

Abstract
Autophagy, the major degradative pathway for organelles and long-lived proteins, is essential for the survival of neurons. Mounting evidence has implicated defective autophagy in the pathogenesis of several major neurodegenerative diseases, particularly Alzheimer's disease (AD). A continuum of abnormalities of the lysosomal system has been identified in neurons of the AD brain, including pathological endocytic pathway responses at the very earliest disease stage and a progressive disruption of autophagy leading to the massive buildup of incompletely digested substrates within dystrophic axons and dendrites. In this review, we examine research on autophagy in AD and evaluate evidence addressing the specific step or steps along the autophagy pathway that may be defective. Current evidence strongly points to disruption of substrate proteolysis within autolysosomes for the principal mechanism underlying autophagy failure in AD. In the most common form of familial early onset AD, mutant presenilin 1 disrupts autophagy directly by impeding lysosomal proteolysis while, in other forms of AD, autophagy impairments may involve different genetic or environmental factors. Attempts to restore more normal lysosomal proteolysis and autophagy efficiency in mouse models of AD pathology have yielded promising therapeutic effects on neuronal function and cognitive performance, demonstrating the relevance of autophagy failure to the pathogenesis of AD and the potential of autophagy modulation as a therapeutic strategy.
Introduction
Alois Alzheimer's discovery of neurofibrillary tangles, and their presence together with senile plaques in the brain of a patient with progressive dementia, established the two neuropathologic features that still define Alzheimer's disease (AD). Coinciding with these seminal studies were more detailed descriptions of senile plaques by Marinesco, Fischer, and even Ramon y Cajal (Garcia-Marin et al., 2007; Goedert, 2009), which emphasized the widespread incidence of gross focal swellings of axons and possibly dendrites, termed dystrophic neurites, that were particularly profuse within senile plaques but were also seen throughout affected regions of the parenchyma. Although dystrophic neurites were originally detected by Alzheimer from their argyrophilia, reflecting pathologically hyperphosphorylated cytoskeletal proteins, dystrophic swellings in the AD brain were subsequently shown to be filled mainly with autophagic vacuoles-vesicular compartments of the autophagic-lysosomal pathway (Nixon et al., 2005). In well-preserved biopsied AD neocortex, ultrastructural and immunogold labeling studies have distinguished not only dense lysosomes (Terry et al., 1964) but also various types of autophagic vacuoles (AVs) including autophagosomes, amphisomes, multilamellar bodies, and autolysosomes, representing “intermediate” stages in the progression of autophagy (Nixon et al., 2005). These observations suggested that the normally efficient autophagic process in neurons is stalled in AD. In recent studies, investigators have begun to pinpoint the site or sites at which autophagy may be disrupted and explore the possibility that modulating specific steps in this process could have a therapeutic impact on pathology and function in AD mouse models. In this review, we consider evidence indicating that neuronal autophagy is defective in AD and evaluate data addressing mechanisms underlying this defect and possible implications for the pathogenesis and therapy of AD.
The stages of autophagy
The term autophagy refers to all processes in which components of the cell are degraded in lysosomes/vacuoles and recycled (Klionsky et al., 2010). Based on how substrates are delivered to the lysosomal compartment, autophagy is classified into three general subtypes in most mammalian cells: chaperone-mediated autophagy (CMA), microautophagy and macroautophagy (Cuervo, 2004; Mizushima et al., 2008). In CMA (Cuervo, 2010), cytosolic proteins containing a KFERQ motif are selectively targeted to the lysosomal lumen for degradation. In a second process called microautophagy, small quantities of cytoplasm are non-selectively introduced into lysosomes when the lysosomal membrane invaginates and pinches off small vesicles for digestion within the lumen. Finally, a third pathway, macroautophagy, conserved from yeast to mammals, mediates large-scale degradation of cytoplasmic constituents including organelles (He and Klionsky, 2009). Macroautophagy, referred to by the general term autophagy in this review, is considered the greatest contributor to the overall autophagy rate under most conditions. This process is regulated by signaling cascades mediated through the liver kinase B1 (LKB1)/AMP-activated protein kinase (AMPK) or the Class I phosphatidylinositol 3-kinase (PI3K)/Akt pathways which converge on the mammalian target of rapamycin (mTOR) kinase through TSC/Rheb (tuberous sclerosis complex/Ras homolog enriched in brain) (Hay and Sonenberg, 2004; He and Klionsky, 2009). The inhibition of mTOR sets in motion a sequence of events coordinated by complexes of autophagy-related (Atg) proteins that initiate the formation of the autophagosome (Diaz-Troya et al., 2008; Levine and Kroemer, 2008).
Autophagy begins when an ‘isolation membrane’ is created from a pre-autophagosomal structure (PAS) or phagophore, such as the ER-derived cup-shaped omegasome, and sequesters a region of cytoplasm to form a double-membrane-limited autophagosome (Axe et al., 2008; Geng et al., 2010; Hailey et al., 2010; Hayashi-Nishino et al., 2009; Itakura and Mizushima, 2010; Ravikumar et al., 2010; Tian et al., 2010; Xie and Klionsky, 2007). The outer membrane of the autophagosome then fuses with a lysosome or a late endosome to form either an autolysosome or an amphisome, respectively, thereby initiating the digestion of sequestered material by a range of acidic hydrolases (Eskelinen, 2005; Fader and Colombo, 2009; Gordon and Seglen, 1988; Liou et al., 1997; Noda et al., 2009). Acidification of autolysosomes by vacuolar [H+] ATPase (v-ATPase), a proton pump assembled on the lysosome membrane, is crucial for activating cathepsins and effecting proteolysis of substrates (Yoshimori et al., 1991). The completion of substrate digestion within autolysosomes ultimately yields lysosomes, which are smaller, less dense vesicles containing mainly lysosomal hydrolases (Figure 1) (Nixon, 2007; Yu et al., 2010). While the term “maturation” is often used to describe the fusion between autophagosomes and lysosomes/late endosomes (Noda et al., 2009), maturation is a continuous process that includes completion of substrate degradation within autolysosomes and the restoration of lysosomes. The functional relationship among different stages of autophagy is underscored by the recent finding that the beginning of this process (i.e., autophagosome formation) and its end (the reformation of lysosomes) are both regulated by mTOR (Yu et al., 2010). In addition to referring to the specific subtypes of autophagy-related vesicular structures, we will also use the term autophagic vacuoles (AVs) as the general term for autophagy-related vesicular structures that include autophagosomes, amphisomes, multilamellar bodies, and autolysosomes.
