

Abstract
The mTORC1 pathway is a central regulator of cell growth, and defective mTORC1 regulation plays a causative role in a variety of human diseases, including cancer, tumor syndromes such as the tuberous sclerosis complex (TSC) and lymphangioleiomyomatosis (LAM), and metabolic diseases such as diabetes and obesity. Given the importance of mTORC1 signaling in these diseases, there has been significant interest in developing screening methods suitable for identifying inhibitors of mTORC1 activation. To this end, we have developed a high-throughput, cell-based assay for the detection of rpS6-phosphorylation as a measure of mTORC1 signaling. This assay takes advantage of the “In Cell Western” (ICW) technique using the Aerius infrared imaging system (LI-COR® Biosciences). The ICW procedure involves fixation and immunostaining of cells in a manner similar to standard immunofluorescence methods but takes advantage of secondary antibodies conjugated to infrared-excitable fluorophores for quantitative detection by the Aerius® scanner. In addition, the cells are stained with an infrared-excitable succinimidyl ester dye, which covalently modifies free amine groups in fixed cells and provides a quantitative measure of cell number. We present validation data and pilot screens in a 384-well format demonstrating that this assay provides a statistically robust method for both small molecule and siRNA screening approaches designed to identify inhibitors of mTORC1 signaling.
INTRODUCTION
The mTor protein Kinase a critical regulator of cell growth, proliferation, and survival, serving as the central integration point for multiple homeostatic inputs, including growth factor availability, energy levels, and amino acid sufficiency (Fig. 1).1 mTOR is a PIKK-family (phosphatidylinositol kinase-related kinase) kinase, first identified in Saccharomyces cerevisae as the cellular target of the immunosuppressant compound rapamycin. It is now appreciated that mTOR is a serine/threonine kinase that functions in 2 distinct macromolecular complexes, the mTORC1 complex (comprising mTOR, raptor, and Lst8) and the mTORC2 complex (comprising mTOR, rictor, Lst8, and the recently identified component Sin1p).2,3 The mTORC1 complex is responsible for the well-characterized role of mTOR in controlling protein translation, achieved in part through the phosphorylation of 2 mTORC1 substrates, the S6-kinases and the eIF4E-binding proteins. Phosphorylation of many mTORC1 substrates is inhibited by rapamycin; however, rapamycin-resistant aspects of mTORC1 signaling have recently been uncovered,4–6 pointing to the need for additional strategies for mTORC1 inhibition. The mTORC2 complex is rapamycin-insensitive and directly phosphorylates Ser473 in the hydrophobic motif of Akt, which in turn regulates phosphorylation of specific Akt substrates with important implications for cell survival and proliferation.3
Fig. 1. .

rpS6 phosphorylation as an endpoint for mTORC1 signaling. (A) A schematic of the signaling events leading to rpS6 phosphorylation. mTORC1 kinase activity is regulated by multiple upstream signals, including growth factors, cellular energy status, and amino acid availability, and phosphorylates S6K1 on T389 in the hydrophobic motif (numbering based on p70 S6K1). PDK1 is regulated by growth factor signaling and phosphorylates S6K1 on T229 in the catalytic domain. Once fully activated by these 2 phosphorylation events, S6K1 then phosphorylates rpS6 on multiple sites. The antibody used for the In Cell Western assay described here recognizes rpS6 phosphorylated on both Ser235 and Ser236. (B) HeLa cells were incubated with the indicated compounds for 3 h, then fixed and processed for immunofluorescence. rpS6-phosphorylation was visualized by staining with the anti-phospho-S6 S235/S236 antibody and an Alexa-488 anti-rabbit secondary antibody (green), actin filaments were stained with Alexa-568-conjugated phalloidin (red), and nuclei were stained with Hoechst (blue). rpS6 phosphorylation is lost following treatment with the pathway-specific inhibitors, rapamycin and LY294002, but not with the MEK inhibitor U0126 or the DMSO vehicle control. (C) HeLa cells were incubated with the indicated compounds for 3 h, then lysed and processed for western blot analysis and probed with the indicated primary antibodies and either the goat anti-rabbit IRDye-800CW or goat anti-mouse IR-Dye700 secondary antibodies. The blots were visualized on the Odyssey infrared imager (LI-COR Biosciences). A reduction in rpS6-phosphorylation was observed upon treatment with the pathway-specific inhibitors, rapamycin and LY294002, but not with the MEK inhibitor U0126 or the DMSO vehicle control. The ability of U0126 to inhibit MEK activity is confirmed by the loss in Erk phosphorylation. GAPDH is shown as a loading control.
Hyperactivation of the mTORC1 signaling network is a common feature of nearly all cancers and is also associated with a variety of other human diseases, including tumor syndromes such as lymphangioleiomyomatosis (LAM) and the tuberous sclerosis complex (TSC), as well as several metabolic disorders.7 The realization that the mutations underlying TSC and LAM, as well as many mutations that contribute to cancer progression, result in activation of mTORC1 has lead to a number of clinical trials evaluating the efficacy of rapamycin analogs for the treatment of these diseases. The initial results from these trials showed regression of tumors in patients treated with rapamycin, but the tumors increased in volume after rapamycin therapy was stopped.8 At a molecular level, the potential contribution of rapamycin-insensitive aspects of mTOR signaling to disease progression,9 the existence of feedback loops that can up-regulate Akt following prolonged rapamycin treatment,10 and the observation that rapamycin monotherapy is often cytostatic rather than cytotoxic in many tumor settings11,12 has generated significant interest in developing alternatives to rapamycin for the inhibition of mTORC1 signaling.
The ribosomal protein S6 (rpS6) is a component of the translational control machinery downstream of mTORC1 and is directly phosphorylated on multiple serine residues by the 40S ribosomal protein S6-kinase in response to mTORC1 activation (Fig. 1A). rpS6 phosphorylation provides a convenient and reliable method for monitoring the activation state of the mTORC1 pathway, owing to the availability of high-quality phospho-specific antibodies. We have developed a high-throughput, cell-based assay for quantitatively measuring phosphorylation of rpS6 at S235/S236 in mammalian tissue culture cells as a readout for the activation state of the mTORC1 signaling network. This assay takes advantage of the In Cell Western (ICW) technique using the Aerius® infrared imaging system (LI-COR® Biosciences).13 The ICW procedure involves fixation and immunostaining of cells in a manner similar to standard immunofluorescence methods, with the primary difference being the requirement for specialized secondary antibodies designed for quantitative detection by the Aerius system. Additionally, in our phospho-rpS6 assay, cells are stained with Alexa-680 succinimidyl ester (Invitrogen, Carlsbad, CA), an infrared-excitable amine-reactive probe that nonspecifically labels the cytoplasm of cells and provides a measure of cell number. Our validation studies show that this Aerius-based protocol has advantages with regard to sensitivity, reproducibility, and throughput when compared to traditional immunofluorescence-based screening techniques that rely on high-throughput microscopy in the visible spectrum and thus provides a powerful tool for high-throughput screening studies.
