

Abstract
Nitric oxide-donating aspirin (NO-ASA) is a promising agent for cancer prevention. Although studied extensively, its molecular targets and mechanism of action are still unclear. S-nitrosylation of signaling proteins is emerging as an important regulatory mechanism by NO. Here, we examined whether S-nitrosylation of the NF-κB, p53, and Wnt signaling proteins by NO-ASA might explain, in part, its mechanism of action in colon cancer. NO-ASA releases significant amounts of NO detected intracellularly in HCT116 and HT-29 colon cells. Using a modified biotin-switch assay we demonstrated that NO-ASA S-nitrosylates the signaling proteins p53, β-catenin, and NF-κB, in colon cancer cells in a time- and concentration-dependent manner. NO-ASA suppresses NF-κB binding to its cognate DNA oligonucleotide, which occurs without changes in the nuclear levels of the NF-κB subunits p65 and p50 and is reversed by dithiothreitol that reduces –S-NO to –SH. In addition to S-nitrosylation, we documented both in vitro and in vivo widespread nitration of tyrosine residues of cellular proteins in response to NO-ASA. Our results suggest that the increased intracellular NO levels following treatment with NO-ASA modulate cell signaling by chemically modifying key protein members of signaling cascades. We speculate that S-nitrosylation and tyrosine nitration are responsible, at least in part, for the inhibitory growth effect of NO-ASA on cancer cell growth and that this may represent a general mechanism of action of NO-releasing agents.
INTRODUCTION
Colon carcinogenesis is often a prolonged process requiring years between the initial cellular change and the development of clinical disease [1]. This process is influenced by diet, environment and genetic factors. Inhibition of the initiation phases and delaying progression could best be achieved through effective cancer prevention approaches [2]. Non-steroidal anti-inflammatory drugs (NSAIDs) show promise as chemopreventive agents against colon cancer [3]. Their use, however, is associated with a wide spectrum of side effects, some of which are life-threatening [1, 4–5]. The need for safer and more effective NSAIDs has led to the synthesis of NO-donating NSAIDs (NO-NSAIDs), which have emerged as safer and highly effective chemopreventive agents [6]. We and others have shown that several NO-NSAIDs are much more effective in reducing the growth of colon cancer cells than the corresponding parent NSAIDs [7–9]. Inhibition of cell growth is linked to inhibition of cell proliferation, induction of apoptosis and a significant block in cell cycle transitions [8]. One of the most promising NO-NSAID to date is NO-aspirin (NO-ASA), which displays a remarkable chemopreventive ability both in vitro and in animal models of colon cancer [10–12]. The mechanism responsible for the enhanced potency of NO-ASA is complex and appears to include effects on cell signaling mediated by the β-catenin/T-cell factor (TCF) [13] and the NF-κB pathways [14].
S-nitrosylation, the formation of S-nitrosothiols by the covalent addition of NO to cysteine residues, has been shown to regulate the function of several cellular proteins [15]. We have previously suggested that S-nitrosylation of β-catenin in response to NO-ASA may explain the inhibitory effect of NO-ASA on the Wnt pathway [16]. In particular, we have proposed that S-nitrosylation of cysteine residues in the binding domains of β-catenin and TCF prevents their association, which is required for the formation of an effective transcriptional outcome. This effect may be due, in part, to steric hindrance or conformational changes, either one being a consequence of S-nitrosylation. A similar mechanism may account for the inhibitory effect of NO-ASA on NF-κB signaling [14].
Given these considerations, we undertook a direct evaluation of S-nitrosylation in the β-catenin/TCF and NF-κB signaling pathways in HT-29 and HCT116 human colon cancer cells. In addition, we evaluated in HCT116 cells whether NO-ASA S-nitrosylates p53, a tetrameric protein that plays a pivotal role in about half of all human cancers, including colon cancer [17–18]. Furthermore, we evaluated both in vitro and in vivo levels of nitrotyrosine, a stable end-product of the nitration of a tyrosine residue. The nitrate moiety for the formation of nitrotyrosine is provided by the reactive nitrating intermediate peroxynitrite (ONOO−), which in turn can result from the reaction of NO and superoxide anion (O2·) [19–21]. Nitrotyrosine, a marker for peroxynitrite and other nitrating species [22], provides a complementary assessment of the effect of NO on cellular proteins.
MATERIALS AND METHODS
Reagents
NO-ASA [NCX4040, 2-(acetyloxy) benzoic acid 4-(nitrooxymethyl) phenyl ester] was synthesized by us [23] and conventional ASA was obtained from Sigma, St. Louis MO. For both we prepared 100 mM stock solutions in DMSO; the final DMSO concentration in all media did not exceed 1%. All general solvents and reagents were of HPLC grade or the highest grade commercially available. All antibodies were from Calbiochem (San Diego, CA), with the exception of the anti-nitro-tyrosine antibody, which was from Chemicon (Temecula, CA).
