

Abstract
The decatenation G2 checkpoint is proposed to delay cellular progression from G2 into mitosis when intertwined daughter chromatids are insufficiently decatenated. Previous studies indicated that the ATM- and Rad3-related (ATR) checkpoint kinase, but not the ataxia telangiectasia-mutated (ATM) kinase, was required for decatenation G2 checkpoint function. Here, we show that the method used to quantify decatenation G2 checkpoint function can influence the identification of genetic requirements for the checkpoint. Normal human diploid fibroblast (NHDF) lines responded to the topoisomerase II (topo II) catalytic inhibitor ICRF-193 with a stringent G2 arrest and a reduction in the mitotic index. While siRNA-mediated depletion of ATR and CHEK1 increased the mitotic index in ICRF-193 treated NHDF lines, depletion of these proteins did not affect the mitotic entry rate, indicating that the decatenation G2 checkpoint was functional. These results suggest that ATR and CHEK1 are not required for the decatenation G2 checkpoint, but may influence mitotic exit after inhibition of topo II. A re-evaluation of ataxia telangiectasia (AT) cell lines using the mitotic entry assay indicated that ATM was required for the decatenation G2 checkpoint. Three NHDF cell lines responded to ICRF-193 with a mean 98% inhibition of the mitotic entry rate. Examination of the mitotic entry rates in AT fibroblasts upon treatment with ICRF-193 revealed a significantly attenuated decatenation G2 checkpoint response, with a mean 59% inhibition of the mitotic entry rate. In addition, a normal lymphoblastoid line exhibited a 95% inhibition of the mitotic entry rate after incubation with ICRF-193, whereas two AT lymphoblastoid lines displayed only 36% and 20% inhibition of the mitotic entry rate. Stable depletion of ATM in normal human fibroblasts with short hairpin RNA also attenuated decatenation G2 checkpoint function by an average of 40%. Western immunoblot analysis demonstrated that treatment with ICRF-193 induced ATM autophosphorylation and ATM-dependent phosphorylation of Ser15-p53 and Thr68 in CHEK2, but no appreciable phosphorylation of Ser139 on H2AX. The results suggest that inhibition of topo II induces ATM to phosphorylate selected targets that contribute to a G2 arrest independently of DNA damage.
Keywords: ATM, ATR, CHK1, CHK2, CHEK1, CHEK2, p53, decatenation G2 checkpoint, ICRF-193
Introduction
Cell cycle checkpoints represent mechanisms of control that 1) ensure the proper order of the required events during the cell division cycle, 2) stabilize stalled or arrested DNA replication forks, 3) provide more time for repair of DNA damage before DNA replication and mitosis, and 4) induce growth arrest or apoptosis in response to genotoxic stress. Some checkpoint genes such as ATR, CHEK1 and BRCA1 are essential, suggesting that checkpoint signaling is required for successful and stable cell division. Other non-essential checkpoint genes are familial cancer genes (e.g., TP53, ATM, CHEK2). Cell cycle checkpoint responses are associated with oncogene-induced senescence, supporting a hypothesis that checkpoints are barriers to the development of cancer. 1, 2 Since cell cycle checkpoints slow growth and stabilize the genome, defects in these checkpoints are thought to enhance clonal growth and genomic instability, thereby accelerating carcinogenesis. 3, 4 It is of great interest to determine the mechanisms of cell cycle checkpoint function, how these mechanisms may be disturbed during the multistage development of cancer, and the consequences of checkpoint dysfunction. This paper specifically addresses the DNA topoisomerase II-dependent decatenation G2 checkpoint.
Type II DNA topoisomerases (topo II) are homodimeric enzymes that can pass one DNA duplex through another using a concerted process of DNA double-strand scission, duplex passage, and double-strand re-ligation to remove DNA knots and separate intertwined or catenated dimers. 5 Human cells express two related topo II enzymatic isoforms, α and β.6 Topo IIβ is not essential and is expressed constitutively, apparently providing a basal level of decatenatory activity in quiescent cells. In contrast, topo IIα is cell cycle-regulated with a pattern of mRNA expression similar to cyclin B1, CDK1 and PLK1, 7-9 all of which regulate the progression from G2 into mitosis. Topo IIα is essential in mammals, 10 suggesting that topo IIβ is unable to compensate for the absence of topo IIα during cell division. Catalytic inhibition of topo II and genetic attenuation or inactivation of topo IIα decatenatory activity produce similar effects in human cells, such as undercondensation of chromosomes and the impairment of segregation of sister chromatids at anaphase of mitosis. 11-15
The decatenation G2 checkpoint was first recognized when catalytic inhibitors of topo II were shown to induce a G2 delay. 16-18 The bisdioxopiperazine ICRF-193 is a potent topo II catalytic inhibitor 19 that holds the enzyme in a closed clamp form tethered to DNA, which is unable to cleave DNA strands due to inhibition of its ATPase activity. 20,21 High concentrations of ICRF-193 (>20 μM) induce a metaphase arrest, apparently by inhibiting decatenation of sister chromatids and triggering a MAD2-dependent checkpoint. 13, 14, 22 Other studies have suggested that topo IIα is targeted to centromeres by the SUMO ligases PIAS-γ and RanBP2, where it removes the final linkages between daughter chromatids at the metaphase-anaphase transition. 14, 22-25 Lower concentrations of ICRF-193 do not induce prolonged metaphase arrest, as most mitotic cells eventually complete mitosis. However, after treatment with these lower concentrations of ICRF-193 (1-4 μM), NHDFs display a severe delay in the progression from G2 into mitosis, 16, 26 causing up to a 95% reduction in the percentage of mitotic cells in asynchronous cultures within 2 h of exposure to the drug. 17, 27 As the topo II catalytic inhibitors do not produce topo II-associated cleaved DNA complexes, nor appreciable numbers of DNA strand breaks, 13, 16, 17, 28 the G2 delay that is induced by such drugs appears to differ from the DNA damage G2 checkpoint response. Previous studies demonstrating that BRCA1 was required for ICRF-193-induced mitotic inhibition suggested that the G2 delay was an active process, and was designated the decatenation G2 checkpoint. 18
A study of human lung cancer cells described three lines that responded to ICRF-193 with a G2 delay, but without inhibiting the kinase activity of the mitosis promoting factor (MPF), which is composed of the cyclin B1 and CDK1 (cdc2) proteins. 28 Since activation of the DNA damage G2 checkpoint inhibits MPF, this result supported the hypothesis that the decatenation G2 checkpoint is distinct from the DNA damage G2 checkpoint and confirms a prior report indicating that the decatenation G2 checkpoint is enforced by retaining active MPF in the cytoplasm where it cannot induce mitosis. 26,29, 30
Although ICRF-193 cannot be consistently shown to induce γ-H2AX foci as a marker of replication stress and DNA double strand breaks (dsbs), 13, 28 the drug did induce phosphorylation of ATM in the lung cancer cell lines with effective decatenation G2 checkpoint function. 28 Canonically, ATM is activated by treatments that induce DNA dsbs. However, it can also be activated by alterations in chromatin structure, such as those produced by incubating cells in high-salt medium, 31 or by treatment with the DNA intercalating drug and topo II catalytic inhibitor chloroquine. 32, 33 The antibiotic ciprofloxacin, which is a topo II inhibitor commonly used to treat mycoplasma contamination in cultured cell lines, has been shown to induce phosphorylation of ATM and trigger G2 arrest without induction of γ-H2AX. 34 These results suggest that ATM may be activated under conditions that trigger the decatenation G2 checkpoint in response to altered states of chromatin that develop upon catalytic inhibition of topo II. In this paper, we have re-examined the roles of ATM, CHEK1, and ATR in the decatenation G2 checkpoint response to the topo II catalytic inhibitor ICRF-193 using assays that monitor mitotic index, mitotic entry rates, and G2 arrest.
