Lett. 2003;191:165–70. doi: 10.1016/s030-43835(02)00693-6. [DOI] [PubMed] [Google Scholar]
- 12.Wheatley DN. Semin Cancer Biol. 2005;15:247–53. doi: 10.1016/j.semcancer.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 13.Wheatley DN. Anticancer Drugs. 2004;15:825–33. doi: 10.1097/00001813-200410000-00002. [DOI] [PubMed] [Google Scholar]
- 14.wheatley DN, Campbell e, Lai PBS, Cheng PNM. Gene Therapy and Molecular Biology. 2005;9:33–40. [Google Scholar]
- 15.Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Cancer Res. 2002;62:5443–50. [PubMed] [Google Scholar]
- 16.Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH, Kim JH, et al. Int J Cancer. 2007;120:897–905. doi: 10.1002/ijc.22322. [DOI] [PubMed] [Google Scholar]
- 17.Szlosarek PW, Klabatsa A, Pallaska A, Sheaff M, Smith P, Crook T, et al. Clin Cancer Res. 2006;12:7126–31. doi: 10.1158/1078-0432.CCR-06-1101. [DOI] [PubMed] [Google Scholar]
- 18.Savaraj N, Wu C, Kuo M, You M, Wangpaichitr M, Robles C, et al. Drug Target Insights. 2007;2:119–128. [PMC free article] [PubMed] [Google Scholar]
- 19.Philip R, Campbell E, Wheatley DN. Br J Cancer. 2003;88:613–23. doi: 10.1038/sj.bjc.6600681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott L, Lamb J, Smith S, Wheatley DN. Br J Cancer. 2000;83:800–10. doi: 10.1054/bjoc.2000.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takaku H, Takase M, Abe S, Hayashi H, Miyazaki K. Int J Cancer. 1992;51:244–9. doi: 10.1002/ijc.2910510213. [DOI] [PubMed] [Google Scholar]
- 22.Takaku H, Misawa S, Hayashi H, Miyazaki K. Jpn J Cancer Res. 1993;84:1195–200. doi: 10.1111/j.1349-7006.1993.tb02821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takaku H, Matsumoto M, Misawa S, Miyazaki K. Jpn J Cancer Res. 1995;86:840–6. doi: 10.1111/j.1349-7006.1995.tb03094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugimura K, Ohno T, Kusuyama T, Azuma I. Melanoma Res. 1992;2:191–6. doi: 10.1097/00008390-199209000-00007. [DOI] [PubMed] [Google Scholar]
- 25.Sugimura K, Ohno T, Fukuda S, Wada Y, Kimura T, Azuma I. Cancer Res. 1990;50:345–9. [PubMed] [Google Scholar]
- 26.Dillon BJ, Holtsberg FW, Ensor CM, Bomalaski JS, Clark MA. Med Sci Monit. 2002;8:BR248–53. [PubMed] [Google Scholar]
- 27.Wheatley DN, Philip R, Campbell E. Mol Cell Biochem. 2003;244:177–85. [PubMed] [Google Scholar]
- 28.Currie GA, Gyure L, Cifuentes L. Br J Cancer. 1979;39:613–20. doi: 10.1038/bjc.1979.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Storr JM, Burton AF. Br J Cancer. 1974;30:50–9. doi: 10.1038/bjc.1974.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng PN, Lam TL, Lam WM, Tsui SM, Cheng AW, Lo WH, et al. Cancer Res. 2007;67:309–17. doi: 10.1158/0008-5472.CAN-06-1945. [DOI] [PubMed] [Google Scholar]
- 31.Gong H, Zolzer F, von Recklinghausen G, Havers W, Schweigerer L. Leukemia. 2000;14:826–9. doi: 10.1038/sj.leu.2401763. [DOI] [PubMed] [Google Scholar]
- 32.Gong H, Zolzer F, von Recklinghausen G, Rossler J, Breit S, Havers W, et al. Biochem Biophys Res Commun. 1999;261:10–4. doi: 10.1006/bbrc.1999.1004. [DOI] [PubMed] [Google Scholar]