Cell Culture
We used two human colon cell lines HT-29 and HCT116 (American Type Culture Collection (ATCC), Manassas, VA). HT-29 is mutant for apc and p53 and wt for cox-2 and β-catenin. HCT116 is wt for apc and p53 and mutant for cox-2 and β-catenin [14, 24–25]. Both cell lines were grown as monolayers in the medium suggested by ATCC and supplemented with 10% FCS (Mediatech, Herndon, VA), penicillin (50 units/ml), and streptomycin (50 μg/ml; Life Technologies, Inc., Grand Island, NY). Cells were incubated at 37°C in 5% CO2. Cell viability/growth was measured using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) colorimetric assay according to the manufacturer’s instructions (Roche Diagnostics, Nutley, NJ).
Assay of Intracellular NO Levels
NO levels were determined using the DAF-FM DA (4-amino-5-methylamino-2′,7′-difluorofluorescein diacetate) molecular probe according to the manufacturer’s instructions (Molecular Probes, Eugene OR). Briefly, after treatment, cells were loaded with 5 μM of DAF-FM DA for 1 hr at room temperature. Excess probe was removed by washing with buffer followed by centrifugation. Fresh buffer was added and de-esterification of the probe was allowed to proceed for 30 min at room temperature. NO detection was achieved with a fluorescent microplate reader (excitation/emission of 495/515 nm).
Confocal Microscopy
Control and NO-ASA-treated colon cancers cells were incubated for 1 hr at 37°C with DAF-FM DA. Excess probe was removed by washing with buffer and cells were scanned using confocal laser scanning microscopy (2 μm sections from the bottom).
Protein Extraction
Whole cell extracts were obtained by lysing cells in RIPA buffer (50 mM Tris-HCl (pH 7.4),150 mM NaCl, 1 mM Na2EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1% NP-40, 0,25% sodium deoxycholate and Protease Inhibitor Cocktail 2 (Sigma-Aldrich, St. Louis, MO)). Cytoplasmic and nuclear extracts were prepared following a standard protocol [26]; trypsinized cells were suspended in lysis buffer to which Nonidet P-40 was added at a subsequent step (the supernatant fraction represented the cytoplasmic extract); nuclei were washed and centrifuged, followed by resuspension in extraction buffer and pelleting. Protein extracts were stored at −80°C until further analysis.
S-nitrosylation
We used a modification of the biotin-switch method by Jaffrey et al. [27]. After treatment with NO-ASA, cells were harvested using a rubber policeman and centrifuged at 1000 rpm for 5 minutes at 4°C. Cells were then washed once with cold PBS (containing 5 mM EDTA, 0.1 mM Neocuprine and 1 mM methyl methanethiosulfonate (MMTS)). Nuclear and whole cell extracts were prepared as described above. The supernatant typically contained 0.8 μg protein per μl assayed with the Bio-Rad protein Assay (BIO-RAD Laboratories, Inc. Hercules, CA). Four volumes of blocking buffer (9 volumes of HEN buffer (250 mM HEPES pH 7.7, 1 mM EDTA, and 0.1 μM neocuproine) plus 1 volume 25% SDS (adjusted to 20 mM MMTS with a 2 M stock prepared in dimethylformamide (DMF)) were added to the supernatant which was then incubated for 20 min at 50°C with frequent vortexing. MMTS was removed by precipitation with ice-cold acetone (4 volumes of acetone were added to one volume of supernatant, mixed gently and stored at −20°C for 30 min followed by centrifugation at 3000× g for 10 min at 4°C). Pellets were resuspended in 0.5 ml HENS buffer (HEN buffer plus 1% SDS) and proteins were biotinylated using biotin–HPD; following the instructions of the manufacturer (Thermo Scientific, Rockford, IL). Biotinylated proteins were isolated by acetone precipitation and centrifugation. The pellet was resuspended in HENS buffer as above. To this point, all steps were carried out in the dark. Two volumes of neutralization buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 1 mM EDTA and 0.5% Triton X-100) were added and any unresolved particulates were pelleted by centrifugation and discarded. Streptavidin–agarose (Sigma, St. Louis, MO) was added to the solution and the mixture was allowed to incubate for 1 h at room temperature. The mixture was subsequently washed 5 times with neutralization buffer with its NaCl concentration adjusted to 600 mM. To recover the bound protein, the mixture was incubated for 20 min at room temperature in elution buffer (20 mM HEPES pH 7.7, 100 mM NaCl, 1 mM EDTA and 100 mM 2-mercaptoethanol). SDS-PAGE sample buffer was added and proteins were fractionated by gel electrophoresis and the presence of S-nitrosylated proteins was accessed by western blot analysis.
Electrophoretic Mobility Shift Assays (EMSAs)
The NF-κB-DNA binding activity was assessed by reacting extracts from control and cells treated with NO-ASA with 33 nmol of a 32P-end-labeled double-stranded consensus oligonucleotides for NF-κB (AGTTGAGGGGACTTTCCCAGGC; Promega Corp., San Luis Obispo, CA) or p53 (GAACATGTCTAAGCATGCTG; Santa Cruz Biotechnology Inc., Santa Cruz CA). The specificity of the binding was tested by competition reactions, using 50-fold molar excess of unlabeled NF-κB or p53 (specific competition). The complexes were separated on a 6% gel following standard protocols.
