

Abstract
Hepatitis B virus (HBV) is a major etiologic agent of chronic liver disease (CLD) and hepatocellular carcinoma (HCC). HBV encoded X antigen, HBx, and pathways implicated in the self-renewal of stem cells contribute to HCC, but it is not clear whether HBx expression promotes “stemness.” Thus, experiments were designed to test the hypothesis that HBx triggers malignant transformation by promoting properties that are characteristic of cancer stem cells (CSCs). To test this hypothesis, HepG2 cells were stably transduced with HBx and then assayed for phenotypic and molecular characteristics of “stemness.” The relationship between HBx and “stemness”-associated markers was also evaluated by immunohistochemical staining of liver and tumor tissue sections from HBV infected patients. The results showed that Oct-4, Nanog, Klf4, β-catenin and EpCAM were activated by HBx in vitro and in vivo. EpCAM was detected in the nuclei of human HCC cells from infected patients. HBx promotes “stemness” by activating β-catenin and epigenetic up-regulation of miR-181, both of which target EpCAM. HBx expression was also associated with depressed levels of E-cadherin. Moreover, HBx stimulated cell migration, growth in soft agar, and spheroid formation. This work is the first to propose that HBV promotes “stemness” in the pathogenesis of HCC. HBx associated up-regulated expression of multiple “stemness” markers support the hypothesis that HBx contributes to hepatocarcinogenesis, at least in part, by promoting changes in gene expression that are characteristics of CSCs.
Keywords: hepatocellular carcinoma, EpCAM, β-catenin, miR181, stemness
Introduction
HBV associated HCC is among the top five most frequent cancers worldwide (1). About 250,000 new cases of HCC are diagnosed yearly, and most people die within a year of diagnosis. The mechanism of HBV mediated HCC is not clear. However, HBV DNA is integrated, often highly rearranged, within host DNA in tumors and tumor-derived cell lines (2). These templates frequently produce HBx (3) that is active in trans-activation assays (4). Sustained production of HBx is associated with hepatocellular transformation, and represents a major contribution of HBV to HCC (5). HBx RNA and protein are highly expressed in chronically infected livers and strongly correlate with CLD (6). HBx has transcriptional trans-regulatory properties that alter patterns of host gene expression (2, 6), inhibits proteasomal degradation of growth regulatory proteins (7), stimulates cellular kinases that alter signal transduction (5), and utilizes epigenetic machinery (8). This suggests that HBx mediated tumor development involves epigenetic mechanisms that become operative during CLD.
Microarray analysis of human HCC specimens, cancer cell lines, and transgenic models established the molecular similarities between CSCs and hepatic stem cells (HSCs), and highlighted the importance of CSCs in the prognosis of liver cancer (9). “Stemness” transcription factors (e.g., Oct-4, Klf-4 and Nanog) re-expressed in cancer cells (10), and their reactivation contributes to tumorigenesis in somatic tissues (11). Epithelial cell adhesion molecule (EpCAM) is a marker of HSCs and CSCs (12) and acts as a mitogenic signal transducer via proteolysis and nuclear translocation of its intracellular domain, EpICD. EpICD binds to DNA in a complex with the scaffolding protein FHL2, β-catenin, and Lef-1, and regulates cell proliferation (13). EpCAM sustains “stemness” through EpICD binding to c-myc, Oct-4, Nanog, and Klf-4 promoters (14). The EpCAM promoter has Tcf-binding sites and is a transcriptional target of β-catenin (15). EpCAM was also regulated by miR-181. Forced expression of miR-181 endowed EpCAM-positive HCC cells with stem cell properties, whereas miR-181 suppression induced differentiation (16).β-catenin, like EpCAM, is important in stem cell self-renewal and in carcinogenesis (17), in part, by binding the promoters of Oct-4 and Nanog (18). Aberrant activation of their self-renewal program by β-catenin may contribute to the appearance of CSCs (19). Importantly, HBx also transcriptionally activates β-catenin in up to 80% of HCCs (20). This underscores the potentially close relationship between HBx, activated β-catenin, and stem cell renewal.
β-catenin acts as a transcriptional activator in the nucleus, but promotes cell adhesion when bound to E-cadherin (21). E-cadherin provides a physical link between adjacent cells and is crucial for cell polarity and the structural integrity of tissue (22). Altered expression and cellular distribution of E-cadherin is frequently associated with invasiveness in human cancers including HCC (23). In this context, HBx suppresses E-cadherin expression by promoting methylation of the E-cadherin promoter (24). This event results in the stabilization of β-catenin through tyrosine phosphorylation mediated by src, which is also activated by HBx (25). Collectively, these findings suggest that HBx promotes the development of HCC by triggering the expression of “stemness” factors in the chronically infected liver. The results of the present study support this hypothesis.
