

Abstract

Functionalized diketopiperazines (a.k.a. dioxopiperazines) are an important class of molecules in medicinal chemistry and material science. Herein we report a diastereoselective synthesis of diketopiperazine bis-α,β-epoxides via the oxidation of exocyclic olefins. Although six diastereomers may be formed by this approach, only one or two of them were observed.
2,5-Diketopiperazines (also known as piperazine-2,5-diones and 2,5-dioxopiperazines) are cyclic dipeptides and part of many biologically active natural products that include polythiodiketopiperazines MPC1001s,1,2 emestrin,3 and bionectin,4 among others.5 The 2,5-diketopiperazine has been an important scaffold in material science6–8 and medicinal chemistry9,10 and is often utilized for biological properties associated with its mimicry of peptidic pharmacophores, conformational rigidity, hydrogen bond donor and acceptor groups, and resistance to proteolysis in vivo.11–13 The structural complexity and the promising biological activities associated with such diketopiperazines continues to inspire synthetic chemists.
While 2,5-diketopiperazine has been the subject of synthetic studies dating back to the 1900s by Emil Fischer et al.,14 it has been studied more intensively in recent years. Typically, chiral amino acid derivatives are dimerized and cyclized, allowing for substitution at the C3 and C6 positions.13,15 As a means to introduce desired substituents at the 3 and 6 positions of the 2,5-diketopiperazines, a majority of synthetic studies have been devoted to the elimination-nucleophilic attack of compounds A in the presence of nucleophiles, often under acidic conditions, to form B (Scheme 1a).16 This approach was employed in the synthetic studies of diketopiperazine natural products,17–21 including the landmark total synthesis by Kishi and his coworkers.22
SCHEME 1.

(a) Approach employed by others to access substituted diketopiperazines. (b) Our proposed approach.
We became interested in alternative approaches toward functionalizing 2,5-diketopiperazines (Scheme 1b). Monoepoxide variants of C have been previously reported as a means to further elaborate the DKP framework.23–25 Our group set out to establish a method for accessing bis-epoxides C, which we thought would serve as a precursor to more complex DKP’s. Specifically, we wished to exploit the dual reactivity of bis-epoxides C under acidic and basic conditions to control the regioselectivity of the subsequent ring-opening. We envisioned that under acidic conditions, compounds D could be formed from C via acyliminium ion intermediates similarly to Scheme 1a. Under basic conditions, compounds E could be formed by nucleophilic attack at the less-hindered carbons of compounds C. In order to test this hypothesis, it was necessary to synthesize compounds C. Herein, we report a diastereoselective method to prepare a variety of bis-epoxides C using a two-step bromohydration-etherification pathway. As we will describe in a separate report in due course, these bis-epoxides are pivotal intermediates for complex 2,5-diketopiperazines.
The attempted condensation of piperazine-2,5-dione 1 with benzaldehyde under various conditions did not yield the corresponding aldol product 5a, which is consistent with the literature (Scheme 2).26 However, the N-acetylated derivative 2 could be coupled with benzaldehyde under basic conditions (Et3N in DMF, 120 °C) to form alkene 5a in 72% yield.26 Other aromatic aldehydes also reacted smoothly to form 5b–5h in 52–96% yields (Table 1). The acetyl group drove the otherwise thermodynamically unfavorable aldol reaction to completion by the aldol addition-acetyl migration-elimination cascade (2 to 5 via 3 and 4) to form exclusively cis products as shown in Scheme 2.26 One plausible explanation for the cis selectivity is an E1cb pathway through the transition state 8β. An alternative transition state, 8α, is presumably disfavored due to the steric repulsion between the ion pair and the aromatic ring. This mechanism would mitigate an undesired stereospecific E2 pathway, accounting for high yields in some cases. As has been observed with similar diketopiperazines, the alkene intermediates 5a–h were readily crystallized and exhibited extraordinarily poor solubility in a variety of protic and aprotic solvents.27–29 This is presumably due to the combination of intermolecular hydrogen bonding and π-π stacking capabilities, which promote tight crystal packing.29,30 Due to the poor solubility, the products from this reaction were crystallized and used in the following step without 13C-NMR analysis.
SCHEME 2.

Preparation of compounds 6a–h.
Conditions: (i) Ac2O (neat), reflux, 5 h, 90%. (ii) ArCHO (2.5 equiv), Et3N (3.0 equiv), DMF, 120 °C, 7–17 h, see Table 1 for isolated yields. (iii) MeI (4.0 equiv), NaH (for 5a–f) or K2CO3 (for 5g and 5h) (3.0 equiv), DMF, 25 °C, 12 h, see Table 1 for isolated yields.
