

Abstract
Angiogenesis, which is the process of sprouting of new blood vessels from pre-existing vessels, is vital for tumor progression. Proteolytic remodeling of extracellular matrix is a key event in vessel sprouting during angiogenesis. Urokinase plasminogen activator receptor (uPAR) and cathepsin B are both known to be overexpressed and implicated in tumor angiogenesis. In the present study, we observed that knockdown of uPAR and cathepsin B using puPAR (pU), pCathepsin B (pC), and a bicistronic construct of uPAR and cathepsin B (pCU) caused significant inhibition of angiogenesis by disrupting the JAK/STAT pathway-dependent expression of VEGF. Further, transcriptional suppression of uPAR and cathepsin B inhibited tumor-induced migration, and proliferation of endothelial cells and decreased tumor-promoted expression of VEGFR-2, Rac1, gp91phox, cyclin D1, Cdk4, and p-Rb in HMEC. Furthermore, U251 and SNB19 xenograft tissue sections from nude mice treated with pCU showed reduced expression of VEGF and CD31, which is a blood vessel visualization marker. Overall, results revealed that knockdown of uPAR and cathepsin B inhibited tumor-induced angiogenesis by disrupting the JAK/STAT pathway-dependent expression of VEGF. These data provide new insight in characterizing the pathways involved in the angiogenic cascade and for the identification of novel target proteins for use in therapeutic intervention for gliomas.
Keywords: VEGF, uPAR, Cathepsin, Angiogenesis, Apoptosis, Glioma
Introduction
Neovascularization, which is the sprouting of new blood vessels from pre-existing microvessels in or at the periphery of a tumor, is vital for tumor survival.1 Angiogenesis is a key regulatory factor in the development, progression and metastasis of tumors, including glioma.2 Further, it is well established that proteolytic remodeling of extracellular matrix is a key event in vessel sprouting during angiogenesis3,4 and is mediated by the interaction of proteases and uPAR.5
Glioblastomas are highly dependent on angiogenesis.6 To progress beyond 1–3 mm3 in size, tumors must be nourished with nutrients and oxygen via newly formed blood vessels.7 Studies from our laboratory and others have reported that urokinase-type plasminogen activator receptor (uPAR) and the cysteine protease cathepsin B are overexpressed in high-grade glioma, which are characterized by aberrant neovascularization.8–11 In tumor angiogenesis, cathepsin B activates soluble or membrane-associated pro-uPA, which in turn, initiates extracellular remodeling and causes the release or formation of pro-angiogenic factors.12–14 In addition, cathepsin B has been reported to regulate the intrinsic angiogenic threshold of endothelial cells.15 Moreover, inhibition of extracellular cathepsin B activity affected tube formation in endothelial cells.16
uPAR is a GPI-anchored glycoprotein localized with proteolytic plasminogen system, caveolin and integrins.17,18 uPAR has been implicated in tumor-associated angiogenesis, growth factor activation and mobilization, ECM remodeling, invasion and metastasis.19–22 In addition, uPAR plays a role in the secretion of procathepsin B in association with caveolae and β1 integrins.22 In addition to its proteolytic activity, uPAR is involved in various intracellular signaling pathways.23–26 Endothelial proliferation is frequently found within and adjacent to high-grade gliomas. Paracrine stimuli derived from tumor cells are the main promoters of angiogenesis. Once activated by these stimuli, endothelial cells begin to proliferate, migrate, and subsequently form capillary-like structures.
Several methods have been developed to specifically target proteases in an attempt to inhibit angiogenesis. Among these methods, the use of small interfering RNA (siRNA) has received much attention because of its therapeutic potential.27 In recent years, RNAi technology has rapidly become the agent of choice in exploring pathways and identifying mediators of the angiogenic cascade.28 Moreover, data from preclinical and clinical studies have suggested that current anti-VEGF therapies are virtually ineffective in glioma-induced angiogenesis.29
Studies from our lab and others have demonstrated the role of uPAR and cathepsin B in tumor-induced angiogenesis using in vitro and in vivo angiogenesis models.30–33 However, the mechanism(s) involved in uPAR and cathepsin B-mediated regulation of angiogenesis is not completely understood. In the present study, we demonstrate that knockdown of uPAR and cathepsin B inhibited glioma-induced angiogenesis by disrupting JAK/STAT-dependent expression of VEGF. We were able to show that downregulation of uPAR and cathepsin B inhibits glioma-induced invasion and proliferation of endothelial cells. The results also demonstrate the role of uPAR and cathepsin B in VEGF-mediated regulation of endothelial cell cycle progression. Overall, results revealed that knockdown of uPAR and cathepsin B inhibited tumor-induced invasion and cell cycle progression of endothelial cells and angiogenesis by disrupting the JAK/STAT pathway-dependent expression of VEGF. The results of the present study suggest that RNAi-mediated gene silencing of uPAR and cathepsin B may prove to be an effective therapeutic application in the treatment of malignant glioma.
Materials and methods
Ethics Statement
The Institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Peoria, Peoria, IL, USA approved all surgical interventions and post-operative animal care. The consent was written and approved. Protocol 851 was approved on November 20, 2009 and protocol 817 was approved on November 1, 2007 and renewed on May 13, 2010.
Cell culture and transfection conditions
U251 and SNB19 cell lines (obtained from American Type Culture Collection, ATCC; Manassas, VA) were cultured in DMEM supplemented with FBS (10%), penicillin/streptomycin (100 units/mL) and maintained in a humidified atmosphere containing 5% CO2 at 37°C. Human dermal microvascular endothelial cell line (HMEC-1) was obtained from Francisco J. Candal (Centers for Disease Control and Prevention, Atlanta, GA). HMEC-1 cells were maintained in advanced DMEM medium containing 10% FBS, 2% hydrocortisone, 0.001% EGF, L-glutamine (200 nM), and penicillin/streptomycin (100 units/mL) at 37°C in a humidified atmosphere of 5% CO2. U251 and SNB19 cells (70–80% confluence) were transfected with scrambled vector (SV), puPAR (pU), pCathepsin B (pC), bicistronic construct of uPAR and cathepsin B (pCU), empty vector (EV), or vectors containing full-length uPAR cDNA (pfU) and cathepsin B (pfC) for 48 hrs using Fugene HD as per the manufacturer’s instructions (Roche, Indianapolis, IN). Single constructs directed against uPAR(pU) and cathepsin B (pC) and the bicistronic construct directed against both cathepsin B and uPAR (pCU) have been described previously.34 Full-length cathepsin B (pfC) and uPAR (pfU) over expressing plasmids were purchased from Origene (Rockville, MD).
Non-contact co-culture of endothelial and glioma cells
To co-culture tumor and endothelial cells, U251 or SNB19 cells (2×105/well) plated in transwell chamber plate (6-well type, Greiner Bio-One Inc., Monroe, NC) were left untreated or transfected with SV, pU, pC and pCU for gene silencing studies or with EV, pfU and pfC for overexpression studies. HMEC (4×105/well) were plated in transwell chamber inserts (6-well type, 0.4 μm pore size), placed in transwell chamber plates and incubated for 48 hrs. After incubation, cells were collected from transwell chamber inserts by trypsinization and lysed in lysis buffer (150 mM NaCl, 50 mM Tris-Hcl, 20 mM EDTA (Ethylenediaminetetraacetic acid), 1% NP-40, pH 7.4) and used for immunoblotting analysis.
Western blotting
Single and co-cultures of cancer cells were harvested and homogenized in lysis buffer and processed for cell lysates. Equal amounts of cellular protein were subjected to SDS-PAGE using appropriate percentage of acrylamide gels. After separation, proteins were transferred to nitrocellulose membrane (Bio-Rad). Membranes were then blocked in 5% non-fat dry milk in PBS-T (PBS containing 0.1% Tween-20) and incubated overnight with primary antibody at 4°C. Membranes were then washed twice with PBS-T at an interval of 15 min and further incubated with suitable HRP (Horseradish peroxidase)-conjugated secondary antibody for 1 hr. Membranes were developed using Pierce ECL Western Blotting Substrate according to manufacturer’s instructions (Thermo Scientific Inc, Rockford, IL). To confirm equal protein loading, blots were stripped and re-probed with GAPDH antibody. The following antibodies were used: anti-uPAR (R&D Systems, Minneapolis, MN), anti-Cathepsin B (Athens Research and Technology Inc., Athens, GA), anti-VEGF, anti-VEGFR-2 or Flt-1, anti-VAV2 (Guanine nucleotide exchange factor), anti-phospho-VEGFR-2, anti-gp91phox(Heme binding subunit of NADPH oxidase), anti-HIF-1α, anti-Cyclin D1, anti-Cdk4, anti-phospho-Rb (Santa Cruz Biotechnology Inc., Santa Cruz, CA), anti-Rac1, anti-JAK1, anti-STAT3, and anti-p-STAT3 (S-727 and Y-705) (Cell Signaling Inc., Danvers, MA).
Reverse transcription PCR
Total RNA was extracted from the transfected cells using TRIZOL reagent (Invitrogen, CA) as per standard protocol. DNase-treated RNA was used as a template for reverse transcription (RT) reaction (Invitrogen, CA) followed by PCR analysis using primers specific for uPAR, cathepsin B, VEGF and GAPDH. The PCR conditions were as follows: 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 58°C for 45 sec and 72°C for 45 sec. GAPDH was used as an internal control.
In vitro angiogenesis assay
In vitro angiogenesis assay was carried out as described previously with some modifications.30 Briefly, U251 or SNB19 cells (5×104 cells/well) were grown to 60–70% confluence in transwell chamber plates (24-well type, Greiner Bio-One Inc., Monroe, NC) and left untreated or transfected with SV, pU, pC and pCU. HMEC (1×104/mL) were grown as a monolayer in collagen-coated porous upper chamber inserts, placed in transwell chamber plates and incubated for 48 hrs. After overnight incubation with serum-free medium, Hema 3 staining was performed, and tube formation was checked under the microscope and photographed. Angiogenic effect was measured by counting the number of branch points in five different fields and expressed as a percentage of the control. Values were expressed as mean and SE of three different experiments.