Xenograft Tumor Model
Male athymic nu/nu mice aged 5 weeks (Harlan Bioproducts, Indianapolis, IN) were maintained in a maximum isolation environment, according to an institutionally approved animal protocol, and given food and water ad libitum. After one week of acclimatization, 106 HT-29 cells suspended in 100 μl of sterile Dulbecco’s phosphate-buffered saline (Mediatech) were implanted subcutaneously into the right flank of each mouse. After 3 weeks, when tumors had reached an average volume of ~300 mm3, intratumoral injections were conducted twice a week for 3 weeks with 200 μl vehicle or NO-ASA 300 μM. One additional control group received no injections. At the end of the study, mice were euthanized and the tumors, cut sagittally, were fixed in 10% buffered formalin. The area of necrosis in each tumor was determined as reported [12].
Immunohistochemistry
Paraffin sections were deparaffinized, rehydrated and antigen retrieval obtained by heating in a microwave for 15 min in 0.01 M citrate buffer (pH 6.0). After 15 min of blocking with serum, the primary antibody nitro-tyrosine () at 1:100 or the control isotype IgG was applied and incubated overnight at 4°C. Slides were washed three times with phosphate buffered saline for 5 min. The biotinylated secondary antibody and the streptavidin biotin complex (Invitrogen Corporation, Carlsbad, CA) were applied, each for 60 min at room temperature with interval washings. The slides were rinsed with distilled water, dehydrated and a coverslip was applied. Two independent evaluators, blinded to sample identity, scored at least 5 randomly selected fields (× 200 magnification) and scored nitro-tyrosine expression as follows: 0 = negative staining for nitro-tyrosine; 1 = weakly positive; 2 = moderately positive; 3 = more than moderate and less than strong; 4= strongly positive (maximal). The average value was calculated for each sample.
Statistical Methods
Data were expressed as mean ± SEM and differences between groups were analyzed by the t-test. P< 0.05 denotes significant differences.
RESULTS
NO-ASA inhibits the growth of human cancer cell lines and releases NO
As expected [8], NO-ASA inhibited the growth of both HT-29 and HCT116 human colon cancer cell lines. The 24-hr IC50s for HT-29 and HCT116 cells were 28±2.2 μM and 92±4.7 μM, respectively. For both cell lines the IC50 of aspirin, the parent compound of NO-ASA, was greater than 5 mM. The reason for the nearly 3-fold difference in potency between these two cell lines is not readily apparent.
The intracellular levels of NO were quantified in HT-29 and HCT116 cells treated with and without NO-ASA for 1 or 3 hr. NO levels were determined in cells preloaded with the NO molecular probe in three ways: a) spectrophotometrically; b) by flow cytometry analysis; and c) by confocal microscopy. As an additional control, we treated these cells with deoxycholate, reported to stimulate the production of NO [28]. After 1 hr of incubation with various concentrations of NO-ASA or 500 μM deoxycholate, we observed a modest elevation of NO levels in both cell lines (data not shown). At 3 hr there was a quantitatively more pronounced increase in NO levels, as detected by all three methods (Fig. 1). The changes in NO levels were concentration-dependent. As can be seen in Fig 1, at 1 and 3 hr of incubation the increase in NO levels ranged between 1.4 and 1.8 fold over control (average of 3 methods), respectively. Incidentally, there was marked increase in NO levels in detached cells (floating in the culture medium), compared to those attached to the culture plate. At the time of the assay, all detached cells were viable (as determined by the trypan blue exclusion method) but the process of cell death had been initiated. This finding is consistent with our previous report on the redox changes in cells dying in response to NO-ASA and related compounds [29].
Fig. 1. NO-ASA increases intracellular NO levels in colon cancer cell lines.

HT-29 and HCT116 colon cancer cells were treated with various concentrations of NO-ASA for 3 hr. Floating cells were harvested from the gently aspirated culture media by low speed centrifugation, whereas adherent cells were harvested following trypsinization and centrifugation. Both non-adherent and adherent cells were kept on ice and washed two times with cold PBS. Cells were then loaded with the molecular probe DCFDA and fluorescence was assayed using a microplate reader (top), imaged by confocal microscopy (bottom left) or quantified by flow cytometry (bottom right). No adherent cells were apparent on the cell culture plate after a 3 hr incubation with 50 μM NO-ASA.
NO-ASA nitrosylates cysteine and nitrates tyrosine residues in proteins
Initially, we assessed the general effect of NO-ASA on S-nitrosylation of proteins in these two cell lines. Cells were treated with various concentrations of NO-ASA for 3 hr, subjected to a modified biotin switch assay and evaluated by Western blot analysis using a primary antibody against biotin. As shown in Fig. 2A, NO-ASA induced S-nitrosylation of several proteins in colon cancer cells. Of interest, among the modified proteins were proteins with sizes similar to those of β-catenin, the p65 and p50 NF-κB subunits, β-catenin and p53. S-nitrosylation occurred in both cytoplasmic and nuclear proteins (data not shown) and was not time-dependent (1 and 3 hr, Fig. 2). A similar effect was demonstrated in BxPC-3 human pancreatic cancer cells treated with NO-ASA (data not shown). A striking change occurs between 1 and 3 hr in the expression of a S-nitrosylated protein of apparent molecular weight slightly larger than 50 kD in the untreated control. The significance of this change is uncertain, especially since this protein has not been identified.