Table 1.
Isolated yields for the formation of compounds 5 and 6.
| Product | Yield (%) | Product | Yield (%) |
|---|---|---|---|
| 5a | 72 | 6a | 60 |
| 5b | 52 | 6b | 61 |
| 5c | 68 | 6c | 89 |
| 5d | 56 | 6d | 78 |
| 5e | 88 | 6e | 45 |
| 5f | 96 | 6f | 46 |
| 5g | 54 | 6g | 60 |
| 5h | 68 | 6h | 50 |
We have been interested in the MPC1001-class of natural products, all of which are N-alkylated in the diketopiperazine rings.1,2 Thus, we proceeded to methylate the secondary amides 5a–h with MeI and NaH to form the tertiary amides 6a–h in 45–89% yields (Table 1).31 The most notable side products from this reaction were the result of O-methylation to form highly conjugated imidates, as represented by 7h. These O-methylated byproducts were significantly more hydrophobic than their N-methylated counterparts and could be readily separated by silica gel chromatography.
With the N-methylated intermediates in hand, our studies continued with an examination of epoxidation conditions for the phenyl-substituted compound 6a. The use of both nucleophilic and electrophilic reagents such as m-CPBA,25 DMDO,24 H2O2/NaOH, and TBHP/VO(acac)2 either showed no reaction or resulted in mono-epoxidation. The desired bis-epoxidation could be accomplished by means of a two-step sequence involving bromohydration with NBS and H2O, followed by epoxide ring closing of the crude bromohydrin under basic conditions.23 Of the six potential diastereomeric epoxides (Scheme 3), only two were observed for the phenyl-substituted analogue. The X-ray crystallographic analysis of these two compounds (Figures S54 and S55 in the Supporting Information) showed that they were the C2-symmetric compound S,S,S,S-9a (and its enantiomer) and the asymmetric compound S,S,S,R-9a (and its enantiomer). The two-step epoxidation strategy was applied to the substrates shown in Table 2. For most substrates, the major product was the C2-symmetric diastereomer S,S,S,S-9 (and its enantiomer) rather than the asymmetric diastereomer S,S,S,R-9 (and their enantiomers). Scheme S1 in the Supporting Information shows the compounds that did not provide the desired bis-epoxides.
SCHEME 3.

Control experiments to examine the reversibility of the 2-step sequence. (a) Possible bromide-promoted conversion of S,S,S,R-9a to S,S,S,S-9a. (b) Possible base-catalyzed epimerization between syn-F and anti-F.
Table 2.
Observed diastereoselectivity in the bis-epoxidation of 6.
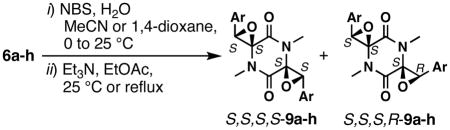 | ||||
|---|---|---|---|---|
| Product | Solvent (step i) | Temp (step ii) | Yield (%)a | |
| SSSS | SSSR | |||
| 9a | MeCN | 25 °C | 34 | 60 |
| 9b | 1,4-dioxane | 25 °C | 62 | <1 |
| 9c | 1,4-dioxane | reflux | 56 | <1 |
| 9d | MeCN | reflux | 51 | <1 |
| 9e | MeCN | 25 °C | 54 | 24 |
| 9fb | MeCN | 25 °C | 6 | 55 |
| 9g | MeCN | 25 °C | 75 | <10 (crude) |
| 9h | MeCN | 25 °C | 42 | 37 |
Isolated yield.
Isolated as a mixture of S,S,S,S- and S,S,S,R-9.
The relative stereochemistry of S,S,S,S-9b-h was determined by 1H NMR spectroscopic analysis. All of these compounds showed one peak for the methine hydrogen atoms, indicating C2 symmetry (S,S,S,S-9b-h or S,R,R,S-9b-h, Figure 1). All of these compounds also exhibited strong NOE signals between the N-methyl groups and methine protons, indicating the cis relationship between them. The only structures that could be consistent with these data were S,S,S,S-9b-h.
Figure 1.

Structures of the six possible bis-epoxide enantiomers.
