

Abstract
The response of transverse (T)-tubules to exercise training in health and disease remains unclear. Therefore, we studied the effect of exercise training on the density and spacing of left ventricle cardiomyocyte T-tubules in normal and remodeled hearts that associate with detubulation, by confocal laser scanning microscopy. First, exercise training in normal rats increased cardiomyocyte volume by 16% (p<0.01), with preserved T-tubule density. Thus, the T-tubules adapted to the physiologic hypertrophy. Next, we studied T-tubules in a rat model of metabolic syndrome with pressure overload-induced concentric left ventricle hypertrophy, evidenced by 15% (p<0.01) increased cardiomyocyte size. These rats had only 85% (p<0.01) of the T-tubule density of control rats. Exercise training further increased cardiomyocyte volume by 8% (p<0.01); half to that in control rats, but the T-tubule density remained unchanged. Finally, post-myocardial infarction heart failure induced severe cardiac pathology, with a 70% (p<0.01) increased cardiomyocyte volume that included both eccentric and concentric hypertrophy and 55% (p<0.01) reduced T-tubule density. Exercise training reversed 50% (p<0.01) of the pathologic hypertrophy, whereas the T-tubule density increased by 40% (p<0.05) compared to sedentary heart failure, but remained at 60% of normal hearts (p<0.01). Physiologic hypertrophy associated with conserved T-tubule spacing (~1.8–1.9 μm), whereas in pathologic hypertrophy, T-tubules appeared disorganized without regular spacing. In conclusion, cardiomyocytes maintain the relative T-tubule density during physiologic hypertrophy and after mild concentric pathologic hypertrophy, whereas after severe pathologic remodeling with a substantial loss of T-tubules; exercise training reverses the remodeling and partly corrects the T-tubule density.
Keywords: Physiologic remodeling, Pathologic remodeling, Reverse Remodeling, Exercise training, Cardiac transverse tubules, Confocal microscopy
INTRODUCTION
Initiation and coordination of the contraction in cardiomyocytes is largely set by depolarization of and Ca2+ entry through the plasma membrane. Transverse (T)-tubules; invaginations of the plasma membrane, allow this to occur uniformly across the whole cell (Brette and Orchard, 2003). This is made possible because (i) the T-tubules occur at ~2 μm intervals at approximately every z line and run deep into the interior of the cell, such that most sarcomeres are individually served by a singular T-tubule (Soeller and Cannell, 1999); (ii) sarcolemmal proteins that traffic ions across the plasma membrane are concentrated in the T-tubules (Brette and Orchard, 2003), and (iii) the T-tubules themselves constitute extracellular ion reservoirs that ensure a rapid access for transmembrane inward transport (Shorten and Soboleva, 2007; Swift et al, 2006). Thus, an intact network of T-tubules supports a rapid and coordinated contraction. However, the majority of studies indicate that T-tubules become disorganized and reduce the density in situations of cardiac dysfunction and pathologic hypertrophy, which contributes to the reduction of excitation-contraction (EC) coupling efficiency and contractile dysfunction (Balijepalli et al, 2003; Cannell et al, 2006; Louch et al, 2006; Quinn et al, 2003; Song et al, 2006). This is also in accordance with acute reduction in contractile performance observed after detubulating cardiomyocytes with the formamide osmotic shock technique (Brette et al, 2005; Thomas et al, 2003).
In contrast to pathologic hypertrophy caused by cardiac injury or chronically increased wall stress due to e.g. pressure overload, physiologic hypertrophy of the cardiomyocyte after exercise training is associated with preserved or improved contractile capacity (Kemi et al, 2004, 2005,Kemi et al, 2007A, 2008; Wisloff et al, 2001), whereas exercise training in models of cardiac and contractile dysfunction such as post-myocardial infarction (MI) heart failure (Kemi et al, 2007B; Wisloff et al, 2002; Zhang et al, 1998) at least partly restores contractile function toward normal levels. However, apart from a study showing that the loss of t-tubules in diabetic cardiomyopathy was partly restored by exercise training (Stolen et al, 2009), the effect of exercise training on the T-tubules remains largely unclear. Therefore, we investigated the effect of exercise training on the density and individual spacing of T-tubules in normal cardiomyocytes that underwent physiologic hypertrophy, and whether exercise training has the potential to reverse the chronic detubulation that occurs during cardiac pathology. For the latter objective, we studied two separate models of cardiac dysfunction and pathology; one with a mild pathologic remodeling and concentric cellular hypertrophy due to the pressure overload that is linked to the metabolic syndrome, and another with a severe pathologic remodeling with evidence of both eccentric and concentric cellular hypertrophy due to post-MI heart failure.
MATERIALS AND METHODS
The animals were kept in a 12:12 hour light:dark cycle with free access to water and a pellet rodent chow diet. At inclusion, animals were 3–4 months old. The study conforms to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23, revised 1996), and was approved by the respective Institutional Animal Research Ethics Committees.
Animals with Mild Cardiac Pathologic Remodeling – Concentric Hypertrophy
As a model of mild pathologic remodeling, we used rats artificially selected for either low or high capacity for running (LCR and HCR, respectively) over 16 generations, initially started from the N:NIH stock from the National Institutes of Health (USA). The model has been described in detail in previous reports (Britton and Koch, 2005; Koch and Britton, 2001; Wisloff et al, 2005). The LCR line has evolved into a model of the metabolic syndrome, as evidenced by visceral obesity and high fat mass, hypertension, insulin resistance, high plasma triglycerides and free fatty acids, depressed cardiac and vascular functions, and reduced work capacity, whereas the HCR line shows no signs of pathology, abnormalities, or dysfunction (Wisloff et al, 2005). Importantly, the LCR also show evidence of a mild degree of pathologic concentric hypertrophy of the heart (see results and Britton and Koch, 2005; Koch and Britton, 2001; Wisloff et al, 2005). 16 LCR and 16 HCR female rats were included in this study, of which 8 rats in each line underwent exercise training and 8 remained sedentary.