Fig. 2. NO-ASA S-nitrosylates proteins and nitrates protein tyrosine residues in human colon cancer cells.

A and B: HCT116 and HT-29 cells were treated with the indicated concentrations of NO-ASA for 1 or 3 hr. Whole cell lysates were isolated and subjected to the modified biotin switch assay (A) or to immunoblot analysis using an anti-nitrotyrosine primary antibody (B). The arrows on the right identify S-nitrosylated proteins or proteins whose nitrotyrosine content changed as a result of treatment with NO-ASA. Loading control: α-tubulin. C: HT-29 human colon cancer xenografts treated with NO-ASA were evaluated by IHC for their nitrotyrosine content as in Methods. Three representative photomicrographs (magnification 200×) are shown on the top. Control, a section treated with non-specific antibody that was isotypic to that specific for nitrotyrosine used in the other two sections. Vehicle, sample from mice treated with vehicle (n=10), showing limited staining for nitrotyrosine; NO-ASA, sample from mice treated with NO-ASA (n=8), strongly positive for nitrotyrosine. The graph on the bottom depicts the mean ± SEM of the nitrotyrosine score for the two study groups.
The cellular production of highly reactive nitrogen species derived from NO, such as peroxynitrite, leads to the nitration of tyrosine residues in tissue proteins [30–31]. Peroxynitrite forms through the reaction of NO with superoxide anion, a reaction considered to mediate cytotoxic effects of NO in the cell [32]. The degree of protein nitrotyrosine formation is considered an indication of the production of reactive nitrogen species and of potential cell damage [21, 33]. Of significance to the present work is the fact that tyrosine nitration of nuclear p65 induces its dissociation from p50 and subsequent association with IκBα and export into the cytoplasm [34]. Consequently, we examined the potential nitration of tyrosine residues of proteins in response to NO-ASA.
HCT116 and HT-29 cells were treated for 3 hr with DMSO or various concentrations of NO-ASA. Cytoplasmic and nuclear fractions were subjected to Western blot analysis using a primary antibody that specifically recognizes proteins with nitrated tyrosine residues. As shown in Figure 2B, NO-ASA caused the formation of nitrotyrosines in several proteins, amongst which members of the NF-κB family have been tentatively identified based on their size.
To evaluate the possibility of tyrosine nitration in vivo, we studied by IHC the levels of tyrosine nitration in HT-29 colon cancer xenografts in nude mice treated with NO-ASA [12]. Mice were injected intratumorally twice a week for 3 weeks with vehicle or NO-ASA, while a third group received no injections. At sacrifice, the necrotic area of tumors, expressed as percentage of total area, was similar in the non-injected and vehicle-injected groups (data not shown). The necrotic area of tumors was higher in the NO-ASA-treated group (65.8±2.4%; mean±SEM for this and all subsequent values), compared with the vehicle group (52.2 ±4.1%; P < 0.001). NO-ASA more than doubled the amount of tyrosine nitration in the xenograft tissues, as shown in Fig. 2C; the tyrosine nitration scores in the two groups were: vehicle = 0.73±0.25 and NO-ASA = 1.82±0.41 (p=0.03).
NO-ASA S-nitrosylates β-catenin, the p65 NF-κB subunit and p53 in HCT116 cells
To assess S-nitrosylation of β-catenin, of the p65 subunit of NF-κB and of p53 in whole cell lysates as well as in cytoplasmic and nuclear factions, HCT116 cells were treated with various concentrations of NO-ASA. As shown in Fig. 3, NO-ASA induced S-nitrosylation of all these three proteins. This effect was clear, regardless of the origin of the proteins (cytoplasmic vs. nuclear). In all instances this effect was concentration-dependent although a plateau seemed to be reached rather quickly. Time dependence was also evident, at least in the case of β–catenin, the only protein for which it was evaluated.
Fig. 3. NO-ASA S-nitrosylates β-catenin, p53 and the NF-κB subunit p65 in HCT 116 cells.

Cells were treated as indicated with NO-ASA for 1 or 3 hr and whole cell, cytoplasmic or nuclear protein extracts, as indicated, were analyzed using the biotin switch method that detects S-nitrosylated proteins. A: Top panel: S-nitrosylated β-catenin levels in protein extracts from HCT 116 cells. Bottom panel: S-nitrosylated β-catenin levels from the cytoplasm or nucleus of cells treated with NO-ASA 80 μM. B: S-nitrosylated p53 levels in protein extracts from whole cells (top panel) or the cytoplasm and nucleus (bottom panel). C: S-nitrosylated p65 levels in protein extracts from whole cells. Loading controls: α-tubulin or β-actin for cytoplasmic or whole cell protein extracts; Sam 68, a nucleus-predominantly expressed protein, for nuclear protein extracts.