Materials and Methods
Cell Culture
The human hepatoblastoma cell line, HepG2, was stably transfected with HBx (HepG2X) or the bacterial chloramphenicol acetyltransferase (control) gene (HepG2CAT) as previously described (26). Cells were cultured in MEM (Invitrogen, Carlsbad, CA) supplemented with 100 mM nonessential amino acids, 100 mM sodium pyruvate, and 10% fetal bovine serum (Invitrogen), at 37°C in a humidified 5% CO2 incubator.
Patient Samples
Forty-three formalin fixed, paraffin embedded paired tumor (HCC)/nontumor (adjacent liver) tissues were obtained from Chinese patients who underwent surgery at the Third Military Medical University, Chongqing, China. All patients were hepatitis B surface antigen (HBsAg) positive in blood; 41 were males, the age range was from 35–69 (average: 48), and all were of Chinese ethnicity (Suppl. Table 1). Additional snap frozen tumor/nontumor pairs from 20 HBV infected patients were obtained from Queen Mary College at the University, Hong Kong, China. All patients were also positive for HBsAg in blood; 18 were male, and their ages ranged from 33–71 years (average: 50.5) (Suppl. Table 2). All samples were used for diagnostic purposes, and then used for this study. Ten uninfected human liver tissues (Abcam, Cambridge, MA) were used as controls. The use of samples was approved by the Institutional Review Boards at all participating universities.
Spheroid Assay
Single cell suspensions of HepG2X and HepG2CAT cells were plated at a density of 1 × 103 cells in 2 ml of medium in 6-well Ultra-Low Attachment Microplates (Corning, Corning, NY) and maintained for up to 16 days. Spheroids were observed and counted using an ECLIPSE Ti inverted microscope operated with a Nikon DS-Fi1 camera and NIS Elements software (Nikon, Melville, NY).
Soft Agar Assay
Single cell suspensions of 1 × 104 cells were mixed with 0.3% agar (Sigma, St. Louis, MO) in complete growth medium and seeded in triplicates into 6-well plates coated with 0.5% hardened agar. Plates were incubated at 37°C for 28 days. Colonies ≥1 mm in diameter were counted under code by light microscopy.
Cell Migration Assay
Single cell suspensions of 1.5 × 105 cells were plated in triplicates into 6-well BD BioCoat™ Matrigel™ Invasion Chambers (BD, Franklin Lakes, NJ) according to enclosed instructions. Cell migration was observed after 24h by hematoxylin and eosin (H & E) staining.
Tumorigenicity in Nude Mice
For tumorigenicity studies, two groups of 10 six-week-old nude mice (Charles River Laboratories, Wilmington, MA) were injected subcutaneously at a single site with 6 × 106 HepG2X or HepG2CAT cells. Tumor onset was scored visually and by palpitation independently by two trained lab personnel. Tumor sizes were determined by wet weight at the time of euthanasia (6 weeks). These experiments were approved by the Institutional Animal Care and Use Committee at Temple University.
Protein Extraction and Western Blotting
Cells were lysed in Cell Lysis Buffer with protease inhibitor cocktail (Cell Signaling, Danvers, MA). Nuclear extracts were prepared using Nuclear Extract Kit (Active Motif, Carlsbad, CA) according to enclosed instructions. Protein extracts were separated by SDS-PAGE and transferred to nitrocellulose membranes (Schleicher & Schuell, Sanford, ME). The membranes were incubated overnight in 5% nonfat milk in Tris Buffered Saline/0.1% Tween-20 with primary antibodies against HBx, Oct-4, E-cadherin, β-catenin, Lamin A and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), Nanog (Cell Signaling, Danvers, MA), Klf-4 and EpCAM (Abcam), Ac-Lys H3 (Active Motif). The blots were developed using the ECL plus detection kit (Amersham, Piscataway, NJ) and exposed to Kodak imaging films (Kodak BioMax, Rochester, NY). Images were processed using ImageJ software (NIH).
Treatment of Cells with Trichostatin A (TSA)
Suspensions of 3.5 × 105 cells were plated in 35-mm culture dishes with or without 350 nM TSA (Sigma) in 2 ml of medium. Extracts were collected at 12 and 24 hours and analyzed by western blotting.