MATERIALS AND METHODS
Cell Lines and Reagents
HeLa cells were obtained from ATCC and maintained in DMEM (CellGro Cat# 10-013-CV) supplemented with 10% fetal bovine serum (FBS; PAA Laboratories Inc., Dartmouth, MA; Cat# A15-301). Rapamycin, LY294002, and UO126 were purchased from Sigma. Hoechst 33342 and phalloidin-568 were purchased from Invitrogen. The phospho-rpS6 Ser235/Ser236 antibody was from Cell Signaling Technologies; the phospho-Erk Thr202/Tyr204 was from Sigma; the total Erk antibody was from Upstate; and the GAPDH antibody was from Ambion.
Western Blot Analysis
Preparation of cell extracts and Western blot analysis was performed using standard protocols similar to those previously described.14 In brief, cells were extracted in modified RIPA buffer (100 mM Tris pH 8.0, 300 mM NaCl, 2% IPEGAL detergent, 1% sodium deoxycholate, and 0.2% sodium dodecyl sulfate) supplemented with 10 mM EDTA, 1 mM EGTA, 1 mM sodium orthovanadate, 1 mM NaF, 2 mM phenylmethylsulfonyl fluoride, 2 mg/mL aprotinin, 2 mg/mL leupeptin, and 1 mg/mL pepstatin. Samples were resuspended in sample buffer (5×: 60 mM Tris–HCl, pH 6.8, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, 0.1% bromophenol blue) and heated to 95°C for 5 min. Samples were electrophoresed by SDS-PAGE and then transferred to nitrocellulose membrane (Whatman). The membranes were blocked with LI-COR blocking buffer diluted 1:1 with PBS (3.8 mM sodium phosphate monobasic, 16.2 mM sodium phosphate dibasic, 150 mM NaCl, pH 7.4) and probed overnight with primary antibodies followed by a 1 h incubation with goat anti-mouse IRDye-680 and goat anti-rabbit IRDye-800CW secondary antibodies (LI-COR). Blots were visualized on the Odyssey® scanner. The phospho-rpS6 Ser(235/S236) antibody was from Cell Signaling Technologies; the phospho-Erk Thr202/Tyr204 was from Sigma; the total Erk antibody was from Upstate; and the GAPDH antibody was from Ambion.
Indirect Immunofluorescence Microscopy
Immunofluorescence was performed using standard procedures similar to those described previously.15 Cells were plated on glass coverslips and incubated in 5% CO2 at 37°C. The cells were fixed with 4% formaldehyde in PBS for 15 min, followed by permeabilization with 0.2% Triton X-100 in PBS for 2 min. The cells were rinsed with PBS 4 times and incubated with a blocking solution containing 3% BSA and 0.2% Triton X-100 in PBS for 15 min. The cells were then incubated with the phospho-rpS6 Ser235/Ser236 antibody in blocking solution for 3 h. The cells were rinsed 4 times with 0.2% Triton X-100 in PBS and incubated with a donkey anti-rabbit Alexa-488 secondary antibody (Invitrogen) in blocking buffer for 1 h. DNA was visualized by Hoechst staining and actin was visualized using phalloidin-568 (Invitrogen). Cells were rinsed with PBS and examined using a fluorescence microscope.
Small Molecule Treatment
Cell dispensing was performed using a Matrix WellMate dispenser under sterile conditions in a standard tissue culture hood. For small molecule studies, HeLa cells (1,000 cells/well) were seeded in 384-well plates (Corning Inc., Corning, NY; Cat# 3712) in 30 µL DMEM/10% FBS and 1% penicillin/streptomycin (CellGro, Cat# 30-002-CI) and cultured in a standard tissue culture incubator at 37°C and 5% CO2. Twenty-four hours after plating, 100 nL of test compounds (solubilized in DMSO) were delivered by pin transfer using an Epson Compound Transfer robot. Small molecule libraries from the Harvard Medical School ICCB-Longwood Screening Facility were screened: NINDS Custom Collection, (MicroSource Discovery Systems, Gaylordsville, CT), BIOMOL ICCB Known Bioactives (BIOMOL), and the Prestwick Collection (Prestwick Chemical). The NINDS library compounds were screened at a final concentration of ∼33 µM and the Prestwick compounds were screened at a final concentration of ∼6.7 µg/mL. The BIOMOL ICCB Known Bioactives was screened at ∼17 µg/mL for most compounds and about 1.7 µg/mL for “potent” compounds. Additional details regarding these libraries can be found on the ICCB Web site (http://iccb.med.harvard.edu/). Compound incubation was carried out for 2 h at 37°C. Following compound incubation, the culture medium was aspirated using a 24-pin manifold (Drummond, Cat# 3-000-102) and the cells were fixed and processed for quantitation of the phospho-S6 signal on the LI-COR Aerius instrument (see below).
For analysis of ERK phosphorylation, cells were plated in a 384-well format as described above and cultured for 24 h in complete media. Cells were then serum-starved for 20 h prior to pin transfer of compounds by washing cells 3 times with 75 µL of starve media (serum-free DMEM supplemented with 10 mM HEPES pH 7.4 and 1% penicillin/streptomycin). After the final wash, the media was aspirated and 30 µL of starve media was added to each well. Following the 20-h serum starvation, compounds were pin-transferred as described above and the cells were incubated with compound for 2 h. Following compound incubation, the cells were then stimulated with 30 µL of 100 ng/mL EGF (Peprotech, Rocky Hill, NJ) for 10 min, fixed, and stained for quantitation of the phospho-ERK signal on the LI-COR Aerius instrument (see below). This stimulation protocol was necessary to achieve a high level of ERK phosphorylation for these control experiments, as basal ERK phosphorylation is relatively low in asynchronously growing cells.
siRNA Reverse Transfection
Cell dispensing was performed using a Matrix WellMate dispenser under sterile conditions in a standard tissue culture hood. siRNAs were SMARTpools targeting protein kinases and phosphatases (Dharmacon siARRAY siRNA Library, Human Genome G-005000-05, Thermo Fisher Scientific, Lafayette, CO; this was obtained by ICCB-L in 2006). For siRNA transfections, 1 µM siRNA stock solutions were arrayed in 384-well source plates (Eppendorf, Cat# 951020745). A sufficient volume of diluted transfection mix containing 0.14 µL Oligofectamine (Invitrogen, 12252-011) transfection reagent diluted in 8.86 µL Opti-MEM (Invitrogen, 11058-021) for each well was prepared 5 min prior to the transfection. Nine microliters of this transfection mix was aliquoted to each well of a 384-well tissue culture plate (Corning, Cat# 3712). The 1.5 µL of siRNA was delivered to each well in the transfection plate from the source plate using a Velocity 11 Bravo robot with a 384-well pipetting head. The siRNA/lipid mix was incubated for 15 min, followed by the addition of HeLa cells suspended at 800 cells/well in 20 µL of DMEM/10% FBS (no antibiotics were included for the transfection step). The final siRNA concentration after addition of cells was 50 nM. The cells were briefly centrifuged at 1,000 rpm in a standard plate centrifuge to ensure all cells and reagents were evenly plated in the wells, and the cells were maintained in a standard tissue culture incubator at 37°C and 5% CO2. Twenty hours after transfection, cells were supplemented with 55 µL of DMEM/10% FBS containing 1% penicillin/streptomycin. Seventy-two hours following transfection, the culture medium was aspirated using the Drummond 24-pin manifold, and the cells were fixed and processed for imaging on the LI-COR Aerius instrument (see below).