In vivo angiogenesis assay
In vivo angiogenesis assay was performed using the dorsal skin-fold chamber model as described previously.30 Briefly, Athymic nude mice (nu/nu; 18 female, 5–7 weeks old) were bred and maintained within a specific pathogen, germ-free environment. The implantation technique of the dorsal skin fold chamber model has been described previously.35 Sterile small-animal surgical techniques were followed. Mice were anesthetized by intraperitoneal injection with ketamine (50mg/kg) and xylazine (10mg/kg). Dorsal air sac was made by injecting 10ml of air in the completely anesthetized mice. Diffusion chambers (Fisher, Hampton, NH) were prepared by aligning 0.45μm Millipore membranes (Fisher) on both sides of the rim of the “O” ring (Fisher) with sealant. Once the chambers were dried (2–3min), they were sterilized by overnight irradiation under UV light. Membranes were wetted with 20μl of PBS. Cancer cells (2×106) transfected with SV, pU, pC, pCU were suspended in 150μl of serum-free medium and injected into the chamber through the opening of the “O” ring. The opening was sealed with a small amount of bone wax. A 1.5–2.0cm superficial incision was made horizontally along the edge of the dorsal air sac, and the air sac was opened. With the help of forceps, the chambers were placed underneath the skin and carefully sutured. After 10 days, the animals were anesthetized with ketamine/xylizine and sacrificed by intracardial perfusion with saline (10ml) followed by 10 ml of 10% formalin/0.1M phosphate solution. The animals were carefully skinned around the implanted chambers, which were removed from the subcutaneous air fascia. The skin-fold covering the chambers was photographed under visible light. The number of blood vessels within the chamber in the area of the air sac fascia was counted and their lengths were measured.
Angiogenesis array
U251 or SNB19 cells (1×105cells/well) were transfected with pCU and co-cultured with HMEC (2×105 cells/well) for 48 hrs. Untreated cells co-cultured with HMEC were maintained to serve as a control. Conditioned media was collected after overnight incubation, exposed to angiogenesis antibody arrays and developed as per manufacturer’s instructions (Ray Biotech, Inc., Norcross, GA). The expression of angiogenic molecules (intensities of signals) was quantified by densitometry, and fold change was calculated by comparison with the control.
Transwell proliferation assay
Cell proliferation analysis was performed by BrdU incorporation assay according to the manufacturer’s protocol (Roche Diagnostics, Indianapolis, IN). Briefly, U251 or SNB19 cells (1×105 cells/well) were grown to 60–70% confluence in transwell chamber plates (12-well type, Greiner Bio-One Inc., Monroe, NC) and transfected with SV, pU, pC and pCU or left untreated. HMEC (5×103 cells/well) were grown as a monolayer in collagen-coated transwell chamber inserts, placed in transwell chamber plates and incubated for 48 hrs. Trypsinized HMEC cells (1×104) were seeded into each well of a 96-well plate and allowed to adhere overnight, and proliferation was evaluated using BrdU incorporation assay. For cell cycle analysis, HMEC were treated with propidium iodide (50 μg/mL) + RNase A (0.001%) solution as per standard protocol. The cells were sorted on a fluorescence-activated cell sorter and quantified (10,000 cells sorted).
Transwell migration assay
To assay for tumor-induced migration of endothelial cells, U251 or SNB19 cells (1×105/well) were grown to 60–70% confluence in transwell chamber plates (12-well type, Greiner Bio-One Inc., Monroe, NC) and left untreated or transfected with SV, pU, pC and pCU. HMEC (5×104cells/well) were grown as a monolayer in collagen-coated transwell chamber inserts, placed in transwell chamber plates and incubated at 37°C for 48 hrs. Following the incubation period, non-migrated cells on the upper surface of the filter were removed. The cells that had migrated onto the filter were stained with Hema 3 stain. For quantification, the mean diameter of randomly selected glioma cells that had migrated from the tumor spheroid was measured manually and expressed as a percent of control.36 Rac1 pull down assay. Active Rac1 from cell lysates (500 μg) was precipitated with GST-PBD (containing amino acids 51–135 of PAK1) according to the manufacturer’s instructions (Cytoskeleton Inc., Denver, CO). The precipitates were washed three times with wash buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 10 mM MgCl2, 0.1 mM PMSF, 10 μg/mL aprotinin, and 10 μg/mL leupeptin), and the bound proteins were eluted by boiling for 5 min. The protein was resolved in 14% polyacrylamide gels, transferred to polyvinylidene difluoride membrane (Bio-Rad) and immunoblotted with anti-Rac1 antibody.
Determination of ROS
Intracellular ROS was determined in endothelial cells co-cultured with glioma cells using 2, 7-Dichlorofluorescien diacetate (DCFH-DA; Sigma, USA). HMEC (2×105 cells/well) and U251 or SNB19 cells (1×105 cell/well) were co-cultured using non-contact transwell co-culture model for 48 hrs. Then, transwell inserts were transferred to another 6-well plate and incubated with serum-free medium containing 20 mM DCFH-DA at 37°C for 30 min. After incubation, inserts were washed with PBS, cells were lifted with trypsin and resuspended in PBS. Fluorescence was quantified by spectrofluorometry (Fluorskan Ascent) using an excitation wavelength of 480 nm and an emission wavelength of 455 nm. Percent of ROS levels were calculated by comparing fluorescence of treated samples to the control (untreated glioma and HMEC co-cultures).
Immunohistochemical analysis of tumor xenografts for VEGF and CD31
Stereotactic implantation of U251 and SNB19 cells, followed by treatment with SV and pCU using Alzet minipumps at the rate of 0.25μL/hr was carried out as previously described.37,38 Briefly, U251 and SNB19 cells (1.0×105) were injected intracranially into anesthetized nude mice (50mg/kg ketamine,10mg/kg xylazine). Tumors were allowed to grow for one week and the animals were divided into five groups with five animals in each group. Alzet mini-osmotic pumps (Durect Corporation, CA) containing 100μl of SV and pCU (1.5μg/μl) were used. Once control animals showed chronic symptoms (3–4weeks), the animals were euthanized by cardiac perfusion using 10% buffered formalin. The brains were removed and stored in 10% formalin and embedded in paraffin following standard protocol. Immunoblot analysis was done on fresh brain tissues. Paraffin-embedded tumors sections were subjected to rehydration by passing through a series of xylene and 100% and 90% ethanol.39,40 Sections were stained with hematoxylin and eosin to characterize tumor growth as described previously.30 The stained sections were blindly reviewed and scored for the size of the tumor in each case semiquantitatively. The average cross sectional diameter measured in sections of each tumor was used to measure tumor size. Immunohistochemical analysis for VEGF and CD31 was performed as described earlier.41 The sections were deparaffinized as described above and antigen retrieval was carried out by boiling sections in 0.01% sodium citrate buffer (pH 6.0) containing 0.05% Triton X-100 for 20 min. Following quenching of endogenous peroxidase activity and blocking of non-specific binding, sections were incubated overnight at 4° C with an anti-VEGF or CD31 antibody (1:100 dilution). Next, sections were treated with HRP-conjugated secondary antibody (1:200 dilution) for 30 min at room temperature. Immunolocalization was accomplished by exposing sections to 0.05% 3,3′-diaminobenzidine tetrahydrochloride (DAB) as the chromogen. The slides were counterstained with Mayer’s hematoxylin and mounted. All microscopy studies were performed using a microscope attached to a CC camera and set to auto.
Statistical analysis
All the western blot, RT-PCR and in vitro angiogenesis experiments were performed three times, and each data point in the results is the mean of three values and expressed as mean ± SE. Statistical significance of the results was analyzed by using 2-tailed Student’s t-test. Statistical differences were set at probability levels of *p<0.05 and **p<0.001.
Results
Inhibition of uPAR and cathepsin B protein and mRNA levels by RNA interference
Small interfering RNA (siRNA) are rapidly becoming prime agents of choice for functional analysis of new target genes and exploring pathways of tumor-induced angiogenesis. Transfection of U251 and SNB19 cells with pU, pC and pCU strongly inhibited the expression of uPAR and cathepsin B mRNA as determined by semi-quantitative RT-PCR analysis (Figs. 1A–B). GAPDH expression demonstrated equal loading. Densitometric analysis revealed that the uPAR mRNA level was decreased to 44.58% with pU transfection (p<0.001) and 28.78% with pCU transfection (p<0.001) in U251 cells; cathepsin B mRNA level was decreased to 35.17% with pC transfection (p<0.001) and 27.14% with pCU transfection (p<0.001) in U251 cells (Fig. 1C). In SNB19 cells, uPAR mRNA expression was decreased to 29.84% with pU transfection (p<0.001) and 20.94% with pCU transfection (p<0.001); cathepsin B mRNA expression was decreased to 38.78% with pC transfection (p<0.001) and 22.81% with pCU transfection (p<0.001) (Fig. 1D). Immunoblot analysis of cell lysates was performed further to analyze uPAR and cathepsin B protein levels in control and treated U251 and SNB19 cells. Individual or simultaneous knockdown of uPAR and cathepsin B significantly inhibited the expression of both the proteins when compared to control or SV-treated cells (Figs. 1E–F). Densitometric analysis revealed that uPAR protein expression was decreased to 30.02% with pU and 15.91% with pCU in U251 cells; cathepsin B protein expression was decreased to 40.22% with pC and 25.23% with pCU (p<0.001) (Fig. 1G). In SNB19 cells, uPAR protein expression was decreased to 34.66% with pU and 20.66% with pCU (p<0.001); cathepsin B protein expression was decreased to 45.02% with pC and 30.12 ± 2.89% with pCU (p<0.001) (Fig. 1H).
Figure 1. RNAi-mediated knockdown of uPAR and cathepsin B expression in U251 and SNB19 cells.