PowerBlot Proteomics Analysis
PowerBlot analysis of HepG2X and HepG2CAT cell lysates was performed by BD Transduction Labs (Franklin Lakes, NJ). Briefly, 400 μg of cell lysates were analyzed by SDS/PAGE, and transferred to Immobilon P (Millipore, Billerica, MA). The membranes were blocked with 5% nonfat milk. Each membrane was cut into vertical strips, and each strip was incubated with a complex antibody cocktail. Signals were developed by chemiluminesence and measured using PDQuest 2-D analysis software (Bio-Rad, Hercules, CA). For each of 3 experiments, 3 gels were run using HepG2X and HepG2CAT cell lysates.
MicroRNA Analysis
Small RNAs were isolated from HepG2X and HepG2CAT cells and 20 pairs of HCC tissue samples using Ambion (Austin, TX) mirVana miRNA isolation kit according to enclosed instructions. Microarray analysis was performed with miRNAs isolated from HepG2X and HepG2CAT cells by LC Sciences (Houston, TX). Differentially expressed miRNAs in cells were identified by Cy3(HepG2X)/Cy5(HepG2CAT) ratio, and those with a p-value < 0.01 were considered for further characterization. Differential expression of selected miRNAs in cells and tissues was validated by qRT-PCR using a Smart Cycler System (Cepheid, Sunnyvale, CA). The ΔΔCt calculation was done as follows: ΔΔCt = ΔCt of miR-181a in tumor–ΔCt of miR-181a in non-tumor tissue. Briefly, the samples with 25 ng small RNA were first denatured at 95 °C for 3 min and then run for 40 cycles (95 °C for 15 sec and 60 °C for 30 sec). U6 was used for normalization.
Immunohistochemistry
Tissue sections were deparaffinized, dehydrated, treated with Uni-TRIEVE antigen retrieval (Innovex, Richmond, CA) and stained using the UltraVision Detection System (Thermo Scientific, Rockford, IL) according to enclosed instructions. Antibodies used for staining were the same as those described above for western blotting except for anti-HBx (anti-99 custom made antibody) (27) and anti-EpCAM (Millipore, Billerica MA). Normal mouse or rabbit IgG (Vector Labs, Burlingame, CA) were used to rule out false-positive responses. Pre-absorption of primary antibodies with corresponding antigens was performed on tissue sections to insure specificity.
Statistics
Statistical values for cell migration, growth in soft agar, and tumorigenesis were defined using an unpaired Student’s t-test, in which P <0.05 was considered significant. The relationship between HBx and EpCAM, β-catenin, and E-cadherin obtained by immunohistochemistry was determined using 2 × 2 comparisons in the Chi square (χ2) test. Statistical significance was considered when P < 0.05.
Results
Relationship between HBx and “stemness”- associated markers
PowerBlot analysis was conducted using whole cell lysates from HepG2X and HepG2CAT cultures. The results showed 4.5 ± 0.5-fold up-regulated expression of β-catenin in HepG2X compared to control cells (Fig. 1A–B). Several β-catenin target genes were also up-regulated, including MDR1 (7.8 ± 1.8-fold; Fig. 1C–D) and c-myc (4.7 ± 0.7-fold; Fig. 1A–B). HepG2X cells expressed 6.7 ± 1.2-fold lower levels of E-cadherin (Fig. 1C–D) compared to HepG2CAT cells. Selected markers were then further characterized by western blotting (Fig. 1E–F). In whole cell extracts, HBx was associated with down-regulation of E-cadherin (3.8-fold), as well as up-regulation of β-catenin (2.5-fold) and EpCAM (3-fold). EpCAM was also up-regulated in HBx-expressing Hep3B and Huh7 cells (Suppl. Fig.1). In HepG2 cells, β-catenin is present as wild type (upper band) and truncated mutant (lower band) (Fig. 1E) due to partial exon 3 deletion (28). HBx up-regulated wild type β-catenin without modifying the levels of truncated β-catenin (Fig. 1E). Previous work confirmed that nuclear β-catenin in HBx positive HCC is wild type by DNA sequence analysis of β-catenin exons obtained from paraffin blocks (29). HBx-associated up-regulation of β-catenin has been demonstrated in Huh7 (30, 31) and Hep3B cells (20, 31). Down-regulation of E-cadherin in Hep3B cells has also been demonstrated previously (24). Moreover, HBx-mediated repression of E-cadherin was associated with activation of β-catenin in Hep3B cells (24). In nuclear extracts, HBx was associated with up-regulation of Oct-4 (1.6-fold), Nanog (3.4-fold), and Klf-4 (3.5-fold) (Fig. 1F). These data suggest that HBx is associated with the re-activation of “stemness” transcription factors Oct-4, Nanog, and Klf-4, and “stemness” associated EpCAM and β-catenin.