NO-ASA inhibits the binding of nuclear NF-κB to its cognate oligonucleotide
HT-29 and HCT 116 cells were treated for 3 hr with NO-ASA and NF-κB activation was evaluated using EMSAs. As expected [14], NO-ASA inhibited NF-κB activation in a concentration-dependent manner in both cell lines, whereas aspirin, the parent compound, exhibited only a marginal inhibitory effect at a concentration of 5 mM. To further evaluate the role of S-nitrosylation in the inhibition of NF-κB-DNA binding, we treated, for 1 hr, the nuclear extracts of these cells with 20 mM dithiotreitol (DTT); an agent that effectively removes the thiol-bound NO groups from proteins [35]. As shown in Fig 4, DTT restored NF-κB-DNA binding. This was also true of untreated HT-29 and HCT 116 cell extracts, demonstrating the baseline formation of nitrosothiols. However, it is important to note that we cannot rule out the possibility that the reversal by DTT is due to disruption of disulfide bonds that may have formed. In addition, HT-29 and HCT 116 cells were treated for 3 hr with NO-ASA and p53 activation was evaluated using EMSAs. Although we have demonstrated S-nitrosylation of p53, NO-ASA did not inhibit the binding of p53 to its cognate oligonucleotide, indicating the specificity of the overall effect. Interestingly, the role of oxidative modifications of p53 for its biological function remains unclear; p53 being subject to more than one level of conformational modulation through oxidation-reduction of cysteines at or near the p53-DNA interface [36–38].
Fig. 4. NO-ASA inhibits DNA binding of NF-κB but not of p53.

Top panel: Nuclear extracts of HT-29 (right) and HCT 116 (left) colon cancer cells treated for 3 hr with NO-ASA at the indicated concentrations were analyzed by EMSA for NF-κB binding. The indicated nuclear extracts were treated with DTT 20 mM for 15 min prior to their reaction with the NF-κB oligonucleotide to determine the effect of the reducing agent on DNA binding. Middle panel: Western blot analysis of the nuclear levels of individual NF-κB signaling proteins and of p53 from cells treated with NO-ASA or ASA as indicated. Loading control: Sam 68. Bottom panel: Nuclear extracts of HT-29 (right) and HCT 116 (left) colon cancer cells treated for 3 hr with NO-ASA at the indicated concentrations were analyzed by EMSA for p53 binding. NE, no extract; Sp, specific competition (unlabeled oligonucleotide); **, sample from cells treated with NO-ASA at a concentration corresponding to IC50 for growth.
DISCUSSION
Modified NSAIDs, such as NO-ASA, hold promise for the control of cancer. Although basic parameters of the mechanism of action of NO-ASA have been defined by us and others [8], very little is known about the underlying molecular mechanism responsible for its enhanced potency and efficacy. The structural hallmark of NO-ASA is its moiety that donates NO, a highly reactive radical modulating various cellular responses. Thiols are a well-recognized target of NO, a reaction that leads to the formation of S-nitrosothiols. Our data demonstrate extensive S-nitrosylation of cellular proteins, some of which play critical roles as signaling molecules. S-nitrosylation of signaling proteins emerges as an important regulatory mechanism [39–40].
Treatment of colon cancer cells with NO-ASA increases significantly their NO levels. Of note, in addition to releasing NO, NO-ASA stimulates the catalytic activity of nitric oxide synthase [41]. The increased intracellular NO levels are accompanied by S-nitrosylation and tyrosine nitration of multiple cellular proteins. Both effects vary in degree with respect to target proteins, reflecting perhaps variations in the number of cysteine and tyrosine residues in each protein. Tyrosine nitration results from the reaction of NO2· (nitrogen dioxide) with tyrosine; NO2· is derived either from peroxynitrite or nitrite, both of which reflect the presence of NO [19, 42]. Tyrosine nitration by NO-ASA occurs also in vivo (>2 fold over controls).
Our work focused on three signaling molecules, NF-κB, p53 and β–catenin that are frequently dysregulated in many cancers. All three interact with other proteins and these interactions either sequester them into an inactive state or are required for their biological activity. For example, p53 is bound to mdm2, preventing its action and transporting it from the nucleus to the cytosol; phosphorylation of p53 disrupts mdm2-binding and releases p53 [43–44]. NF-κB, itself a dimer, is also sequestered in the cytoplasm by binding to IκB, whereas β-catenin interacts with cytoplasmic proteins and, released from them, reaches the nucleus and becomes transcriptionally active only when it binds to other proteins [45].
We have speculated that S-nitrosylation may induce steric hindrance in Wnt signaling proteins [16] and the same is conceivable for NF-κB and p53 proteins. NF-κB, electrophoretic mobility shift assays demonstrated that S-nitrosylation was responsible of its reduced binding to DNA; chemical reduction of NF-κB thiols restored the binding of NF-κB to DNA. It is likely that the S-nitrosylation of NF-κB and perhaps of other proteins affects their signaling functions. Thus S-nitrosylation may mediate part of the anticancer effect of NO-ASA. It should be kept in mind, however, that our experimental method did not allow quantification of the extent of S-nitrosylation of a given protein(s), e.g. NF-κB. It is likely that only a fraction of the molecules are S-nitrosylated. Apparently, in our studies the extent of S-nitrosylation exceeds a threshold that allows a functional effect.