Animals with Severe Cardiac Pathologic Remodeling – Eccentric/Concentric Hypertrophy
As a model of severe pathologic remodeling, we used the female Sprague-Dawley rat model (Mollegaards Breeding Center, Lille Skensved, Denmark). Permanent left coronary artery ligation during thoracotomy inducing MI and subsequent heart failure or sham-operation was performed during anesthesia (1% isoflurane mixed with 30% O2/70% N2O). Buprenorphine (0.05 mg Temgesic, Reckitt and Coleman, Hull, UK) was given subcutaneously immediately and 8 hours after surgery. This model shows evidence of substantial pathologic remodeling within 1 week of MI, and severe dysfunction, heart failure and evidence of both eccentric and concentric hypertrophy of the myocardium after 4 weeks (Kemi et al, 2007B; Loennechen et al, 2002A). This was also confirmed in the current study; see results. For this study, we included 16 post-MI heart failure and 16 sham-operated rats, of which 8 from each group started exercise training 4 weeks post-surgery and 8 remained sedentary. In addition, to study post-MI heart failure hypertrophied myocardium that was unloaded by pharmacological treatment, we also included 8 sedentary and 8 exercise trained post-MI heart failure rats that were treated with the angiotensin II type 1 (AT1) receptor antagonist losartan (MSD, Whitehouse Station, NJ). Losartan, initiated one week post-MI, was administered through the drinking water (2 g/L, ad libitum).
To confirm MI after coronary artery ligation, echocardiography was performed during sedation (40 mg/kg ketamine hydrochloride and 8 mg/kg xylazine intraperitoneally) 7 days post-surgery. The left ventricle endocardial circumference was traced in 2-dimensional short axis imaging mode, whereupon infarct size was estimated as percentage infarcted (non-contracting) part of the total circumference (Litwin et al, 1994). Infarct sizes were estimated to 40±2%.
To confirm dysfunction and heart failure after coronary artery ligation, a pressure microtip catheter (size 2-Fr, Millar Instruments, Houston, TX) was inserted through the right carotid artery and into the left ventricle to record pressure. The means of 10 consecutive cardiac cycles were used in analysis. Subcutaneous anesthesia (in mL/kg: 0.33 haloperidol, 0.5 fentanyl, 0.5 midazolam, 1.3 ketamine hydrochloride and 0.6 H2O) was given prior to the procedure.
Exercise Training
Endurance exercise training by treadmill running was performed 1 hour per day, 5 days per week for 8 weeks, at a relative exercise intensity of 85–90% of maximal oxygen uptake (VO2max). The relative exercise intensity was determined and kept constant by measuring VO2max at the start of every exercise training week, by an incremental treadmill running test in a metabolic chamber that allows for measurements of VO2max. The exercise training protocols have been previously published in detail (Kemi et al, 2004, 2005,Kemi et al, 2007A, 2007B, 2008; Wisloff et al, 2001, 2002).
Left Ventricle Cardiomyocyte Isolation and Size
The hearts were excised during anesthesia as described above for pressure recordings, whereupon single cardiomyocytes from the left ventricle were isolated with a Ca2+-free Krebs-Henseleit tyrode and collagenase type-2 (250 IU/mL; Worthington, Freeland, NJ). In post-MI heart failure rats, cardiomyocytes were isolated from the remote region of the left ventricle. During the protocol, CaCl2 was added stepwise to 1.2 mM. After HEPES-resting for ~1 hour at 37° C, cells were placed in a cell chamber at 37° C on an inverted microscope (Diaphot-TMD, Nikon, Tokyo, Japan). From each animal, 100 cells without obvious cellular damage were measured for length and midpoint width. On the basis of these measurements, cardiomyocyte volume was calculated as cell length × width × 0.00759 (Satoh et al, 1996).
Confocal Laser Scanning Microscopy
Cardiomyocytes suspended in HEPES were incubated for 20 minutes with 10 μM Di-8-ANEPPS (Molecular Probes, Eugene, OR) for T-tubule visualization, whereupon cells were scanned at 37° C with an inverted microscope (Axiovert 100-M, Carl Zeiss, Jena, Germany) equipped with a LSM 510 laser scanner unit and a plan-apochromat 63x/1.4 NA oil-immersion objective lens. Di-8-ANEPPS was excited with a 488 nm Argon laser, whereas 505–530 nm fluorescence emission was collected with a photomultiplier tube. Confocal images were generated with a 1.0 airy unit pinhole diameter, yielding an XYZ resolution of ~0.5×0.5×0.8 μm. XY image-mode (512×512 pixels) Z-stacks throughout the cell were obtained, with 0.5 μm spacing between each slice (Z-axis); each slice Kalman averaged twice. 6–10 cells from each animal were scanned.
The relative density of T-tubules was obtained from 15–25 images from the center of the cell, analyzed with custom-made applications in the IDL programming language (ITT Visual Informations Solutions, Boulder, CO). Pixels associated with the non-T-tubular sarcolemma were removed, and binary images were generated in which pixels stained positively with Di-8-ANEPPS were counted relative to the total number of pixels within the cell boundary, such that the relative density of the T-tubule membranes to the cell size was identified. Also, based upon the original cell boundaries, the cell was split into 10 equally long sections along the longitudinal axis, in order to identify regional differences along the cardiomyocyte (Quinn et al, 2003). Finally, spacing between individual T-tubules was assessed by the power spectrum of each image achieved by Fast-Fourier Transforms (FFT) of the signal after baseline subtraction, along the longitudinal axis of the cell adjacent to, but not across, the nuclei. The power spike as a function of the spatial frequency identifies the average distance between individual T-tubules in the cell.
Statistics
Data are expressed as mean±SEM, with significance level p<0.05. VO2max was allometrically scaled by body mass raised to the power of 0.75 to neutralize any effects of changes in body mass (Kemi et al, 2004, 2005). The univariate repeated-measures General Linear Model (GLM) with the Scheffe post hoc test was used to determine changes in VO2max, whereas the one-way ANOVA with the Scheffe post hoc test evaluated unrelated observations between groups. For comparisons between different sections within the cell, we used the univariate repeated-measures ANOVA with the Scheffe post-hoc test.
RESULTS
T-Tubules in Exercise Training-Induced Physiologic Hypertrophy
First, we studied the T-tubule response during exercise training-induced physiologic hypertrophy in healthy rats, as these were also independently interesting because T-tubule responses to exercise training in normal individuals remain poorly understood. No abnormalities with regard to cardiac pressure or contractile characteristics were observed in the healthy rats (Table 1). High-intensity exercise training (85%–90% of VO2max) increased VO2max by ~40% (Figure 1A white symbols) and induced physiologic hypertrophy of the cardiomyocytes, evidenced by 10% and 6% increases in cell length and width, respectively, and a 16% increase in cell volume (Table 1).
Table 1.