Results
ATR and CHEK1 are not required for decatenation G2 checkpoint function
Due to the recent reports suggesting a role for topo II in both G2 arrest and mitotic arrest, decatenation G2 checkpoint function was quantified using two distinct flow cytometry methods. The first method, termed the mitotic index assay, quantifies the reduction in mitotic index two hours after a15-min incubation with 4 μM ICRF-193 relative to vehicle control-treated cells. Inhibition of topo II blocks decatenatory activity in G2 cells and activates a checkpoint that delays progression of G2 cells into mitosis. Since cells already in mitosis divide within two hours after the drug treatment, catalytic inhibition of topo II should cause the mitotic cell compartment to empty. However, depletion of topo IIα or inhibition of topo II activity with catalytic inhibitors can block centromere separation, even in cells with a disruption or depletion of cohesion, and delay the completion of mitosis. 14, 22-24 Since topo IIα appears to be required at the metaphase-to-anaphase transition to decatenate centromeric DNA and may influence the length of mitosis, a second method, referred to as the mitotic entry assay, was employed to determine decatenation G2 checkpoint function. The mitotic entry assay relies on the microtubule depolymerization inhibitor colcemid to prevent mitotic exit, allowing a strict measure of the progression from G2 into mitosis and eliminating confounding factors, such as the rate of mitotic exit, from the analysis. Cells are accumulated in colcemid over a period of two, four, or six hours in the presence or absence of the catalytic topo II inhibitor ICRF-193, and a mitotic entry rate is calculated from the slope of the resulting line by linear regression.
ATR and CHEK1 have previously been reported to be required for decatenation G2 checkpoint function. 18, 43 Studies investigating the role of ATR have relied on an SV40-transformed human fibroblast line over-expressing a kinase-inactive ATR protein.18 The role of CHEK1 was previously examined by genetic inactivation in DT40 chicken erythroleukemia cells.43 To determine the role of ATR and CHEK1 in decatenation G2 checkpoint function in NHDFs, non-targeting control (NTC) siRNA, ATR siRNA, or CHEK1 siRNA were electroporated to deplete protein expression. Electroporation of siRNA was effective in depleting the ATR and CHEK1 proteins (Fig. 1A and 2A), producing mean reductions of 90% and 95%, respectively.
Figure 1. ATR is not required for decatenation G2 checkpoint function.
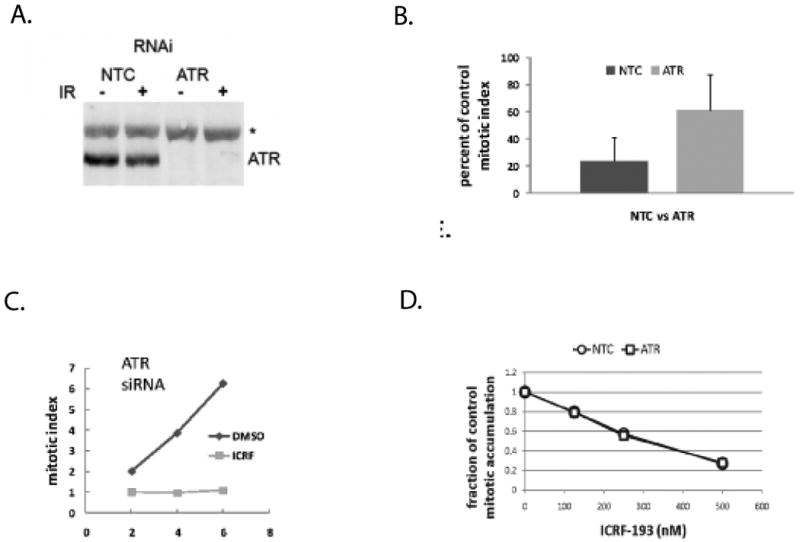
Diploid fibroblast lines were electroporated with non-targeting control (NTC) siRNA or siRNA directed towards ATR. A. Cells were harvested for immunoblot analysis of ATR protein levels at 48 h after electroporation when protein depletion was maximal. Treatment with 1.5 Gy IR 30 min before harvest did not affect expression of ATR. B. NTC-treated and ATR-depleted NHDFs were incubated with 4 μM ICRF-193 for 15 min, then further incubated in drug-free medium for 2 h before cell harvest for quantification of phospho-histone H3+ mitotic cells by flow cytometry. Results show the mean reductions in the mitotic index in ICRF-193-treated NHF1hTERT cells relative to DMSO-treated controls (+sd, n=4). Depletion of ATR appeared to attenuate the ICRF-193-induced inhibition of the emptying of the mitotic compartment. C. NTC-treated and ATR-depleted NHF1hTERT cells were incubated with 100 ng/ml colcemid for 0-6 hours and the percentage of mitotic cells was measured by flow cytometry. Inclusion of 4 μM ICRF-193 during incubation with colcemid blocked the accumulation of mitotic cells in ATR-depleted fibroblasts, suggesting that ATR is not required to prevent mitotic entry in the presence of catalytically inactive topo II. Results are representative of three independent experiments. D. ATR-depleted NHF1hTERT fibroblasts were incubated for 6 h with colcemid and increasing concentrations of ICRF-193. The percentage of mitotic cells that accumulated with colcemid alone was determined, and the mitotic index in ICRF-193-treated cells is expressed as a fraction of the DMSO-treated control value. ATR-depleted cells displayed the same sensitivity to ICRF-193-induced G2 delay as the NTC-treated counterparts, suggesting that ATR does not play a role in decatenation G2 checkpoint function, even at lower doses of ICRF-193 exposure.
Figure 2. CHEK1 is not required for decatenation G2 checkpoint function.
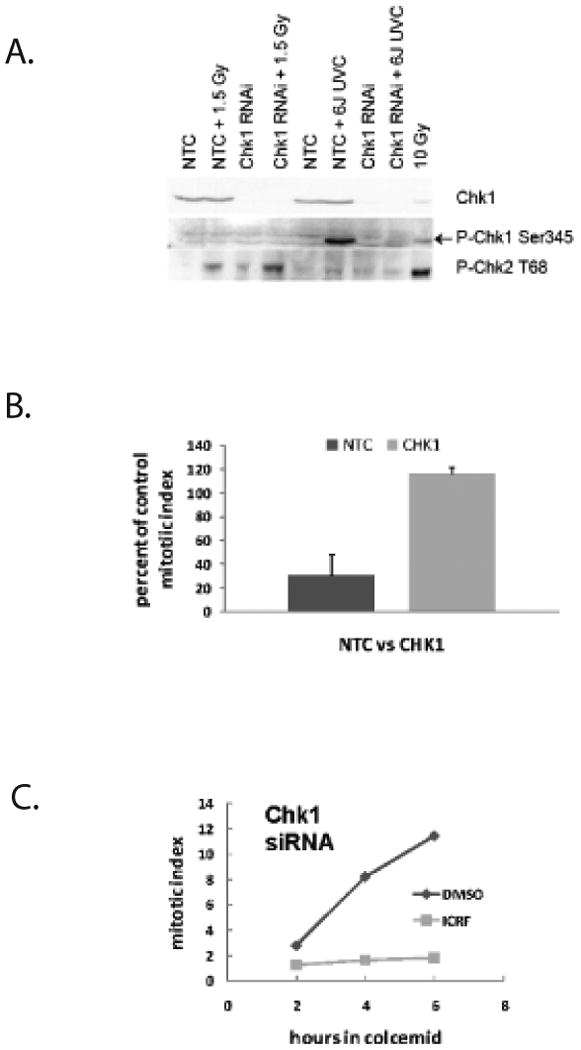
Diploid fibroblast lines were electroporated with non-targeting control (NTC) siRNA or siRNA directed towards CHEK1. A. Cells were harvested for immunoblot analysis of CHEK1 protein levels at 48 h after electroporation when protein depletion was maximal. Control and CHEK1-depleted cells were treated with 1.5 Gy IR or 6 J/m2 UVC 30 min before cell harvest to test for ATM-dependent phosphorylation of CHEK2 and ATR-dependent phosphorylation of CHEK1. B. NTC-treated and CHEK1-depleted NHDFs were subjected to the mitotic index reduction assay as described in Figure 1B. (+sd, n=3). Depletion of CHEK1 also appeared to attenuate the ICRF-193-induced inhibition of the mitotic compartment emptying. C. NTC-treated and CHEK1-depleted NHF1hTERT cells were subjected to the mitotic entry assay described in Figure 1C. Similar to the ATR-depleted fibroblasts, 4 μM ICRF-193 blocked the accumulation of mitotic cells in the CHEK1-depleted fibroblasts, suggesting that CHEK1 is not required for decatenation G2 checkpoint function. Results are representative of three independent experiments.