Figure 1.
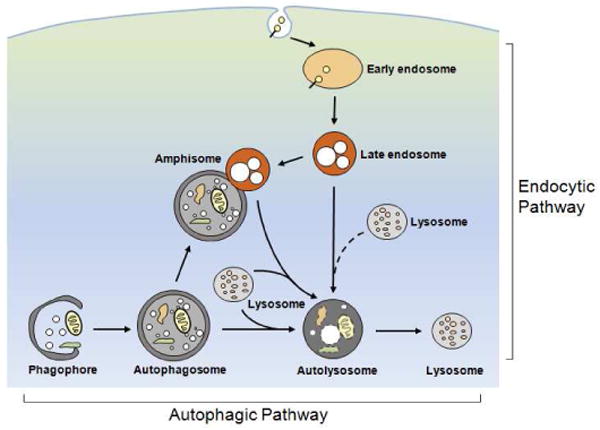
Schematic illustrating the endocytic and the autophagic pathways to the lysosome. Internalized materials entering the endocytic pathway are directed to early endosomes, which mature to late endosomes/multivesicular bodies. In the autophagic pathway, a phagophore sequesters an area of cytoplasm containing organelles to form a double membrane-limited autophagosome. The formation of amphisomes via the fusion between autophagosomes and late endosomes/multivesicular bodies provides an interactive point between the two pathways. Autophagosomes, and amphisomes in the two pathways receive hydrolases by fusing with lysosomes to form autolysosomes or late endosomes/multivesicular bodies to form amphisomes. Efficient digestion of substrates within these compartments yields lysosomes containing mainly acid hydrolases.
Since its very early descriptions, autophagy has been defined as the lysosomal digestion of a cell's own cytoplasmic material, and not simply as the sequestration of these components. Appreciation of this notion has become particularly critical to understanding how autophagy may be involved in disease states because impairments of any of the steps in autophagy may result in a diminished turnover of specific autophagy substrates. Identifying which step along the autophagy pathway may be defective in a given neurodegenerative disease, therefore, requires an evaluation of autophagosome formation, autophagosome clearance by lysosomes, and autophagic flux—the rate at which a given substrate cycles through the entire autophagy process. Autophagic flux reflects a dynamic balance between the rates of substrate sequestration and degradation (Figure 2) (Chu, 2006; Wong and Cuervo, 2010).
Figure 2.
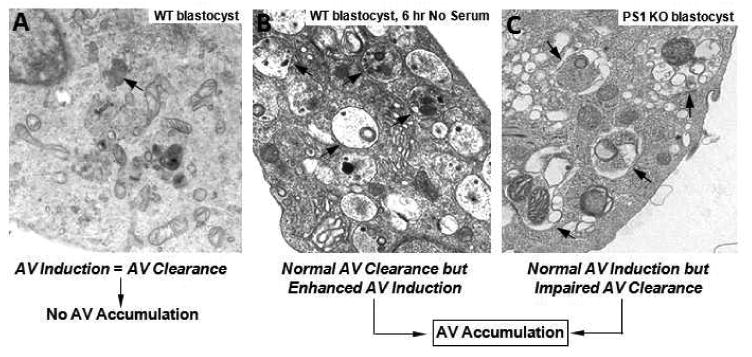
Autophagy homeostasis requires a delicate balance between autophagosome formation and clearance. Despite a constitutive level of autophagic protein turnover, normal cells in their basal state display dense lysosomes (arrow) but relatively few autophagic vacuoles (A). Strong autophagy induction, in this case by acute serum deprivation, elicits the robust formation of autophagic vacuoles (AVs)(arrows) because autophagosome formation now exceeds the rate of AV clearance (B). The loss of the capacity to properly acidify lysosomes, as it exists in the blastocyst-lacking presenilin 1, markedly slows lysosomal proteolysis and AV clearance without altering autophagy induction (Lee et al., 2010), and AVs (arrows) robustly accumulate (C). Although both experimental conditions induce AV accumulation, albeit with somewhat different composition, the rate of autophagic protein turnover is much higher than normal in the serum-deprived cells and much lower than normal in the PS1 KO cells.
Autophagy pathology in the AD brain is extensive
Abnormalities of lysosomal system function in the AD brain range from the earliest known disease-specific pathology in the disease, which involves the endocytic pathway (Nixon and Cataldo, 2006), to the most abundant pathology in the AD brain involving the autophagic pathway, which is the main focus of this review. The autophagy pathology of AD, including accumulated AVs of all types and the numbers of dystrophic neurites containing these AVs, is uniquely extensive when compared to that in other aging-related neurodegenerative diseases (Nixon and Cataldo, 2006; Nixon et al., 2005). The near complete replacement of normal cytoplasmic contents by AVs in dystrophic neurites and the increased frequency of AVs in less extensively affected neurites (Figure 3), together with the uniquely high number of dystrophic neurites in senile plaques and elsewhere throughout the AD brain, represents an enormous “burden” of undigested or partially digested proteins. This burden of waste proteins is comparable to that seen in certain primary lysosome storage disorders (LSDs), which are associated with severe neurodegeneration in early life (Nixon et al., 2008). Interestingly, neurofibrillary tangles and increased amyloidogenic processing of amyloid precursor protein (APP) as well as neuritic dystrophy, have been identified in certain primary LSDs as well as AD and potentially represent clues about common pathogenic mechanisms (see below). Autophagy-related pathology, including lesser degrees of AV “storage” in neurons, has also been increasingly recognized in other late-onset neurodegenerative diseases (Anglade et al., 1997; Liberski et al., 1995; Rudnicki et al., 2008; Yue et al., 2002; Zhou et al., 1998), although the severity and extent of the neuritic dystrophy in AD (Figure 3) (Masliah et al., 1993; Nixon et al., 2005; Schmidt et al., 1994; Suzuki and Terry, 1967) distinguishes it from these other aging-related neurodegenerative diseases (Benzing et al., 1993).
Figure 3.