Myocardial function and cardiomyocyte morphology in sedentary (SED) and exercise trained (TR) sham-operated (SHAM) and post-myocardial infarction (MI) rats, including MI rats treated with the angiotensin II type 1 receptor antagonist losartan.
| SHAM | MI | MI Losartan | ||||
|---|---|---|---|---|---|---|
| SED | TR | SED | TR | SED | TR | |
| LVEDP (mmHg) | 3.8±2.4 | 4.1±2.1 | 25.7±1.3a | 21.9±1.5a | 18.2±1.2ad | 17.7±1.1ad |
| LVPSP (mmHg) | 114.3±5.8 | 120.0±5.1 | 100.3±3.1a | 98.3±3.1a | 87.6±4.5a | 88.1±5.2a |
| +dP/dtmax (mmHg · ms−1) | 8.9±1.1 | 8.4±1.1 | 3.4±1.2a | 3.9±1.2a | 4.0±0.9a | 3.7±1.4a |
| −dP/dtmax (mmHg · ms−1) | 8.8±1.3 | 8.7±1.1 | 3.2±1.2a | 3.5±1.1a | 4.3±1.3a | 3.8±1.3a |
| Cell Length (μm) | 100.9±2.6 | 110.8±4.2b | 130.1±8.1a | 115.0±4.5ac | 117.5±3.2ac | 113.2±3.1ac |
| Cell Width (μm) | 22.8±1.3 | 24.4±0.7b | 30.7±2.6a | 26.7±1.6ac | 28.0±1.7ac | 26.9±1.9ac |
| Cell Volume (pL) | 17.7±1.3 | 20.5±0.8b | 30.4±2.9a | 23.4±2.1ac | 25.5±1.9ac | 23.3±1.8ac |
LVEDP: left ventricle end-diastolic pressure, LVPSP: left ventricle peak systolic pressure, (+dP/dtmax: peak rate of left ventricle pressure rise, −dP/dtmax: peak rate of left ventricle pressure fall. Note that SHAM TR rats developed physiologic hypertrophy, and that post-MI rats developed heart failure as defined by LVEDP >15 mmHg; presented with reduced LVPSP and rates of contraction and relaxation of the heart, and developed pathologic hypertrophy, which was reversed by exercise training as well as losartan.
difference from SHAM groups, p<0.01;
difference between SHAM SED and SHAM TR, p<0.01;
difference from MI SED, p<0.01.
difference from MI SED without losartan, p<0.01.
Data are mean±SEM with 8 animals/group.
Figure 1.

Maximal oxygen uptake (VO2max; A), representative examples of cardiomyocytes stained by Di-8-ANEPPS for imaging transverse (T)-tubules (B), and relative T-tubule density in whole cardiomyocytes (C) and in sections along the longitudinal cells (D; inlet illustrates the cardiomyocyte sectioning) from sedentary (SED) and exercise trained (TR) sham-operated (SHAM) and post-myocardial infarction (MI) rats. Note that SHAM TR cardiomyocytes showed sustained T-tubules despite the physiologic hypertrophy; that post-MI cardiomyocytes showed reduced T-tubules amid pathologic hypertrophy, and that TR in post-MI rats partly restored T-tubules and reversed the pathologic remodeling. a: difference from other groups, p<0.01; b: difference between MI groups and SHAM groups, p<0.01; c: difference between MI TR and MI SED, p<0.05; d: difference from middle section (20–80%) of SHAM cardiomyocytes to the end sections (0–10% and 90–100%), p<0.05 (not in MI cardiomyocytes). Data are mean±SEM with 8 animals/group.
Representative examples of confocal images depicting cardiomyocyte Di-8-ANEPPs-stained T-tubules are shown in Figure 1B. These studies showed that cardiomyocytes isolated from exercise trained normal rats had a relative density of T-tubules similar to sedentary control rats (Figure 1C white bars), despite exercise training also inducing physiologic hypertrophy. This suggests that the T-tubules adapted accordingly with the increase in cell size during exercise training. When studying the T-tubules in sections along the longitudinal cell, it became clear that the middle 20–80% sections of the cardiomyocytes from normal rats had denser networks of T-tubules compared to the two ends of the same cell (0–10% and 90–100%, respectively; p<0.05; Figure 1D white symbols), and that the training-induced adaptation occurred throughout the length of the cell. Finally, the FFT analysis indicated that spacing between individual T-tubules remained constant during physiologic hypertrophy, and that the distance between individual T-tubules was ~1.8–1.9 μm both before and after exercise training (Figure 2A, B).
Figure 2.

Representative examples of the Fast-Fourier Transform (FFT) analysis along the longitudinal cardiomyocyte (A), and distance between individual transverse (T)-tubules derived from the FFT power versus spatial frequency spectrum (B) in cardiomyocytes from sedentary (SED) and exercise trained (TR) sham-operated (SHAM) rats. T-tubule spacing derivation was not possible in post-myocardial infarction (MI) rats, as cardiomyocytes from those animals did not produce consistent peaks, indicating disorganized T-tubule spacing. The scattering of power across the spatial frequency spectrum observed in MI, in contrast to SHAM rats, also indicates disorganized T-tubules after MI. Data are mean±SEM with 8 animals/group.
For simplicity, only data from sham-operated Sprague-Dawley rats were highlighted above, but the corresponding data for HCR; the control group for LCR rats with mild pathologic concentric remodeling, show similar results. Exercise training induced physiologic hypertrophy of the cardiomyocyte (Table 2), but the relative density of the T-tubules and the distance between individual T-tubules were sustained (Figure 3C, F white bars), and moreover, the T-tubule characteristics appeared similar between cardiomyocytes from sham-operated Sprague-Dawley and HCR rats. Finally, T-tubule density and spacing was similar between sham-operated rats and rats undergoing no surgical or invasive procedures prior to data collection, including the effect of exercise training on both cardiomyocyte morphology and T-tubules (data not shown).
Table 2.
Cardiomyocyte morphology in sedentary (SED) and exercise trained (TR) rats with a high or low capacity for running (HCR and LCR, respectively).
| HCR |
LCR |
|||
|---|---|---|---|---|
| SED | TR | SED | TR | |
| Cell Length (μm) | 124.2±2.4 | 141.5±3.3a | 118.2±2.6ab | 125.5±2.7c |
| Cell Width (μm) | 19.2±1.6 | 19.5±1.5 | 23.1±2.0ab | 23.5±2.5 |
| Cell Volume (pL) | 17.9±0.7 | 20.9±1.0a | 20.6±0.9ab | 22.3±0.7c |
Note that LCR rats developed mild concentric pathologic cellular hypertrophy, and that exercise training increased cardiomyocyte length and volume.
difference between HCR SED and HCR TR, p<0.01;
difference between HCR SED and LCR SED, p<0.01;
difference between LCR SED and LCR TR, p<0.01.