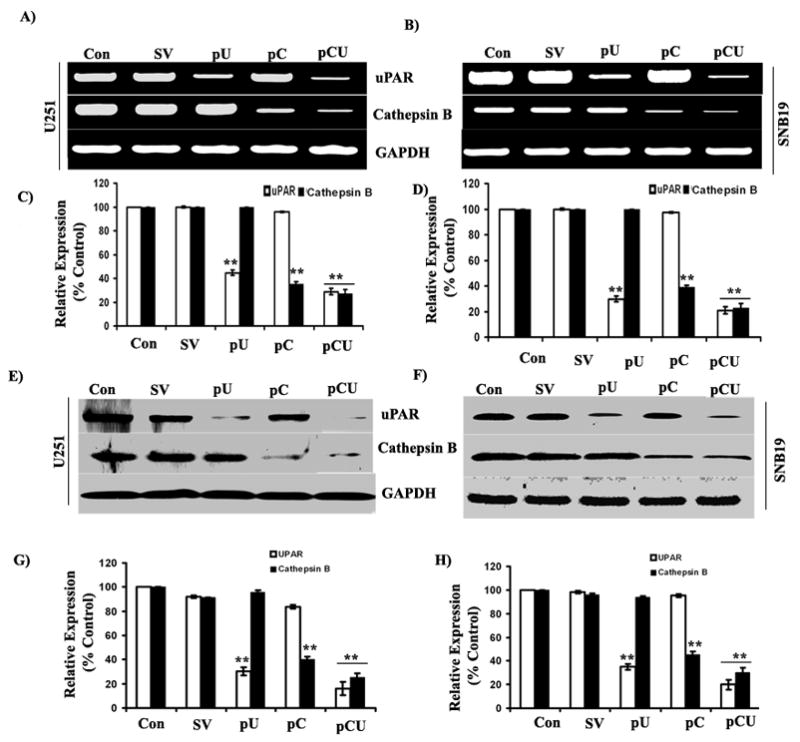
(A–B) Semi-quantitative RT-PCR analysis of RNA extracted from U251 and SNB19 cells transfected with SV, pU, pC, pCU and control cells with primers specific for uPAR, cathepsin B and GAPDH. (C–D) Densitometric analysis of relative expression of uPAR and cathepsin B mRNA levels in U251 and SNB19 cells. (E–F) Western blot analysis of uPAR and cathepsin B expression in control and SV-, pU-, pC-and pCU-transfected U251 and SNB19 cells using specific antibodies. (G–H) Quantitative analysis of uPAR and cathepsin B expression in U251 and SNB19 by densitometry. Results from three independent experiments are shown as mean ± SE (*p<0.05, **p<0.001). GAPDH served as a loading control.
siRNA targeting uPAR and cathepsin B inhibits tumor-induced angiogenesis in vitro and in vivo
To assess the effect of uPAR and cathepsin B knockdown on tumor-induced vessel formation, transfected or untreated U251 or SNB19 cells were co-cultured with HMEC using non-direct contact model. Figure 2A shows that endothelial cells co-cultured with untreated or SV-transfected U251 and SNB19 cells formed capillary-like networks after 48 hrs. In contrast, transfection with pU, pC and pCU significantly inhibited capillary network formation in both U251 and SNB19 cells. pU-transfected U251 and SNB19 cells showed that endothelial capillary formation was decreased to 22.61% and 21.47% (p<0.001), respectively, as compared to controls. Moreover, U251 and SNB19 cells transfected with pC decreased endothelial capillary formation to 31.64% and 32.27% (p<0.001), respectively, as compared to controls (Fig. 2B). Interestingly, pCU-transfected U251 and SNB19 cells decreased endothelial capillary formation to 11.82 and 12.51 (p<0.001), respectively, as compared to controls.
Figure 2. Effect of uPAR and cathepsin B knockdown on tumor-induced angiogenesis and expression of angiogenic molecules.
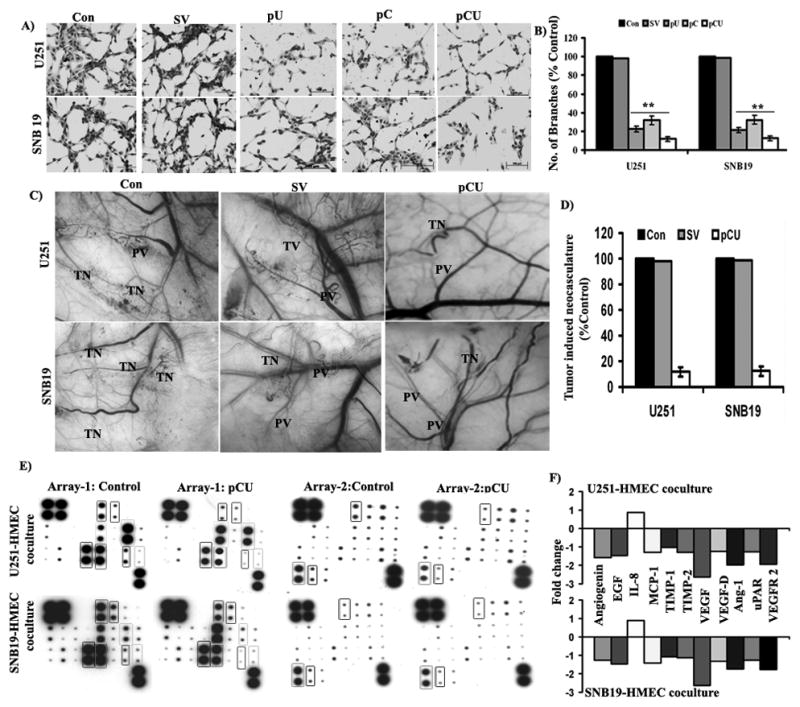
(A) In vitro angiogenesis in U251 and SNB cells. Tumor-induced tube formation in HMEC cells was carried out as described in Materials and Methods. The tube formation was observed under the bright field microscope and number of branch points was calculated. (B) Graphical representation of relative branch points in U251 and SNB19 cells transfected with SV, pU, pC and pCU. Bars represents the means ± SE of three different experiments. The asterisk (*) represents statistically different compared to control (**p<0.001). (C) Inhibition of tumor-induced angiogenesis in U251 and SNB19 cells transfected with pU, pC and pCU as assessed by dorsal skin-fold chamber assay. Tiny, zigzag-shaped microvessels showing irregular arrangement were recorded as tumor-induced neovasculature (TN) and more organized vessels were designated as pre-existing vasculature (PV). (D) In vivo angiogenesis was quantified by measuring the length of TN in control and transfected cells and expressed as a percentage of control. Values were mean ± SD from five animals per treatment group. (E) Expression of pro and anti-angiogenic molecules in HMEC and U251 or SNB19 co-cultures. Conditioned media from HMEC and U251 or SNB19 co-cultures, exposed to angiogenesis antibody arrays and processed as per manufacturer’s instructions. (F) Graphical representation of fold change of angiogenin, EGF, IL-8, MCP-1, TIMP-1, TIMP-2, VEGF, VEGF-D, angiopoietin-1, uPAR and VEGFR-2 expression in HMEC and U251 or SNB19 co-cultures.
To further confirm the in vitro angiogenesis results, we used the dorsal skin-fold chamber assay to model the in vivo angiogenesis system. Implantation of chambers containing untreated or SV-transfected U251 or SNB19 cells underneath the dorsal skin of athymic mice resulted in the development of numerous microvessels, which are observed as tiny and curved structures (Fig. 2C). In contrast, microvessel formation was significantly decreased with pCU transfection in both U251 and SNB19 cells. Quantification of tumor-induced neovasculature revealed that microvessel formation was decreased to 8.84% and 10.52% with pCU transfected U251 and SNB19 cells, respectively, as compared to controls (Fig. 2D). Our results suggest that both uPAR and cathepsin B play prominent roles in tumor-induced angiogenesis.
Effect of uPAR and cathepsin B knockdown on expression of angiogenic molecules in co-cultures
To evaluate the impact of siRNA-mediated downregulation of uPAR and cathepsin B on the spectrum of angiogenic modulators, antibody arrays were incubated with conditioned media from untreated or pCU-treated U251 or SNB19 and HMEC co-cultures. The results indicate that expression levels of VEGF, VEGFR-2 and angiopoietin-1 were significantly decreased, and angiogenin, EGF, MCP-1, TIMP-1, TIMP-2, VEGF-D and uPAR were decreased in both U251 and SNB19 co-cultures (Fig. 2E). Densitometric analysis of signals revealed that VEGF expression was significantly decreased by 2.6-fold in both U251 and SNB19 co-cultures. VEGFR-2 and angiopoietin-1 were appreciably decreased by 2.0-fold in U251 and HMEC co-cultures and 1.75-fold in SNB19 and HMEC co-cultures as compared to controls (Fig. 2F).
siRNA targeting uPAR and cathepsin B inhibits expression of VEGF protein and mRNA
VEGF is a widely studied growth factor for neovascularization. To check the expression of VEGF, conditioned media was collected from control and treated U251 and SNB19 cells and concentrated using centrifuge filter (Cutoff 3K, Millipore, Cat # UFC900396). The results show that individual or simultaneous knockdown of uPAR and cathepsin B decreased VEGF secretion into tumor-conditioned media as compared to control or SV-transfected cells (Fig. 3A). Densitometric analysis revealed that VEGF secretion was decreased to 35.11% and 36.72% (p<0.001) in pU-transfected U251 and SNB19 cells, respectively, as compared to controls. Likewise, VEGF secretion was decreased to 29.68% and 41.04% (p<0.001) in pC-transfected U251 and SNB19 cells, respectively. VEGF secretion was further decreased to 18.51% and 20.19% (p<0.001) as compared to controls in pCU-transfected U251 and SNB19 cells, respectively (Fig. 3B).
Figure 3. Downregulation of uPAR and cathepsin B using siRNA inhibits VEGF expression.