NTC siRNA-treated NHDFs responded to the topo II inhibitor with a significant reduction in mitotic index (Fig. 1B and 2B); two hours after drug treatment the mitotic index was reduced by about 80% compared to the vehicle control-treated cells. This value is similar to values previously observed for the NHF1hTERT fibroblast line,18 suggesting that the conditions of electroporation of siRNA did not affect the determination of mitotic inhibition after treatment with ICRF-193. In contrast, ATR-depleted NHDFs displayed an attenuated reduction of the mitotic index; an average 40% reduction in mitotic index was observed after treatment with ICRF-193 (Fig. 1B). Due to large inter-experimental variability, the difference between the NTC-treated and ATR-depleted NHDF mean mitotic index reduction was not statistically significant. In contrast, depletion of CHEK1 produced a significant increase in the mitotic index in ICRF-193-treated cells when compared to vehicle control-treated cells (Fig. 2B). These results suggested that ATR and CHEK1 were influencing the number of mitotic cells after topo II catalytic inhibition.
In contrast to the mitotic index assay results, depletion of ATR and CHEK1 did not affect the ICRF-193-induced G2 arrest (Fig. 1C and 2C). As previously described, the increment of mitotic NHDFs upon addition of colcemid was delayed by about 2 h. 44 Thereafter, mitotic indices of ATR- and CHEK1-depleted NHDFs increased by 1-3% per hour during incubation with colcemid and the drug solvent DMSO. Addition of colcemid and 4 μM ICRF-193 to NHDFs severely delayed the accumulation of mitotic cells with an average inhibition of 95% in three different ATR-depleted NHDF lines (Fig. 1C and data not shown). The average inhibition of mitotic entry relative to DMSO-treated controls was 97% in three different NHDF lines that had been depleted of CHEK1 (Fig. 2C and data not shown). Since NHDFs depleted of ATR and CHEK1 responded to ICRF-193 with a stringent G2 arrest equivalent to that seen in NTC siRNA-treated cells, we concluded that ATR and CHEK1 were not required for a functional decatenation G2 checkpoint.
It was possible that the 4 μM concentration of ICRF-193 saturated signaling of the decatenation G2 checkpoint and thereby obscured a partial contribution of ATR. To test for more subtle effects of ATR on the decatenation G2 checkpoint, cells were depleted of ATR and the mitotic entry assay was performed with increasing concentrations of ICRF-193 in the range of 125-500 nM. As shown in Figure 1D, ATR-depleted cells displayed the same sensitivities to ICRF-193-induced G2 delay as NTC siRNA-treated cells at all tested concentrations of the drug. A similar result was obtained with CHEK1-depleted cells (data not shown).
ATM is required for decatenation G2 checkpoint function
Given the previous results that ATR was not required for decatenation G2 checkpoint function, and earlier studies suggested that the checkpoint could be overridden by the general kinase inhibitor caffeine, it was important to reassess the role of ATM in the decatenation G2 checkpoint.18 Three NHDF lines were compared to three AT fibroblast lines using the mitotic index reduction assay and the mitotic entry assay. NHDFs displayed a significant 63-83% reduction in the mitotic index upon treatment with ICRF-193 with an average reduction of 75% (Fig. 3A and 3B). The AT fibroblast lines displayed less mitotic inhibition after ICRF-193 treatment with an average inhibition of mitosis of 15% (Fig. 3A and 3B), indicating that the AT fibroblasts displayed a significant attenuation in mitotic reduction upon drug treatment.
Figure 3. AT fibroblasts display an attenuated decatenation G2 checkpoint.
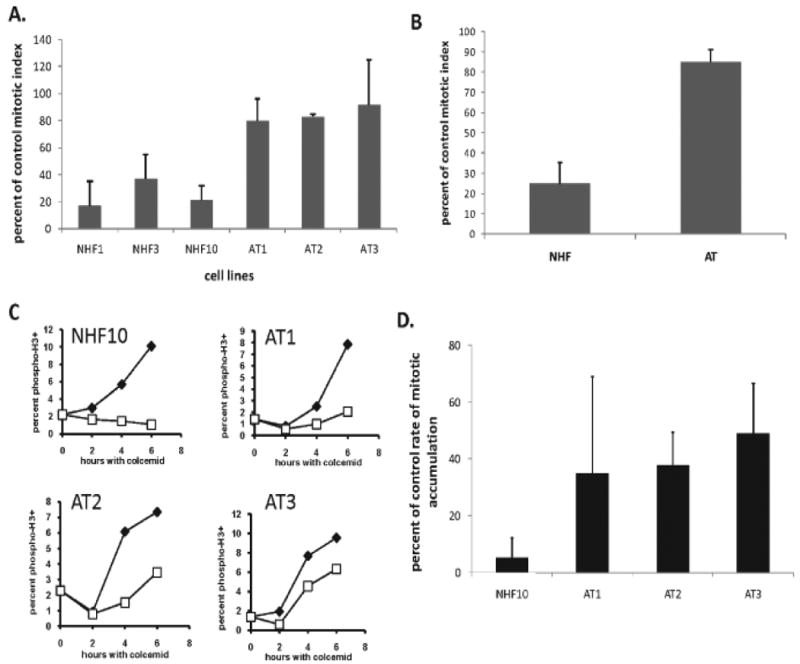
A. Normal and AT fibroblast lines were incubated with 4 μM ICRF-193 for 15 min, and for an additional 2 h in drug-free medium before quantification of mitotic index by flow cytometry. The mitotic index in ICRF-193-treated cells was expressed as a percent of the mitotic index in DMSO-treated controls (+sd; NHF1, n=5; NHF3, n=4; NHF10, n=7; AT1, n=8; AT2, n=3; AT3, n=3). B. Mean inhibitions of mitotic index after treatment with ICRF-193 among normal and AT fibroblast lines (+sd, n=3). The attenuated inhibition of mitotic emptying in the AT fibroblasts suggested that ATM may play a role in mitotic entry or mitotic exit. C. Normal and AT fibroblasts were incubated with colcemid for 0-6 h with 0.1% DMSO (closed symbols) or 4 μM ICRF-193 (open symbols) and harvested for flow cytometric analysis of mitotic cells. Results are representative of at least four independent experiments. D. Bar graphs representing the mean fraction of cells evading the ICRF-193-induced G2 arrest as compared to DMSO-treated controls (NHF10, n=5; AT1, n=5; AT2, n=4; AT3, n=4). AT fibroblasts exhibited an attenuated response to treatment with ICRF-193, indicating that ATM may play a role in the decatenation G2 checkpoint.
As previously demonstrated for NHDFs, AT fibroblasts accumulated in mitosis in the presence of colcemid; the mitotic index increased from 1-2% at the time of addition of colcemid to 6-10% after 6 h. In the NHF10hTERT line depicted in Figure 3C, the accumulation of mitotic cells in colcemid was abolished during incubation with ICRF-193. Under the same conditions, ICRF-193-treated AT fibroblasts exhibited an accumulation of mitotic cells over time (Fig. 3C). Although there was substantial experimental variability in the AT1 line, the mean reduction in the rate of progression from G2 to M in the presence of ICRF-193 was 65%, similar to the mean values acquired for the AT2 and AT3 lines (Fig. 3D). Among the three different AT fibroblast lines, the average inhibition of the mitotic entry rate upon ICRF-193 treatment was 59%, as compared to an average 98% inhibition of mitotic entry rate in three NHDFs. These results suggested that the AT lines displayed a significant (∼40%) attenuation of decatenation G2 checkpoint function.
Our prior analysis of AT cells, which showed an average of 60% inhibition in response to ICRF-193 treatment, employed a set of lymphoblastoid lines and was measured with the mitotic index assay.18 It was therefore necessary to re-examine those lines with the mitotic entry rate assay. Two AT lymphoblastoid lines and one normal lymphoblastoid line that were included in the prior analysis were analyzed. The normal and AT lymphoblastoid lines both responded to colcemid with a steady increase in mitotic cells, and the accumulation of mitotic cells was 95% blocked in the normal line upon addition of ICRF-193 (Fig. 4A). In contrast, the two AT lymphoblastoid lines allowed the accumulation of mitotic cells during incubation with ICRF-193 (Fig. 4A). Rates of mitotic entry in the ICRF-193-treated AT cells were reduced by 36% and 20% in comparison to values obtained in the DMSO-treated controls (Fig. 4B), indicating that the AT lymphoblastoid lines also displayed a severe defect in decatenation G2 checkpoint function.
Figure 4. AT lymphoblasts exhibit a defective decatenation G2 checkpoint.