Data are mean±SEM with 8 animals/group.
Figure 3.

Maximal oxygen uptake (VO2max; A), representative examples of cardiomyocytes stained by Di-8-ANEPPS for imaging transverse (T)-tubules in HCR and LCR rats (B), relative T-tubule density in the whole cardiomyocyte (C) and in sections along the longitudinal cell (D), representative examples of the Fast-Fourier Transform (FFT) analysis along the longitudinal cardiomyocyte (E), and the distance between individual T-tubules derived from the FFT power versus spatial frequency spectrum (F) in sedentary (SED) and exercise trained (TR) rats with a high or low capacity for running (HCR and LCR, respectively). Note that LCR rats present with a reduced T-tubule density; greater scattering of power across the spatial frequency spectrum of the FFT analysis indication greater T-tubule disorganization, and that exercise training maintained the T-tubule density amid physiologic hypertrophy. a: difference from other groups, p<0.01; b: difference between LCR SED and HCR SED, p<0.01; c: difference from middle section (30–70%) of HCR cardiomyocytes to the end sections (0–20% and 80–100%), p<0.05 (not in LCR cardiomyocytes). Data are mean±SEM with 8 animals/group.
Transverse Tubules in Mild Cardiac Pathologic Remodeling – Concentric Hypertrophy
To study a scenario with mild cardiac pathology, we used rats that were artificially selected and bred for low fitness (LCR) over 16 generations. This breeding strategy produces rats that present with evidence of cardiac dysfunction and pathology, most likely due to chronic hypertension (Wisloff et al, 2005), albeit the pathologic remodeling is less severe compared to e.g. post-MI heart failure rats. Rats selected and bred for high fitness (HCR) over the same period constitute the control group. Reduced functionality in LCR was indicated by lower VO2max; LCR only obtained ~60% of the corresponding value in HCR (Figure 3A), whereas cardiac pathologic remodeling was evidenced by the concentric hypertrophy of the cardiomyocyte, as the LCR cells were 5% shorter and 20% wider than the HCR cells, whereas the cell volume was 15% greater (Table 2).
Representative examples of T-tubules in cardiomyocytes from LCR and HCR rats are shown in Figure 3B. Compared to HCR, LCR cardiomyocytes had a reduced relative density of T-tubules, amounting to only 85% of the density in HCR (Figure 3C). On closer inspection, LCR showed reduced T-tubule density along the whole length of the cell, but the difference between LCR and HCR was especially prevalent toward the center of the cell (Figure 3D). This was explained by the HCR cardiomyocytes presenting with a higher T-tubule density in the middle 30–70% (p<0.05) section of the cell compared to the two ends, which in contrast did not occur in the LCR cells, which had a more even distribution along the cell. This suggests that at least in this model, mild cardiac pathology and concentric hypertrophy was associated with a 15% reduction in T-tubule density. However, no clear disruption to spacing of individual T-tubules was observed despite the reduction in the relative density (Figure 3E, F), as cardiomyocytes from HCR and LCR presented with similar distances between individual T-tubules, though a small, but insignificant increase in the distance between T-tubules was noted in LCR cardiomyocytes (p=0.097, HCR SED versus LCR SED). In line with this, we also observed that the regularity of the spacing was less obvious in the LCR cardiomyocytes compared to HCR, as illustrated by the increased variability around the mean (Figure 3F black bars).
Transverse Tubules after Exercise Training in Mild Cardiac Pathologic Remodeling – Concentric Hypertrophy
Next, we performed 8 weeks of exercise training in both LCR and HCR rats. The effect of exercise training was comparable between LCR and HCR; both groups increased VO2max by ~45% (Figure 3A). In contrast, exercise training induced an increase in HCR cardiomyocyte length by 14%, but only a 6% increase in LCR (Table 2). No exercise training-induced changes were observed in cell width (Table 2). Thus, the increase in cardiomyocyte volume amounted to 17% in HCR and 8% in LCR (Table 2).
The exercise training-induced hypertrophy of the cardiomyocyte was associated with maintenance of the relative T-tubule density in LCR both globally (Figure 3C) and along sections of the longitudinal cell (Figure 3D), suggesting that the T-tubules adapt to exercise training by retaining the same relative density throughout the full length of the cell, despite the mild pathology and concentric hypertrophy. Moreover, spacing between individual T-tubules also remained intact during exercise training (Figure 3E, F). Thus, this suggests that cardiomyocytes with a mild degree of pathologic hypertrophy have a reduced network of T-tubules, but this network is capable of responding to exercise training to maintain the relative T-tubule density through physiologic hypertrophy.
Transverse Tubules in Severe Cardiac Pathologic Remodeling – Eccentric/Concentric Hypertrophy
The next experiments were performed in a post-MI heart failure model with severe cardiac pathology and dysfunction (Kemi et al, 2007B; Loennechen et al, 2002A; Wisloff et al, 2002). Here, MI was confirmed 1 week after coronary artery ligation by echocardiography, and only rats with confirmed MI proceeded to the next stages of the study. Overall, 40±2% of the left ventricle was infarcted. Heart failure was defined at the time of sacrifice by intra-left ventricular pressure recordings. Left ventricle end-diastolic pressure (LVEDP) of >15 mmHg was used to define heart failure (Sjaastad et al, 2000), and only rats with heart failure were included in the study. As seen in Table 1, the MI-procedure elevated LVEDP well above 15 mmHg. Furthermore, post-MI heart failure was also accompanied by reduced left ventricle peak systolic pressure (LVPSP), reduced rates of contraction and relaxation (Table 1), and reduced exercise capacity (VO2max; Figure 1A).
Severe pathologic hypertrophy of the individual cardiomyocytes in post-MI heart failure was confirmed by 30% and 35% increased cell length and width, respectively, resulting in a 70% increase in cell volume (Table 1). Thus, this phenotype differs from the concentric cellular hypertrophy in LCR rats; the hypertrophy is more pronounced with both eccentric and concentric cellular growth.
Consistent with pathologic remodeling, post-MI heart failure rats also presented with reduced cardiomyocyte T-tubule densities (Figure 1C); with a density corresponding to 45% of that observed in sham-operated control rats, a reduction that occurred along the whole length of the cell (Figure 2D). However, while cardiomyocytes from sham-operated rats had a denser network of T-tubules in the middle sections of the cell, the post-MI heart failure cells had more evenly distributed T-tubules. Further FFT analysis of the individual T-tubules also indicated that normal spacing was lost in post-MI heart failure and that the T-tubule signal was disorganized with no clear power peaks versus spatial frequency occurring in post-MI heart failure rats (Figure 2A). As such, no consistent distances between individual T-tubules could be established for post-MI heart failure cardiomyocytes.