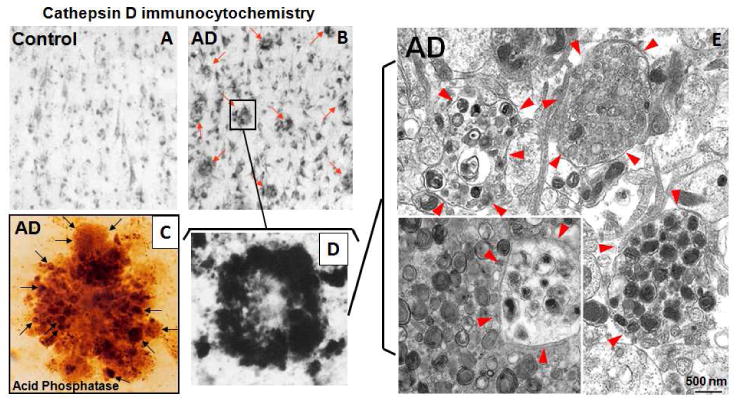
The “burden” of AVs containing incompletely digested proteins stored in dystrophic neurites of the AD brain is great. At the light microscopic levels, CatD immunostaining in the AD brain (B) but not in the control brain (A) reveals a high density of senile plaques (B, arrows) each containing dozens of dystrophic neurites visible after histochemical staining for acid phosphatase (C, arrows) or by CatD immunocytochemistry (D). (E) Ultrastructural analysis of dystrophic neurites (circled by arrowheads) demonstrates that dystrophic neurites predominantly contain hundreds of AVs of distinct subtypes but the majority have double membranes with electron dense lumens, most consistent with autolysosomes and amphisomes in which proteolysis of substrates, including inner membrane, has stalled. Given the number of dystrophic neurites in the AD brain, the amount of “stored” wasted proteins is enormous, rivaling that in certain primary lysosomal storage disorders.
It is noteworthy that the composition of organelles within dystrophic swellings in the AD brain also differs from that seen in the many other disorders characterized by neuroaxonal dystrophy. In these other conditions vesicles and cytoskeletal elements of all types are relatively abundant, resembling the pattern seen after axonal transport is focally interrupted by nerve ligation or toxic agents (Griffin and Watson, 1988; Schmidt and Plurad, 1985; Wirtschafter et al., 1977), suggesting a general failure of the axonal transport system. By contrast, a more selective transport deficit affecting mainly autophagy-related compartments is seen in mouse and cell models of AD pathology. Indeed, recent live-imaging studies of cortical neurons from our laboratory demonstrate that inhibiting lysosomal proteolysis, as believed to occur in AD neurons, selectively disrupts the axonal transport of autophagy-related compartments, causing an AD-like axonal dystrophy characterized by selective AV accumulation (Sooyeon Lee and Ralph Nixon, unpublished results). Therefore, a general disruption of axonal transport, as often accompanies neurodegeneration, cannot account for the particular pattern of organelle accumulation within dystrophic neurites in AD. Defective autophagic lysosomal proteolysis, as discussed below, is likely to contribute to the development of this pathology.
Autophagy in normal neurons is constituently active, inducible and highly efficient
Autophagic vacuoles are uncommon in neurons of the healthy brain (Cota et al., 2006; Mizushima et al., 2008; Mizushima et al., 2004; Nixon et al., 2005). These observations initially led some investigators to assume that autophagy is relatively inactive in neurons unless it is induced, although this now seems unlikely. Autophagosomes are “intermediates” in a multi-step pathway and their presence in cells depends on both their rate of formation and rate of clearance by lysosomal degradation (Figure 2). As long as their clearance matches or exceeds production, autophagosomes or other AV “intermediates” may be rarely detected, even though autophagy flux—the net rate of formation and clearance of autophagosomes—could be very rapid (Boland et al., 2008; Chu, 2006). Illustrative of this point is the observation in cultured primary neurons that blocking autophagosome clearance by inhibiting cathepsins causes rapid AV accumulation without altering autophagy induction (Boland et al., 2008). These observations indicate that autophagy is constitutively active in neurons and that efficient clearance of autophagosomes keeps their numbers low. Consistent with this concept, deletion of genes essential for autophagy results in the accumulation of protein aggregates and neuronal cell death implying that constitutive autophagy is essential for both normal protein turnover and for neuron survival (Hara et al., 2006; Komatsu et al., 2006).
Recent studies suggest that both autophagic flux and the magnitude of the autophagy induction in response to classical autophagy inducers may vary in different types of neurons. In primary cortical neurons (Boland et al., 2008), LC3 and AVs accumulate when lysosomal proteolysis is inhibited but this accumulation is dramatically increased when autophagy is also induced by rapamycin or starvation. By contrast, primary striatal neurons exhibit a significant level of constitutive autophagic flux but little additional response to rapamycin (Tsvetkov et al., 2010; Tsvetkov et al., 2009). Based on these observations, Tsvetkov et al. have suggested that neurons may be poorly responsive to rapamycin and related conventional autophagy inducers (Tsvetkov et al., 2010). Other investigators, however, have shown that neurons form autophagosomes and, more importantly, accelerate autophagic flux (Rubinsztein and Nixon, 2010) after rapamycin (Alirezaei et al., 2008; Boland et al., 2008; Crews et al., 2010; Rose et al., 2010) and starvation (Du et al., 2009). At least a partial explanation for the different conclusions may lie in observations that the clearance of LC3-positive autophagosomes in cortical neurons is very efficient even after strong induction (Boland et al., 2008). Neurons exposed to rapamycin or starvation alone accumulate mainly late AVs (i.e., autolysosomes) containing extensively digested substrates and therefore exhibit only modest elevations of LC3. However, when autophagic flux is measured in the presence of the cathepsin inhibitor leupeptin, LC3 compartments accumulate robustly, showing that induction of autophagy is, indeed, quite robust (Boland et al., 2008). It is likely, however, that autophagy is regulated differently in neuronal subtypes, glial cells and non-neuronal cells and future analyses of this issue, which require assessments of autophagy with and without lysosomal inhibitors, will be instructive to elucidate canonical and non-canonical autophagy pathways and their relative activities.
Autophagy in AD is principally defective at the stage of autolysosomal proteolysis
Converging lines of evidence indicate that a disruption of the proteolytic clearance of AVs by lysosomes is the principal basis for the massive neuronal accumulation of AVs in the AD brain. Even when autophagy is strongly induced in primary cortical neurons by rapamycin, autophagosomes can rapidly mature to autolysosomes and then to electron-translucent lysosomes without a major buildup of “intermediate” AVs (Boland et al., 2008). By contrast, the AVs accumulating in the AD brain are electron-dense autolysosomes and autophagosomes filled with undigested or partially digested substrates (Nixon et al., 2005). These substrate-filled intermediates accumulate in AD neurons even though the expression of lysosomal hydrolases is strongly upregulated (Cataldo et al., 1995; Cataldo et al., 1996) and the abundant lysosomes in dystrophic neurites are able to fuse with accumulating AVs. A similar pattern of AV accumulation and neuritic dystrophy is observed when lysosomal degradation is inhibited by deleting one or more cathepsins (Felbor et al., 2002; Koike et al., 2000; Koike et al., 2005) or by using cysteine protease inhibitors or general lysosomal enzyme inhibitors in vivo (Bednarski et al., 1997; Boland et al., 2008; Ivy et al., 1989; Ivy et al., 1984; Takeuchi and Takeuchi, 2001; Yang et al., 2008a). In cathepsin L deficient cells, for example, electron-dense autolysosomes accumulate even though autophagy is not induced and the formation of autophagosomes and fusion with lysosomes are minimally affected (Dennemarker et al., 2010), consistent with the results of Boland et al. (2008) in neurons.