The relative stereochemistry of S,S,S,R-9b–h was determined in a slightly different manner. These compounds showed a significant difference on the chemical shift of the two N-methyl groups (Δδ = ~0.7 ppm in all of the cases; Table S1, Supporting Information), indicating that the two N-methyl groups in each compound were in different proximity to the aromatic rings. This excluded S,R,S,R-9 and S,S,R,R-9. NOE experiments would not distinguish between S,S,S,R-9 and S,S,R,S-9. However, the 1H NMR analysis of a derivative (cf. compound D in Scheme 1b) of compound S,S,S,R-9a revealed that the relative stereochemistry at the two benzylic positions after epoxide opening was opposite.32 All together, the structures that are most consistent with these spectroscopic data are S,S,S,R-9b–h. To further support the structures, we compared the 1H NMR spectra of these compounds with that of S,S,S,R-9a (confirmed by x-ray crystallographic analysis) as shown in Table S1 (Supporting Information).
The observed diastereoselectivity clearly demonstrates that there is some steric or electronic bias directing the stereochemical outcome. Although we cannot rule out the possibility that some of the six possible diastereomers (see Scheme S2 in the Supporting Information) decomposed in a stereospecific manner, it is surprising that only one or two diastereomers were isolated. When the aromatic moieties were phenyl groups, the asymmetric compound S,S,S,R-9a was the major product (60% yield). The minor and symmetric product, S,S,S,S-9a might be more thermodynamically favored because neither of the phenyl groups is proximal to the N-methyl group. Therefore, with the phenyl group, the reaction might be kinetically controlled.
With a π-donor (Br or O) at the ortho or para position (compounds 6b, 6c, 6d, 6e, and 6g), the symmetric compounds S,S,S,S-9 were the major products (Table 2). This suggested that the two-step process might be reversible with these π-donors at the ortho or para position, which would enable isomerization from the asymmetric S,S,S,R-9 to symmetric S,S,S,S-9. The reversibility appeared to stem from the enhanced reactivity of the benzylic epoxides toward Et3NH+Br− (Scheme 3a, S,S,S,R-9a to S,S,R,S-10a and S,S,R,S-10a to S,S,R,R-10a). To test this hypothesis, asymmetric compound S,S,S,R-9a was treated with Et3NH+Br− and Et3N in refluxing MeCN. Under these conditions, no reaction appeared to take place, invalidating our hypothesis, at least when Ar = Ph.
Another mechanistic possibility was that Et3N-catalyzed epimerizations of carbinol carbon stereocenters were involved in the stereocontrol (Scheme 3b). Thus, we treated 12a with Ag2O in the absence of base. In this reaction, bromide ions should be sequestered from the reaction mixture as insoluble AgBr, excluding the transformations shown in Scheme 3a. Under these conditions, S,S,S,S-9a and S,S,S,R-9a were formed in a 1:2 ratio.33 Because the Ag2O- and Et3N-promoted reactions provided the two epoxides in similar ratios, the origin of diastereoselectivity using Et3N is unlikely to involve the aforementioned epimerizations.
The preference for a syn relationship between the two hydroxy groups is a well-documented phenomenon in the synthesis of similar diketopiperazines.18,34,35 Therefore, our working hypothesis is that syn diols are the reactive intermediates after the bromohydration step. Because SN2 reactions are stereospecific, the origin of stereoselectivity would be the formation of bromohydrins 12 (Scheme 4). More specifically, it would be during the second bromohydration that the syn- or anti-relationship between the two bromide groups would be established (11 to 12). Further studies are needed to thoroughly understand the origin of the diastereoselectivity in this step.
SCHEME 4.

Possible pathways leading to S,S,S,S-9 and S,S,S,R-9
In this study, we achieved the synthesis of diketopiperazine bis-α,β-epoxides 9 in four steps from inexpensive glycine anhydride 1. The final bromohydration-cyclization sequence favored the formation of the C2-symmetric diastereomer. Although other methods for the preparation of piperazine-2,5-diones have been reported, our route is notable for its ability to produce a variety of aromatic analogues and promises accessibility to more complex α- and β-substituted substrates. Our progress toward achieving the goal depicted in Scheme 1b will be reported in due course.