Transverse Tubules after Exercise Training in Severe Cardiac Pathologic Remodeling – Eccentric/Concentric Hypertrophy
Exercise training was also introduced to post-MI heart failure rats, after the pathologic remodeling was established, a process that in this model takes 3–4 weeks (Loennechen et al, 2002A). Therefore, we initiated daily exercise training at 85%–90% of VO2max 4 weeks after the induction of an MI. 8 weeks of exercise training increased VO2max by 38%, comparable to the increase in sham-operated rats (Figure 1A). Exercise training in post-MI heart failure also induced reverse remodeling, i.e. a reversal of the pathologic hypertrophy, observed as 12% and 13% reductions in cardiomyocyte length and width, compared to sedentary post-MI heart failure cells, whereas cell volume was reduced by 23% (Table 1). Thus, ~50% of the pathologic hypertrophy was reversed by the exercise training program, in line with previous reports (Wisloff et al, 2002; Zhang et al, 1998). The reverse remodeling was also associated with a 40% increase in the relative T-tubule density, such that after exercise training, post-MI heart failure cardiomyocytes had a T-tubule network corresponding to 60% of sham-operated rats (Figure 1C). The increased T-tubule density after exercise training occurred along the full length of the cell (Figure 1D). However, exercise training did not observably affect T-tubule spacing (Figure 2A).
The Effect of Cardiac Unloading on the Transverse Tubules in Severe Cardiac Pathologic Remodeling – Eccentric/Concentric Hypertrophy
Finally, we repeated the experiments in post-MI heart failure rats with severe cardiac pathology and eccentric/concentric hypertrophy, but after treatment with the AT1 receptor antagonist losartan that started 1 week post-MI. Losartan ameliorates the hypertension and thereby unloads the heart (Kemi et al, 2007B; Pitt et al, 1997), which therefore may also contribute toward reversing the pathologic hypertrophy. In this study, losartan reduced LVEDP by 8 mmHg (Table 1) in both sedentary and exercise trained post-MI heart failure rats compared to equivalent rats not on losartan, and improved exercise capacity (VO2max) by 20%, from 28±2 mL·kg−0.75·min−1 to 33±2 mL·kg−0.75·min−1 (p<0.01; data not shown) in sedentary post-MI heart failure rats, but not in exercise trained post-MI heart failure rats. These results are in line with previous studies in which losartan was similarly administered to post-MI heart failure rats (Kemi et al, 2007B, Loennechen et al, 2002B).
Similar to exercise training, losartan also reversed the post-MI heart failure-associated hypertrophy of the cardiomyocyte; both cell length and width by 10% and cell volume by 17% (Table 1), whereas when combined with exercise training, no additional effect was observed. Losartan did not have any effects on the T-tubule density (Figure 4), or spacing (data not shown) of cardiomyocytes. This suggests that exercise training and not losartan was the component that partly reversed the post-MI heart failure effect on the T-tubule network in cardiomyocytes.
Figure 4.
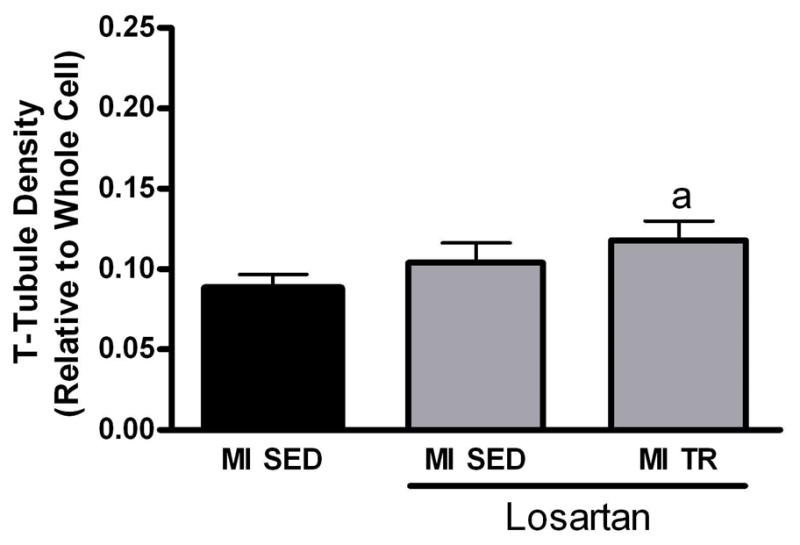
Cardiomyocyte transverse (T)-tubules in sedentary (SED) and exercise trained (TR) post-myocardial infarction (MI) heart failure rats treated with the angiotensin II type 1 receptor antagonist losartan by administration through the drinking water (2 g/L, ad libitum). Note that losartan alone did not restore T-tubules in post-MI heart failure rats. a: difference from MI SED without losartan, p<0.05. Data are mean±SEM with 8 animals/group. For control comparison, we also included data from Figure 1.
DISCUSSION
Here, we report the results from the first extensive studies of the effect of exercise training on T-tubules in cardiac muscle cells under (i) normal conditions and after the cardiomyocytes had undergone either (ii) mild concentric pathologic hypertrophy due to the metabolic syndrome or (iii) severe eccentric/concentric pathologic hypertrophy due to post-MI heart failure after coronary artery ligation. Disadvantageous T-tubule remodeling is prevalent during pathologic conditions, which may be ameliorated by exercise training. Several novel aspects were identified in this study. First, physiologic hypertrophy after exercise training in normal cardiomyocytes was associated with a comparative adaptation of the T-tubule network such that the relative density was maintained. Secondly, also cardiomyocytes undergoing pathologic hypertrophy responded to exercise training by adapting the T-tubule density. In fact, under conditions of severe cardiac pathologic remodeling with eccentric/concentric hypertrophy, the net effect of exercise training was to partially correct for the loss of t-tubules by increasing the relative density, although a complete restoration was not observed.