A critical role for lysosomal proteolytic failure in the development of AD-related neurodegeneration is further supported by studies of primary LSDs – a group of more than 50 genetic disorders caused by defective lysosomal and non-lysosomal proteins, resulting in accumulation of autophagic and endosomal substrates. While the effects resulting from the deficiency of a specific lysosomal protein in a given LSD are ubiquitous, some LSDs, such as GM1 and GM2 gangliosidoses, Niemann Pick type C disease (NPC) and neuronal ceroid-lipofuscinosis (NCL), are associated with prominent nervous system degeneration each involving disruption of the internal environment of the lysosome by dissimilar lysosomal factors (Jeyakumar et al., 2005; Walkley, 1998; Walkley, 2009). Moreover, similarities are being identified between the cellular pathologies of AD and of certain LSDs (Chevrier et al., 2010; Eckhardt, 2010; Fukuda et al., 2006; Koike et al., 2005; Settembre et al., 2008). For instance, neurofibrillary tangles, a diagnostic hallmark of AD, are seen in NPC and mucopolysaccharidosis type IIB (Ohmi et al., 2009) and in a mouse model of this disease (Ryazantsev et al., 2007), in addition to the profuse AV accumulations seen also in AD brains. In NPC, which arises from a defect in endosomal trafficking of cholesterol and the accumulation of unesterified cholesterol in late endosomes/lysosomes, additional features overlap with those in AD, including highly disease-selective abnormalities of endosomes, elevated levels of the beta-site APP-cleaving enzyme (BACE), and BACE-cleaved carboxyl-terminal fragments of APP (βCTF), and mild deposition of the amyloid-β peptide (Aβ). Allele-selective influences of apolipoprotein E (APOE) genotype on pathology are also common to both NPC and AD (Nixon, 2004).
AD genetics directly support the pathogenic importance of lysosomal proteolytic failure in AD
a. Role of presenilin 1 (PS1) in autophagic-lysosomal proteolysis
Presenilin 1 (PS1) mutations are the most common cause of early-onset familial AD (FAD) (Sherrington et al., 1995). PS1, a ubiquitous transmembrane protein, has diverse biological roles in cell adhesion, apoptosis, neurite outgrowth, calcium homeostasis and synaptic plasticity (Shen and Kelleher, 2007). Most of these roles involve PS1 as the catalytic subunit of the γ-secretase complex that mediates intramembrane cleavage of over 20 known substrates, including APP. However, PS1 also has γ-secretase-independent roles, which notably include a critical role in lysosome acidification that is essential for the activation of lysosomal proteases during autophagy. A failure to degrade autophagy substrates and clear AVs in PS1-null blastocysts and neurons of mice conditionally depleted of PS1 was recently established on the basis of abnormally elevated levels of autophagy substrates (Esselens et al., 2004; Wilson et al., 2004), inhibited macroautophagic turnover of radiolabeled proteins, impaired cathepsin maturation, and reduced cathepsin-specific activities. All of these effects of PS1 deletion are the outcomes of the loss of a specific role of PS1 in the acidification of autolysosomes/lysosomes. PS1 deficiency is associated with the failure of the V0a1 subunit of v-ATPase to become N-glycosylated in the ER, rendering it susceptible to increased degradation before it can be delivered to autolysosomes/lysosomes at levels sufficient to support normal lysosomal acidification (Lee et al., 2010).
Of particular clinical importance, fibroblasts from patients with FAD caused by PS1 mutations also exhibit markedly defective lysosome acidification and autolysosome maturation, similar in mechanism to that seen in PS1-null cells (Lee et al., 2010). Given that autophagy is essential for neuron survival (Mizushima et al., 2008), the marked autophagy impairment in PS1-null or PS-FAD fibroblasts can account for observations that PS1 mutations promote cell death in injured neurons (Chui et al., 1999; Guo et al., 1997). This defect of lysosomal function leading to inefficient protein/peptide clearance can also provide a basis for earlier observations that PS1 mutations in the brains of patients with PS-FAD or mouse models potentiate autophagic-lysosomal, amyloid, and tau pathologies as well as accelerate neuronal cell death (Cataldo et al., 2004).
b. Amyloid Precursor Protein (APP) and ApoE
Additional AD-related genes and environmental risk factors disrupt lysosomal system function and also support a critical role of lysosomal failure in the progression of AD. Severe autophagy neuropathology develops in all forms of AD and also in mouse models of AD where only FAD-related mutant forms of APP are overexpressed, although the onset is often later than that in corresponding disease variants involving PS1 mutations. For example, autophagiclysosomal pathology in TgCRND8 mice overexpressing human APP with two FAD mutations (Chishti et al., 2001; Yang et al., 2008b; Yang et al., 2011) is nearly as florid as that seen in the PS/APP mouse model of AD (Cataldo et al., 2004; Yu et al., 2005), indicating that mutant APP overexpression alone can lead to autophagic-lysosomal pathology. Hippocampal and cortical neurons in these mice develop strikingly enlarged autolysosomes containing incompletely digested materials, including membranous structures and a minor lipopigment component, resembling the ceroid-containing AVs seen in the brain when one or more cathepsins are deleted (Koike et al., 2005) or inactivated pharmacologically (Bednarski et al., 1997; Ivy et al., 1989; Takeuchi and Takeuchi, 2001). Failed autophagic digestion is also evidenced by the accumulation of undigested autophagy substrates including LC3-II, ubiquitinated proteins, and Aβ peptides in AVs and lysosomes isolated from the brains of these mice. Moreover, stimulating lysosomal proteolytic efficiency in the TgCRND8 mice by deleting an endogenous inhibitor of lysosomal cysteine proteases (cystatin B), rescues the lysosomal pathology, eliminates abnormal autolysosomal accumulation of autophagy substrates including Aβ and ubiquitinated proteins, decreases extracellular amyloid deposition and total brain Aβ40/42 levels, and ameliorates learning and memory deficits (Yang et al., 2011). These observations support the pathogenic significance of autophagic-lysosomal dysfunction in AD and specifically the importance of deficient lysosomal proteolysis. Therapeutic effects have also been reported in another AD model of β-amyloidosis after over-expressing cathepsin B or deleting cystatin C (Mueller-Steiner et al., 2006; Sun et al., 2008). These findings suggest the potential value of targeting impaired lysosomal proteolysis as a therapeutic strategy for AD.