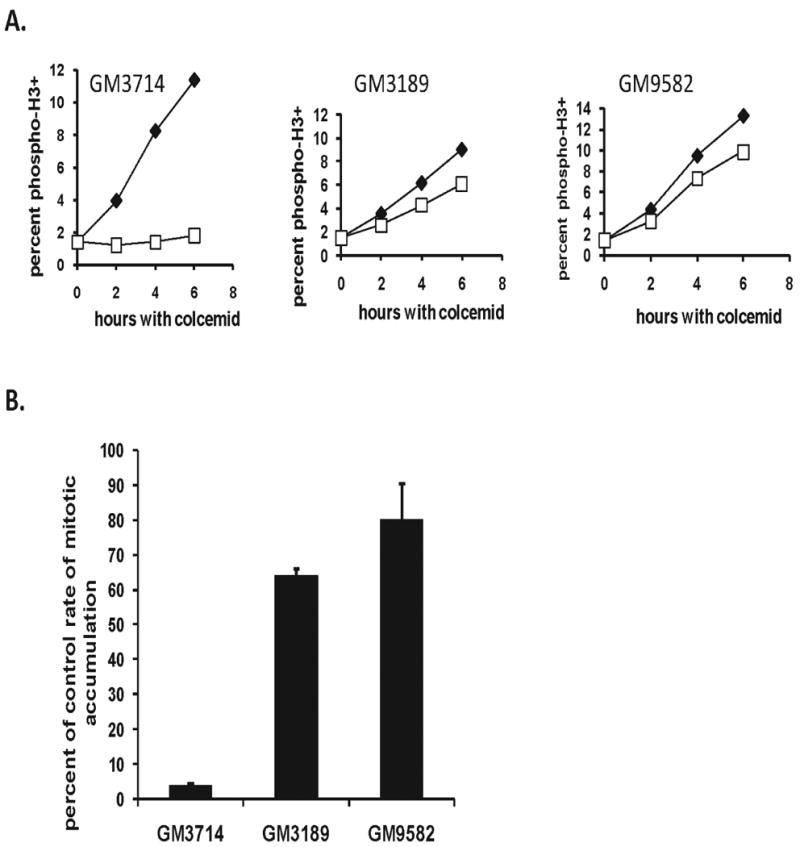
A. Normal (GM3714) and AT lymphoblasts (GM3189, GM9582) were incubated with colcemid for 0-6 h with or without 4 μM ICRF-193 then harvested for quantification of mitotic entry rates by flow cytometry. Results are representative of three independent experiments. B. Bar graphs depicting the average percentages of cells evading the ICRF-193-induced G2 arrest (+sd, n=2).
Although several studies have indicated that ICRF-193 does not induce topo II-associated DNA cleavage complexes, 16, 17, 45 other studies have demonstrated that ICRF-193 can activate DNA damage response elements. 28, 46 To explore this issue further, the fibroblast and lymphoblastoid lines were treated with ICRF-193 and examined for the expression of several markers of the DNA damage response. Lymphoblasts were incubated for 3 or 6 h with colcemid with and without ICRF-193 to explore the activation of the canonical DNA damage checkpoint proteins CHEK2, CHEK1, p53, and H2AX. As a positive control for activation of DNA damage checkpoint signaling, normal lymphoblasts were treated with 12 μM etoposide, and the induction of γH2AX, phospoho-Ser15 p53, and phospho-Thr68 CHEK2 was observed (Figure 5A). In the normal lymphoblasts there was no increment of phospho-Ser345 CHEK1 after 3 h of ICRF-193 treatment, and only a small increment of CHEK1 activation over colcemid alone after 6 h. No increment of phospho-CHEK1 was seen in AT cells under any treatment condition. Two downstream targets of activated ATM are CHEK2 and p53, and treatment with ICRF-193 induced phosphorylation of both in normal lymphoblasts, but not those collected from AT patients. These results indicate that ICRF-193 induced a selective phosphorylation of ATM substrates CHEK2 and p53, but not H2AX in lymphoblastoid cell lines. Very similar results were obtained with the NHDFs incubated with 4 μM ICRF-193 for three hours (Fig. 5B.).
Figure 5. ICRF-193 induces ATM-dependent checkpoint signaling, but not DNA damage in human fibroblasts and lymphoblasts.
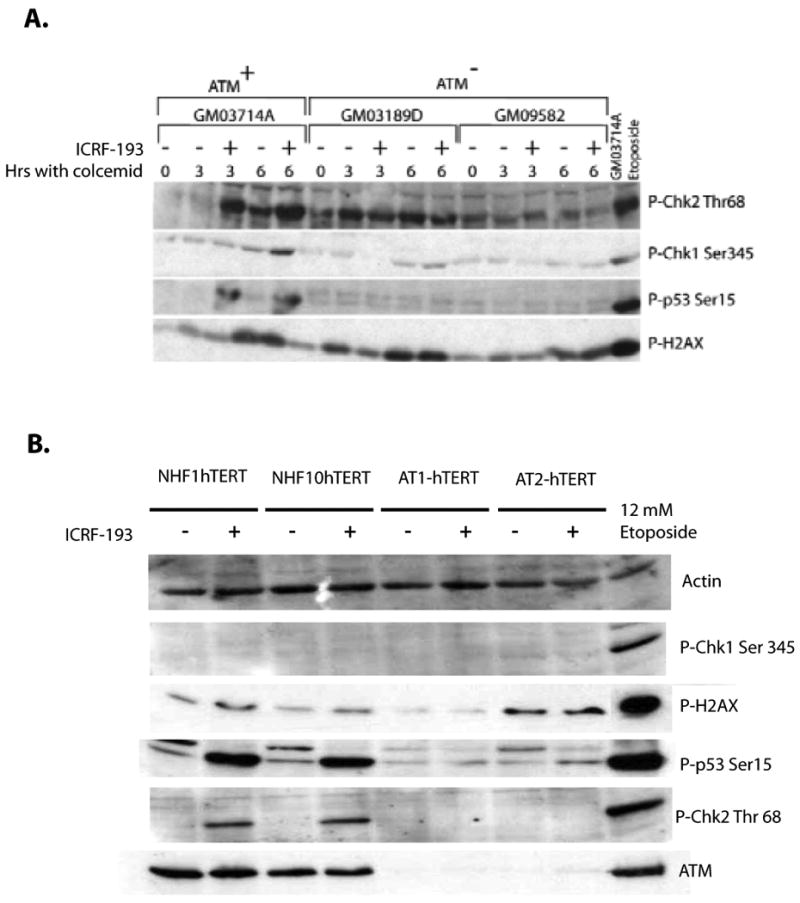
A. Normal and AT lymphoblasts were treated with colcemid and DMSO or ICRF-193 for 3 or 6 h and harvested for western immuno-blot analysis. Equal amounts of cell lysate protein were analyzed for phospho-Ser 345 CHEK1, phospho-Thr 68 CHEK2, and phospho-Ser 15 p53. For γH2AXdetection, equal numbers of cells were boiled in gel loading buffer before SDS-PAGE analysis. ICRF-193 treatment induced ATM-dependent phosphorylation of Thr 68-Chk2 and Ser 15-p53, but not γH2AX. A small increment of phophorylated Ser345-Chk1 is observed in the normal lymphoblasts upon 6 hr of ICRF-193 treatment, suggesting that lymphoblasts may activate low levels of Chk1 after a prolonged exposure to ICRF-193. Normal lymphoblasts treated with 12 μM etoposide for 6 h were analyzed as a positive control for DNA damage-induced signaling responses. B. Normal and AT fibroblast lines were treated with DMSO or 4 μM ICRF-193 for 3 h and harvested for western immunoblot analysis. Levels of γH2AX and phospho-Ser345 Chk1 remained low in all fibroblast lines examined. Similar to the normal lymphoblasts described in Fig. 1A, an ATM-dependent phosphorylation of Thr 68-Chk2 and Ser15-p53 were detected upon treatment with ICRF-193. NHF1hTERTs treated with 12 μM etoposide for 6 h were analyzed as a positive control for DNA damage.
Given that AT cells may harbor genetic mutations other than ATM, we further explored the role of ATM in the decatenation G2 checkpoint in an NHF1hTERT fibroblast line with stable depletion of ATM protein using the mitotic entry rate assay. The control NHF1hTERTs (expressing a short hairpin directed towards LacZ) responded to ICRF-193 with 97% inhibition of mitotic entry (Fig. 6A). The ATM-depleted fibroblasts displayed a significant attenuation of the ICRF-193-induced G2 arrest with a rate of mitotic entry approximately 40% of the control, similar to the values recorded with AT fibroblast and AT lymphoblast lines (Fig. 3 and Fig. 4). Furthermore, the NHF1hTERT-shLacZ fibroblast line responded to the topo II poison etoposide with substantial induction of phospho-Ser1981 ATM (Fig. 6B). Treatment with ICRF-193 produced a modest increase in phospho-Ser1981 ATM in NHDF and lymphoblastoid lines, but not in ATM-defective or ATM-depleted fibroblast or lymphoblastoid lines.