Our findings also establish widespread modification of cellular proteins through nitration, another result of increased NO levels in the cell. This finding suggests that NO may affect proteins beyond those that are S-nitrosylated and that nitration of tyrosines may further modulate cell signaling in response to NO-ASA. Indeed, tyrosine nitration of p65 has been recognized to rapidly inactivate NF-κB [34, 46].
Fig. 5 offers a plausible mechanism of action of NO-ASA that integrates S-nitrosylation and tyrosine nitration. Specifically, NO-ASA increases the intracellular levels of NO, both directly and through its stimulatory effect on NOS. The first result is S-nitrosylation of proteins. NO-ASA also increases the production of superoxide anion from mitochondria [47], which can react with NO to form peroxynitrite, which can then be converted to NO2· (which can also be produced by the reaction of NO with O2 [19]). NO2· nitrates protein tyrosine residues. This overall mechanism of action may be applicable to other NO-releasing compounds.
Fig. 5. Proposed model of protein S-nitrosylation and tyrosine nitration by NO-ASA.
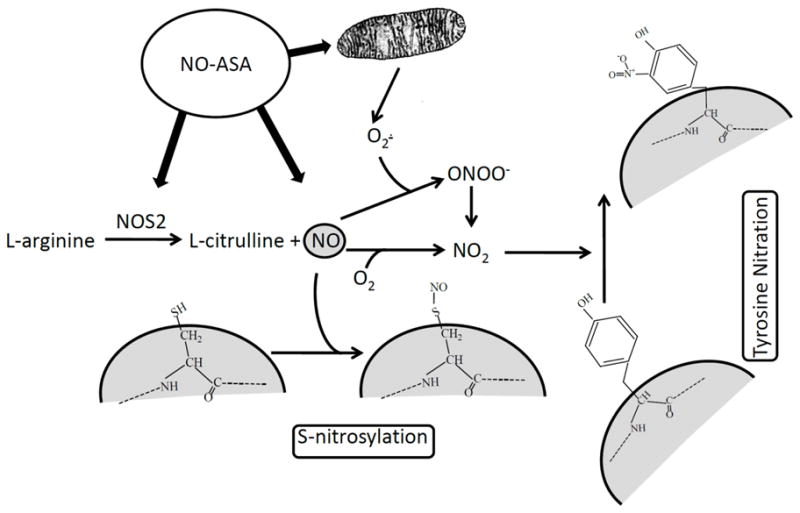
NO-ASA a) stimulates NOS2 activity increasing NO production form L-arginine, b) releases NO, and c) stimulates O2· production by the mitochondria. NO is shown here to nitrosylate a cysteine residue of a protein. NO2·, forming a) from peroxynitrite (ONOO−) or b) from the reaction of NO with O2, is shown to nitrate a protein tyrosine residue.
In conclusion, our findings establish extensive and functionally significant S-nitrosylation and nitration of protein tyrosine residues in response to NO-ASA, an agent whose cardinal feature is the release of NO. These findings suggest a mechanism of action that may also apply other members of this class of promising anti-cancer and anti-inflammatory compounds.
Acknowledgments
Grant support: NIH CN43302WMC08WA7 and NIH KO1 CA106604-05
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Refrences
- 1.Arber N, Levin B. Chemoprevention of colorectal neoplasia: the potential for personalized medicine. Gastroenterology. 2008;134:1224–1237. doi: 10.1053/j.gastro.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Half E, Arber N. Colon cancer: preventive agents and the present status of chemoprevention. Expert Opin Pharmacother. 2009;10:211–219. doi: 10.1517/14656560802560153. [DOI] [PubMed] [Google Scholar]
- 3.Cuzick J, Otto F, Baron JA, Brown PH, Burn J, Greenwald P, Jankowski J, La Vecchia C, Meyskens F, Senn HJ, Thun M. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: an international consensus statement. Lancet Oncol. 2009;10:501–507. doi: 10.1016/S1470-2045(09)70035-X. [DOI] [PubMed] [Google Scholar]
- 4.Fiorucci S, Santucci L, Gresele P, Faccino RM, Del Soldato P, Morelli A. Gastrointestinal safety of NO-aspirin (NCX-4016) in healthy human volunteers: a proof of concept endoscopic study. Gastroenterology. 2003;124:600–607. doi: 10.1053/gast.2003.50096. [DOI] [PubMed] [Google Scholar]
- 5.Cryer B, Feldman M. Effects of very low dose daily, long-term aspirin therapy on gastric, duodenal, and rectal prostaglandin levels and on mucosal injury in healthy humans. Gastroenterology. 1999;117:17–25. doi: 10.1016/s0016-5085(99)70545-7. [DOI] [PubMed] [Google Scholar]
- 6.Rigas B. The use of nitric oxide-donating nonsteroidal anti-inflammatory drugs in the chemoprevention of colorectal neoplasia. Curr Opin Gastroenterol. 2007;23:55–59. doi: 10.1097/MOG.0b013e32801145b0. [DOI] [PubMed] [Google Scholar]
- 7.Yeh RK, Chen J, Williams JL, Baluch M, Hundley TR, Rosenbaum RE, Kalala S, Traganos F, Benardini F, del Soldato P, Kashfi K, Rigas B. NO-donating nonsteroidal antiinflammatory drugs (NSAIDs) inhibit colon cancer cell growth more potently than traditional NSAIDs: a general pharmacological property? Biochem Pharmacol. 2004;67:2197–2205. doi: 10.1016/j.bcp.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 8.Williams JL, Borgo S, Hasan I, Castillo E, Traganos F, Rigas B. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–3289. [PubMed] [Google Scholar]