The mechanism by which overexpression of mutant APP may lead to impaired autophagy and neuritic dystrophy, is not well understood. One possible contributor may be the toxic actions of the βCTF of APP on the endosomal-lysosomal system, which is also a possible basis for other AD subtypes, including sporadic AD. The earliest disease-specific pathologic change in sporadic AD—appearing before amyloid is deposited in the neocortex—is the enlargement of Rab5- and Rab7-positive endosomes (Cataldo et al., 2008; Cataldo et al., 2000), which reflects a pathological acceleration of endocytosis (Cataldo et al., 2008; Ginsberg et al., 2010; Grbovic et al., 2003; Jiang et al., 2010). This pattern is specific for AD among the aging-related neurodegenerative diseases studied and is exacerbated by inheritance of the ε4 allele of APOE, the major genetic risk factor for late onset AD (Cataldo et al., 2000). The same pathological endosomal response is also seen in Down's syndrome (trisomy 21), a cause of early onset AD, where it is linked specifically to an extra copy of App on the trisomic region of chromosome 21 and is mediated by βCTF (Cataldo et al., 2003; Jiang et al., 2010). The acceleration of endocytosis in cells of individuals with Down syndrome also causes increased protein and lipid accumulation in endosomes and slowed lysosomal degradation of endocytic cargoes (Cataldo et al., 2008). By upregulating endocytosis, elevated dietary cholesterol and over-expression of its receptor ApoE (particularly ApoE4) elevate βCTF levels and also lead to increased delivery of Aβ1-42 to lysosomes (Cossec et al., 2010; Ji et al., 2006). Expression of an APOE ε4 allele, but not the APOE ε3 allele, in a mouse AD model increases levels of intracellular Aβ in lysosomes, altering their function and causing neurodegeneration of hippocampal CA1, entorhinal, and septal neurons (Belinson et al., 2008). Thus, in sporadic AD and under conditions of increased genetic and environmental AD risk, accelerated endocytosis mediated by Rab5 and βCTF seems to be a common pathway in AD leading to increased delivery of substrates into degradative compartments. The reduced efficiency of lysosomal degradation combines with additional effects of oxidative damage (Kurz et al., 2008; Terman and Brunk, 2006) and other toxic factors, including Aβ, to impair lysosomal and autophagy function. Impaired lysosomal degradation, in turn, promotes accumulation of proteins and peptide fragments (e.g., Aβ, tau) that can further accelerate this toxic cascade.
It has been proposed that the low pH of lysosomes accentuates the conversion of ApoE4 to a molten globule, induces reactive intermediates capable of destabilizing cellular membranes leading to lysosomal leakage and apoptosis (Ji et al., 2002; Ji et al., 2006; Mahley and Huang, 2006). Consistent with these findings, strong overexpression of human Aβ42, but not Aβ40, in Drosophila neurons induces age-related autophagic-lysosomal dysfunction characterized by autolysosome accumulation and neurotoxicity (Ling et al., 2009). Aβ42-induced neurotoxicity is further enhanced by autophagy activation and is partially rescued by autophagy inhibition. The authors propose that the structural integrity of post-fusion AVs may be compromised in Aβ42 affected neurons, leading to subcellular damage and loss of neuronal integrity in the Aβ42 flies, as originally demonstrated by Glabe, Yang and colleagues in studies of Aβ uptake in neuronal cells. In these cells, internalized Aβ1-42 is slowly degraded and therefore accumulates in lysosomes, which is followed by leakage of lysosomes enzymes into the cytosol, preceding morphological evidence for cellular toxicity (Glabe, 2001; Yang et al., 1998).
Evidence for altered autophagy induction in AD is conflicting
Evidence supporting additional abnormalities of autophagy at the level of induction or autophagosome formation as a possible basis for autophagy dysfunction in AD is equivocal so far. Although autophagosomes are more numerous in the AD brain and in the PS1/APP mouse model of AD pathology (Yu et al., 2005), this cannot be considered strong support specifically for autophagy induction if AV clearance is also defective as previously discussed. Strong autophagy induction in association with neurodegeneration and autophagic cell death is so far uncommon compared to neurological responses associated with autophagy deficiency (Nixon, 2006; Wong and Cuervo, 2010).
Supporting the possibility that autophagy induction could contribute to AD pathology is recent evidence that the synthesis of many components of the lysosome is upregulated at the transcriptional and translational levels in the AD brain and AD mouse models (Cataldo et al., 1995; Cataldo et al., 1996; Cataldo et al., 2004; Ginsberg et al., 2010; Ginsberg et al., 2000; Mufson et al., 2002; Nixon and Cataldo, 2006). Moreover, Lipinski et al. recently reported that the transcription of factors promoting autophagy is up-regulated in the brains of AD patients, while negative regulators of autophagy as a group are down-regulated (Lipinski et al., 2010). Oxidative stress and production, an early pathological event in AD (Perry et al., 2002; Zhu et al., 2007), contributes to this apparent induction of autophagy by activating type III PI3 kinase (Lipinski et al., 2010). Reduced mTOR kinase activity, reflected in lowered levels of phosphorylated p70S6 kinase, has also been seen in cells treated with Aβ1-42 and in brains of PS1/APP mice, although not all studies are consistent with a stimulatory effect of Aβ on induction. For example, Aβ oligomers stimulated mTOR activity in cultured neurons while promoting cell cycle events associated with later apoptosis (Bhaskar et al., 2009). Moreover, mTOR enzymatic activity and phosphorylated p70S6K levels were also elevated in CHO cells stably transfected with FAD-related mutant APP and in the cortex and hippocampus of 3xTg-AD mice (Caccamo et al., 2010). In these mice, rapamycin administered beginning early in life restored mTOR signaling, decreased Aβ and tau pathology, and ameliorated cognitive deficits. Activated forms of mTOR and downstream mTOR-dependent factors promoting mRNA translation were detected in the AD brain within neurons exhibiting neurofibrillary degeneration, suggesting increased translation of certain transcripts and implying that autophagy may be down-regulated (Li et al., 2005).