Cell Staining and Quantitation on the LI-COR Aerius Infrared Scanner
Media was aspirated from the 384-well plates using a Drummond 24-pin manifold that was modified to leave ∼10 µL of liquid in the well in order to prevent disruption of the cell monolayer. Cells were fixed for 20 min by the addition of 75 µL of 4% EM-grade formaldehyde (Polysciences Inc., Warrington, PA; Cat# 04018) diluted in PBS, using a Matrix WellMate dispenser. Samples were washed 4 times with 75 µL wash buffer (PBS + 0.2% Triton-X 100) using the Bio-Tek ELx405 plate washer. The cells were then stained for 15 min with 20 ng/mL Alexa-680 succinimidyl ester (Invitrogen, Cat# A-20108), an amine reactive probe that labels the cytoplasm of fixed cells. We have also used IRDye-800CW succinimidyl ester (LI-COR Biosciences, Lincoln, NE; Cat# 929-70020) for cell number staining with equivalent results. Following cell number staining, the cells were washed 4 times with wash buffer as above. Subsequently, the cells were incubated in Odyssey Blocking Buffer (LI-COR Biosciences, Cat# 927-40000) diluted 1:1 with PBS plus 0.2% Triton X-100, followed by overnight incubation with 20 µL of anti-rpS6 phospho-Ser235/Ser236 antibody (Cell Signaling Technologies, Danvers, MA; Cat# 2211) diluted 1:1,000 in blocking buffer. Following the primary antibody incubation, cells were washed 4 times with wash buffer, incubated for 1 h with an IRDye-800CW-conjugated goat anti-rabbit secondary antibody (LI-COR Biosciences; Cat# 926-32211) diluted 1:1,000 in blocking buffer, and again washed 4 times with wash buffer. A final wash step with PBS lacking detergents was performed prior to imaging the plates. In pilot studies, the following alternative cell number counter stains were used in place of the Alexa-680 succinimidyl ester staining: TO-PRO3 iodide and Alexa-680 phalloidin (Invitrogen), DRAQ5 (Cell Signaling Technology), and the goat anti-mouse IRDye-700 secondary antibody (LI-COR Biosciences). A similar procedure was used for quantitation of the phospho-ERK staining shown in Figure 2 using a monoclonal anti-MAP Kinase, activated (diphosphorylated ERK-1/2) antibody (Sigma; Cat# M9692) detected with the IRDye-800CW-conjugated goat anti-mouse secondary antibody (LI-COR Biosciences; Cat# 926-32210).
Fig. 2. .
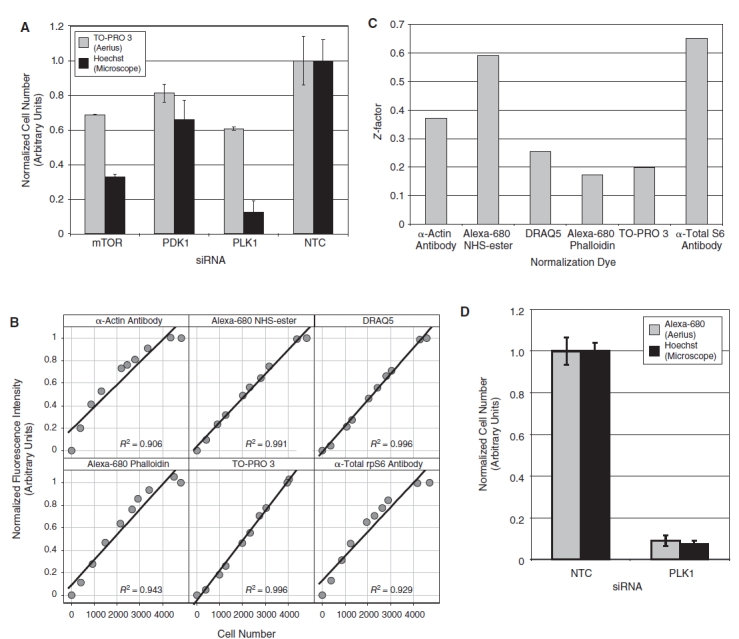
Cell number quantitation. (A) HeLa cells were transfected with the indicated siRNA, fixed, and stained with TO-PRO 3 and Hoechst. The fluorescence intensity of the TO-PRO 3 stain was measured on the 700 nm channel of the LI-COR Aerius infrared scanner (gray bars). In the same wells, Hoechst-stained nuclei were imaged on the CellWorx high-content screening microscope, and the number of cells was counted directly using the “count nuclei” module in the Metamorph image analysis software (black bars). For each staining method, the data were normalized relative to the value of the nontargeting control siRNA (n = 4, error bars show 1 standard deviation). (B) The relative fluorescence intensity of candidate cell number dyes was quantified in a 384-well format over a range of increasing cell number using the LI-COR Aerius infrared imager and plotted against cell number measured in the same samples by direct counting of Hoechst-stained nuclei as described above. Each data point represents an average of 4 replicates. Each plot is annotated with the R2 value from a linear regression of the raw data. (C) The fluorescence intensity of phospho-rpS6 staining was quantified on the 800 nm channel of the LI-COR Aerius infrared imager and normalized using the indicated counter stain as a measure of cell number. The Z′ for the normalized phospho-rpS6 stain was calculated using HeLa cells transfected with either nontargeting control siRNA or PDK1 siRNA (n = 12). The normalized phospho-rpS6 signal was used to calculate a Z′ for the assay using each of the different normalization approaches. (D) HeLa cells were transfected with either a nontargeting control siRNA or the PLK1 siRNA, fixed, and stained with Alexa-680 succinimidyl ester and Hoechst. The fluorescence intensity of the Alexa-680 succinimidyl ester stain was measured on the 700 nm channel of the LI-COR Aerius infrared scanner (gray bars). In the same wells, Hoechst-stained nuclei were imaged on the CellWorx high-content screening microscope, and the number of cells was counted directly using the “count nuclei” module in the Metamorph image analysis software (black bars). For each staining method, the data were normalized relative to the value of the nontargeting control siRNA (n = 4, error bars show 1 standard deviation).