Figure 6. ATM is required for decatenation G2 checkpoint function.
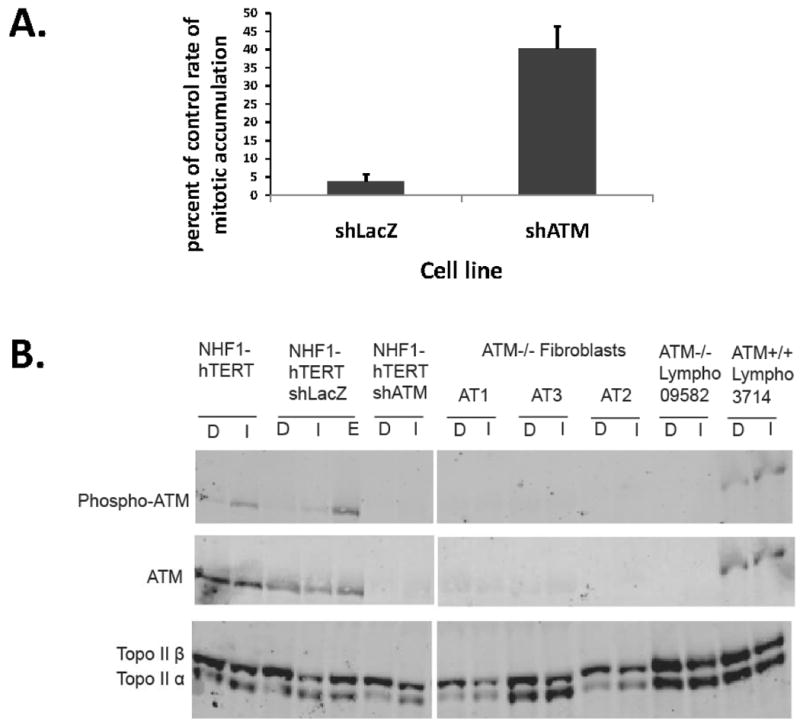
The NHF1hTERT fibroblast line was infected with a retrovirus to express LacZ or ATM shRNA. 38 A. ICRF-193-induced G2 arrest was quantified using the mitotic entry assay (described in Fig. 1 and 2). ATM-depleted cells and LacZ-transduced control cells were incubated with colcemid for 0-6 h with DMSO or 4 μM ICRF-193 and harvested for quantification of mitotic cells by flow cytometry. The average percentages of cells evading the ICRF-193-induced G2 arrest are shown (+sd, n=3). B. Western immunoblot showing expression of ATM, phospho-ATM, topo IIα and topo IIβ in human fibroblasts and lymphoblasts. Cells were treated with 0.1% DMSO (D), 4 μM ICRF-193 (I) or 4 μM etoposide (E) for three hours before harvest. Equal amounts of cell protein were loaded for electrophoretic separation and immunoblot analysis. ICRF-193 induced phosphorylation of ATM in the shLacZ NHDFs, but not the shATM NHDFs.
It has recently been suggested that depletion of topo IIα severely attenuates the ICRF-193-induced decatenation G2 checkpoint (Bower and Kaufmann, unpublished data).47 Thus, it was possible that the attenuation of decatenation G2 checkpoint function in AT cells was related to a reduction in the expression of topo IIα. Western immunoblot analysis confirmed that AT fibroblasts, AT lymphoblasts, and ATM-depleted fibroblasts expressed both topo IIα and topo IIβ (Fig. 6B). Although the levels of topo IIα varied among the cell lines, AT cells displayed similar or higher levels of topo IIα than the NHDFs, indicating that the significant attenuation of decatenation G2 checkpoint function in AT cells and ATM-depleted cells was not associated with reduced expression of topo IIα. However, we cannot rule out the possibility that truncation or depletion of ATM may influence one of the many post-translational modifications found on the topo IIα protein.
Discussion
The results presented here suggest that decatenation G2 checkpoint function is more accurately quantified by measuring the rate of mitotic entry, rather than measuring the mitotic index at a single time point. The mitotic index assay relies upon two events to detect decatenation G2 checkpoint function: G2 arrest and mitotic exit. Given that topo IIα contributes to a decatenation reaction at centromeres that suppresses a MAD2-dependent metaphase checkpoint, 13, 22 treatment with ICRF-193 can induce a metaphase delay that blocks mitotic exit. The mitotic entry rate assay quantifies the rate of G2/M progression, and is not influenced by the ICRF-193-induced metaphase arrest or mitotic exit rates. By applying the improved mitotic entry rate assay, it was clear that ATR and CHEK1 are not required for decatenation G2 checkpoint function, whereas depletion or truncation of ATM significantly attenuated the checkpoint response in NHDFs and lymphoblasts, respectively.
The prior study of ATR function in the decatenation G2 checkpoint utilized an SV40-transformed human fibroblast line with inducible expression of kinase-inactive ATR.18 SV40-transformed human fibroblast lines typically display highly aneuploid genomes due to a phase of severe telomere crisis before establishment of immortality. 49 DNA damage checkpoints acting in G1, S, and G2 may be defective in SV40-transformed and large T antigen-transformed lines. 50, 51 It is possible that the over-expression of kinase-inactive ATR and incubation with ICRF-193 in the SV40-transformed line disrupted centromeric decatenation and induced a metaphase arrest.
The complicated biology of SV40-transformed immortal human cell lines with aneuploid genomes requires that caution should be applied when interpreting features of cell cycle regulation in such immortalized lines. The telomerase-expressing diploid human fibroblast lines were developed to overcome this problem. Transduction of hTERT to immortalize human fibroblasts generates a cell culture model with extended-lifespan, stable diploid genomes, and retention of all DNA damage checkpoint functions at levels equivalent to those measured in telomerase-negative, parental fibroblast strains.30, 35 Severe depletion of ATR in three diploid normal human fibroblast lines had no effect on ICRF-193-induced G2 arrest (Fig.1 and data not shown). As depletion of protein expression does not always produce the same phenotype as drug-induced over-expression of kinase-inactive mutants, 52 it is conceivable that over-expression of the kinase-inactive ATR allele produced biochemical alterations within SV40-transformed cells that affected mitotic entry or exit after treatment with ICRF-193.
The prior analysis in DT40 cells showed that 1 μM ICRF-193 reduced the rate of accumulation of mitotic cells during incubation with nocodazole by 15% relative to control but, in cells with genetic depletion of CHEK1, ICRF-193 had no effect on mitotic accumulation.43 The DT40 cells are known to display defects in DNA damage G1 checkpoint function, 53 and their modest response to ICRF-193 suggests they may express a defective decatenation G2 checkpoint. In diploid human fibroblast lines, 0.5 μM ICRF-193 reduced the rate of mitotic entry by 75% relative to control (Fig. 1D) and 4 μM produced a 97-99% inhibition. Fibroblasts depleted of CHEK1 responded to 4 μM ICRF-193 with a 97% reduction in the mitotic entry rate, indicating that CHEK1 was not required for decatenation G2 checkpoint function. Equivalent depletion of CHEK1 in normal fibroblasts produced a severe attenuation of the intra-S checkpoint response to UV irradiation, 54 demonstrating that the degree of depletion of CHEK1 protein was sufficient to inhibit DNA damage checkpoint signaling.
The prior studies of ATM and decatenation G2 checkpoint function used either lymphoblastoid lines with the mitotic index assay, 18 or an SV40-transformed AT fibroblast line with the mitotic entry rate assay. 40 The telomerase-expressing AT fibroblast lines display canonical defects in DNA damage checkpoint functions, hypersensitivity to ionizing radiation, 36 and significantly less mitotic inhibition and G2 delay than normal fibroblast lines when treated with ICRF-193 (Fig. 3). In addition, AT lymphoblastoid and ATM-depleted NHF1hTERTs displayed similar defects in decatenation G2 checkpoint function (Fig. 4A and Fig. 6A). Taken together, the biological data presented here strongly support the hypothesis that ATM contributes to decatenation G2 checkpoint function.