In further support for impairment in the early stages of autophagy, levels of the autophagy-related protein beclin-1, a component of the PI3 kinase complex essential for autophagosome formation (Funderburk et al., 2010; He and Levine, 2010), have been reported to be reduced, along with PI3K, in brains from individuals with AD, although not in brains from two different mouse models of AD in which the App gene was overexpressed (Pickford et al., 2008). The deletion of beclin-1 in an APP transgenic mouse model decreased neuronal autophagy, increased intraneuronal Aβ and induced neurodegeneration associated with significant accumulations of abnormal lysosomal compartments. By contrast, overexpressing beclin-1 in these mice decreased the extent of amyloid pathology. A reduction in beclin-1 levels in AD, if confirmed, would be expected to reduce autophagosome formation, although given the multiplicity of actions of Beclin-Vsp34 core complexes on vesicular trafficking, the expected effects of lowering beclin-1 on AD pathology in the mouse model are not straightforward. Indeed, beclin-1 deficiency in this study also leads to impaired AV clearance by lysosomes, which may explain why amyloid pathology was exacerbated even though autophagosome formation was apparently reduced.
In summary, upregulation of autophagy induction in AD is supported by some, but not all analyses of mTOR activation state and is consistent with the autophagy response that might be expected in the face of increased numbers of damaged proteins and organelles in aging and diseased neurons. An increased delivery of substrates into the autophagy pathway would be expected, in turn, to compound the problem presented by the impaired AV clearance seen in the AD brain.
Conclusion
The pathology of the autophagic-lysosomal system in the AD brain and in AD models is uniquely extensive among late age onset neurological disorders, strongly suggesting a profound disturbance of autophagy-related functions. Multiple lines of evidence support the conclusion that the efficiency of lysosomal/autolysosomal proteolysis of autophagy substrates is markedly impaired by possibly multiple factors, which include the direct disruption of lysosomal acidification/proteolysis by PS1 mutations that cause early onset AD. Although a defect in AV and autophagic substrate clearance is sufficient to account for the robust accumulation of AVs and incompletely digested proteins in AD, there is additional evidence that autophagy may be upregulated and that increased levels of substrates may reach autolysosomes due to a pathological acceleration of endocytosis driven by genetic and environmental risk factors for AD. These are conditions that could compound the primary defect in clearance. This is a rapidly emerging field and new findings will further illuminate the underlying mechanisms of autophagy failure and no doubt identify additional contributing factors. A more complete understanding of autophagy regulation in the coming years will yield innovative approaches to modulate autophagy and evaluate their promise as therapies for AD and possibly other neurodegenerative diseases.
Acknowledgments
We are grateful to Nicole Piorkowski for assistance in the preparation of this manuscript. Studies from these laboratories have been supported by the National Institute on Aging and the Alzheimer's Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alirezaei M, et al. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One. 2008;3:e2906. doi: 10.1371/journal.pone.0002906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglade P, et al. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarski E, et al. Suppression of cathepsins B and L causes a proliferation of lysosomes and the formation of meganeurites in hippocampus. J Neurosci. 1997;17:4006–21. doi: 10.1523/JNEUROSCI.17-11-04006.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belinson H, et al. Activation of the amyloid cascade in apolipoprotein E4 transgenic mice induces lysosomal activation and neurodegeneration resulting in marked cognitive deficits. J Neurosci. 2008;28:4690–701. doi: 10.1523/JNEUROSCI.5633-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benzing WC, et al. Alzheimer's disease-like dystrophic neurites characteristically associated with senile plaques are not found within other neurodegenerative diseases unless amyloid beta-protein deposition is present. Brain Res. 1993;606:10–18. doi: 10.1016/0006-8993(93)91563-8. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, et al. The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14. doi: 10.1186/1750-1326-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, et al. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, et al. Gene expression and cellular content of cathepsin D in Alzheimer's disease brain: evidence for early up-regulation of the endosomal-lysosomal system. Neuron. 1995;14:671–680. doi: 10.1016/0896-6273(95)90324-0. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, et al. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci. 1996;16:186–99. doi: 10.1523/JNEUROSCI.16-01-00186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, et al. Down syndrome fibroblast model of Alzheimer-related endosome pathology. Accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008;173:370–384. doi: 10.2353/ajpath.2008.071053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, et al. App gene dosage modulates endosomal abnormalities of Alzheimer's disease in a segmental trisomy 16 mouse model of down syndrome. J Neurosci. 2003;23:6788–92. doi: 10.1523/JNEUROSCI.23-17-06788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, et al. Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol. 2004;63:821–830. doi: 10.1093/jnen/63.8.821. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–86. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevrier M, et al. Autophagosome maturation is impaired in Fabry disease. Autophagy. 2010;6 doi: 10.4161/auto.6.5.11943. [DOI] [PubMed] [Google Scholar]
- Chishti MA, et al. Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem. 2001;276:21562–70. doi: 10.1074/jbc.M100710200. [DOI] [PubMed] [Google Scholar]
- Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–32. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui DH, et al. Transgenic mice with Alzheimer presenilin 1 mutations show accelerated neurodegeneration without amyloid plaque formation. Nat Med. 1999;5:560–564. doi: 10.1038/8438. [DOI] [PubMed] [Google Scholar]
- Cossec JC, et al. Clathrin-dependent APP endocytosis and Abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim Biophys Acta. 2010;1801:846–52. doi: 10.1016/j.bbalip.2010.05.010. [DOI] [PubMed] [Google Scholar]
- Cota D, et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312:927–30. doi: 10.1126/science.1124147. [DOI] [PubMed] [Google Scholar]
- Crews L, et al. Selective molecular alterations in the autophagy pathway in patients with Lewy body disease and in models of alpha-synucleinopathy. PLoS One. 2010;5:e9313. doi: 10.1371/journal.pone.0009313. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263:55–72. doi: 10.1023/B:MCBI.0000041848.57020.57. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21:142–50. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennemarker J, et al. Impaired turnover of autophagolysosomes in cathepsin L deficiency. Biol Chem. 2010;397:913–22. doi: 10.1515/BC.2010.097. [DOI] [PubMed] [Google Scholar]