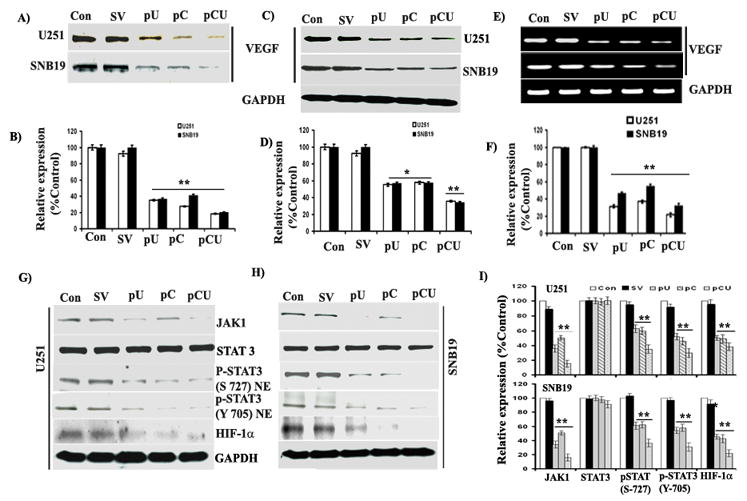
U251 and SNB19 cells were transfected with SV, pU, pC and pCU for 48 hrs. Conditioned media was collected after overnight incubation with serum-free medium. (A) Immunoblot analysis of VEGF expression in conditioned media from control and transfected U251 and SNB19 cells (B) Quantification of VEGF expression in conditioned media by densitometry. (C) Western blot analysis of VEGF expression in U251 and SNB19 cell lysates, which were prepared 48 hrs after transfection. The blots were stripped and re-probed with GAPDH antibody to verify equal loading. The experiments were performed three times and representative blots are shown. (D) Densitometric analysis of VEGF expression in cell lysates from U251 and SNB19 cells. Columns: mean of triplicate experiments; bars: SE; *p<0.05 and **p<0.001. (E) Semi-quantitative RT-PCR analysis of VEGF mRNA transcription in U251 and SNB19 cells. Total RNA was extracted and PCR was set up as described in Materials and Methods for VEGF. (F) Densitometric analysis of VEGF mRNA expression. (G–H) Western blot analysis of JAK1, p-STAT3 (S-727 and Y-705), and HIF-1α expression in U251 and SNB19 cell lysates. Cell lysates were collected from SNB19 and U251 after transfection with SV, pU, pC or pCU. (I) Quantification of protein band intensities by densitometric analysis using NIH ImageJ software. Columns: mean of experiments performed in triplicate; bars: SE; *p<005 and **p<0.001, significant difference from untreated control or SV transfected control.
Finally, expression of VEGF was prominently decreased in pU-, pC- and pCU-transfected U251 and SNB19 cell lysates (Fig. 3C). Densitometric analysis revealed that VEGF expression was decreased to 54.82 and 55.73% (p<0.05) in pU-transfected U251 and SNB19 cell lysates, respectively, as compared to controls. In addition, VEGF expression was decreased to 56.55% and 54.47% (p<0.05) in pC-transfected U251 and SNB19 cell lysates, respectively. Notably, VEGF expression was significantly decreased to 34.81% and 33.44% (p<0.001) in U251 and SNB19 cells, respectively, with pCU transfection (Fig. 3D).
To test whether uPAR and cathepsin B knockdown inhibited VEGF mRNA transcription, we performed semi-quantitative RT-PCR analysis using total RNA. The results showed that U251 and SNB19 cells treated with pCU have significantly lower levels of VEGF mRNA than control or SV-treated cells. However, pU- and pC-transfected U251 and SNB19 cells expressed moderate levels of VEGF mRNA as compared to controls (Fig. 3E). Densitometric analysis indicates that expression of VEGF mRNA was decreased to 31.29% and 46.29% in pU-transfected U251 and SNB19 cells (p<0.001), respectively, as compared to controls. Moreover, expression of VEGF mRNA was decreased to 36.92% and 54.61% in pC-transfected U251 and SNB19 cells (p<0.001), respectively, as compared to controls. Furthermore, VEGF mRNA expression was decreased to 21.63% and 32.11% in pCU-transfected U251 and SNB19 cells (p<0.001), respectively, as compared to controls (Fig. 3E).
Effect of uPAR and cathepsin B overexpression on VEGF expression
To test the hypothesis whether overexpression of uPAR and cathepsin B increases the expression of VEGF, U251 and SNB19 cells were transfected with EV, pfU or pfC, and expression of uPAR, cathepsin B and VEGF was determined by western blotting. The results show that expression of VEGF was significantly elevated in cells overexpressing uPAR and cathepsin B (Supplementary Figs. 1A–B). Densitometric analysis of western blots reveals that uPAR was increased to 226.6% in U251 cells and 260.3% in SNB19 cells with pfU. In addition, cathepsin B was increased to 391.2% in U251 cells and 225% in SNB19 cells with pfC. Interestingly, expression of VEGF was increased to 346% with uPAR and cathepsin B overexpression in both U251 and SNB19 cells when compared to controls (Supplementary Figs. 1C–D). These results strongly support the roles of uPAR and cathepsin B in the regulation of VEGF.
Downregulation of uPAR and cathepsin B inhibits expression of JAK1, STAT3 and HIF-1α
To identify possible signaling molecules in uPAR and cathepsin B-mediated regulation of VEGF, we examined the expression of JAK1, STAT3 and HIF-1α in U251 and SNB19 cell lysates. The results show that expression of JAK1, p-STAT3 and HIF-1α were significantly decreased with pU, pC and pCU treatment in both U251 and SNB19 cells (Figs. 3G–H). Densitometric analysis revealed that expression of JAK1, p-STAT3 and HIF-1α proteins were decreased to 35.9%, 62.5% (S-727) and 51.9% (Y-705), 50.2%, respectively, in pU-treated cells and 50.3%, 60.2% (S-727) and 45.8%(Y-705),49.1%, respectively, in pC-transfected U251 cells as compared to controls. Likewise, expression of JAK1, p-STAT3 and HIF-1α were decreased to 34.4%, 61.1% (S-727) and 54.3% (Y-705), 45.3%, respectively, in pU-transfected cells and 50.9%, 62.3% (S-727) and 57.9% (Y-705), 42.7%, respectively in pC-transfected SNB19 cells. Notably, expression of JAK1 was significantly decreased in pCU-transfected U251 cells (15.3%) and SNB19 cells (16.12%) when compared to controls. Moreover, expression of phosphorylated STAT3 was considerably decreased in the nuclear fractions of pCU-transfected U251 cells (S-727: 35.1% and Y-705: 29.8%) and SNB19 cells (S-727: 36.5% and Y-705: 30.8%). Further, expression of HIF-1α was predominantly less in pCU-treated U251 cells (38.42 %) and SNB19 cells (21.9%) (Fig. 3I). These results indicate that uPAR and cathepsin B either alone or in combination regulate VEGF expression through the JAK/STAT pathway and also by modulating HIF-1α expression.
Effect of uPAR and cathepsin B knockdown on tumor-induced expression of VEGFR-2, active Rac1 and gp91phox
Evidence supporting the role of gp91phox and Rac1 in VEGF-mediated proliferation of endothelial cells already exists.42 To identify downstream molecules of VEGF/VEGFR-2 signaling, endothelial cells were co-cultured with SV-, pU-, pC- and pCU-transfected U251 or SNB19 cells using the non-contact co-culture model. Expression of VEGFR-2 and gp91phox was determined in HMEC by western blotting. Active Rac1 (GTP form) was analyzed by immunoprecipitation. Results show that expression of p-VEGFR-2, VAV2, active Rac1 and gp91 phox were considerably decreased with pU and pC transfection while a more significant decrease was observed with pCU treatment in both U251 and SNB19 cells (Figs. 4A–B). Densitometric analysis of western blots indicated that expression levels of p-VEGFR-2, VAV2, active Rac1, and gp91phox were decreased to 49.44%, 88.71%, 57.66%, and 65.72%, respectively, with pU; decreased to 55.11%, 87.26%, 67.68%, and 52.81%, respectively, with pC; and decreased to 41.81%, 62.12%, 55.21%, and 35.44%, respectively, with pCU in U251 cells. Further, in SNB19 cells, the expression levels of p-VEGFR-2, VAV2, active Rac1, and gp91phox were decreased to 51.78%, 55.25%, 45.65%, and 41.24%, respectively, with pU; decreased to 41.15%, 41.39%, 61.10%, and 74.49%, respectively, with pC; and decreased to 41.81%, 62.12%, 55.21%, and 35.44%, respectively, with pCU (Fig. 4C).
Figure 4. Effect of uPAR and cathepsin B knockdown on tumor-induced expression of VEGFR-2, Rac1 and gp91phox.
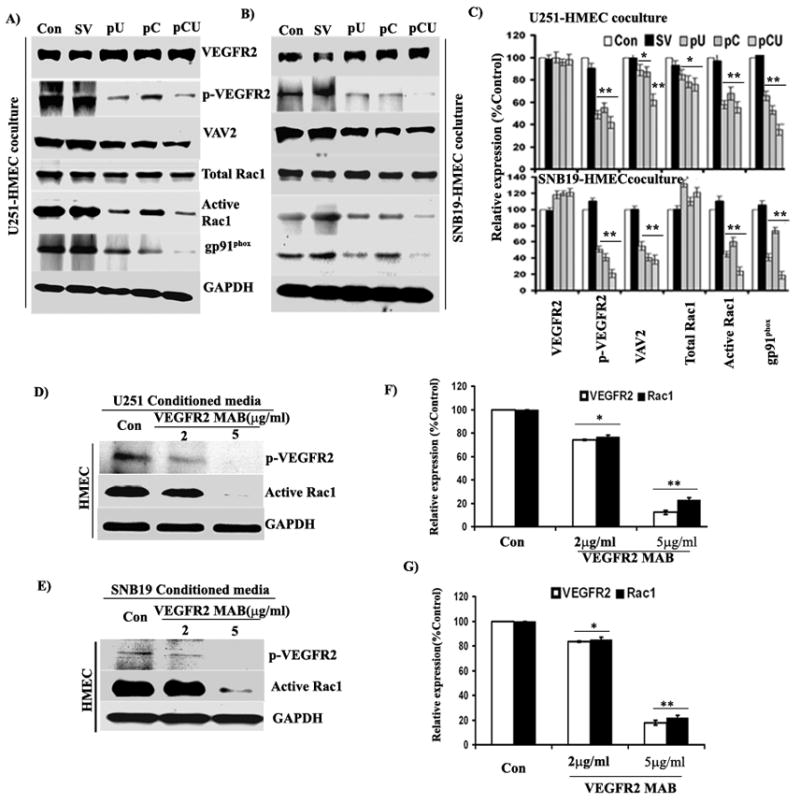
HMECs were co-cultured with U251 or SNB19 as described in Materials and Methods for 48 hrs. Cell lysates were collected and used for immunoblot analysis. (A–B) Immunoblot analysis of VEGFR-2, p-VEGFR-2, VAV2 and gp91phox expression in HMEC co-cultured with control and U251 and SNB19 cells transfected with SV, pU, pC and pCU using specific antibodies. Active Rac1 was selectively isolated by immunoprecipitation using GST-PBD (containing amino acids 51–135 of PAK1) and analyzed by western blotting. (C) Densitometric analysis of VEGFR-2, p-VEGFR-2, VAV2, Rac1 and gp91phox western blots. Bars represents the mean and SE of three experiments (*p<0.05, **p<0.001). (D–E) VEGFR-2 blocking inhibits tumor-induced expression of p-VEGFR-2 and Rac1 in HMEC. U251 and SNB19 cells were grown to 80% confluence, and conditioned media was collected after overnight incubation with serum-free medium. HMEC were grown as a monolayer and incubated with tumor-conditioned media containing VEGFR-2 monoclonal antibody (2 μg/mL and 5 μg/mL) for 48 hrs. After incubation, cells were harvested and cell lysates were used to determine the expression of p-VEGFR-2 and active Rac1 by western blotting. (F–G) Mean densitometric values ± SE values were calculated and plotted as a histogram. *Statistically different as compared to respective controls and treated groups (*p<0.05 and **p<0.001).