- 9.Tesei A, Rosetti M, Ulivi P, Fabbri F, Medri L, Vannini I, Bolla M, Amadori D, Zoli W. Study of molecular mechanisms of pro-apoptotic activity of NCX 4040, a novel nitric oxide-releasing aspirin, in colon cancer cell lines. J Transl Med. 2007;5:52. doi: 10.1186/1479-5876-5-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rao CV, Reddy BS, Steele VE, Wang CX, Liu X, Ouyang N, Patlolla JM, Simi B, Kopelovich L, Rigas B. Nitric oxide-releasing aspirin and indomethacin are potent inhibitors against colon cancer in azoxymethane-treated rats: effects on molecular targets. Mol Cancer Ther. 2006;5:1530–1538. doi: 10.1158/1535-7163.MCT-06-0061. [DOI] [PubMed] [Google Scholar]
- 11.Ouyang N, Williams JL, Rigas B. NO-donating aspirin isomers downregulate peroxisome proliferator-activated receptor (PPAR){delta} expression in APCmin/+ mice proportionally to their tumor inhibitory effect: Implications for the role of PPAR{delta} in carcinogenesis. Carcinogenesis. 2006;27:232–239. doi: 10.1093/carcin/bgi221. [DOI] [PubMed] [Google Scholar]
- 12.Ouyang N, Williams JL, Rigas B. NO-donating aspirin inhibits angiogenesis by suppressing VEGF expression in HT-29 human colon cancer mouse xenografts. Carcinogenesis. 2008 doi: 10.1093/carcin/bgn127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams JL, Nath N, Chen J, Hundley TR, Gao J, Kopelovich L, Kashfi K, Rigas B. Growth inhibition of human colon cancer cells by nitric oxide (NO)-donating aspirin is associated with cyclooxygenase-2 induction and beta-catenin/T-cell factor signaling, nuclear factor-kappaB, and NO synthase 2 inhibition: implications for chemoprevention. Cancer Res. 2003;63:7613–7618. [PubMed] [Google Scholar]
- 14.Williams JL, Ji P, Ouyang N, Liu X, Rigas B. NO-donating aspirin inhibits the activation of NF-kappaB in human cancer cell lines and Min mice. Carcinogenesis. 2008;29:390–397. doi: 10.1093/carcin/bgm275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 16.Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci U S A. 2003;100:12584–12589. doi: 10.1073/pnas.2134840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodrigues NR, Rowan A, Smith ME, Kerr IB, Bodmer WF, Gannon JV, Lane DP. p53 mutations in colorectal cancer. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:7555–7559. doi: 10.1073/pnas.87.19.7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nigro JM, Baker SJ, Preisinger AC, Jessup JM, Hostetter R, Cleary K, Bigner SH, Davidson N, Baylin S, Devilee P, Glover T, Collins FS, Weston A, Modali R, Harris CC, Vogelstein B. Mutations in the p53 gene occur in diverse human tumour types. Nature. 1989;342:705–708. doi: 10.1038/342705a0. [DOI] [PubMed] [Google Scholar]
- 19.Mohiuddin I, Chai H, Lin PH, Lumsden AB, Yao Q, Chen C. Nitrotyrosine and chlorotyrosine: clinical significance and biological functions in the vascular system. J Surg Res. 2006;133:143–149. doi: 10.1016/j.jss.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 20.Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63:854–865. [PubMed] [Google Scholar]
- 21.Kamat JP. Peroxynitrite: a potent oxidizing and nitrating agent. Indian J Exp Biol. 2006;44:436–447. [PubMed] [Google Scholar]
- 22.Gal A, Tamir S, Kennedy LJ, Tannenbaum SR, Wogan GN. Nitrotyrosine formation, apoptosis, and oxidative damage: relationships to nitric oxide production in SJL mice bearing the RcsX tumor. Cancer Res. 1997;57:1823–1828. [PubMed] [Google Scholar]
- 23.Penning TD, Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Lee LF, Malecha JW, Miyashiro JM, Rogers RS, Rogier DJ, Yu SS, Anderson Gd, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veenhuizen AW, Zhang YY, Isakson PC. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib) J Med Chem. 1997;40:1347–1365. doi: 10.1021/jm960803q. [DOI] [PubMed] [Google Scholar]
- 24.Kapitanovic S, Cacev T, Antica M, Kralj M, Cavric G, Pavelic K, Spaventi R. Effect of indomethacin on E-cadherin and beta-catenin expression in HT-29 colon cancer cells. Exp Mol Pathol. 2006;80:91–96. doi: 10.1016/j.yexmp.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 25.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci U S A. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Natarajan K, Singh S, Burke TR, Jr, Grunberger D, Aggarwal BB. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-kappa B. Proc Natl Acad Sci U S A. 1996;93:9090–9095. doi: 10.1073/pnas.93.17.9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 28.Powolny A, Xu J, Loo G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int J Biochem Cell Biol. 2001;33:193–203. doi: 10.1016/s1357-2725(00)00080-7. [DOI] [PubMed] [Google Scholar]