Plates were scanned on the LI-COR Aerius infrared scanner, staining was quantified using the scanner software, and the data were exported into MS Excel worksheets for further analysis. The normalized phospho-rpS6 intensity was calculated by dividing the trimmed mean intensity for the phospho-S6 staining on the 800 nm channel by the trimmed mean intensity of the Alexa-680 stain quantified on the 700 nm channel. Dose–response data was fit to a standard 4-parameter model to determine IC50 using the MATLAB software package. Data from the cell number staining experiment as well as the small molecule and siRNA pilot screens were visualized and analyzed with the Spotfire software package. For the pilot screens, a cell viability Z-score was calculated relative to the plate median and standard deviation using the Alexa-680 trimmed mean intensity as a measure of cell number. A Z-score for rpS6-phosphorylation was calculated based on the plate median and standard deviation for the normalized phospho-rpS6 staining intensity, omitting the data from treatments that induced cytotoxicity. Cytotoxic compounds were eliminated for practical reasons; specifically, the normalization procedure involves dividing the signal on the 800 nm channel by the signal on the 700 nm channel. In wells with highly cytotoxic compounds that contain few or no cells, the intensity of the signal on the 2 channels is dominated by noise. Taking the ratio of 2 noisy signals amplifies this noise and leads to spurious results. For this reason, we applied fairly strict cytotoxicity criteria and did not calculate the normalized phospho-S6 signal for the cytotoxic wells as this value is unreliable. Regarding our goal of identifying alternative inhibitors of mTORC1 signaling that may induce apoptosis, we anticipate that such induction of apoptosis will require a much longer incubation time than the 2-h incubation time used here and will be assessed with a detailed time course of apoptosis induction in future work. All plates in the pilot screens were assayed in duplicate and results reported as the average Z-score from the 2 replicates.
RESULTS
Phosphorylation of rpS6 Provides a Robust Endpoint for Monitoring the Activation State of the mTORC1 Signaling Network
We have used an anti-rpS6 phospho-S235/S236 antibody, which reliably detects changes in the activation state of mTORC1 signaling in both immunofluorescence- and Western blot-based detection methods (Fig. 1B and 1C, respectively). The ICW method using the LI-COR Aerius scanner provides a robust high-throughput method for quantitation of immunoflourescent staining and has advantages over microscopy-based screening approaches with regard to throughput and sensitivity (see Discussion).13 For this reason, we set out to develop an ICW-based screening assay to detect rpS6 S235/S236 phosphorylation as a method for high-throughput screening for regulators of the mTORC1 signaling network.
Development of an Infrared Protocol for Cell Number Enumeration
In our ICW rpS6-phosphorylation assay, HeLa cells were fixed and processed for quantitation of cell number and phospho-rpS6 intensity (see Materials and Methods) following siRNA or small molecule treatment. Accurate determination of cell number is a critical feature of this method. Specifically, the phospho-rpS6 staining measured on the 800 nm channel of the Aerius instrument must be normalized to total cell number. Cell number is measured on the LI-COR instrument using a fluorescent probe that is detected on the 700 nm channel and used to control for differences in cell number, conceptually similar to using a loading control in a traditional western blot. In previous work, DNA-binding dyes that fluoresce in the infrared spectrum, such as TO-PRO3, have been used as normalization controls for the ICW approach.13 During the early phases of assay development, we found that the intensity of many DNA-binding dyes did not correlate well with cell number. These problems were particularly pronounced when cells were transfected with siRNAs that induced apoptotic cell death (Fig. 2A). For example, siRNA-mediated knockdown of PLK1 induced apoptosis and lead to a large reduction in cell number as measured by direct counting of Hoechst-stained nuclei, but this reduction in cell number did not result in a similar reduction in TO-PRO 3 intensity. We speculate that these discrepancies are because dye bound to the fragmented DNA characteristic of apoptotic cells exhibited dramatically different fluorescent properties than dye bound to intact chromatin of nonapoptotic cells.
To identify a more suitable counter stain for cell number normalization, we explored a number of alternative dyes with spectral properties compatible with the 700 nm channel of the Aerius instrument, including DNA-binding dyes, actin-binding dyes, nonspecific cytoplasmic dyes, and additional antibody-based stains visualized with a goat anti-mouse IRDye-700-conjugated secondary antibody. We first determined whether the intensity of these stains provided a linear measure of cell number. Increasing numbers of cells were plated in a 384-well format, fixed 24 h later, and co-stained with the candidate normalization dye and 0.5 µg/mL Hoechst. The intensity of the counter stain was quantified for each cell density using the LI-COR Aerius scanner. The number of cells in each well was also measured directly by imaging the Hoechst-stained nuclei on a Cellworx high-throughput microscope (Applied Precision Inc., Issaquah, WA) and counting the cells with the count nuclei module of the MetaExpress image analysis software (Molecular Devices, Sunnyvale, CA). The results of this analysis are plotted in Figure 2B, showing that each of these techniques provides a linear measure of cell number over a range of cell densities typically encountered in screening experiments.
The Z′ provides a statistical measure of the reproducibility of an assay, with a Z′ of 0.5 or greater generally considered suitable for high-throughput screening.16 We then calculated a Z′ for an siRNA transfection experiment in which we compared cell number normalization for a nontargeting control siRNA to an siRNA targeting PDK1 (Fig. 2C). The total S6 antibody provided the most statistically robust normalization, with a Z′ of 0.65 in this experiment but was deemed impractical for large-scale screening efforts due to the high cost of the antibody. Direct labeling of fixed cells with an infrared-excitable succinimidyl ester dye, which directly labels reactive lysine residues and other primary amines in the fixed cells, gave a Z′ of 0.59 in the experiment shown in Figure 3C. For normalization, direct labeling of fixed cells with an infrared-excitable succinimidyl ester dye is a significantly less expensive and shorter protocol than using a separate primary and secondary antibody to detect total S6 protein levels.
Fig. 3. .