A few studies have questioned the existence of the decatenation G2 checkpoint. One study showed that high concentrations of ICRF-193 caused Indian muntjac cells first to delay in an early phase of mitosis called antephase and then to fall back into a G2–like state.55 ICRF-193-treated muntjac cells also expressed γH2AX as a putative marker of DNA dsbs, 55 although recent studies suggest that γH2AX may not be specific for DNA dsbs. 56, 57 Another study examined DNA damage response markers in diploid human fibroblasts and HeLa cells after treatment with ICRF-193. 46 Elements of ATM-dependent DNA damage response were recognized, including activation of CHEK2 and expression of γH2AX. The demonstration of γH2AX in ICRF-193-treated cells has not been reproducible, and HeLa cells examined by the mitotic entry rate assay display an attenuated decatenation G2 checkpoint (Bower and Kaufmann, unpublished data). In addition, Skoufias et al. found that while both IR and etoposide induced γH2AX in HeLa cells as expected, ICRF-193 did not,13 and neither Nakagawa et al. nor Luo et al. were able to detect γH2AX in human cancer lines after treatment with ICRF-193. 28, 47 The ultrasensitive method of alkaline elution chromatography did not detect DNA damage in ICRF-193-treated diploid human fibroblasts, 17 and we have been unable to detect appreciable activation of γH2AX in ICRF-193-treated NHDFs by flow cytometry or immunofluorescence (Bower and Kaufmann, in preparation). However, cells that evaded the decatenation G2 checkpoint after treatment with ICRF-193 displayed chromosomal aberrations, 26 which may be recognized as DNA dsbs upon mitotic exit and return to interphase.
The analysis of DNA damage response biomarkers reported here indicated that ATM signaling was induced by ICRF-193. Remarkably, treatment with ICRF-193 induced an ATM-dependent phosphorylation of p53 and CHEK2, but not H2AX. This result is reminiscent of recent reports showing that the topo II catalytic inhibitors chloroquine and ciprofloxacin can activate ATM without phosphorylation of H2AX. 33, 34 Thus, it appears that catalytic inhibitors of topo II may induce a conformational change in chromatin that activates ATM. This method of activation of ATM differs from the response to DNA dsbs, as γH2AX is not induced.
It has been recently demonstrated that Ser1524 in topo IIα is phosphorylated in HT1080 fibrosarcoma cells, and this phosphorylation is required for ICRF-193-induced G2 arrest. 47 Cells in which endogenous topo IIα was replaced with ectopic topo IIα containing a non-phosphorylatable alanine at this position failed to arrest in G2 when treated with ICRF-193, and entered mitosis with severely entangled sister chromatids. The phospho-Ser1524 in topo IIα was bound by the BRCT repeat of the checkpoint mediator protein MDC1 and this binding was stimulated by inhibition of topo IIα with ICRF-193. Furthermore, depletion of MDC1 attenuated the ICRF-193-induced G2 arrest. It is interesting to note that MDC1 is phosphorylated by ATM and serves to recruit ATM to γH2AX on chromatin at the sites of DNA dsbs. 58 As ICRF-193 does not induce γH2AX, MDC1 may recruit ATM to catalytically inactive topo IIα, upon exposure to ICRF-193, chloroquine, and/or ciprofloxacin.
Materials and Methods
Cell lines and cell culture
To avoid idiosyncratic biological responses in cell culture models, three lines of NHDFs (NHF1hTERT, NHF3hTERT, and NHF10hTERT) and three lines of AT fibroblasts (designated AT1, AT2 and AT3) were analyzed. The properties of these lines have been described. 35, 36 NHDFs were isolated from neonatal foreskin and immortalized by the overexpression of hTERT as described previously.35 Each fibroblast line was isolated from a different individual and each experiment was performed in all three NHDF or AT fibroblast lines to assess biological reproducibility as well as inter-individual genetic variation. The three telomerase-expressing normal lines displayed diploid karyotypes without chromosomal aberrations and produced highly stereotypic patterns of response to DNA damage, with effective DNA damage G1 and G2 checkpoints. 35 The three telomerase-expressing AT lines displayed marker chromosomal aberrations but retained near diploid karyotypes. The AT lines also displayed significant defects in DNA damage G1 and G2 checkpoint responses to ionizing radiation-induced DNA dsbs. 36 Human lymphoblastoid lines were obtained from the Coriell Institute including two lines from AT patients (GM3189 and GM9582) and one line from a normal healthy donor (GM3714).37 The NHF1hTERT fibroblast line with stable expression of ATM shRNA or LacZ shRNA were a kind gift from Richard Paules and were established as described.38 NHDF and AT fibroblast lines were grown in DMEM (Invitrogen, Carlsbad, CA) and supplemented with 15% FBS (Sigma, St. Louis, MO) and 2 mM L-Glutamine (Invitrogen, Carlsbad, CA). Lymphoblastoid lines were grown in RPMI medium and supplemented with 15% FBS. Cultures were maintained at 5% CO2 and 37°C. All experiments were performed on fibroblast cultures that had undergone less than 100 population doublings. Periodic tests for mycoplasma contamination using a commercial kit (Gen-Probe, San Diego, CA) were negative.
Depletion of ATR and CHEK1 by siRNA
The non-targeting control (NTC) siRNA sequence directed towards the firefly luciferase mRNA, the ATR siRNA SMARTpool, and the CHEK1 siRNA were obtained from Dharmacon (Dharmacon, Inc., Lafayette, CO). NHDFs were electroporated using an NHDF Nucleofector Kit (Amaxa, Inc., Gaithersburg, MD), and allowed to recover for 48 hours before assaying the biological effects, when depletion of the target protein was maximal. Western immunoblot analysis was used to confirm protein depletion for each individual experiment at the time of assay.
Mitotic inhibition assay of decatenation G2 checkpoint
Asynchronous cultures of diploid human fibroblasts were treated briefly (15 min) with 4 μM ICRF-193 then incubated in drug-free reserved medium for 2 h before cell harvest.27 Mitotic cells were quantified by flow cytometry using a primary mouse monoclonal antibody to mitosis-associated phospho-histone H3 and a fluorescein isothiocyanate-labeled secondary rabbit antibody to the mouse primary antibody. 39
Mitotic entry assay of decatenation G2 checkpoint
Electroporated fibroblasts were plated at a density of 106 cells per 10 cm2 dish (day 0), and fed with fresh medium on day 1. On day 2, cells were treated with 100 ng/mL colcemid (to collect cells in mitosis), and either 0.1% DMSO or 4 μM ICRF-193 for 2, 4, and 6 hours. Fibroblasts were then trypsinized into a single cell suspension and fixed with 95% ethanol: 5% acetic acid overnight at 4° C. An anti-phospho Ser 10 histone H3 primary antibody (Millipore, Billerica, MA) and an anti-mouse FITC labeled secondary (Santa Cruz Biotechnology, Santa Cruz, CA) were used to identify mitotic cells. Propidium iodide (Sigma, St. Louis, MO) was used to measure DNA content. Samples were measured on a Dako CyAN ADP instrument at the Flow Cytometry Core Facility at UNC-CH. Flow cytometry samples were analyzed using Summit 4.3 plots to quantify the percentage of fibroblasts with 4N DNA content that also were labeled with phospho-Ser10 histone H3, a specific marker of mitosis. The percentage of mitotic cells for each sample was plotted against time and the resulting slope of the line was used to measure the rate of entry into mitosis (% of fibroblasts entering mitosis per hour).
Western immunoblot analysis
Antibodies were obtained that recognized phospho-Ser1981 ATM (Epitomics, Burlingame, CA or Rockland Immunochemicals, Gilbertsville, PA), phospho-Ser139 H2AX (Millipore, Billerica, MA), phospho-Ser345 CHEK1 (Cell Signaling Technology, Danvers, MA), phospho-Ser15 p53 (Cell Signaling Technology, Danvers, MA), ATR (Santa Cruz, Santa Cruz, CA), total ATM (Bethyl, Montgomery, TX), topo IIα (BD Biosciences, San Jose, CA), topoII β (BD Biosciences, San Jose, CA), and phospho-Thr68 CHEK2 (Cell Signaling Technology, Danvers, MA). Conditions for protein extraction, polyacrylamide gel electrophoresis, and immunoblot analysis using enhanced chemiluminescence and X-ray film exposure were as previously described. 39, 41 For histone analyses, cells were heated to 100°C in gel-loading buffer before electrophoresis. For fluorescence detection of western immunoblots, a secondary anti-mouse Cy3or Cy5 labeled antibody was obtained from GE Healthcare (Piscataway, NJ) and fluorescence was measured on a Typhoon 9400 (GE Healthcare, Piscataway, NJ). Pixel intensity values from scanned X-ray film or fluorescence intensities were used to quantify protein depletion by siRNA. 42
Acknowledgments
Supported in part by PHS grants CA81343, ES10126, ES07017, and by the Intramural Research Program of the National Institute of Environmental Health Sciences, NIH (SJA and RSP).