Experimental Section
Preparation of compounds S,S,S,S-9a and S,S,S,R-9a (racemic)
NBS (78 mg, 0.44 mmol) was added to a suspension of compound 6a (64 mg, 0.20 mmol) in H2O/MeCN (2.2 mL of 1:10 mixture) while on an ice bath with stirring. The resulting mixture was then allowed to warm to 25 °C and stirred for 18 h at the same temperature. EtOAc (4.0 mL) was added to the reaction mixture, and the resulting solution was dried over Na2SO4, filtered, and concentrated under reduced pressure. The mixture of diastereomers was used for the next step. The crude residue was dissolved in EtOAc (2.0 mL), and Et3N (0.20 mL, 1.4 mmol) was added to the resulting mixture at 25 °C under a nitrogen atmosphere. After stirring for 23 h at 25 °C, the reaction mixture was filtered to remove the insoluble salts. The filtrate was concentrated under reduced pressure, and the crude residue was purified by flash chromatography (10 to 40% EtOAc in hexanes) on silica gel (12 mL) to afford compound S,S,S,S-9a (racemic mixture, 25 mg, 36%) as a white solid and compound S,S,S,R-9a (racemic mixture, 42 mg, 60%) as a colorless crystalline. To prepare samples for X-ray crystallography, compound S,S,S,S-9a (racemic) was recrystallized from EtOAc, and compound S,S,S,R-9a (racemic) was recrystallized from hexanes-EtOAc. Data for compound S,S,S,S-9a (racemic): m.p. = 140–142 °C; Rf = 0.29 (30% EtOAc in hexanes); IR (KBr pellet): νmax = 3063, 2935, 1694 (C=O), 1455, 1429, 1378, 1271, 1167, 1072, 762, 726 cm−1; 1H NMR (300 MHz, CDCl3, 293 K, Figure S23): δ = 7.49–7.38 (m, 10H), 4.02 (s, 2H), 2.89 (s, 6H); 13C NMR (75 MHz, CDCl3, 293 K, Figure S24): δ = 160.7, 130.4, 129.1, 128.4, 126.8, 71.4, 63.0, 26.3; HRMS (EI+) calcd. for C20H18N2O4 [M]+ 350.1267, found 350.1264. Data for compound S,S,S,R-9a (racemic): m.p. = 179–181 °C; Rf = 0.20 (30% EtOAc in hexanes); IR (KBr pellet): νmax = 3055, 2925, 2854, 1693 (C=O), 1453, 1427, 1373, 1272, 1160, 933, 877, 742 cm−1; 1H NMR (300 MHz, CDCl3, 293 K, Figure S25): δ = 7.57–7.52 (m, 2H), 7.43–7.38 (m, 3H), 7.21 (dd, J = 7.2, 7.2 Hz, 1H), 7.07 (dd, J = 7.5, 7.5 Hz, 2H), 7.02 (dd, J = 7.2, 7.2 Hz, 2H), 4.38 (s, 1H), 4.33 (s, 1H), 3.10 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3, 293 K, Figure S26): δ = 164.3, 160.6, 131.1, 130.5, 129.4, 129.1, 128.8, 128.5, 127.5, 127.0, 71.8, 7.05, 62.8, 62.6, 29.7, 26.5; HRMS (EI+) calcd. for C20H18N2O4 [M]+ 350.1267, found 350.1265.
Supplementary Material
Acknowledgments
This work was supported by the US National Institutes of Health (R01 CA120792). We thank Dr. Damodaran Krishnan, Dr. Steve Geib, Dr. John Williams, and Dr. Bhaskar Godugu for assisting with NMR, X-ray and mass spectroscopic analyses, respectively. A.L.G. is a recipient of the Arts and Sciences Fellowship and the Mary E. Warga Predoctoral Fellowship from the University of Pittsburgh.
Footnotes
Supporting Information Available. Experimental procedures and spectroscopic data for all new compounds, and additional figures, schemes, and X-ray crystal structures are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Onodera H, Hasegawa A, Tsumagari N, Nakai R, Ogawa T, Kanda Y. Org Lett. 2004;6:4101–4104. doi: 10.1021/ol048202d. [DOI] [PubMed] [Google Scholar]
- 2.Tsumagari N, Nakai R, Onodera H, Hasegawa A, Rahayu ES, Ando K, Yamashita Y. J Antibiot. 2004;57:532–534. doi: 10.7164/antibiotics.57.532. [DOI] [PubMed] [Google Scholar]
- 3.Seya H, Nozawa K, Nakajima S, Kawai K-i, Udagawa S-i. J Chem Soc Perkin Trans 1. 1986:109–116. [Google Scholar]
- 4.Zheng CJ, Kim CJ, Bae KS, Kim YH, Kim WG. J Nat Prod. 2006;69:1816–1819. doi: 10.1021/np060348t. [DOI] [PubMed] [Google Scholar]