- 29.Sun Y, Chen J, Rigas B. Chemopreventive agents induce oxidative stress in cancer cells leading to COX-2 overexpression and COX-2-independent cell death. Carcinogenesis. 2009;30:93–100. doi: 10.1093/carcin/bgn242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanafy KA, Krumenacker JS, Murad F. NO, nitrotyrosine, and cyclic GMP in signal transduction. Med Sci Monit. 2001;7:801–819. [PubMed] [Google Scholar]
- 31.Krumenacker JS, Hanafy KA, Murad F. Regulation of nitric oxide and soluble guanylyl cyclase. Brain research bulletin. 2004;62:505–515. doi: 10.1016/S0361-9230(03)00102-3. [DOI] [PubMed] [Google Scholar]
- 32.Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J Physiol Pharmacol. 2003;54:469–487. [PubMed] [Google Scholar]
- 33.Lohinai Z, Stachlewitz R, Virag L, Szekely AD, Hasko G, Szabo C. Evidence for reactive nitrogen species formation in the gingivomucosal tissue. J Dent Res. 2001;80:470–475. doi: 10.1177/00220345010800021401. [DOI] [PubMed] [Google Scholar]
- 34.Park SW, Huq MD, Hu X, Wei LN. Tyrosine nitration on p65: a novel mechanism to rapidly inactivate nuclear factor-kappaB. Mol Cell Proteomics. 2005;4:300–309. doi: 10.1074/mcp.M400195-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Fiorucci S, Santucci L, Cirino G, Mencarelli A, Familiari L, Soldato PD, Morelli A. IL-1 beta converting enzyme is a target for nitric oxide-releasing aspirin: new insights in the antiinflammatory mechanism of nitric oxide-releasing nonsteroidal antiinflammatory drugs. J Immunol. 2000;165:5245–5254. doi: 10.4049/jimmunol.165.9.5245. [DOI] [PubMed] [Google Scholar]
- 36.Stoner CS, Pearson GD, Koc A, Merwin JR, Lopez NI, Merrill GF. Effect of thioredoxin deletion and p53 cysteine replacement on human p53 activity in wild-type and thioredoxin reductase null yeast. Biochemistry. 2009;48:9156–9169. doi: 10.1021/bi900757q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, Srivenugopal KS. Human p53 is inhibited by glutathionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry. 2007;46:7765–7780. doi: 10.1021/bi700425y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rainwater R, Parks D, Anderson ME, Tegtmeyer P, Mann K. Role of cysteine residues in regulation of p53 function. Mol Cell Biol. 1995;15:3892–3903. doi: 10.1128/mcb.15.7.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foster MW, Hess DT, Stamler JS. Protein S-nitrosylation in health and disease: a current perspective. Trends Mol Med. 2009;15:391–404. doi: 10.1016/j.molmed.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spadaro D, Yun BW, Spoel SH, Chu C, Wang YQ, Loake GJ. The redox switch: dynamic regulation of protein function by cysteine modifications. Physiol Plant. 2010;138:360–371. doi: 10.1111/j.1399-3054.2009.01307.x. [DOI] [PubMed] [Google Scholar]
- 41.Spiegel A, Hundley TR, Chen J, Gao J, Ouyang N, Liu X, Go MF, Tsioulias GJ, Kashfi K, Rigas B. NO-donating aspirin inhibits both the expression and catalytic activity of inducible nitric oxide synthase in HT-29 human colon cancer cells. Biochem Pharmacol. 2005;70:993–1000. doi: 10.1016/j.bcp.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 42.Gu Z, Nakamura T, Lipton SA. Redox reactions induced by nitrosative stress mediate protein misfolding and mitochondrial dysfunction in neurodegenerative diseases. Mol Neurobiol. 2010;41:55–72. doi: 10.1007/s12035-010-8113-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sebastian S, Azzariti A, Silvestris N, Porcelli L, Russo A, Paradiso A. p53 as the main traffic controller of the cell signaling network. Front Biosci. 2010;15:1172–1190. doi: 10.2741/3669. [DOI] [PubMed] [Google Scholar]
- 44.Waning DL, Lehman JA, Batuello CN, Mayo LD. Controlling the Mdm2-Mdmx-p53 Circuit. Pharmaceuticals (Basel) 2010;3:1576–1593. doi: 10.3390/ph3051576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 46.Bar-Shai M, Reznick AZ. Peroxynitrite induces an alternative NF-kappaB activation pathway in L8 rat myoblasts. Antioxid Redox Signal. 2006;8:639–652. doi: 10.1089/ars.2006.8.639. [DOI] [PubMed] [Google Scholar]
- 47.Sun Y, Rigas B. The Thioredoxin System Mediates Redox-Induced Cell Death in Human Colon Cancer Cells: Implications for the Mechanism of Action of Anticancer Agents. Cancer Res. 2008;68:8269–8277. doi: 10.1158/0008-5472.CAN-08-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]