- Diaz-Troya S, et al. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–65. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- Du L, et al. Starving neurons show sex difference in autophagy. J Biol Chem. 2009;284:2383–96. doi: 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M. Pathology and Current Treatment of Neurodegenerative Sphingolipidoses. Neuromolecular Med. 2010 doi: 10.1007/s12017-010-8133-7. Aug 22 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Eskelinen EL. Maturation of autophagic vacuoles in Mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- Esselens C, et al. Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J Cell Biol. 2004;166:1041–54. doi: 10.1083/jcb.200406060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fader CM, Colombo MI. Autophagy and multivesicular bodies: two closely related partners. Cell Death Differ. 2009;16:70–8. doi: 10.1038/cdd.2008.168. [DOI] [PubMed] [Google Scholar]
- Felbor U, et al. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc Natl Acad Sci U S A. 2002;99:7883–7888. doi: 10.1073/pnas.112632299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda T, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59:700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- Funderburk SF, et al. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–62. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Marin V, et al. Cajal's contributions to the study of Alzheimer's disease. J Alzheimers Dis. 2007;12:161–74. doi: 10.3233/jad-2007-12206. [DOI] [PubMed] [Google Scholar]
- Geng J, et al. Post-golgi sec proteins are required for autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2010;21:2257–69. doi: 10.1091/mbc.E09-11-0969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, et al. Microarray Analysis of Hippocampal CA1 Neurons Implicates Early Endosomal Dysfunction During Alzheimer's Disease Progression. Biol Psychiatry. 2010;68:885–93. doi: 10.1016/j.biopsych.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, et al. Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- Glabe C. Intracellular mechanisms of amyloid accumulation and pathogenesis in Alzheimer's disease. J Mol Neurosci. 2001;17:137–45. doi: 10.1385/JMN:17:2:137. [DOI] [PubMed] [Google Scholar]
- Goedert M. Brain. Vol. 132. 2009. Oskar Fischer and the study of dementia; pp. 1102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PB, Seglen PO. Prelysosomal convergence of autophagic and endocytic pathways. Biochem Biophys Res Commun. 1988;151:40–7. doi: 10.1016/0006-291x(88)90556-6. [DOI] [PubMed] [Google Scholar]
- Grbovic OM, et al. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J Biol Chem. 2003;278:31261–8. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Griffin JW, Watson DF. Axonal transport in neurological disease. Ann Neurol. 1988;23:3–13. doi: 10.1002/ana.410230103. [DOI] [PubMed] [Google Scholar]
- Guo Q, et al. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–22. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–67. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–45. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–7. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–9. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy GO, et al. Lipofuscin-like substances accumulate rapidly in brain, retina and internal organs with cysteine protease inhibition. Adv Exp Med Biol. 1989;266:31–45. doi: 10.1007/978-1-4899-5339-1_3. [DOI] [PubMed] [Google Scholar]
- Ivy GO, et al. Inhibitors of lysosomal enzymes: accumulation of lipofuscin-like dense bodies in the brain. Science. 1984;226:985–7. doi: 10.1126/science.6505679. [DOI] [PubMed] [Google Scholar]
- Jeyakumar M, et al. Storage solutions: treating lysosomal disorders of the brain. Nat Rev Neurosci. 2005;6:713–25. doi: 10.1038/nrn1725. [DOI] [PubMed] [Google Scholar]
- Ji ZS, et al. Apolipoprotein E4 potentiates amyloid beta peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem. 2002;277:21821–21828. doi: 10.1074/jbc.M112109200. [DOI] [PubMed] [Google Scholar]
- Ji ZS, et al. Reactivity of apolipoprotein E4 and amyloid beta peptide: lysosomal stability and neurodegeneration. J Biol Chem. 2006;281:2683–2692. doi: 10.1074/jbc.M506646200. [DOI] [PubMed] [Google Scholar]
- Jiang Y, et al. Alzheimer's-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A. 2010;107:1630–5. doi: 10.1073/pnas.0908953107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy. 2010;6:438–448. doi: 10.4161/auto.6.4.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike M, et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–28. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kurz T, et al. Lysosomes in iron metabolism, ageing and apoptosis. Histochem Cell Biol. 2008;129:389–406. doi: 10.1007/s00418-008-0394-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, et al. Presenilin 1 (PS1) is required for v-ATPase targeting and autolysosome acidification: PS1 mutations in Alzheimer's disease disrupt lysosomal proteolysis and autophagy. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the Pathogenesis of Disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer's disease brain. FEBS J. 2005;272:4211–20. doi: 10.1111/j.1742-4658.2005.04833.x. [DOI] [PubMed] [Google Scholar]
- Liberski PP, et al. Neuroaxonal dystrophy in experimental Creutzfeldt-Jakob disease: electron microscopical and immunohistochemical demonstration of neurofilament accumulations within affected neurites. J Comp Pathol. 1995;112:243–55. doi: 10.1016/s0021-9975(05)80078-7. [DOI] [PubMed] [Google Scholar]
- Ling D, et al. Abeta42-induced neurodegeneration via an age-dependent autophagic-lysosomal injury in Drosophila. PLoS One. 2009;4:e4201. doi: 10.1371/journal.pone.0004201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou W, et al. The autophagic and endocytic pathways converge at the nascent autophagic vacuoles. J Cell Biol. 1997;136:61–70. doi: 10.1083/jcb.136.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:14164–9. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahley RW, Huang Y. Apolipoprotein (apo) E4 and Alzheimer's disease: unique conformational and biophysical properties of apoE4 can modulate neuropathology. Acta Neurol Scand Suppl. 2006;185:8–14. doi: 10.1111/j.1600-0404.2006.00679.x. [DOI] [PubMed] [Google Scholar]
- Masliah E, et al. Re-evaluation of the structural organization of the neuritic plaques in Alzheimer's disease. J Neuropathology and Experimental Neurology. 1993;52:619–632. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- Mizushima N, et al. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–75. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–11. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller-Steiner S, et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron. 2006;51:703–714. doi: 10.1016/j.neuron.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, et al. Gene expression profiles of cholinergic nucleus basalis neurons in Alzheimer's disease. Neurochem Res. 2002;27:1035–48. doi: 10.1023/a:1020952704398. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Niemann-Pick Type C disease and Alzheimer's disease: the APP-endosome connection fattens up. Am J Pathol. 2004;164:757–61. doi: 10.1016/S0002-9440(10)63163-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. Autophagy in neurodegenerative disease: friend, foe or turncoat? Trends Neurosci. 2006;29:528–35. doi: 10.1016/j.tins.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- Nixon RA, Cataldo AM. Lysosomal system pathways: genes to neurodegeneration in Alzheimer's disease. J Alzheimers Dis. 2006;9:277–89. doi: 10.3233/jad-2006-9s331. [DOI] [PubMed] [Google Scholar]