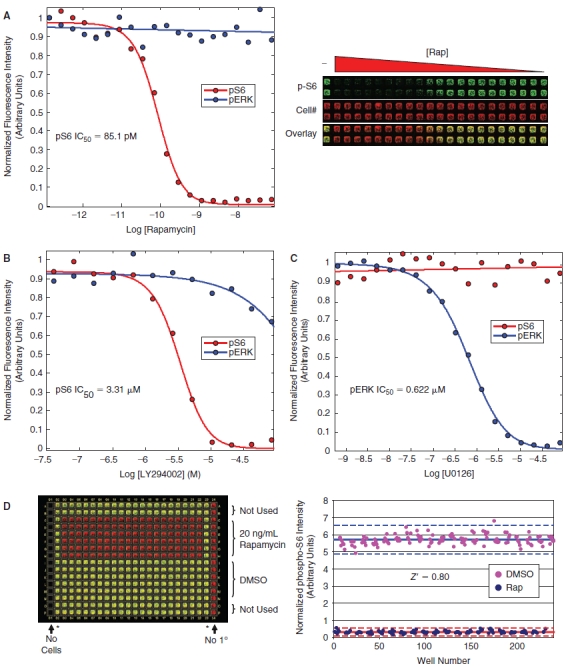
Small molecule validation data. (A) HeLa cells were treated with increasing amounts of rapamycin in a 384-well format, fixed, and stained for analysis on the LI-COR Aerius instrument using the Alexa-680 succinimidyl ester as a counter stain for cell number and the phospho-rpS6 antibody followed by an IRDye-800CW secondary antibody for quantitation of rpS6 phosphorylation. The dose-dependent inhibition of rpS6-staining was fit with a standard 4-parameter model giving an IC50 of 85.1 pM (red curve). In a parallel experiment, cells were serum-starved overnight, treated with increasing amounts of rapamycin, stimulated with EGF, fixed, and stained using the Alexa-680 succinimidyl ester and the phospho-ERK antibody followed by an IRDye-800CW secondary antibody for quantitation of ERK phosphorylation (blue curve), showing that rapamycin does not inhibit ERK phosphorylation. Each data point is an average of 4 independent experiments. The raw image from the LI-COR Aerius scanner is shown on the right. (B) A similar dose–response experiment was performed using the PI3-kinase inhibitor, LY294002, showing an IC50 of 3.31 µM for the phospho-S6 assay (red curve) (LY294002 was significantly less active toward the MAPK pathway showing an IC50 > 0.25 mM (blue curve). (C) A dose response using the MEK inhibitor, UO126, was performed showing no inhibition of rpS6-phosphorylation across a range of concentrations (red curve). In a parallel experiment, the activity of the UO126 compound was confirmed, showing a dose-dependent inhibition of ERK phosphorylation in response to EGF stimulation with an IC50 of 0.622 µM (blue curve). (D) The statistical reproducibility of the assay for small molecule screening was determined by calculating a Z′ for the assay comparing 20 ng/mL rapamycin to DMSO control in a 384-well format. The raw data for this assay is shown in the left panel, with the phospho-rpS6 signal (green) overlaid with the Alexa-680 succinimidyl ester cell number stain (red). The quantitation of the raw data is shown in the right panel and was used to calculate a Z′ of 0.80 for this experiment. The asterisks (*) denote untreated wells that were not used in the Z′ determination.
To confirm that the Alexa-680 succinimidyl ester staining correlates well with cell number, even under conditions of apoptotic cell death, and does not exhibit the problems we encountered with the upward bias of the TO-PRO 3 signal in apoptotic cells shown, we performed a similar experiment to compare the relative cell number measured by the Alexa-680 succinimidyl ester staining to the number of cells measured by the microscopy-based counting of Hoechst-stained nuclei under conditions of apoptotic cell death. The results of this experiment are shown in Figure 2D and demonstrate that the reduction in cell number induced by apoptosis in response to PLK1 knockdown as measured by direct counting of Hoechst-stained nuclei also leads to a similar change in Alexa-680 NHS-ester fluorescence intensity as measured on the LI-COR Aerius instrument. Based on these results, and the additional siRNA validation studies described below, we conclude that Alexa-680 succinimidyl ester staining provides an inexpensive and statistically robust measure of cell number and was therefore used as the cell number dye for our screening experiments. We have used both the 700DX (LI-COR) and the Alexa-680 succinimidyl ester dyes with similar results (data not shown).
Small Molecule Validation Data
In order to validate the phospho-rpS6 ICW assay for small molecule-based high-throughput screening approaches, we generated dose–response curves for known inhibitors of mTORC1 signaling, including rapamycin and LY294002, and calculated IC50 values of 224 pM and 2.5 µM, respectively (Fig. 3A and 3B). Importantly, the MEK inhibitor UO126 showed no effect on rpS6 phospho-S235/S236 staining across a range of concentrations where this compound is reported to be active against MEK (Fig. 3C). To confirm the activity of the U0126 compound and to demonstrate the specificity of rapamycin and LY294002, we performed a parallel experiment monitoring the dose-dependent effects of these compounds on EGF-stimulated ERK phosphorylation by the ICW assay. We show that the U0126 used in these experiments is able to potently inhibit the MEK-dependent, EGF-stimulated phosphorylation of ERK, with an IC50 of 0.622 µM in our ICW assay. Thus, the inhibition of rpS6-phosphorylation observed for rapamycin and LY294002 is specifically due to activity of the mTORC1 pathway, and this data along with the results in Figure 1B and 1C demonstrate that ERK/MAPK signaling does not appreciably affect the phospho-rpS6 endpoint under our assay conditions as has been shown in other cell systems.17 To demonstrate the reproducibility of the assay, we compared a 20 ng/mL (21.9 nM) rapamycin treatment to a DMSO vehicle control treatment across an entire 384-well plate as shown in Figure 3D. These data were used to calculate a Z′ of 0.80 and 0.77 in independent experiments comparing inhibition by rapamycin to DMSO, demonstrating that the ICW-based phospho-rpS6 assay is statistically robust and suitable for small molecule high-throughput screening applications.