- 5.Gardiner DM, Waring P, Howlett BJ. Microbiology. 2005;151:1021–1032. doi: 10.1099/mic.0.27847-0. [DOI] [PubMed] [Google Scholar]
- 6.Levins CG, Schafmeister CE. J Am Chem Soc. 2003;125:4702–4703. doi: 10.1021/ja0293958. [DOI] [PubMed] [Google Scholar]
- 7.Brown ZZ, Schafmeister CE. Org Lett. 2010;12:1436–1439. doi: 10.1021/ol100048g. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S, Macala M, Schafmeister CE. J Org Chem. 2006;71:8691–8695. doi: 10.1021/jo0609125. [DOI] [PubMed] [Google Scholar]
- 9.McCleland K, Milne PJ, Lucieto FR, Frost C, Brauns SC, Van De Venter M, Du Plessis J, Dyason K. J Pharm Pharmacol. 2004;56:1143–1153. doi: 10.1211/0022357044139. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Brahimi F, Saragovi HU, Burgess K. J Med Chem. 2010;53:5044–5048. doi: 10.1021/jm100148d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton DA, Bourne GT, Smythe ML. Mol Diversity. 2000;5:289–304. doi: 10.1023/a:1021365402751. [DOI] [PubMed] [Google Scholar]
- 12.Mitova M, Tutino M, Infusini G, Marino G, Rosa S. Mar Biotechnol. 2005;7:523–531. doi: 10.1007/s10126-004-5098-2. [DOI] [PubMed] [Google Scholar]
- 13.Martins MB, Carvalho I. Tetrahedron. 2007;63:9923–9932. [Google Scholar]
- 14.Fischer E, Raske K. Ber Dtsch Chem Ges. 1907;39:3981–3995. [Google Scholar]
- 15.Fischer PM. J Pept Sci. 2003;9:9–35. doi: 10.1002/psc.446. [DOI] [PubMed] [Google Scholar]
- 16.Avendaño C, de la Cuesta E. Curr Org Synth. 2009;6:143–168. [Google Scholar]
- 17.Miknis GF, Williams RM. J Am Chem Soc. 1993;115:536–547. [Google Scholar]
- 18.Kim J, Ashenhurst JA, Movassaghi M. Science. 2009;324:238–241. doi: 10.1126/science.1170777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overman LE, Sato T. Org Lett. 2007;9:5267–5270. doi: 10.1021/ol702518t. [DOI] [PubMed] [Google Scholar]
- 20.Aliev AE, Hilton ST, Motherwell WB, Selwood DL. Tetrahedron Lett. 2006;47:2387–2390. [Google Scholar]
- 21.Kim J, Movassaghi M. J Am Chem Soc. 2010:14376–14378. doi: 10.1021/ja106869s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kishi Y, Fukuyama T, Nakatsuka S. J Am Chem Soc. 1973;95:6492–6493. doi: 10.1021/ja00800a078. [DOI] [PubMed] [Google Scholar]
- 23.Marcuccio SM, Elix JA. Aust J Chem. 1985;38:1785–1796. [Google Scholar]
- 24.Bartels A, Jones PG, Liebscher Jr. Tetrahedron Lett. 1995;36:3673–3674. [Google Scholar]
- 25.Hoare JH, Yates P. J Chem Soc Chem Comm. 1981:1126–1128. [Google Scholar]
- 26.Gallina C, Liberatori A. Tetrahedron. 1974;30:667–673. [Google Scholar]
- 27.Ajo D, Casarin M, Bertoncello R, Busetti V, Ottenheijm HCJ, Plate R. Tetrahedron. 1985;41:5543–5552. [Google Scholar]
- 28.Ongania KH, Granozzi G, Busetti V, Casarin M, Ajò D. Tetrahedron. 1985;41:2015–2018. [Google Scholar]
- 29.Williams LJ, Jagadish B, Lyon SR, Kloster RA, Carducci MD, Mash EA. Tetrahedron. 1999;55:14281–14300. [Google Scholar]
- 30.Palacin S, Chin DN, Simanek EE, MacDonald JC, Whitesides GM, McBride MT, Palmore GTR. J Am Chem Soc. 1997;119:11807–11816. [Google Scholar]
- 31.Kubo A, Saito N, Yamoto H, Yamauchi R, Hiruma K, Inoue S. Chem Pharm Bull. 1988;36:2607–2614. doi: 10.1248/cpb.36.2607. [DOI] [PubMed] [Google Scholar]
- 32.Ando S, Koide K. Manuscript in preparation. [Google Scholar]
- 33.See the Supporting Information for the details of this experiment.
- 34.Iwasa E, Hamashima Y, Fujishiro S, Higuchi E, Ito A, Yoshida M, Sodeoka M. J Am Chem Soc. 2010;132:4078–4079. doi: 10.1021/ja101280p. [DOI] [PubMed] [Google Scholar]
- 35.Öhler E, Tataruch F, Schmidt U. Chem Ber. 1973;106:396–398. doi: 10.1002/cber.19731060205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.