- Nixon RA, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- Nixon RA, et al. Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy. 2008;4:590–9. doi: 10.4161/auto.6259. [DOI] [PubMed] [Google Scholar]
- Noda T, et al. The late stages of autophagy: how does the end begin? Cell Death Differ. 2009;16:984–90. doi: 10.1038/cdd.2009.54. [DOI] [PubMed] [Google Scholar]
- Ohmi K, et al. Sanfilippo syndrome type B, a lysosomal storage disease, is also a tauopathy. Proc Natl Acad Sci U S A. 2009;106:8332–7. doi: 10.1073/pnas.0903223106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry G, et al. Is oxidative damage the fundamental pathogenic mechanism of Alzheimer's and other neurodegenerative diseases? Free Radic Biol Med. 2002;33:1475–9. doi: 10.1016/s0891-5849(02)01113-9. [DOI] [PubMed] [Google Scholar]
- Pickford F, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–9. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravikumar B, et al. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–57. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington's disease. Hum Mol Genet. 2010;19:2144–53. doi: 10.1093/hmg/ddq093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Nixon RA. Rapamycin induces autophagic flux in neurons. Proc Nat Acad Sci. 2010;107:E181. doi: 10.1073/pnas.1014633107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudnicki DD, et al. A comparison of huntington disease and huntington disease-like 2 neuropathology. J Neuropathol Exp Neurol. 2008;67:366–74. doi: 10.1097/NEN.0b013e31816b4aee. [DOI] [PubMed] [Google Scholar]
- Ryazantsev S, et al. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol Genet Metab. 2007;90:393–401. doi: 10.1016/j.ymgme.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt ML, et al. An extensive network of PHF tau-rich dystrophic neurites permeates neocortex and nearly all neuritic and diffuse amyloid plaques in Alzheimer disease. FEBS Lett. 1994;344:69–73. doi: 10.1016/0014-5793(94)00259-2. [DOI] [PubMed] [Google Scholar]
- Schmidt RE, Plurad SB. Ultrastructural appearance of intentionally frustrated axonal regeneration in rat sciatic nerve. J Neuropathol Exp Neurol. 1985;44:130–46. doi: 10.1097/00005072-198503000-00002. [DOI] [PubMed] [Google Scholar]
- Settembre C, et al. Proteoglycan desulfation determines the efficiency of chondrocyte autophagy and the extent of FGF signaling during endochondral ossification. Genes Dev. 2008;22:2645–50. doi: 10.1101/gad.1711308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Kelleher RJ., 3rd The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A. 2007;104:403–409. doi: 10.1073/pnas.0608332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R, et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease [see comments] Nature. 1995;375:754–60. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Sun B, et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer's disease. Neuron. 2008;60:247–57. doi: 10.1016/j.neuron.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer's presenile dementia. Acta Neuropathol (Berl) 1967;8:276–84. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- Takeuchi IK, Takeuchi YK. Transient accumulation of Gallyas-Braak-positive and phosphorylated tau-immunopositive substances in neuronal lipofuscin granules in the amygdala, hippocampus and entorhinal cortex of rats during long-term chloroquine intoxication. Acta Neuropathol. 2001;102:191–194. doi: 10.1007/s004010100360. [DOI] [PubMed] [Google Scholar]
- Terman A, Brunk UT. Oxidative stress, accumulation of biological ‘garbage’, and aging. Antioxid Redox Signal. 2006;8:197–204. doi: 10.1089/ars.2006.8.197. [DOI] [PubMed] [Google Scholar]
- Terry RD, et al. The ultrastructure of the cerebral cortex in Alzheimer's disease. Trans Am Neurol Assoc. 1964;89:12. [PubMed] [Google Scholar]
- Tian Y, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell. 2010;141:1042–55. doi: 10.1016/j.cell.2010.04.034. [DOI] [PubMed] [Google Scholar]
- Tsvetkov AS, et al. A small-molecule scaffold induces autophagy in primary neurons and protects against toxicity in a Huntington disease model. Proc Natl Acad Sci U S A. 2010;107:16982–7. doi: 10.1073/pnas.1004498107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvetkov AS, et al. Protein turnover differences between neurons and other cells. Autophagy. 2009;5 [Google Scholar]
- Walkley SU. Cellular pathology of lysosomal storage disorders. Brain Pathol. 1998;8:175–93. doi: 10.1111/j.1750-3639.1998.tb00144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley SU. Pathogenic cascades in lysosomal disease-Why so complex? J Inherit Metab Dis. 2009;32:181–9. doi: 10.1007/s10545-008-1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, et al. Degradative organelles containing mislocalized α- and β-synuclein proliferate in presenilin-1 null neurons. J Cell Biol. 2004;165:335–346. doi: 10.1083/jcb.200403061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtschafter JD, et al. Intraocular axonal swelling produced by partial, immediately retrobulbar ligature of optic nerve. Invest Ophthalmol Vis Sci. 1977;16:537–41. [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2010;13:805–11. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–9. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Yang AJ, et al. Loss of endosomal/lysosomal membrane impermeability is an early event in amyloid Abeta1-42 pathogenesis. J Neurosci Res. 1998;52:691–8. doi: 10.1002/(SICI)1097-4547(19980615)52:6<691::AID-JNR8>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Yang DS, et al. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer's disease. Am J Pathol. 2008a;173:665–81. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DS, et al. Cystatin B deletion in a mouse model of Alzheimer's disease, TgCRND8, ameliorates both autophagiclysosomal and amyloid pathologies. International Conference on Alzheimer's Disease (ICAD); Chicago. 2008b. [Google Scholar]
- Yang DS, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011;134:258–277. doi: 10.1093/brain/awq341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, et al. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
- Yu L, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–6. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu WH, et al. Macroautophagy--a novel β-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Z, et al. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–33. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- Zhou L, et al. Frontotemporal dementia: neuropil spheroids and presynaptic terminal degeneration. Ann Neurol. 1998;44:99–109. doi: 10.1002/ana.410440116. [DOI] [PubMed] [Google Scholar]
- Zhu X, et al. Alzheimer disease, the two-hit hypothesis: an update. Biochim Biophys Acta. 2007;1772:494–502. doi: 10.1016/j.bbadis.2006.10.014. [DOI] [PubMed] [Google Scholar]