Following validation of the phospho-rpS6 ICW assay, we performed a pilot small molecule screen using a library of 2,640 known bioactive molecules available through the Harvard Medical School ICCB-Longwood Screening Facility (ICCB-L). Each library plate was screened in duplicate and the average Z-score for both cell viability and phospho-S6 staining was calculated (see Materials and Methods). The results of this screen are summarized in Figure 4A, which plots the average Z-score measured in the phospho-S6 assay for each compound in the known bioactives library. In the pilot screen, cytotoxicity was defined as a reduction in the Alexa-680 succinimidyl ester cell number staining >3 standard deviations relative to the plate median (cell viability Z-score < −3). Twenty-seven compounds (1.0%) scored as cytotoxic on both replicate plates in the pilot small molecule screen using these criteria (yellow symbols in Fig. 4A). Inhibition of rpS6-phosphorylation was defined as a reduction in normalized phospho-S6 staining of >3 standard deviations relative to the plate median (the phospho-rpS6 Z-score < −3 cutoff is indicated by the red line in Fig. 4A). Eighteen compounds (0.68%) exhibited a Z-score < −3 for the phospho-S6 assay on both replicate plates without significantly affecting cell viability and were scored as hits (red symbols in Fig. 4A). An additional 4 compounds scored with a phospho-S6 Z-score < −3, but were not counted as hits because these compounds also scored as cytotoxic. Five compounds had an average Z-score below the −3 cutoff, but did not score as a hit on both replicate plates (accounting for the blue symbols below the −3 cutoff in Fig. 4A). Known inhibitors of the mTORC1 pathway, including the pan-kinase inhibitor staurosporine, the mTORC1 inhibitor rapamycin, and the dual PI3-kinase and mTOR inhibitor LY294002, scored among the most potent inhibitors of the pathway (green symbols in Fig. 4A), validating the ability of this method to identify known inhibitors of the mTORC1 pathway in an unbiased screen. Additional targets identified in this screen with novel activity toward the mTORC1 pathway will be characterized further in a future publication. These data are also represented as a correlation plot in Figure 4B, comparing the raw intensity of the Alexa-680 succinamidyl ester staining to the raw intensity of phospho-S6 staining. This analysis shows that staurosporine and LY294002 do not induce apoptosis under short 2 h incubation time used in the experiments and that inhibitors of mTORC1 signaling were not generally associated with a reduction in cell number. The correlation plot also shows that compounds that reduce cell number do not score as false positives in the screen, demonstrating that the normalization strategy accurately corrected for changes in the phospho-S6 signal caused by changes in cell number. The reproducibility of the ICW assay is demonstrated by plotting of the Z-score for the phospho-S6 staining from the 2 independent replicates a representative plate from the small molecule library as shown in Figure 4C (the symbols are colored as in Fig. 4A). A linear fit of the data gives an R2 of 0.91, indicative of excellent plate-to-plate reproducibility.
Fig. 4. .
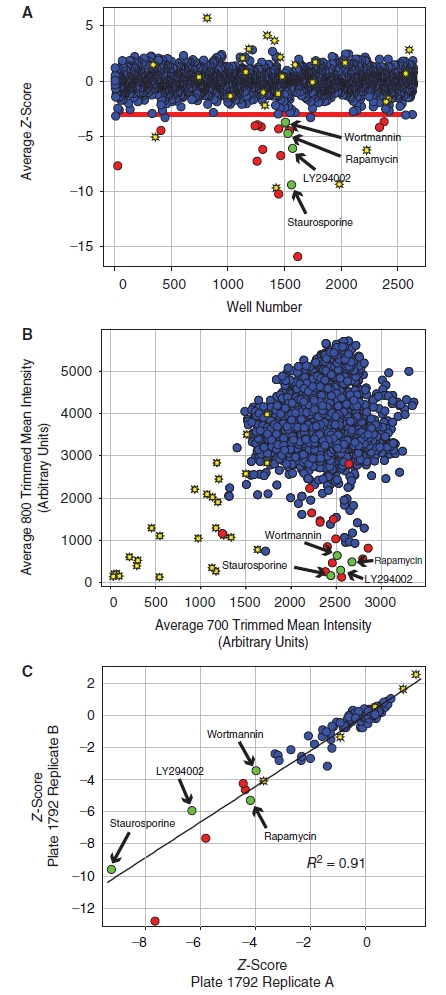
Small molecule pilot screen. (A) A library of 2,640 compounds from the ICCB-L Known Bioactives Collection was screened to identify inhibitors of rpS6-phosphorylation. The average Z-score for each compound is shown as a blue circle. The viability of cells was also measured using the Alexa-680 cell number stain, and compounds with cell numbers at least 3 standard deviations below the plate mean (Zcell# < −3) were scored as cytotoxic (yellow stars). Hits were defined as compounds that reduced normalized phospho-rpS6 staining at least 3 standard deviations below the plate mean (ZrpS6 < −3) without cytotoxicity (red circles). Known inhibitors of the pathway, including wortmannin, rapamycin, LY294002, and staurosporine, scored as hits in the screen demonstrating the validity of the approach (green circles). (B) The data from the small molecule screen is represented as a correlation plot where the x-axis shows the raw trimmed mean intensity for the Alexa-680 NHS-ester cell number staining measured on the 700 nm channel and the y-axis shows the raw trimmed mean intensity for the phospho-S6 staining measured on the 800 nm channel. The symbols are defined as in panel A. The correlation plot shows that the inhibitors identified in the screen, including the known inhibitors of mTORC1 signaling, did not generally lead to a reduction in cell number, demonstrating that the normalization strategy used for data analysis accurately corrected for changes in the phospho-S6 signal caused by changes in cell number. (C) The plate to plate reproducibility of the assay is shown for a representative plate from the screen (Plate# 1792). The Z-score for each compound on 2 independent replicate plates is shown and labeled as above. A linear fit of the data gives an R2 of 0.91, indicating good agreement between the 2 replicate plates.
siRNA Validation Data
In order to validate the phospho-rpS6 ICW assay for siRNA-based high-throughput screening approaches, we tested the ability of siRNA-mediated knockdown of known pathway components to block signaling through the pathway. Using a reverse transfection protocol in a 384-well format, we transfected a nontargeting control siRNA, siRNA pools targeting known components of the mTORC1 pathway, and an siRNA pool targeting PLK1 that is required for cell viability. The raw image from the Aerius scanner is shown on the left in Figure 5A, demonstrating a significant reduction in phospho-rpS6 staining in wells transfected with PDK1 or a pool of S6K1 and S6K2 siRNAs relative to control siRNA. The phospho-S6 staining was measured on the 800 nm channel and is shown in green, the Alexa-680 succinamidyl ester cell number staining was measured on the 700 nm channel and shown in red, and an overlay of the 2 channels is also shown. The plates were also stained with Hoechst, nuclei were imaged on the CellWorx high-content screening microscope, and the number of cells was counted directly using the “count nuclei” module in the Metamorph image analysis software package. PLK1 knockdown is well established to induce apoptosis in cancer cell lines,18 and we used PLK1 siRNA as a positive control for the Alexa-680 succinamidyl ester cell number staining. Comparison of the relative cell number measured by the Alexa-680 succinamidyl ester staining and by direct counting of the Hoechst-stained nuclei is shown on the lower right panel of Figure 5A, demonstrating a >80% reduction in cell number in response to PLK1 knockdown by both methods, again demonstrating that the Alexa-680 succinamidyl ester staining provides an accurate measure of cell number even under conditions of apoptotic cell death.
Fig. 5. .