Abbreviations
- NHDFs
normal human diploid fibroblasts
- topo II
topoisomerase II
- ATM
ataxia telangiectasia mutated kinase
- ATR
ATM and Rad3-related kinase
- CHEK1
checkpoint kinase 1
- CHEK2
checkpoint kinase 2
References
- 1.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 2.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 3.Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1992;71:543–6. doi: 10.1016/0092-8674(92)90586-2. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann WK, Kaufman DG. Cell cycle control, DNA repair and initiation of carcinogenesis. Faseb J. 1993;7:1188–91. doi: 10.1096/fasebj.7.12.8375618. [DOI] [PubMed] [Google Scholar]
- 5.Roca J, Berger JM, Harrison SC, Wang JC. DNA transport by a type II topoisomerase: direct evidence for a two- gate mechanism. Proc Natl Acad Sci U S A. 1996;93:4057–62. doi: 10.1073/pnas.93.9.4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lang AJ, Mirski SE, Cummings HJ, Yu Q, Gerlach JH, Cole SP. Structural organization of the human TOP2A and TOP2B genes. Gene. 1998;221:255–66. doi: 10.1016/s0378-1119(98)00468-5. [DOI] [PubMed] [Google Scholar]
- 7.de Toledo SM, Azzam EI, Keng P, Laffrenier S, Little JB. Regulation by ionizing radiation of CDC2, cyclin A, cyclin B, thymidine kinase, topoisomerase IIalpha, and RAD51 expression in normal human diploid fibroblasts is dependent on p53/p21Waf1. Cell Growth Differ. 1998;9:887–96. [PubMed] [Google Scholar]
- 8.Zhu H, Chang BD, Uchiumi T, Roninson IB. Identification of Promoter Elements Responsible for Transcriptional Inhibition of Polo-like Kinase 1 and Topoisomerase IIalpha Genes by p21(WAF1/CIP1/SDI1) Cell Cycle. 2002;1:59–66. [PubMed] [Google Scholar]
- 9.Zhou T, Chou J, Mullen TE, Elkon R, Zhou Y, Simpson DA, Bushel PR, Paules RS, Lobenhofer EK, Hurban P, Kaufmann WK. Identification of primary transcriptional regulation of cell cycle-regulated genes upon DNA damage. Cell Cycle. 2007;6:972–81. doi: 10.4161/cc.6.8.4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akimitsu N, Adachi N, Hirai H, Hossain MS, Hamamoto H, Kobayashi M, Aratani Y, Koyama H, Sekimizu K. Enforced cytokinesis without complete nuclear division in embryonic cells depleting the activity of DNA topoisomerase IIalpha. Genes Cells. 2003;8:393–402. doi: 10.1046/j.1365-2443.2003.00643.x. [DOI] [PubMed] [Google Scholar]
- 11.Clarke DJ, Johnson RT, Downes CS. Topoisomerase II inhibition prevents anaphase chromatid segregation in mammalian cells independently of the generation of DNA strand breaks. J Cell Sci. 1993;105:563–9. doi: 10.1242/jcs.105.2.563. [DOI] [PubMed] [Google Scholar]
- 12.Carpenter AJ, Porter AC. Construction, characterization, and complementation of a conditional-lethal DNA topoisomerase IIalpha mutant human cell line. Mol Biol Cell. 2004;15:5700–11. doi: 10.1091/mbc.E04-08-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skoufias DA, Lacroix FB, Andreassen PR, Wilson L, Margolis RL. Inhibition of DNA decatenation, but not DNA damage, arrests cells at metaphase. Mol Cell. 2004;15:977–90. doi: 10.1016/j.molcel.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Diaz-Martinez LA, Gimenez-Abian JF, Azuma Y, Guacci V, Gimenez-Martin G, Lanier LM, Clarke DJ. PIASgamma is required for faithful chromosome segregation in human cells. PLoS ONE. 2006;1:e53. doi: 10.1371/journal.pone.0000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H, Wang Y, Liu X. Plk1-dependent phosphorylation regulates functions of DNA topoisomerase IIalpha in cell cycle progression. J Biol Chem. 2008;283:6209–21. doi: 10.1074/jbc.M709007200. [DOI] [PubMed] [Google Scholar]
- 16.Downes CS, Clarke DJ, Mullinger AM, Gimenez-Abian JF, Creighton AM, Johnson RT. A topoisomerase II-dependent G2 cycle checkpoint in mammalian cells/[published erratum appears in Nature 1994 Dec 15;372(6507):710] Nature. 1994;372:467–70. doi: 10.1038/372467a0. [DOI] [PubMed] [Google Scholar]
- 17.Kaufmann WK, Kies PE. DNA signals for G2 checkpoint response in diploid human fibroblasts. Mutation Research. 1998;400:153–67. doi: 10.1016/s0027-5107(98)00041-4. [DOI] [PubMed] [Google Scholar]
- 18.Deming PB, Cistulli CA, Zhao H, Graves PR, Piwnica-Worms H, Paules RS, Downes CS, Kaufmann WK. The human decatenation checkpoint. Proc Natl Acad Sci U S A. 2001;98:12044–9. doi: 10.1073/pnas.221430898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tanabe K, Ikegami Y, Ishida R, Andoh T. Inhibition of topoisomerase II by antitumor agents bis(2,6- dioxopiperazine) derivatives. Cancer Res. 1991;51:4903–8. [PubMed] [Google Scholar]
- 20.Olland S, Wang JC. Catalysis of ATP hydrolysis by two NH(2)-terminal fragments of yeast DNA topoisomerase II. J Biol Chem. 1999;274:21688–94. doi: 10.1074/jbc.274.31.21688. [DOI] [PubMed] [Google Scholar]
- 21.Jensen LH, Nitiss KC, Rose A, Dong J, Zhou J, Hu T, Osheroff N, Jensen PB, Sehested M, Nitiss JL. A novel mechanism of cell killing by anti-topoisomerase II bisdioxopiperazines. J Biol Chem. 2000;275:2137–46. doi: 10.1074/jbc.275.3.2137. [DOI] [PubMed] [Google Scholar]
- 22.Toyoda Y, Yanagida M. Coordinated requirements of human topo II and cohesin for metaphase centromere alignment under Mad2-dependent spindle checkpoint surveillance. Mol Biol Cell. 2006;17:2287–302. doi: 10.1091/mbc.E05-11-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Spence JM, Phua HH, Mills W, Carpenter AJ, Porter AC, Farr CJ. Depletion of topoisomerase II{alpha} leads to shortening of the metaphase interkinetochore distance and abnormal persistence of PICH-coated anaphase threads. J Cell Sci. 2007;120:3952–64. doi: 10.1242/jcs.013730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang LH, Schwarzbraun T, Speicher MR, Nigg EA. Persistence of DNA threads in human anaphase cells suggests late completion of sister chromatid decatenation. Chromosoma. 2008;117:123–35. doi: 10.1007/s00412-007-0131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawlaty MM, Malureanu L, Jeganathan KB, Kao E, Sustmann C, Tahk S, Shuai K, Grosschedl R, van Deursen JM. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell. 2008;133:103–15. doi: 10.1016/j.cell.2008.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deming PB, Flores KG, Downes CS, Paules RS, Kaufmann WK. ATR enforces the topoisomerase II-dependent G2 checkpoint through inhibition of Plk1 kinase. J Biol Chem. 2002;277:36832–8. doi: 10.1074/jbc.M206109200. [DOI] [PubMed] [Google Scholar]
- 27.Doherty SC, McKeown SR, McKelvey-Martin V, Downes CS, Atala A, Yoo JJ, Simpson DA, Kaufmann WK. Cell cycle checkpoint function in bladder cancer. J Natl Cancer Inst. 2003;95:1859–68. doi: 10.1093/jnci/djg120. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Hayashita Y, Maeno K, Masuda A, Sugito N, Osada H, Yanagisawa K, Ebi H, Shimokata K, Takahashi T. Identification of decatenation G2 checkpoint impairment independently of DNA damage G2 checkpoint in human lung cancer cell lines. Cancer Res. 2004;64:4826–32. doi: 10.1158/0008-5472.CAN-04-0871. [DOI] [PubMed] [Google Scholar]
- 29.Paules RS, Levedakou EN, Wilson SJ, Innes CL, Rhodes N, Tlsty TD, Galloway DA, Donehower LA, Tainsky MA, Kaufmann WK. Defective G2 checkpoint function in cell from individuals with familial cancer syndromes. Cancer Research. 1995;55:1763–73. [PubMed] [Google Scholar]