Transverse Tubules in Physiologic Hypertrophy
The exercise training-induced physiologic hypertrophy has also previously been reported in both cardiomyocytes and whole hearts, as a mechanism for supporting cardiac pump function (Kemi et al, 2004, 2005,Kemi et al, 2007A, 2008; Wisloff et al, 2001, 2005). The maintenance of the T-tubule network during this scenario contributes likely to the improved contractile function, by preserving intact inter-T-tubule distances and T-tubule-to-sarcomere spacing across the cell, which would support a uniform and coordinated depolarization. Thus, this suggests that the T-tubules constitute a part of the cardiomyocyte response to exercise training to support EC coupling, Ca2+ handling, and contractility (Kemi et al, 2004, 2005,Kemi et al, 2007A; Wisloff et al, 2001). This is also consistent with studies showing that detubulation is associated with asynchronous intracellular Ca2+ release and inefficient EC coupling, leading to mechanical dysfunction (Brette et al, 2005; Heinzel et al, 2008; Louch et al, 2004; Stolen et al, 2009; Thomas et al, 2003).
Transverse Tubules in Pathologic Remodeling
Consistent with most studies (Balijepalli et al, 2003; Cannell et al, 2006; Louch et al, 2006; Quinn et al, 2003; Song et al, 2006); pathologic remodeling was associated with a loss of cardiomyocyte T-tubules. The magnitude of detubulation coincided with the magnitude of pathologic remodeling; mild pathologic concentric remodeling had a relatively mild effect on the T-tubules, whereas severe pathologic eccentric/concentric remodeling was associated with a relative loss of ~half the T-tubules and complete loss of normal spacing. The inverse relationship between cell size and T-tubule density during pathologic remodeling is plotted in Figure 5. Similar to normal cardiomyocytes, exercise training was associated with sustained T-tubule density also after pathologic remodeling with concentric hypertrophy; indicating the occurrence of an exercise training-induced adaptation. In severe pathology with eccentric/concentric hypertrophy, exercise training partly corrected the abnormal T-tubule architecture. These findings are novel and indicate that exercise training may correct abnormal T-tubule architecture, at least in cells originating from the area remote to the infarct. Whether this is true in cells bordering the infarct remains unknown. The possibility remains that the repair ability of exercise training on the T-tubules is governed by the severity of the T-tubule loss and by the extent and/or nature of the pathologic hypertrophy, which may differ between peri-infarct and non-ischemic areas.
Figure 5.

The relationship between cardiomyocyte volume and transverse (T)-tubule density in cardiomyocytes from sham-operated (SHAM) or post-myocardial infarction (MI) heart failure rats and rats with either a high or low capacity for running (HCR and LCR, respectively) that remained either sedentary (SED) or underwent exercise training (TR). LCR and post-MI heart failure rats developed respectively mild concentric and severe eccentric/concentric pathologic remodeling. Moreover, SED and TR post-MI heart failure rats that also received the angiotensin II type 1 receptor antagonist losartan were also included. Note that the cardiomyocyte volume-T-tubule density relationship is right-shifted after TR-induced physiologic hypertrophy in normal and mild cardiac pathology, but left-shifted after TR-induced reverse remodeling in post-MI heart failure rats with severe cardiac pathology, indicating that T-tubule density at least partly is dissociated from cell size. Data are mean±SEM with 8 animals/group.
A possibility has been that the T-tubules remain static during the development of the pathologic remodeling, and that it is the increased cell size that leads to the reduction in the relative T-tubule density. This would also explain why exercise training in post-MI heart failure increased the relative density of T-tubules, because it reduced the pathologic hypertrophy. By extension, this would suggest that the T-tubules are secondary to cell size. However, the current results indicate that cell size alone cannot regulate T-tubules. First, re-plotting the data from Figures 1 and 3 shows that the density of T-tubules was sustained despite the fact that cell size increased during exercise training in normal (SHAM and HCR) rats and in rats with mild pathologic remodeling (LCR) (Figure 5). If T-tubules had not adapted, physiologic hypertrophy would also have reduced the relative T-tubule density. Secondly, the FFT analysis of T-tubule spacing showed that the distance between individual T-tubules remained constant during physiologic hypertrophy and was not significantly different between cardiomyocytes from normal and mild pathology hearts, albeit a small trend toward a difference was noted together with a greater variability of T-tubule distances and indications of reduced T-tubule organization in mild pathology. Furthermore, whereas exercise trained post-MI heart failure cardiomyocytes had a cell size close to exercise trained sham-operated cardiomyocytes; the differences in T-tubule density and spacing between those cell populations were considerable. Thus, under at least certain conditions, T-tubule density was independent of cell size. To which degree detubulation was explained by disorganization, detachment, and degradation of individual T-tubules, or by altered cell size remains unknown, but it seems likely that both aspects may independently contribute to the net effect. It also remains unknown how the loss and disorganization of T-tubules affects adjacent intracellular structures, such as those sarcomeres that loose a closely linked functional T-tubule, though previous results indicate that EC coupling becomes asynchronous within the cell under circumstances that at least partly resemble those reported here (Heinzel et al; 2008; Louch et al, 2004, 2006; Stolen et al, 2009).
As exercise training only partially corrected T-tubules in post-MI heart failure cardiomyocytes, two possibilities for further correction arise. First, it is possible that exercise training lasting longer than 8 weeks may restore a normal T-tubule network, and secondly, exercise training starting early after the cardiac injury and hence before the pathologic remodeling settles, may also counteract detubulation at an earlier stage. In contrast, pharmacologic treatment with losartan did not associate with T-tubule correction, despite its unloading and reverse remodeling actions. This may be explained by losartan inhibiting AT1-transforming growth factor-β1, which reduces fibrosis, but has its action mainly on extra-cardiomyocyte tissue (Rosenkranz, 2004). Finally, pathologic hypertrophy and heart failure also associate with disruptions to intracellular morphology, myofilaments, and sarcomere spacing (Heineke et al, 2005), such that correction of T-tubules alone may not be sufficient to restore contractile function.
The Functional Consequences of Loss and Restoration of Transverse Tubules
This study aimed to clarify T-tubule remodeling and was not designed to determine any secondary consequences of the T-tubule changes. However, as the only transmembrane ion transporter, the cardiac Na+-Ca2+ exchanger (NCX) has been studied in similar sedentary and exercise trained sham-operated and post-MI heart failure rats (Brette and Orchard, 2003; Satoh et al, 1996; Soeller and Cannell, 1999; Wisloff et al, 2002). This allowed us to model the effect of loss and restoration of T-tubules on NCX function, by taking into account a 3:1 T-tubule:surface membrane distribution ratio of NCX in the rat cardiomyocyte (Brette and Orchard, 2003; Kieval et al, 1992). Based on this, we asserted that:
This was then normalized to current density using existing factors for cell capacitance from volume (8.88 pF/pL; Satoh et al, 1996; Soeller and Cannell, 1999). Loss of capacitance was calculated on the basis that 49% of the capacitance is contained in the T-tubules (Soeller and Cannell, 1999). This indicated a reduction of NCX current in post-MI heart failure rats to 46% of normal levels, with exercise training improving it to 66%. The reduced NCX current would be further compounded by the reduced surface:volume ratio (−26%) in post-MI heart failure, which would further reduce NCX effectiveness. This was improved by exercise training to −7%.