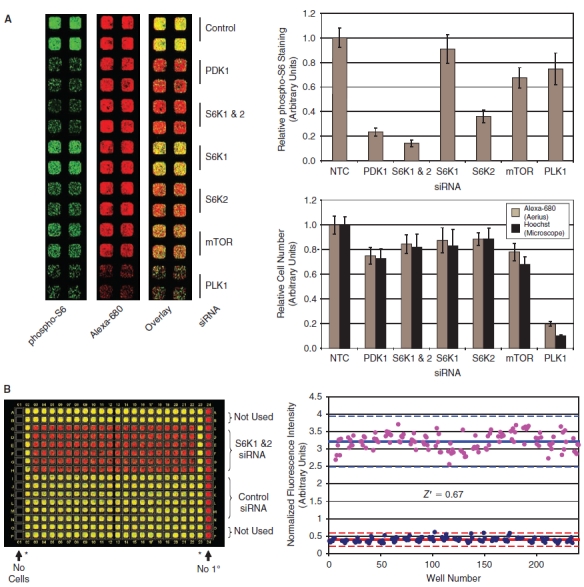
siRNA validation data. (A) HeLa cells were transfected with the indicated siRNAs. Seventy-two hours post-transfection, the cells were fixed and stained for analysis on the LI-COR Aerius instrument to quantify cell number and rpS6 phosphorylation. The raw data from the LI-COR Aerius scanner is shown for representative wells in the panel on the left. Quantitation of the normalized phospho-S6 data is shown in the graph on the upper right panel (for each treatment n = 10, error bars show 1 standard deviation). Quantitation of the Alexa-680 succinamidyl ester cell number data is shown in the graph on the lower right panel (gray bars). The plates were also stained with Hoechst; nuclei were imaged on the CellWorx high-content screening microscope; and the number of cells was also counted directly using the “count nuclei” module in the Metamorph image analysis software package (black bars). (B) The statistical reproducibility of the In Cell Western assay for siRNA screening was determined by calculating a Z′ comparing S6K1/2 siRNA to nontargeting siRNA control in a 384-well format. Quantitation of the raw data is shown and used to calculate a Z′ of 0.67 for this experiment. The asterisks (*) denote untreated wells that were not used in the Z′ determination.
PDK1 and the 2 S6-kinase isoforms (S6K1 and S6K2) are both required for phosphorylation of the rpS6 S235/S236 site (see Fig. 1A). Quantitation of the rpS6 phosphorylation data is shown in the upper right panel of Figure 5A, demonstrating a >80% reduction in the phospho-S6 staining in response to knockdown of either PDK1 or both S6-kinase isoforms. Knockdown of S6K1 or S6K2 individually results in a more modest decrease in S6-phosphorylation, demonstrating that both isoforms contribute to the S6-phosphorylation observed in HeLa cells, with S6K2 apparently playing a more significant role. Finally, an siRNA pool targeting the mTOR kinase leads to an ∼40% reduction in S6-phosphorylation, consistent with the incomplete knockdown of mTOR protein achieved by this siRNA (data not shown).
To demonstrate the reproducibility of the assay for siRNA screening in a 384-well format, we compared knockdown of S6K1 and S6K2 to transfection of a nontargeting control siRNA across an entire 384-well plate as shown in Figure 5B. These data were used to calculate a Z′ of 0.67 and 0.65 in independent experiments, demonstrating that the ICW-based phospho-rpS6 assay is statistically robust and suitable for siRNA screening applications. Following validation of the phospho-rpS6 ICW assay for siRNA screening, we performed a pilot screen using a library of 779 siRNA pools targeting protein kinases and phosphatases (see Materials and Methods). The results of this pilot screen are shown in Figure 6, which shows the average Z-score calculated form the phospho-S6 assay for each siRNA in the screen arranged in order of hit strength. siRNAs that inhibit in the phospho-S6 assay were defined as those that result in a change in the normalized rpS6-phosphorylation of ±2 standard deviations relative to the plate median on both replicate plates (the phospho-rpS6 cutoff is indicated by the red lines at a Z-score of ±2 SD in Figure 6; hits are indicated with red symbols). Using these criteria, 38 genes (4.9%) of the siRNAs in the kinase/phosphatase siRNA library score as inhibitors in the screen and are expected to target genes required for S6-phosphorylation. Importantly, siRNAs targeting known regulators of mTORC1 signaling, including PDK1, S6K2, Akt2, and IGF1R, scored among the most potent inhibitors of the pathway in this pilot screen (Fig. 6), validating the ability of this method to identify known components of the mTORC1 pathway in an unbiased screen. Similar to the small molecule screen, cytotoxic siRNAs were defined as targets with a reduction of cell number of >3 standard deviations below the plate median on both replicate plates (cell viability Z-score < −3, yellow symbols in Fig. 6). Fourteen genes (1.8%) in the kinase/phosphatase library were required for cell viability as measured by a statistically significant reduction in the Alexa-680 succinamidyl ester cell number staining. To confirm the hits identified in our primary screen, a secondary assay was preformed using the ICW phospho-S6 assay to test 4 individual siRNAs against each target identified in the primary screen. The results of this validation experiment for siRNAs that lead to inhibition of rpS6 phosphorylation are shown in Figure 6B. siRNAs that result in a >2-fold reduction in rpS6-phosphorylation relative to the nontargeting control siRNA were scored as hits. Based on this analysis, we identified 15 high confidence targets that retested with 2 or more individual siRNAs. Further characterization of novel targets will be described in a subsequent publication.
FIG. 6. .
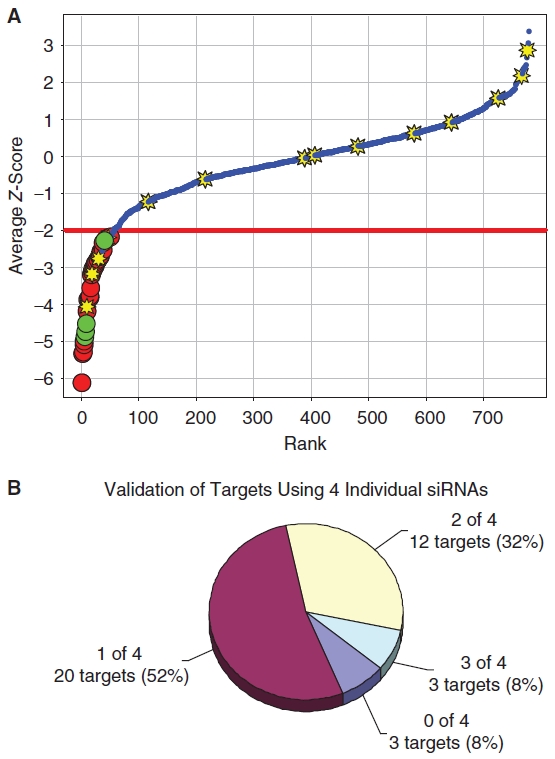
Kinase and phosphatase siRNA library screen. (A) The Dharmacon Human Kinase and Phosphatase library of 779 siRNAs targeting all known kinases and phosphatases was screened to identify inhibitors of rpS6-phosphorylation. The average Z-score for each siRNA is shown as a blue circle. The viability of cells was also measured using the Alexa-680 cell number stain, and compounds with cell numbers at least 3 standard deviations below the plate mean (Zcell# < −3) were scored as cytotoxic (yellow stars). Hits were defined as compounds that reduced normalized phospho-rpS6 staining at least 2 standard deviations below the plate mean (ZrpS6 < −2) without cytotoxicity (red circles). Known components of the mTORC1 signaling pathway (green circles), including PDK1, S6K2, Akt2, and IGF1R, scored as hits, demonstrating the ability of the approach to identify known regulators of mTORC1 signaling in an unbiased screen. (B) To confirm the hits identified in our primary screen, a secondary assay was preformed using the ICW phospho-S6 assay to test 4 individual siRNAs against each target. The chart shows the number of hits from the primary screen that lead to a >2-fold reduction in phospho-S6 staining relative to the negative control with either 0, 1, 2, or 3 individual siRNAs (none of the targets retested with 4 of 4 individual siRNAs). Based on these results, we identified 15 high-confidence targets (40% of the hits from the primary screen) that retested with at least 2 or more individual siRNAs.