- 30.Kaufmann WK, Schwartz JL, Hurt JC, Byrd LL, Galloway DA, Levedakou E, Paules RS. Inactivation of G2 checkpoint function and chromosomal destabilization are linked in human fibroblasts expressing human papillomavirus type 16 E6. Cell Growth & Differentiation. 1997;8:1105–14. [PubMed] [Google Scholar]
- 31.Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 32.Snyder RD. Use of catalytic topoisomerase II inhibitors to probe mechanisms of chemical-induced clastogenicity in Chinese hamster V79 cells. Environ Mol Mutagen. 2000;35:13–21. [PubMed] [Google Scholar]
- 33.Loehberg CR, Thompson T, Kastan MB, Maclean KH, Edwards DG, Kittrell FS, Medina D, Conneely OM, O'Malley BW. Ataxia telangiectasia-mutated and p53 are potential mediators of chloroquine-induced resistance to mammary carcinogenesis. Cancer Res. 2007;67:12026–33. doi: 10.1158/0008-5472.CAN-07-3058. [DOI] [PubMed] [Google Scholar]
- 34.Smart DJ, Halicka HD, Traganos F, Darzynkiewicz Z, Williams GM. Ciprofloxacin-induced G(2) arrest and apoptosis in TK6 lymphoblastoid cells is not dependent on DNA double-strand break formation. Cancer Biol Ther. 2007;7 doi: 10.4161/cbt.7.1.5136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou T, Chou JW, Simpson DA, Zhou Y, Mullen TE, Medeiros M, Bushel PR, Paules RS, Yang X, Hurban P, Lobenhofer EK, Kaufmann WK. Profiles of global gene expression in ionizing-radiation-damaged human diploid fibroblasts reveal synchronization behind the G1 checkpoint in a G0-like state of quiescence. Environ Health Perspect. 2006;114:553–9. doi: 10.1289/ehp.8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou T, Chou J, Zhou Y, Simpson DA, Cao F, Bushel PR, Paules RS, Kaufmann WK. Ataxia telangiectasia-mutated dependent DNA damage checkpoint functions regulate gene expression in human fibroblasts. Mol Cancer Res. 2007;5:813–22. doi: 10.1158/1541-7786.MCR-07-0104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Innes CL, Heinloth AN, Flores KG, Sieber SO, Deming PB, Bushel PR, Kaufmann WK, Paules RS. ATM Requirement in Gene Expression Responses to Ionizing Radiation in Human Lymphoblasts and Fibroblasts. Mol Cancer Res. 2006;4:197–207. doi: 10.1158/1541-7786.MCR-05-0154. [DOI] [PubMed] [Google Scholar]
- 38.Arlander SJ, Greene BT, Innes CL, Paules RS. DNA protein kinase-dependent G2 checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res. 2008;68:89–97. doi: 10.1158/0008-5472.CAN-07-0675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaufmann WK, Heffernan TP, Beaulieu LM, Doherty S, Frank AR, Zhou Y, Bryant MF, Zhou T, Luche DD, Nikolaishvili-Feinberg N, Simpson DA, Cordeiro-Stone M. Caffeine and human DNA metabolism: the magic and the mystery. Mutat Res. 2003;532:85–102. doi: 10.1016/j.mrfmmm.2003.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kaufmann WK, Campbell CB, Simpson DA, Deming PB, Filatov L, Galloway DA, Zhao XJ, Creighton AM, Downes CS. Degradation of ATM-independent decatenation checkpoint function in human cells is secondary to inactivation of p53 and correlated with chromosomal destabilization. Cell Cycle. 2002;1:210–9. [PubMed] [Google Scholar]
- 41.Heffernan TP, Unsal-Kacmaz K, Heinloth AN, Simpson DA, Paules RS, Sancar A, Cordeiro-Stone M, Kaufmann WK. Cdc7-Dbf4 and the human S checkpoint response to UVC. J Biol Chem. 2007;282:9458–68. doi: 10.1074/jbc.M611292200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaufmann WK, Nevis KR, Qu P, Ibrahim JG, Zhou T, Zhou Y, Simpson DA, Helms-Deaton J, Cordeiro-Stone M, Moore DT, Thomas NE, Hao H, Liu Z, Shields JM, Scott GA, Sharpless NE. Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J Invest Dermatol. 2008;128:175–87. doi: 10.1038/sj.jid.5700935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robinson HM, Bratlie-Thoresen S, Brown R, Gillespie DA. Chk1 is required for G2/M checkpoint response induced by the catalytic topoisomerase II inhibitor ICRF-193. Cell Cycle. 2007;6:1265–7. doi: 10.4161/cc.6.10.4225. [DOI] [PubMed] [Google Scholar]
- 44.Jha MN, Bamburg JR, Bedford JS. Cell cycle arrest by Colcemid differs in human normal and tumor cells. Cancer Res. 1994;54:5011–5. [PubMed] [Google Scholar]
- 45.Roca J, Ishida R, Berger JM, Andoh T, Wang JC. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci U S A. 1994;91:1781–5. doi: 10.1073/pnas.91.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park I, Avraham HK. Cell cycle-dependent DNA damage signaling induced by ICRF-193 involves ATM, ATR, CHK2, and BRCA1. Exp Cell Res. 2006;312:1996–2008. doi: 10.1016/j.yexcr.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 47.Luo K, Yuan J, Chen J, Lou Z. Topoisomerase IIalpha controls the decatenation checkpoint. Nat Cell Biol. 2009;11:204–10. doi: 10.1038/ncb1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao H, Mao Y, Desai SD, Zhou N, Ting CY, Hwang J, Liu LF. The topoisomerase IIbeta circular clamp arrests transcription and signals a 26S proteasome pathway. Proc Natl Acad Sci U S A. 2003;100:3239–44. doi: 10.1073/pnas.0736401100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ducray C, Pommier JP, Martins L, Boussin FD, Sabatier L. Telomere dynamics, end-to-end fusions and telomerase activation during the human fibroblast immortalization process. Oncogene. 1999;18:4211–23. doi: 10.1038/sj.onc.1202797. [DOI] [PubMed] [Google Scholar]
- 50.Bullock SK, Kaufmann WK, Cordeiro-Stone M. Enhanced S phase delay and inhibition of replication of an undamaged shuttle vector in UVC-irradiated xeroderma pigmentosum variant. Carcinogenesis. 2001;22:233–41. doi: 10.1093/carcin/22.2.233. [DOI] [PubMed] [Google Scholar]
- 51.Kaufmann WK, Levedakou EN, Grady HL, Paules RS, Stein GH. Attenuation of G2 checkpoint function precedes human cell immortalization. Cancer Res. 1995;55:7–11. [PubMed] [Google Scholar]
- 52.Anantha RW, Sokolova E, Borowiec JA. RPA phosphorylation facilitates mitotic exit in response to mitotic DNA damage. Proc Natl Acad Sci U S A. 2008;105:12903–8. doi: 10.1073/pnas.0803001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sonoda E, Sasaki MS, Buerstedde JM, Bezzubova O, Shinohara A, Ogawa H, Takata M, Yamaguchi-Iwai Y, Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. Embo J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen B, Simpson DA, Zhou Y, Mitra A, Mitchell DL, Cordeiro-Stone M, Kaufmann WK. Human papilloma virus type16 E6 deregulates CHK1 and sensitizes human fibroblasts to environmental carcinogens independently of its effect on p53. Cell Cycle. 2009;8:1775–87. doi: 10.4161/cc.8.11.8724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mikhailov A, Shinohara M, Rieder CL. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J Cell Biol. 2004;166:517–26. doi: 10.1083/jcb.200405167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hunt CR, Pandita RK, Laszlo A, Higashikubo R, Agarwal M, Kitamura T, Gupta A, Rief N, Horikoshi N, Baskaran R, Lee JH, Lobrich M, Paull TT, Roti Roti JL, Pandita TK. Hyperthermia activates a subset of ataxia-telangiectasia mutated effectors independent of DNA strand breaks and heat shock protein 70 status. Cancer Res. 2007;67:3010–7. doi: 10.1158/0008-5472.CAN-06-4328. [DOI] [PubMed] [Google Scholar]
- 57.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc Natl Acad Sci U S A. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]