Although simplistic, the calculated values for the NCX current provide an initial working hypothesis for the contractile effect of the T-tubule changes provided by exercise training, and are in agreement with previous observations of contractility during these conditions (Wisloff et al, 2002). For a final analysis, other transmembrane currents should also be incorporated once sufficient experimental data become available. For instance, since both the L-type Ca2+ channel and the plasma membrane Ca2+ ATPase concentrate to the T-tubules (Brette and Orchard, 2003); in fact, a recent report suggests that Ca2+ efflux through this route solely occurs in the T-tubule and not across the sarcolemmal plasma membrane in rat cardiomyocytes (Chase and Orchard, 2010), it is clear that T-tubule alterations may profoundly affect different aspect of EC coupling, especially under circumstances of substantial loss such as in post-MI heart failure.
Reverse remodeling by Exercise Training
The existence of exercise-induced reverse remodeling is still being debated. This study confirms the existence of this phenomenon at the cellular level, at least in models of post-MI heart failure rats associated with severe pathologic remodeling and eccentric/concentric cellular hypertrophy, and at least if the exercise carried out has a high aerobic intensity. In contrast, the opposite was observed after exercise training in mild pathologic concentric hypertrophy, under which conditions exercise induced a further elongation of the cell, albeit of smaller magnitude than in controls. Although the mechanism allowing differential effects remains unknown, it suggests that the degree of pathology affects the response to exercise training. Furthermore, reverse remodeling of severe pathologic hypertrophy may also be achieved by pharmacologically unloading the heart, although this does not correct abnormal T-tubules.
Conclusions
This study provides new insight into the effects of exercise training on cardiomyocyte T-tubules under various conditions. First, we show that exercise training under healthy conditions induces an adaptation of the T-tubules whereby the physiologic hypertrophy of the cell does not translate into a reduction of the T-tubule density. Secondly, pathologic hypertrophy and remodeling of the cardiomyocyte is associated with a loss of T-tubules. The magnitude of the reduction depends on the severity of the remodeling. Exercise training under these circumstances is also associated with maintenance of the T-tubule density. If the severity of the pathologic remodeling is mild, exercise training induces adaptive cell growth with a sustained T-tubule density. In contrast, if the pathologic remodeling is severe, such as after post-MI heart failure, exercise training induces reverse remodeling of the cardiomyocyte, which associates with an increased T-tubule density. Thus, these results suggest that the beneficial effects of exercise training encompass the T-tubule network of the cardiomyocyte in both health and disease.
Acknowledgments
Contract grant sponsor: Norwegian University of Science and Technology; St. Olavs Hospital; National Center for Research Resources (National Institutes of Health); British Heart Foundation.
Contract grant number: R24RR017718, National Institutes of Health.
This study was supported by grants from the Norwegian University of Science and Technology and the St. Olavs Hospital, Trondheim, Norway; the National Center for Research Resources (National Institutes of Health), MD, USA (Award R24RR017718); and the British Heart Foundation. We also acknowledge MSD for providing us with losartan, and the respective Institutional Animal Resource Units for expert care of the rat populations.
References
- Balijepalli RC, Lokuta AJ, Maertz NA, Buck JM, Haworth RA, Valdivia HH, Kamp DJ. Depletion of t-tubules and specific subcellular changes in sarcolemmal proteins in tachycardia-induced heart failure. Cardiovasc Res. 2003;59:67–77. doi: 10.1016/s0008-6363(03)00325-0. [DOI] [PubMed] [Google Scholar]
- Brette F, Despa S, Bers DM, Orchard CH. Spatiotemporal characteristics of SR Ca2+ uptake and release in detubulated rat ventricular myocytes. J Mol Cell Cardiol. 2005;39:804–812. doi: 10.1016/j.yjmcc.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Brette F, Orchard C. T-tubule function in mammalian cardiac myocytes. Circ Res. 2003;92:1182–1192. doi: 10.1161/01.RES.0000074908.17214.FD. [DOI] [PubMed] [Google Scholar]
- Britton SL, Koch LG. Animal models of complex diseases: an initial strategy. IUBMB Life. 2005;57:631–638. doi: 10.1080/15216540500251684. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Crossman DJ, Soeller C. Effect of changes in action potential spike configuration, junctional sarcoplasmic reticulum micro-architecture and altered t-tubule structure in human heart failure. J Muscle Res Cell Motil. 2006;27:297–306. doi: 10.1007/s10974-006-9089-y. [DOI] [PubMed] [Google Scholar]
- Chase A, Orchard CH. Ca efflux via the sarcolemmal Ca ATPase occurs only in the t-tubules of rat ventricular myocytes. J Mol Cell Cardiol. 2010 doi: 10.1016/j.yjmcc.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Heineke J, Ruetten H, Willenbockel C, Gross SC, Naguib M, Schaefer A, Kempf T, Hilfiker-Kleiner D, Caroni P, Kraft T, Kaiser RA, Molkentin JD, Drexler H, Wollert KC. Attenuation of cardiac remodeling after myocardial infarction by muscle LIM protein-calcineurin signaling at the sarcomeric Z-disc. Proc Natl Acad Sci USA. 2005;102:1655–1660. doi: 10.1073/pnas.0405488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel FR, Bito V, Biesmans L, Wu M, Detre E, von Wegner F, Claus P, Dymarkowski S, Maes F, Bogaert J, Rademakers F, D’hooge J, Sipido K. Remodeling of T-tubules and reduced synchrony of Ca2+ release in myocytes from chronically ischemic myocardium. Circ Res. 2008;102:338–346. doi: 10.1161/CIRCRESAHA.107.160085. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, Condorelli G, Ellingsen O. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy. J Cell Physiol. 2008;214:316–321. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Ellingsen O, Ceci M, Grimaldi S, Smith GL, Condorelli G, Wisloff U. Aerobic interval training enhances cardiomyocyte contractility and Ca2+ cycling by phosphorylation of CaMKII and Thr-17 of phospholamban. J Mol Cell Cardiol. 2007A;43:354–361. doi: 10.1016/j.yjmcc.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemi OJ, Haram PM, Loennechen JP, Osnes J, Skomedal T, Wisloff U, Ellingsen O. Moderate vs. high exercise intensity: differential effects on aerobic fitness, cardiomyocyte contractility, and endothelial function. Cardiovasc Res. 2005;67:161–172. doi: 10.1016/j.cardiores.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Haram PM, Wisloff U, Ellingsen O. Aerobic fitness is associated with cardiomyocyte contractile capacity and endothelial function in exercise training and detraining. Circulation. 2004;109:2897–2904. doi: 10.1161/01.CIR.0000129308.04757.72. [DOI] [PubMed] [Google Scholar]