DISCUSSION
The results presented here demonstrate that the ICW assay provides a robust high-throughput method for the quantitative measurement of rpS6-phosphorylation in response to mTORC1 activation that can be applied to both small molecule- and siRNA-based screening approaches to identify inhibitors of mTORC1 signaling. The cell-based ICW method described here provides a complimentary approach to previously published biochemical approaches that rely on in vitro kinase assays to identify inhibitors of the purified mTORC1 complex.19 From the perspective of small molecule screening, an advantage of the cell-based ICW assay described here is the potential to identify inhibitors of upstream signaling pathways required for mTORC1 activation, including the growth factor and amino acid signaling pathways, as well as inhibitors of mTORC1 catalytic activity. This may be particularly useful for identifying inhibitors of poorly characterized aspects of the mTORC1 network including components of the amino acid sensing pathway. Cell-based approaches also have the advantage of identifying inhibitors that are cell permeable and can be used to eliminate compounds that have undesirable cytotoxic effects. As demonstrated in the current manuscript, an additional advantage of cell-based approaches is their applicability to both small molecule- and siRNA-based screens.
The ICW-based approach provides an attractive alternative to high-throughput microscopy for applications involving immunoflouresence-based detection. The most significant advantage over high-throughput microscopy is the rapid throughput of the Aerius scanner used for the ICW method. In our experience, using the phospho-rpS6 ICW assay, acquisition and quantitation of an entire 384-well plate can be accomplished in 5–10 min on the Aerius scanner. This is in contrast to acquisition times of 2–3 h for a similar experiment using an Alexa-488 conjugated secondary antibody for detection of the phospho-S6 signal on a high-throughput microscope in the visible spectrum. In addition, the high sensitivity of the Aerius scanner can reliably detect signals using lower primary antibody concentrations than are required for a microscopy-based system, significantly reducing the cost associated with a large-scale screen. Microscopy- and ICW-based detection of rpS6-phosphorylation are similarly robust statistically (data not shown). Near-IR dyes have a number of advantages over shorter wavelength dyes that fluoresce in the visible spectrum including less light scattering, reduced fluorescence form autofluorescence from cellular components, and less signal loss due to absorbance. We found that these advantages of the near-IR dyes provided robust signals at lower antibody dilutions than were possible using dyes in the visible spectrum. Moreover, the ICW approach provides well-based, rather than cell-based data, which dramatically reduces time required for image acquisition and analysis as compared to high-resolution imaging-based approaches. Given the advantages with regard to throughput and antibody concentration, we find the ICW assay is superior for applications where high-resolution information is not a central feature of the assay. The ICW method can readily be adapted to any antibody that is suitable for immunofluorescence-based detection and provides a powerful tool for cell-based high-throughput screening applications using activation state-specific antibodies.
ACKNOWLEDGMENTS
The authors would like to thank Stewart Rudnicki, Sean Johnston, and the entire staff of the ICCB-L for their outstanding technical support. We thank Peter Johnson of LI-COR Biosciences for outstanding technical support and valuable discussions during the assay development. We also thank the members of the Blenis laboratory for advice and support as well as Stephanie Walker for helpful discussions during preparation of the manuscript. Greg Hoffman was supported by fellowships from the Helen Hay Whitney Foundation and the LAM Foundation. This work was supported by NIH Grants CA46595 and GM51405 to John Blenis and through a User Innovation Grant from LI-COR Biosciences.
ABBREVIATIONS:
- ICW
In Cell Western
- LAM
lymphangioleiomyomatosis
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- mTORC2
mammalian target of rapamycin complex 2
- rpS6
ribosomal protein S6
- TSC
tuberous sclerosis complex.
Contributor Information
Gregory R. Hoffman, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts.
Nathan J. Moerke, ICCB–Longwood Screening Facility, Harvard Medical School, Boston, Massachusetts.
Max Hsia, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts..
Caroline E. Shamu, ICCB–Longwood Screening Facility, Harvard Medical School, Boston, Massachusetts.
John Blenis, Department of Cell Biology, Harvard Medical School, Boston, Massachusetts..
REFERENCES
- 1.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–484. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 2.Frias MA, Thoreen CC, Jaffe JD, Schroder W, Sculley T, Carr SA, et al. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol. 2006;16:1865–1870. doi: 10.1016/j.cub.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 3.Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, et al. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 4.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci USA. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choo AY, Blenis J. TORgeting oncogene addiction for cancer therapy. Cancer Cell. 2006;9:77–79. doi: 10.1016/j.ccr.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 8.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 10.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 11.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 12.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 13.Chen H, Kovar J, Sissons S, Cox K, Matter W, Chadwell F, et al. A cell-based immunocytochemical assay for monitoring kinase signaling pathways and drug efficacy. Anal Biochem. 2005;338:136–142. doi: 10.1016/j.ab.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 14.Yoon SO, Shin S, Liu Y, Ballif BA, Woo MS, Gygi SP, et al. Ran-binding protein 3 phosphorylation links the Ras and PI3-kinase pathways to nucleocytoplasmic transport. Mol Cell. 2008;29:362–375. doi: 10.1016/j.molcel.2007.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murphy LO, Smith S, Chen RH, Fingar DC, Blenis J. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol. 2002;4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 16.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 17.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Erikson RL. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA. 2003;100:5789–5794. doi: 10.1073/pnas.1031523100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toral-Barza L, Zhang WG, Lamison C, Larocque J, Gibbons J, Yu K. Characterization of the cloned full-length and a truncated human target of rapamycin: activity, specificity, and enzyme inhibition as studied by a high capacity assay. Biochem Biophys Res Commun. 2005;332:304–310. doi: 10.1016/j.bbrc.2005.04.117. [DOI] [PubMed] [Google Scholar]