- 33.Noh EJ, Kang SW, Shin YJ, Choi SH, Kim CG, Park IS, et al. Int J Cancer. 2004;112:502–8. doi: 10.1002/ijc.20435. [DOI] [PubMed] [Google Scholar]
- 34.Shen LJ, Shen WC. Curr Opin Mol Ther. 2006;8:240–8. [PubMed] [Google Scholar]
- 35.Holtsberg FW, Ensor CM, Steiner MR, Bomalaski JS, Clark MA. J Control Release. 2002;80:259–71. doi: 10.1016/s0168-3659(02)00042-1. [DOI] [PubMed] [Google Scholar]
- 36.Dillon BJ, Prieto VG, Curley SA, Ensor CM, Holtsberg FW, Bomalaski JS, et al. Cancer. 2004;100:826–33. doi: 10.1002/cncr.20057. [DOI] [PubMed] [Google Scholar]
- 37.Gong H, Pottgen C, Stuben G, Havers W, Stuschke M, Schweigerer L. Int J Cancer. 2003;106:723–8. doi: 10.1002/ijc.11298. [DOI] [PubMed] [Google Scholar]
- 38.Kang SW, Kang H, Park IS, Choi SH, Shin KH, Chun YS, et al. Mol Cells. 2000;10:331–7. [PubMed] [Google Scholar]
- 39.Szlosarek P, Klabatsa A, Pallaska A, Sheaf fM, Balkwell F, Fennell D. Proc Am Assoc Can Res. Washington D. C, USA: 2006. [Google Scholar]
- 40.Shen LJ, Lin WC, Beloussow K, Hosoya K, Terasaki T, Ann DK, et al. Biochem Pharmacol. 2003;66:1945–52. doi: 10.1016/s0006-2952(03)00555-0. [DOI] [PubMed] [Google Scholar]
- 41.Hao G, Xie L, Gross SS. J Biol Chem. 2004;279:36192–200. doi: 10.1074/jbc.M404866200. [DOI] [PubMed] [Google Scholar]
- 42.Beloussow K, Wang L, Wu J, Ann D, Shen WC. Cancer Lett. 2002;183:155–62. doi: 10.1016/s0304-3835(01)00793-5. [DOI] [PubMed] [Google Scholar]
- 43.Park IS, Kang SW, Shin YJ, Chae KY, Park MO, Kim MY, et al. Br J Cancer. 2003;89:907–14. doi: 10.1038/sj.bjc.6601181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Izzo F, Montella M, Orlando AP, Nasti G, Beneduce G, Castello G, et al. J Gastroenterol Hepatol. 2007;22:86–91. doi: 10.1111/j.1440-1746.2006.04463.x. [DOI] [PubMed] [Google Scholar]
- 45.Sarbassov DD, Ali SM, Sabatini DM. Curr Opin Cell Biol. 2005;17:596–603. doi: 10.1016/j.ceb.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 46.Tokunaga C, Yoshino K, Yonezawa K. Biochem Biophys Res Commun. 2004;313:443–6. doi: 10.1016/j.bbrc.2003.07.019. [DOI] [PubMed] [Google Scholar]
- 47.Guertin DA, Sabatini DM. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Kimura N, Tokunaga C, Dalal S, Richardson C, Yoshino K, Hara K, et al. Genes Cells. 2003;8:65–79. doi: 10.1046/j.1365-2443.2003.00615.x. [DOI] [PubMed] [Google Scholar]
- 49.Smith EM, Finn SG, Tee AR, Browne GJ, Proud CG. J Biol Chem. 2005;280:18717–27. doi: 10.1074/jbc.M414499200. [DOI] [PubMed] [Google Scholar]
- 50.Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, et al. Proc Natl Acad Sci U S A. 2005;102:14238–43. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Biochimie. 2007. [DOI] [PubMed] [Google Scholar]
- 52.Lenaerts K, Renes J, Bouwman FG, Noben JP, Robben J, Smit E, et al. Proteomics. 2007;7:565–77. doi: 10.1002/pmic.200600715. [DOI] [PubMed] [Google Scholar]
- 53.Grabon W. Postepy Hig Med Dosw (Online) 2006;60:483–9. [PubMed] [Google Scholar]