Effect of VEGFR-2 blockage on expression of p-VEGFR-2 and active Rac1
To confirm uPAR and cathepsin B-mediated VEGF/VEGFR-2 signaling, tumor-induced expression of VEGFR-2 was blocked by pretreatment of HMEC with neutralizing antibody for VEGFR-2 (MAB3571, R&D Systems, Minneapolis, MN). The results show that glioma-induced expression of p-VEGFR-2 and active Rac1 were decreased significantly with 5 μg/mL of VEGFR-2 antibody (Figs. 4D–E). Quantification of protein intensities by densitometry reveals that both U251 and SNB19 induced expression of p-VEGFR-2 was decreased to ~13% and active Rac1 decreased to ~22% with 5 μg/mL of VEGFR-2 antibody (Fig. 4F).
Knockdown of uPAR and cathepsin B inhibits tumor-induced endothelial cell migration
To investigate the effect of uPAR and cathepsin B knockdown on tumor induced migration of endothelial cells, untreated or pU-, pC- and pCU-treated U251 or SNB19 cells were co-cultured with HMEC for 48 hrs. Cells that migrated though the membrane were then stained. Results show that migration capability of HMEC was significantly inhibited by pCU and considerably with pU- and pC-transfected U251 and SNB19 cells (Supplementary Fig. 2A). Further, quantification reveals that endothelia cell migration was decreased to 48.63% and 35.51% by pU-transfected U251 cells and SNB19 cells, respectively, as compared to controls. In pC-transfected U251 and SNB19 cells, migration was decreased to 58.38% and 41.78% (p<0.05), respectively, as compared to controls. However, endothelial cell migration was significantly decreased to 66.72% and 72.19% by pCU-transfected U251 cells and SNB19 cells (p<0.001), respectively, as compared to controls (Supplementary Fig. 2B).
uPAR and cathepsin B knockdown inhibits tumor-induced endothelial cell proliferation
To study the effect of uPAR and cathepsin B knockdown on tumor-induced proliferation of endothelial cells, untreated or SV-, pU-, pC- or pCU-transfected U251 or SNB19 cells were co-cultured with HMEC, and proliferation was determined by BrdU incorporation assay. The results show that HMEC proliferation was decreased to 32.58% and 30.79% (p<0.05) by pU-transfected U251 and SNB19 cells, respectively, as compared to controls. Moreover, pC-transfected U251 and SNB19 cells decreased HMEC proliferation to 38.76% and 36.38% (p<0.001), respectively. Notably, HMEC proliferation was decreased to 28.68% and 26.56% (p<0.001) with pCU-transfected U251 and SNB19 cells, respectively, as compared to controls (Supplementary Fig. 2C).
uPAR and cathepsin B knockdown inhibits tumor-induced endothelial cell cycle progression
Proliferation of cells occurs through different phases of the cell cycle and is regulated by both positive as well as negative regulators. To study the effect of uPAR and cathepsin B downregulation on tumor-induced endothelial cell cycle progression and distribution of endothelial cell population in different phases of cell cycle, HMEC and transfected U251 or SNB19 cells were co-cultured for 48 hrs, and cell cycle analysis was carried by flow cytometry. The results show that pU-, pC- and pCU-transfected U251 and SNB19 cells significantly arrested HMEC cell cycle at G1 phase when compared to controls (Fig. 5A). The HMEC population was increased 1.63 and 1.65 fold with pU-transfected U251 and SNB19 cells, respectively as compared to controls at the G1 phase. However, both pC-treated U251 and SNB19 cells increased 1.65 fold of HMEC population at the G1 phase. The HMEC population was increased 1.7 fold in pCU-transfected U251 and SNB19 cells as compared to controls (Fig. 5B).
Figure 5. Effect of uPAR and cathepsin B knockdown on tumor-induced cell cycle progression.
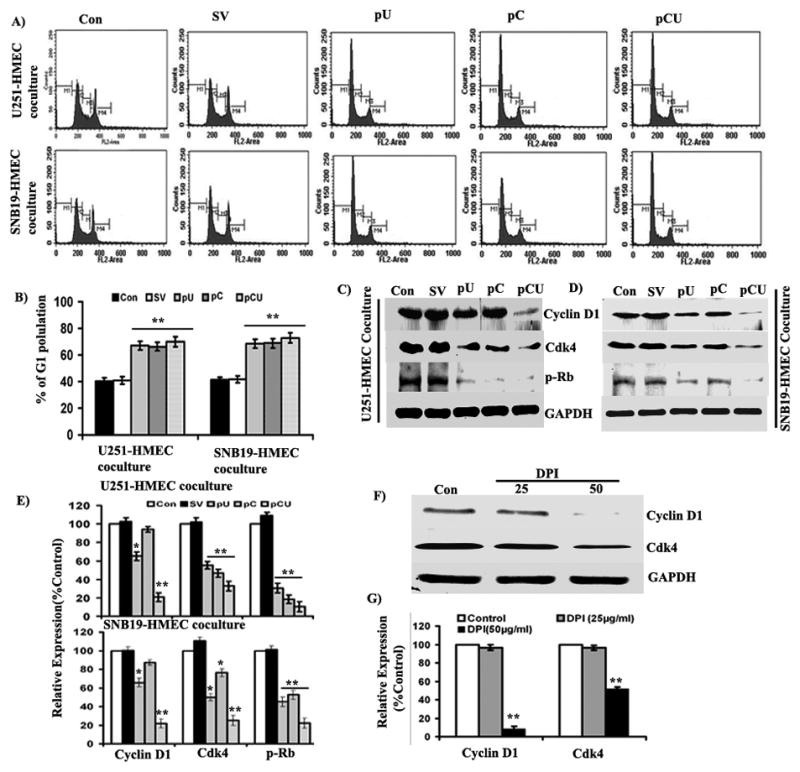
HMEC were co-cultured with U251 or SNB19 cells transfected with SV, pU, pC, and pCU for 48 hrs using a non-direct contact model. Propidium iodide-stained HMEC were analyzed for DNA content using flow cytometry to determine the fate of the cell cycle. (A) FACS analysis of HMEC co-cultured with U251 or SNB19 cells. (B) Graphical representation of cell population in G1 phase (M2) of HMEC. Values are mean ± SE from three different experiments (**p<0.001). (C–D) Western blot analysis of cyclin D1, Cdk4 and p-Rb in HMEC co-cultured with U251 or SNB19 cells. Cell lysates were collected from HMEC and U251 or SNB19 cocultures and fifty micrograms of total cell lysate were used to check the expression of cyclin D1, CDK4 and p-Rb by western blotting using specific antibodies. GAPDH served as a loading control. (E) Densitometric analysis of cyclin D1, Cdk4 and p-Rb western blots. Bars represents the mean and SE of three experiments (*p<0.05, **p<0.001). Diphenyleneiodonium ion (DPI), a specific inhibitor of gp91phox, inhibits expression of cyclin D1 and Cdk4 in HMEC. Cells were grown as a monolayer and treated with 25 μg/mL and 50 μg/mL of DPI for 48 hrs. (F) Cell lysates were used to determine the expression of cyclin D1 and Cdk4 by western blotting using specific antibodies. (G) Quantification of cyclin D1and Cdk4 bands by densitometry. Mean densitometric values ± SE values were calculated and plotted as a histogram. *Statistically different as compared to respective control and treated groups (**p<0.001).
As knockdown of uPAR and cathepsin B caused cell cycle arrest at the G1-S transition, we further determined glioma-induced expression of key G1-S transition positive modulators such as cyclin D1, cdk4, and p-Rb in HMEC. Here, we observed significant downregulation of cyclin D1, Cdk4 and p-Rb in HMEC with pCU-transfected U251 and SNB19 cells (Figs. 5C–D). Quantification of protein band intensities revealed that expression of cyclin D1, Cdk4 and p-Rb was decreased to 35–70%, 50–80% and 10–20% in both pU-, pC- and pCU-transfected U251 and SNB19 cells, respectively (Fig. 5E).
Effect of uPAR and cathepsin B on intracellular ROS
Several studies have reported the role of Rac1 and gp91phox in ROS generation.42–45 Emerging evidence indicates that intracellular ROS are involved in proliferation of endothelial cells.46 To know the effect of uPAR and cathepsin B knockdown on tumor-induced intracellular ROS formation, untreated or treated U251 or SNB19 cells were co-cultured with HMEC for 48 hrs and intracellular ROS levels were measured. The results show that pU transfection of U251 and SNB19 cells inhibited ROS to 55.45% and 50.34%, respectively, as compared to controls. In addition, pC treatment of U251 and SNB19 cells decreased intracellular ROS to 61.38% and 59.74%, respectively, in HMEC. Finally, pCU-transfected U251 and SNB19 cells inhibited intracellular ROS to 41.76% and 39.19%, respectively, in HMEC (Supplementary Fig. 2D).
Effect of gp91phox inhibitor (DPI) on expression of cyclin D1 and Cdk4
To confirm ROS-mediated regulation of cyclin D1 and Cdk4 (Cyclin dependent kinase), HMEC were treated with diphenyleneiodonium ion (DPI) (Sigma, Minneapolis, MN), a specific inhibitor of gp91phox, for 48 hrs. The results show that expression of cyclin D1 and Cdk4 were significantly downregulated at a 50 μM concentration of DPI (Fig. 5F). Densitometric analysis of protein bands indicate that expression of cyclin D1 was decreased to ~10% and Cdk4 to ~55% with DPI (50 μM) (Fig. 5G).