- Kemi OJ, Hoydal MA, Haram PM, Garnier A, Fortin D, Ventura-Clapier R, Ellingsen O. Exercise training restores aerobic capacity and energy transfer in heart failure treated with losartan. Cardiovasc Res. 2007B;76:91–99. doi: 10.1016/j.cardiores.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Kieval RS, Bloch RJ, Lindenmayer GE, Ambesi A, Lederer WJ. Immunofluorescence localization of the Na-Ca exchanger in heart cells. Am J Physiol Cell Physiol. 1992;263:C545–C550. doi: 10.1152/ajpcell.1992.263.2.C545. [DOI] [PubMed] [Google Scholar]
- Koch LG, Britton SL. Artificial selection for intrinsic aerobic endurance running capacity in rats. Physiol Genomics. 2001;5:45–52. doi: 10.1152/physiolgenomics.2001.5.1.45. [DOI] [PubMed] [Google Scholar]
- Litwin SE, Katz SE, Morgan JP, Douglas PS. Serial echocardiographic assessment of left ventricular geometry and function after large myocardial infarction in the rat. Circulation. 1994;89:345–354. doi: 10.1161/01.cir.89.1.345. [DOI] [PubMed] [Google Scholar]
- Loennechen JP, Wisloff U, Falck G, Ellingsen O. Cardiomyocyte contractility and calcium handling partially recover after early deterioration during post-infarction failure in rats. Acta Physiol Scand. 2002A;176:17–26. doi: 10.1046/j.1365-201X.2002.01011.x. [DOI] [PubMed] [Google Scholar]
- Loennechen JP, Wisloff U, Falck G, Ellingsen O. Effects of cariporide and losartan on hypertrophy, calcium transients, contractility, and gene expression in congestive heart failure. Circulation. 2002B;105:1380–1386. doi: 10.1161/hc1102.105258. [DOI] [PubMed] [Google Scholar]
- Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- Louch WE, Mork HK, Sexton J, Stromme TA, Laake P, Sjaastad I, Sejersted OM. T-tubule disorganization and reduced synchrony of Ca2+ release in murine cardiomyocytes following myocardial infarction. J Physiol. 2006;574:519–533. doi: 10.1113/jphysiol.2006.107227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt B, Segal R, Martinez FA, Meurers G, Cowley AJ, Thomas I, Deedwania PC, Ney DE, Snavely DB, Chang PI. Randomised trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly Study, ELITE) Lancet. 1997;349:747–752. doi: 10.1016/s0140-6736(97)01187-2. [DOI] [PubMed] [Google Scholar]
- Quinn FR, Currie S, Duncan AM, Miller S, Sayeed R, Cobbe SM, Smith GL. Myocardial infarction causes increased expression but decreased activity of the myocardial Na+-Ca2+ exchanger in the rabbit. J Physiol. 2003;553:229–242. doi: 10.1113/jphysiol.2003.050716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenkranz S. TGF-beta1 and angiotensin networking in cardiac remodeling. Cardiovasc Res. 2004;63:423–432. doi: 10.1016/j.cardiores.2004.04.030. [DOI] [PubMed] [Google Scholar]
- Satoh H, Delbridge LMD, Blatter LA, Bers DM. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J. 1996;70:1494–1504. doi: 10.1016/S0006-3495(96)79711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorten PR, Soboleva TK. Anomalous ion diffusion within skeletal muscle transverse tubule networks. Theor Biol Med Model. 2007;4:18. doi: 10.1186/1742-4682-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjaastad I, Sejersted OM, Ilebekk A, Bjornerheim R. Echocardiographic criteria for detection of postinfarction congestive heart failure in rats. J Appl Physiol. 2000;89:1445–1454. doi: 10.1152/jappl.2000.89.4.1445. [DOI] [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- Song L-S, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolen TO, Hoydal MA, Kemi OJ, Catalucci D, Ceci M, Aasum E, Larsen T, Rolim N, Condorelli G, Smith GL, Wisloff U. Interval training normalizes cardiomyocyte function, diastolic Ca2+ control, and SR Ca2+ release synchronicity in a mouse model of diabetic cardiomyopathy. Circ Res. 2009;105:527–536. doi: 10.1161/CIRCRESAHA.109.199810. [DOI] [PubMed] [Google Scholar]
- Swift F, Stromme TA, Amundsen B, Sejersted OM, Sjaastad I. Slow diffusion of K+ in the T tubules of rat cardiomyocytes. J Appl Physiol. 2006;101:1170–1176. doi: 10.1152/japplphysiol.00297.2006. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Sjaastad I, Andersen K, Helm PJ, Wasserstrom JA, Sejersted OM, Ottersen OP. Localization and function of the Na+/Ca2+-exchanger in normal and detubulated rat cardiomyocytes. J Mol Cell Cardiol. 2003;35:1325–1337. doi: 10.1016/j.yjmcc.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Loennechen JP, Currie S, Smith GL, Ellingsen O. Aerobic exercise reduces cardiomyocyte hypertrophy and increases contractility, Ca2+ sensitivity and SERCA-2 in rat after myocardial infarction. Cardiovasc Res. 2002;54:162–174. doi: 10.1016/s0008-6363(01)00565-x. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Loennechen JP, Falck G, Beisvag V, Currie S, Smith G, Ellingsen O. Increased contractility and calcium sensitivity in cardiac myocytes isolated from endurance trained rats. Cardiovasc Res. 2001;50:495–508. doi: 10.1016/s0008-6363(01)00210-3. [DOI] [PubMed] [Google Scholar]
- Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernstrom M, Rezaei K, Lee SJ, Koch LG, Britton SL. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005;307:418–420. doi: 10.1126/science.1108177. [DOI] [PubMed] [Google Scholar]
- Zhang XQ, NGYC, Musch TI, Moore RL, Zelis R, Cheung JY. Sprint training attenuates myocyte hypertrophy and improves Ca2+ homeostasis in postinfarction myocytes. J Appl Physiol. 1998;84:544–552. doi: 10.1152/jappl.1998.84.2.544. [DOI] [PubMed] [Google Scholar]