- 54.Carritt B, Povey S. Cytogenet Cell Genet. 1979;23:171–81. doi: 10.1159/000131323. [DOI] [PubMed] [Google Scholar]
- 55.Su TS, Bock HG, O’Brien WE, Beaudet AL. J Biol Chem. 1981;256:11826–31. [PubMed] [Google Scholar]
- 56.Jinno Y, Matuo S, Nomiyama H, Shimada K, Matsuda I. J Biochem (Tokyo) 1985;98:1395–403. doi: 10.1093/oxfordjournals.jbchem.a135407. [DOI] [PubMed] [Google Scholar]
- 57.Anderson GM, Freytag SO. Mol Cell Biol. 1991;11:1935–43. doi: 10.1128/mcb.11.4.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haggerty DF, Spector EB, Lynch M, Kern R, Frank LB, Cederbaum SD. J Biol Chem. 1982;257:2246–53. [PubMed] [Google Scholar]
- 59.Gebhardt R, Mecke D. Eur J Biochem. 1979;97:29–35. doi: 10.1111/j.1432-1033.1979.tb13082.x. [DOI] [PubMed] [Google Scholar]
- 60.Lin RC, Snodgrass PJ, Rabier D. J Biol Chem. 1982;257:5061–7. [PubMed] [Google Scholar]
- 61.Ulbright C, Snodgrass PJ. Arch Biochem Biophys. 1993;301:237–43. doi: 10.1006/abbi.1993.1139. [DOI] [PubMed] [Google Scholar]
- 62.Morris SM, Jr, Moncman CL, Rand KD, Dizikes GJ, Cederbaum SD, O’Brien WE. Arch Biochem Biophys. 1987;256:343–53. doi: 10.1016/0003-9861(87)90455-3. [DOI] [PubMed] [Google Scholar]
- 63.Nebes VL, Morris SM., Jr Mol Endocrinol. 1988;2:444–51. doi: 10.1210/mend-2-5-444. [DOI] [PubMed] [Google Scholar]
- 64.Husson A, Bouazza M, Buquet C, Vaillant R. Biochem J. 1983;216:281–5. doi: 10.1042/bj2160281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tomomura M, Tomomura A, Dewan MA, Saheki T. FEBS Lett. 1996;399:310–2. doi: 10.1016/s0014-5793(96)01344-0. [DOI] [PubMed] [Google Scholar]
- 66.Grofte T, Wolthers T, Jensen SA, Moller N, Jorgensen JO, Tygstrup N, et al. Hepatology. 1997;25:964–9. doi: 10.1002/hep.510250429. [DOI] [PubMed] [Google Scholar]
- 67.McLean P, Novello F. Biochem J. 1965;94:410–22. doi: 10.1042/bj0940410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grofte T, Jensen DS, Gronbaek H, Wolthers T, Jensen SA, Tygstrup N, et al. Am J Physiol. 1998;275:E79–86. doi: 10.1152/ajpendo.1998.275.1.E79. [DOI] [PubMed] [Google Scholar]
- 69.Hattori Y, Shimoda S, Gross SS. Biochem Biophys Res Commun. 1995;215:148–53. doi: 10.1006/bbrc.1995.2445. [DOI] [PubMed] [Google Scholar]
- 70.Zhang WY, Gotoh T, Oyadomari S, Mori M. Brain Res Mol Brain Res. 2000;83:1–8. doi: 10.1016/s0169-328x(00)00154-6. [DOI] [PubMed] [Google Scholar]
- 71.Brasse-Lagnel C, Fairand A, Lavoinne A, Husson A. J Biol Chem. 2003;278:52504–10. doi: 10.1074/jbc.M306752200. [DOI] [PubMed] [Google Scholar]
- 72.Jackson MJ, Allen SJ, Beaudet AL, O’Brien WE. J Biol Chem. 1988;263:16388–94. [PubMed] [Google Scholar]
- 73.Ascierto PA, Scala S, Castello G, Daponte A, Simeone E, Ottaiano A, et al. J Clin Oncol. 2005;23:7660–8. doi: 10.1200/JCO.2005.02.0933. [DOI] [PubMed] [Google Scholar]
- 74.Curley SA, Bomalaski JS, Ensor CM, Holtsberg FW, Clark MA. Hepatogastroenterology. 2003;50:1214–6. [PubMed] [Google Scholar]
- 75.Izzo F, Marra P, Beneduce G, Castello G, Vallone P, De Rosa V, et al. J Clin Oncol. 2004;22:1815–22. doi: 10.1200/JCO.2004.11.120. [DOI] [PubMed] [Google Scholar]