Immunohistochemical staining of VEGF and CD31 in intracranial tumor sections
To correlate the in vitro and in vivo results, we analyzed tumor growth and expression of VEGF and CD31 in intracranial tumor sections by immunohistochemical analysis. Mock and SV-treated brain sections had a large spread of tumor cells, whereas pCU-treated brain sections had a small number of tumor cells as illustrated by H&E staining (Fig. 6A–B). Semiquantification of H&E stained sections revealed ~80–90% reduction of tumor growth with pCU treatment (Fig. 6C). To determine whether the knockdown of uPAR and cathepsin B affected angiogenesis in intracranial tumors, the sections were stained with CD31 antibody, which specifically stains blood vessels. The results show that significant reduction of CD31 was observed in pCU-treated brain sections as compared to controls. Similarly, VEGF expression was significantly decreased in pCU-transfected brain sections as compared to controls (Fig. 6). Irrespective of cells used (U251 and SNB19), the effect of pCU treatment on tumor growth and the expression of CD31 and VEGF was the same.
Figure 6. uPAR and cathepsin B knockdown inhibits expression of VEGF and CD31 in tumor xenografts.
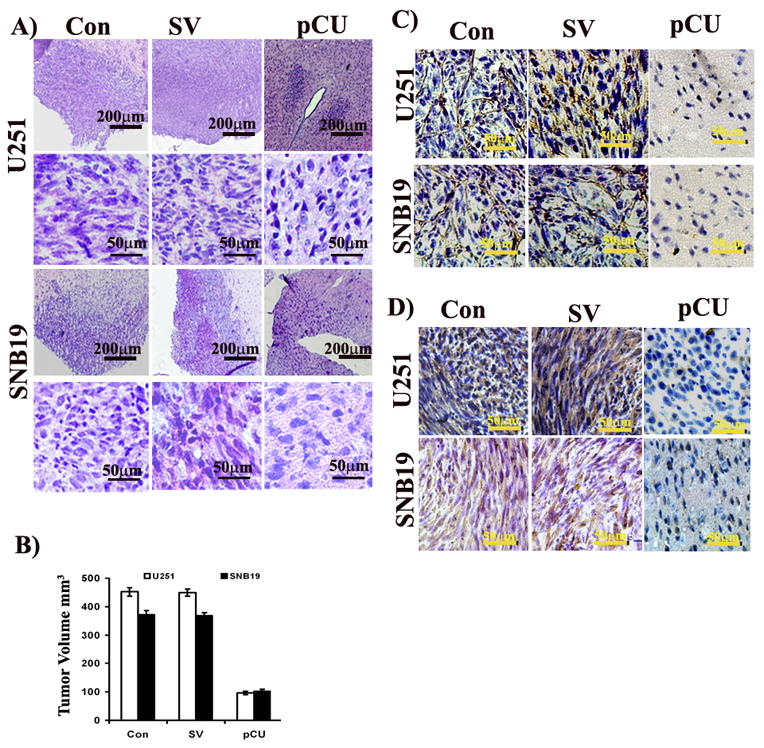
A) siRNA mediated regression of tumor growth. Hematoxylin and eosin staining performed on the brain sectioned to reveal tumor growth. The stained sections were blindly reviewed and scored for the size of the tumor in each case semiquantitatively. B) Semiquantification of tumor volume in control, SV and pCU-treated U251 and SNB19 brain sections. Immunohistochemical analysis of CD31 (C) and VEGF (D) in control, SV and pCU-treated U251 and SNB19 brain sections. To visualize VEGF and CD31 expression, immunohistochemical analysis was performed using specific antibodies as described in Materials and Methods.
Discussion
Neovascularization is fundamental in tumor growth, progression, and metastasis and requires growth factor driven recruitment, migration, proliferation, and differentiation of endothelial cells.47,48 Previous studies from our lab and others have shown that SNB19 and U251 glioma cell lines overexpress uPAR and cathepsin B.6–9 Gondi et al.19 have demonstrated the role of uPAR and cathepsin B in angiogenesis using tumor-conditioned media from SNB19 cells. However, the underlying mechanism of action is poorly understood. In this report, we have employed an endothelial/glioma co-culture model system to explore the role of uPAR and cathepsin B with regard to VEGF expression at cellular and molecular levels in tumor angiogenesis.
In this study, we demonstrated that downregulation of uPAR and cathepsin B using puPAR, pCathepsin B and pCU (a bicistronic construct against Cathepsin B and uPAR) decreased in vitro and in vivo tumor-induced angiogenesis. Gondi, et al. reported similar results in SNB19 glioma cells30,40 In the present study, we also demonstrated that downregulation of uPAR and cathepsin B in endothelial and U251 or SNB19 co-cultures significantly decreased VEGF and moderately decreased angiogenin, EGF, MCP-1, TIMP-1, TIMP-2, VEGF-D, Ang-1 (Angiopoietin), uPAR and VEGFR2 levels in tumor-conditioned media. Recently, Raghu et al. 49 reported that simultaneous downregulation of uPA and uPAR decreased IL-6, VEGF, Ang-1, VEGFR2 in endothelial and glioma co-cultures.
As a mechanism in support of the observed effect of in vitro and in vivo tumor-induced angiogenesis, we found that transcriptional suppression of uPAR and cathepsin B decreased VEGF expression at the protein and mRNA levels. Interestingly, upregulation of uPAR and cathepsin B further increased the VEGF expression. These results suggest that uPAR and cathepsin B might play an important role in the regulation of VEGF expression in glioma. Yanamandra, et al reported that downregulation of cathepsin B decreased expression of VEGF protein in glioblastoma.33 Nalabothula, et al. reported that transduction of glioblastoma cells with adenovirus vector containing the cDNA of wild-type p16 and antisense RNA of uPAR significantly inhibited capillary formation and VEGF expression.32
Interestingly, suppression of uPAR and cathepsin B at the transcriptional level with siRNA decreased JAK1, p-STAT3 (S727) and HIF-1α expression. Association of uPAR with some components of the JAK/STAT pathway was recently demonstrated in the human cancer cell line TCL-598.50 Niu, et al. reported that STAT3 is required for VEGF-mediated tumor progression.51 Joo et al. reported that binding of both STAT3 and HIF-1α to the VEGF promoter was required for maximum induction of VEGF expression.52 In contrast, Schindler, et al. reported that c-terminal region and phosphorylation at serine −727 are critically required for transcriptional activity of STAT3 with regard to VEGF expression.53 In either the case, STAT3 and HIF-1α or STAT3 alone is responsible for VEGF expression. Results from the present study revealed that uPAR and cathepsin B may modulate transcriptional regulation of VEGF expression through the JAK/STAT pathway.
We observed that the downregulation of uPAR and cathepsin B decreased tumor-induced migration and proliferation of endothelial cells. It is well known that VEGF plays a key role in the regulation of proliferation and migration of endothelial cells.54,55 VEGF was originally discovered as a potent vascular permeability factor that enhances proliferation of endothelial cells and tube formation after degradation of ECM by uPAR and cathepsin B.20,56
Angiogenesis is a highly complex process involving multiple interactions of pro-angiogenic, anti-angiogenic, angiostatic factors.57,58 Even though, numerous pro-angiogenic factors have been characterized, VEGF has been identified as the predominant regulator of tumor angiogenesis.59 However, the complex role of other pro-angiogenic and anti-angiogenic factors in the regulation of angiogenesis could not be ruled out. VEGF exerts its angiogenic effect by coupling to the VEGFR.60 The downstream effect of binding, dimerization and activation of the VEGF/VEGFR-2 complex includes migration and proliferation of endothelial cells.61 Like other tyrosine kinase receptors, VEGFR-2 triggers signal transduction by promoting receptor phosphorylation and the recruitment of specific downstream mediators.62,63 An interesting finding of the present study is that silencing of uPAR and cathepsin B genes either individually or simultaneously significantly decreased tumor-induced expression of p-VEGFR-2, Rac1, gp91phox and intracellular ROS levels. VEGFR2-Previous reports demonstrate that VEGF induces NADPH oxidase activity and ROS formation in endothelial cells.64,65 However, blockage of VEGFR-2 with VEGFR-2 monoclonal antibody decreased tumor-induced expression of p-VEGFR and active Rac1 in HMEC. The effect of VEGFR-2 blockage on VEGF-induced proliferation and migration has been reported in cultured normal human epidermal keratinocytes and orthotropic human breast cancer models.66,67 These results highlight the complexity and interconnectedness of angiogenic signaling pathways.
We also observed that uPAR and cathepsin B knockdown suppressed tumor-promoted endothelial cell cycle progression and expression of cyclin D1, Cdk4 and p-Rb. Recently, VEGF and bFGF-mediated regulation of cyclin D1 was reported in endothelial cells, which is central for the G1 to S transition in the cell cycle.68–70 DPI, a specific inhibitor of gp91phox, decreased the expression of cyclin D1 and Cdk4 in HMEC. Previous reports say that gp91-derived ROS is critically important in VEGF-induced angiogenesis and regulation of the G1 to S transition.42,71
In summary, our study demonstrates that knockdown of uPAR and cathepsin B may attenuate tumor-induced angiogenesis by decreasing VEGF expression and also affecting endothelial cell migration and proliferation. Notably, our results further indicate that simultaneous downregulation of uPAR and cathepsin B effectively inhibits glioma angiogenesis. In addition, the results of the present study reveal that uPAR and cathepsin B regulates tumor-induced angiogenesis through the VEGF/VEGFR-2 and ROS signaling pathways (Fig. 7). Taken together, our results show that siRNA-mediated silencing of uPAR and cathepsin B exhibits significant anti-angiogenic potential and may be an effective therapeutic agent for treatment of malignant glioma.
Figure 7.
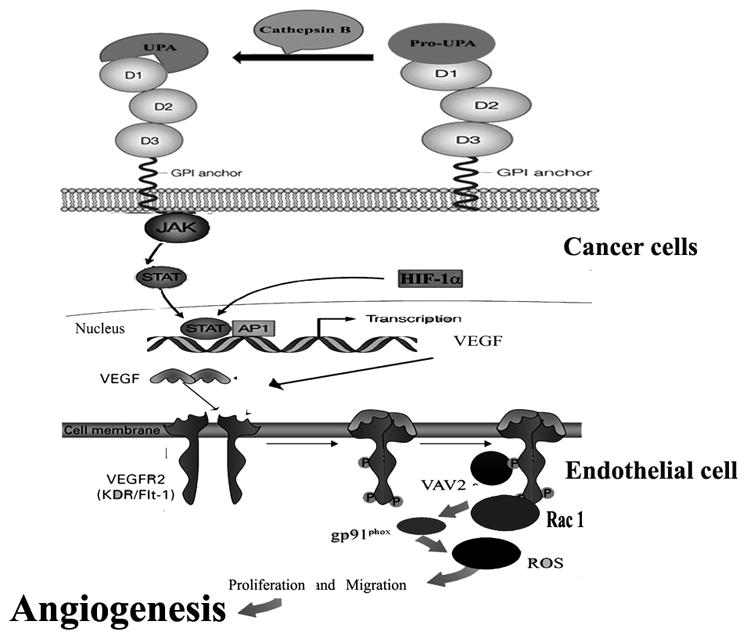
Schematic representation of the proposed molecular mechanism of uPAR and cathepsin B-mediated regulation of angiogenesis in glioma.
Supplementary Material
Table 1.
Genes analyzed by reverse transcriptase – PCR
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| uPAR | 5′GCCTTACCGAGGTTGTGTGT3′ | 5′CATCCAGGCACTGTTCTTCA3′ |
| Cathepsin B | 5′GCTACAGCCCGACCTACAAA3′ | 5′CCAGTAGGGTGTGCCATTCT3′ |
| VEGF | 5′CTACCTCCACCATGCCAAGT3′ | 5′CACACAGGATGGCTTGAAGA3′ |
| GAPDH | 5′CGGAGTCAACGGATTTGGTCGTAT3′ | 5′AGCCTTCTCCATGGTGGTGAAGAC3′ |
Acknowledgments
We thank Noorjehan Ali for technical assistance, Shellee Abraham for manuscript preparation, and Diana Meister and Sushma Jasti for manuscript review.
This research was supported by National Cancer Institute Grant CA116708. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of NIH.
Abbreviation
- uPAR
urokinase-type plasminogen activator receptor
- ECM
extra cellular matrix
- RT-PCR
reverse transcription polymerase chain reaction
- SDS-PAGE
sodium dodecyl sulphate-polyacrlyamide gel electrophoresis
- PBS
phosphate buffered saline
- HMEC
human dermal microvascular endothelial cells
- EGF
Epidermal growth factor
- MCP
Monocyte chemotactic protein
- TIMP
Tissue Inhibitor of Metalloproteinases
- JAK
Janus Kinase
- STAT
Signal Transducer and Activator of Transcription
- VEGF
Vascular endothelial growth factor
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenease
Footnotes
Supplementary information is available at Cancer Gene Therapy’s website.
Conflict of Interest:
RamaRao Malla This author declares no conflict of interest.
Sreelatha Gopinath This author declares no conflict of interest.
Christopher S. Gondi This author declares no conflict of interest.
Kiranmai Alapati This author declares no conflict of interest.
Dzung H. Dinh This author declares no conflict of interest.
Meena Gujrati This author declares no conflict of interest.
Jasti S. Rao Dr. Rao’s work has been funded by NIH.
Reference List
- 1.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 2.Bello L, Giussani C, Carrabba G, Pluderi M, Costa F, Bikfalvi A. Angiogenesis and invasion in gliomas. Cancer Treat Res. 2004;117:263–284. doi: 10.1007/978-1-4419-8871-3_16. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 4.Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–433. doi: 10.1038/nrc1094. [DOI] [PubMed] [Google Scholar]
- 5.Lakka SS, Gondi CS, Rao JS. Proteases and glioma angiogenesis. Brain Pathol. 2005;15:327–341. doi: 10.1111/j.1750-3639.2005.tb00118.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamberlain MC, Raizer J. Antiangiogenic therapy for high-grade gliomas. CNS Neurol Disord Drug Targets. 2009;8:184–194. doi: 10.2174/187152709788680706. [DOI] [PubMed] [Google Scholar]
- 7.Folkman J. What is the evidence that tumors are angiogenesis dependent? J Natl Cancer Inst. 1990;82:4–6. doi: 10.1093/jnci/82.1.4. [DOI] [PubMed] [Google Scholar]
- 8.Mohanam S, Sawaya R, McCutcheon I, Ali-Osman F, Boyd D, Rao JS. Modulation of in vitro invasion of human glioblastoma cells by urokinase-type plasminogen activator receptor antibody. Cancer Res. 1993;53:4143–4147. [PubMed] [Google Scholar]
- 9.Yamamoto M, Sawaya R, Mohanam S, Bindal AK, Bruner JM, Oka K, et al. Expression and localization of urokinase-type plasminogen activator in human astrocytomas in vivo. Cancer Res. 1994;54:3656–3661. [PubMed] [Google Scholar]
- 10.Gladson CL, Pijuan-Thompson V, Olman MA, Gillespie GY, Yacoub IZ. Up-regulation of urokinase and urokinase receptor genes in malignant astrocytoma. Am J Pathol. 1995;146:1150–1160. [PMC free article] [PubMed] [Google Scholar]
- 11.Sivaparvathi M, Sawaya R, Wang SW, Rayford A, Yamamoto M, Liotta LA, et al. Overexpression and localization of cathepsin B during the progression of human gliomas. Clin Exp Metastasis. 1995;13:49–56. doi: 10.1007/BF00144018. [DOI] [PubMed] [Google Scholar]
- 12.Pepper MS. Extracellular proteolysis and angiogenesis. Thromb Haemost. 2001;86:346–355. [PubMed] [Google Scholar]
- 13.Mai J, Sameni M, Mikkelsen T, Sloane BF. Degradation of extracellular matrix protein tenascin-C by cathepsin B: an interaction involved in the progression of gliomas. Biol Chem. 2002;383:1407–1413. doi: 10.1515/BC.2002.159. [DOI] [PubMed] [Google Scholar]
- 14.Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J. 1992;282:273–278. doi: 10.1042/bj2820273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Im E, Venkatakrishnan A, Kazlauskas A. Cathepsin B regulates the intrinsic angiogenic threshold of endothelial cells. Mol Biol Cell. 2005;16:3488–3500. doi: 10.1091/mbc.E04-11-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Premzl A, Turk V, Kos J. Intracellular proteolytic activity of cathepsin B is associated with capillary-like tube formation by endothelial cells in vitro. J Cell Biochem. 2006;97:1230–1240. doi: 10.1002/jcb.20720. [DOI] [PubMed] [Google Scholar]
- 17.Mazar AP. The urokinase plasminogen activator receptor (uPAR) as a target for the diagnosis and therapy of cancer. Anticancer Drugs. 2001;12:387–400. doi: 10.1097/00001813-200106000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Chapman HA, Wei Y, Simon DI, Waltz DA. Role of urokinase receptor and caveolin in regulation of integrin signaling. Thromb Haemost. 1999;82:291–297. [PubMed] [Google Scholar]
- 19.Preissner KT, Kanse SM, May AE. Urokinase receptor: a molecular organizer in cellular communication. Curr Opin Cell Biol. 2000;12:621–628. doi: 10.1016/s0955-0674(00)00141-1. [DOI] [PubMed] [Google Scholar]
- 20.Andreasen PA, Egelund R, Petersen HH. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell Mol Life Sci. 2000;57:25–40. doi: 10.1007/s000180050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3:15–32. doi: 10.1023/a:1009095825561. [DOI] [PubMed] [Google Scholar]
- 22.Obermajer N, Jevnikar Z, Doljak B, Kos J. Role of cysteine cathepsins in matrix degradation and cell signalling. Connect Tissue Res. 2008;49:193–196. doi: 10.1080/03008200802143158. [DOI] [PubMed] [Google Scholar]
- 23.Busso N, Masur SK, Lazega D, Waxman S, Ossowski L. Induction of cell migration by pro-urokinase binding to its receptor: possible mechanism for signal transduction in human epithelial cells. J Cell Biol. 1994;126:259–270. doi: 10.1083/jcb.126.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumler I, Petri T, Schleuning WD. Induction of c-fos gene expression by urokinase-type plasminogen activator in human ovarian cancer cells. FEBS Lett. 1994;343:103–106. doi: 10.1016/0014-5793(94)80298-x. [DOI] [PubMed] [Google Scholar]
- 25.Bohuslav J, Horejsi V, Hansmann C, Stockl J, Weidle UH, Majdic O, et al. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med. 1995;181:1381–1390. doi: 10.1084/jem.181.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Resnati M, Guttinger M, Valcamonica S, Sidenius N, Blasi F, Fazioli F. Proteolytic cleavage of the urokinase receptor substitutes for the agonist-induced chemotactic effect. EMBO J. 1996;15:1572–1582. [PMC free article] [PubMed] [Google Scholar]
- 27.Gondi CS, Rao JS. Therapeutic potential of siRNA-mediated targeting of urokinase plasminogen activator, its receptor, and matrix metalloproteinases. In: Sioud M, editor. siRNA and miRNA gene Silencing: From Bench to Bedside. Humana Press/Springer; 2008. pp. 267–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiffelers RM, van Roo I, Storm G. siRNA-mediated inhibition of angiogenesis. Expert Opin Biol Ther. 2005;5:359–368. doi: 10.1517/14712598.5.3.359. [DOI] [PubMed] [Google Scholar]
- 29.Jain RK, di TE, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8:610–622. doi: 10.1038/nrn2175. [DOI] [PubMed] [Google Scholar]
- 30.Gondi CS, Lakka SS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, et al. Adenovirus-mediated expression of antisense urokinase plasminogen activator receptor and antisense cathepsin B inhibits tumor growth, invasion, and angiogenesis in gliomas. Cancer Res. 2004;64:4069–4077. doi: 10.1158/0008-5472.CAN-04-1243. [DOI] [PubMed] [Google Scholar]
- 31.Rao JS, Gondi CS, Chittivelu S, Joseph PA, Lakka SS. Inhibition of invasion, angiogenesis, tumor growth and metastasis by adenovirus-mediated transfer of antisense uPAR and MMP-9 in non-small cell lung cancer cells. Mol Cancer Ther. 2005;4:1399–1408. doi: 10.1158/1535-7163.MCT-05-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nalabothula N, Lakka SS, Dinh DH, Gujrati M, Olivero WC, Rao JS. Sense p16 and antisense uPAR bicistronic construct inhibits angiogenesis and induces glioma cell death. Int J Oncol. 2007;30:669–678. [PMC free article] [PubMed] [Google Scholar]
- 33.Yanamandra N, Gumidyala KV, Waldron KG, Gujrati M, Olivero WC, Dinh DH, et al. Blockade of cathepsin B expression in human glioblastoma cells is associated with suppression of angiogenesis. Oncogene. 2004;23:2224–2230. doi: 10.1038/sj.onc.1207338. [DOI] [PubMed] [Google Scholar]
- 34.Gondi CS, Kandhukuri N, Kondraganti S, Gujrati M, Olivero WC, Dinh DH, et al. RNA interference-mediated simultaneous down-regulation of urokinase-type plasminogen activator receptor and cathepsin B induces caspase-8-mediated apoptosis in SNB19 human glioma cells. Mol Cancer Ther. 2006;5:3197–3208. doi: 10.1158/1535-7163.MCT-05-0531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leunig M, Yuan F, Menger MD, Boucher Y, Goetz AE, Messmer K, et al. Angiogenesis, microvascular architecture, microhemodynamics, and interstitial fluid pressure during early growth of human adenocarcinoma LS174T in SCID mice. Cancer Res. 1992;52:6553–6560. [PubMed] [Google Scholar]
- 36.Gondi CS, Lakka SS, Yanamandra N, Siddique K, Dinh DH, Olivero WC, et al. Expression of antisense uPAR and antisense uPA from a bicistronic adenoviral construct inhibits glioma cell invasion, tumor growth, and angiogenesis. Oncogene. 2003;22:5967–5975. doi: 10.1038/sj.onc.1206535. [DOI] [PubMed] [Google Scholar]
- 37.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. Intraperitoneal injection of an hpRNA-expressing plasmid targeting uPAR and uPA retards angiogenesis and inhibits intracranial tumor growth in nude mice. Clin Cancer Res. 2007;13:4051–4060. doi: 10.1158/1078-0432.CCR-06-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakka SS, Gondi CS, Yanamandra N, Olivero WC, Dinh DH, Gujrati M, et al. Inhibition of cathepsin B and MMP-9 gene expression in glioblastoma cell line via RNA interference reduces tumor cell invasion, tumor growth and angiogenesis. Oncogene. 2004;23:4681–4689. doi: 10.1038/sj.onc.1207616. [DOI] [PubMed] [Google Scholar]
- 39.Gondi CS, Lakka SS, Dinh D, Olivero W, Gujrati M, Rao JS. Downregulation of uPA, uPAR and MMP-9 using small, interfering, hairpin RNA (siRNA) inhibits glioma cell invasion, angiogenesis and tumor growth. Neuron Glia Biology. 2004;1:165–176. doi: 10.1017/s1740925x04000237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gondi CS, Lakka SS, Dinh DH, Olivero WC, Gujrati M, Rao JS. RNAi-mediated inhibition of cathepsin B and uPAR leads to decreased cell invasion, angiogenesis and tumor growth in gliomas. Oncogene. 2004;23:8486–8496. doi: 10.1038/sj.onc.1207879. [DOI] [PubMed] [Google Scholar]
- 41.Kargiotis O, Chetty C, Gogineni V, Gondi CS, Pulukuri SM, Kyritsis AP, et al. uPA/uPAR downregulation inhibits radiation-induced migration, invasion and angiogenesis in IOMM-Lee meningioma cells and decreases tumor growth in vivo. Int J Oncol. 2008;33:937–947. [PMC free article] [PubMed] [Google Scholar]
- 42.Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, et al. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- 43.Garrett TA, Van Buul JD, Burridge K. VEGF-induced Rac1 activation in endothelial cells is regulated by the guanine nucleotide exchange factor Vav2. Exp Cell Res. 2007;313:3285–3297. doi: 10.1016/j.yexcr.2007.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 45.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 46.Peshavariya H, Dusting GJ, Jiang F, Halmos LR, Sobey CG, Drummond GR, et al. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:193–204. doi: 10.1007/s00210-009-0413-0. [DOI] [PubMed] [Google Scholar]
- 47.Tandle A, Blazer DG, III, Libutti SK. Antiangiogenic gene therapy of cancer: recent developments. J Transl Med. 2004;2:22. doi: 10.1186/1479-5876-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 49.Raghu H, Lakka SS, Gondi CS, Mohanam S, Dinh DH, Gujrati M, et al. Suppression of uPA and uPAR attenuates angiogenin mediated angiogenesis in endothelial and glioblastoma cell lines. PLoS One. 2010;5:e12458. doi: 10.1371/journal.pone.0012458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koshelnick Y, Ehart M, Hufnagl P, Heinrich PC, Binder BR. Urokinase receptor is associated with the components of the JAK1/STAT1 signaling pathway and leads to activation of this pathway upon receptor clustering in the human kidney epithelial tumor cell line TCL-598. J Biol Chem. 1997;272:28563–28567. doi: 10.1074/jbc.272.45.28563. [DOI] [PubMed] [Google Scholar]
- 51.Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene. 2002;21:2000–2008. doi: 10.1038/sj.onc.1205260. [DOI] [PubMed] [Google Scholar]
- 52.Jung JE, Lee HG, Cho IH, Chung DH, Yoon SH, Yang YM, et al. STAT3 is a potential modulator of HIF-1-mediated VEGF expression in human renal carcinoma cells. FASEB J. 2005;19:1296–1298. doi: 10.1096/fj.04-3099fje. [DOI] [PubMed] [Google Scholar]
- 53.Schindler C, Levy DE, Decker T. JAK-STAT signaling: from interferons to cytokines. J Biol Chem. 2007;282:20059–20063. doi: 10.1074/jbc.R700016200. [DOI] [PubMed] [Google Scholar]
- 54.Wang S, Li X, Parra M, Verdin E, Bassel-Duby R, Olson EN. Control of endothelial cell proliferation and migration by VEGF signaling to histone deacetylase 7. Proc Natl Acad Sci U S A. 2008;105:7738–7743. doi: 10.1073/pnas.0802857105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science. 1983;219:983–985. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 56.Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer Metastasis Rev. 2003;22:271–286. doi: 10.1023/a:1023007717757. [DOI] [PubMed] [Google Scholar]
- 57.Chen Y, Wei T, Yan L, Lawrence F, Qian HR, Burkholder TP, et al. Developing and applying a gene functional association network for anti-angiogenic kinase inhibitor activity assessment in an angiogenesis co-culture model. BMC Genomics. 2008;9:264. doi: 10.1186/1471-2164-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tokumo K, Kodama J, Seki N, Nakanishi Y, Miyagi Y, Kamimura S, et al. Different angiogenic pathways in human cervical cancers. Gynecol Oncol. 1998;68:38–44. doi: 10.1006/gyno.1997.4876. [DOI] [PubMed] [Google Scholar]
- 59.Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. doi: 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- 60.Milkiewicz M, Hudlicka O, Brown MD, Silgram H. Nitric oxide, VEGF, and VEGFR-2: interactions in activity-induced angiogenesis in rat skeletal muscle. Am J Physiol Heart Circ Physiol. 2005;289:H336–H343. doi: 10.1152/ajpheart.01105.2004. [DOI] [PubMed] [Google Scholar]
- 61.Rini BI, Small EJ. Biology and clinical development of vascular endothelial growth factor-targeted therapy in renal cell carcinoma. J Clin Oncol. 2005;23:1028–1043. doi: 10.1200/JCO.2005.01.186. [DOI] [PubMed] [Google Scholar]
- 62.Thomas KA. Vascular endothelial growth factor, a potent and selective angiogenic agent. J Biol Chem. 1996;271:603–606. doi: 10.1074/jbc.271.2.603. [DOI] [PubMed] [Google Scholar]
- 63.Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69 (Suppl 3):4–10. doi: 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- 64.Abid MR, Kachra Z, Spokes KC, Aird WC. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000;486:252–256. doi: 10.1016/s0014-5793(00)02305-x. [DOI] [PubMed] [Google Scholar]
- 65.Abid MR, Tsai JC, Spokes KC, Deshpande SS, Irani K, Aird WC. Vascular endothelial growth factor induces manganese-superoxide dismutase expression in endothelial cells by a Rac1-regulated NADPH oxidase-dependent mechanism. FASEB J. 2001;15:2548–2550. doi: 10.1096/fj.01-0338fje. [DOI] [PubMed] [Google Scholar]
- 66.Man XY, Yang XH, Cai SQ, Yao YG, Zheng M. Immunolocalization and expression of vascular endothelial growth factor receptors (VEGFRs) and neuropilins (NRPs) on keratinocytes in human epidermis. Mol Med. 2006;12:127–136. doi: 10.2119/2006-00024.Man. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang W, Ran S, Sambade M, Huang X, Thorpe PE. A monoclonal antibody that blocks VEGF binding to VEGFR2 (KDR/Flk-1) inhibits vascular expression of Flk-1 and tumor growth in an orthotopic human breast cancer model. Angiogenesis. 2002;5:35–44. doi: 10.1023/a:1021540120521. [DOI] [PubMed] [Google Scholar]
- 68.Pai R, Szabo IL, Kawanaka H, Soreghan BA, Jones MK, Tarnawski AS. Indomethacin inhibits endothelial cell proliferation by suppressing cell cycle proteins and PRB phosphorylation: a key to its antiangiogenic action? Mol Cell Biol Res Commun. 2000;4:111–116. doi: 10.1006/mcbr.2000.0260. [DOI] [PubMed] [Google Scholar]
- 69.Pedram A, Razandi M, Levin ER. Natriuretic peptides suppress vascular endothelial cell growth factor signaling to angiogenesis. Endocrinology. 2001;142:1578–1586. doi: 10.1210/endo.142.4.8099. [DOI] [PubMed] [Google Scholar]
- 70.Li A, Li H, Jin G, Xiu R. A proteomic study on cell cycle progression of endothelium exposed to tumor conditioned medium and the possible role of cyclin D1/E. Clin Hemorheol Microcirc. 2003;29:383–390. [PubMed] [Google Scholar]
- 71.Havens CG, Ho A, Yoshioka N, Dowdy SF. Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol Cell Biol. 2006;26:4701–4711. doi: 10.1128/MCB.00303-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.