Fig. 1.

PowerBlot analysis of (A, C) HepG2X and (B, D) HepG2CAT cells. Selected differentially expressed genes are highlighted. Representative western blots with HepG2X and HepG2CAT cells using (E) total (50 μg) or (F) nuclear (100 μg) extracts. β-actin and Lamin A are loading controls.
Relationship between HBx, “stemness-” associated markers, and E-cadherin in vivo
Additional experiments were conducted to determine whether the results of PowerBlot analyses and western blotting could be validated in vivo. Thus, clinical samples containing HCC and nontumor liver were stained for HBx, Oct-4, Nanog, β-catenin, EpCAM and E-cadherin (Suppl. Table 1). Among 43 patients who underwent surgical resection for HBV associated HCC, 31 had both tumor and adjacent nontumor liver, 9 had only tumor in their blocks, and 3 patients had only nontumor liver. Among these, HBx staining was observed in 26 of 40 tumors (65%) and in all 34 nontumor livers (100%). In 82% of cases with tumor and nontumor tissues, HBx staining was stronger and more widespread in liver compared to tumor, as previously reported (32). Ten commercially available liver sections from uninfected individuals were uniformly negative for HBx. HBx staining was cytoplasmic in all cases (Suppl. Fig. 2), and one case showed nuclear staining in nontumor liver (data not shown). EpCAM was present in 26 cases in the tumor compartment (66%). Membranous EpCAM staining (Fig. 2A) was observed in the nontumor liver among 21 patients (60%) (Fig. 2C), even though EpCAM is not expressed in uninfected, healthy liver (12). In tumors from 3 patients, nuclear EpCAM was also detected (Fig. 2B). With regard to β-catenin, staining was observed in the tumor compartment from 28 patients (74%), and in the nontumor compartment from 19 patients (61%). Although membranous β-catenin is characteristic of normal, uninfected liver, all cases of tumor and nontumor tissues showed both membranous and cytoplasmic staining for β-catenin, consistent with β-catenin activation, as previously shown (29). β-catenin was also observed in the nuclei of two tumors (data not shown). In contrast, E-cadherin staining, which was exclusively membranous, was detected in only 7 tumors (18%), but was present in 20 cases where nontumor liver was available (74%). These results are similar to those previously published (24). When staining was performed for Nanog and Oct-4, six and two tumor samples were positive in scattered nuclei, respectively (Fig. 2D and E). Hence, “stemness” and “stemness”-like markers were observed in HBV associated HCC.
Fig. 2.

Staining for EpCAM in (A) tumor (×400), (B) tumor (×200) and in (C) nontumor (×200). All panels show membranous staining and (C) also shows mixed membranous and nuclear localization. Detection of (D) Oct-4 and (E) Nanog in human HCC tissue sections from HBV carriers (×200).
Additional analyses were conducted to determine the relationship between HBx and these markers. In the tumor compartment, co-staining between HBx and EpCAM was seen in only 16 cases (Fig. 3A–B) (χ2 = 2.03; P > 0.1), between HBx and β-catenin in 19 cases (χ2 = 0.23; P > 0.5), and between HBx and E-cadherin in 6 cases (χ2 = 0.88; P > 0.3). Hence, there appears to be no correlation between HBx and these markers in tumors. In contrast, in the nontumor compartment, where HBx is strongly expressed, co-staining between HBx and EpCAM was seen in 21 cases (χ2 = 11.8; P < 0.001), between HBx and β-catenin in 19 cases (χ2 = 5.88; P < 0.02), while an inverse correlation was seen between HBx and E-cadherin in 20 cases (χ2 = 6.04; P < 0.02), suggesting a strong correlation between HBx and these markers. Other blocks of chronically infected liver from patients without HCC were also evaluated, but the results obtained were the same as in adjacent liver tissues from HCC patients (data not shown).
Fig. 3.

Staining for (A) HBx and (B) EpCAM in consecutive HCC sections from a representative patient (×200).
Analysis of relationship between β-catenin and EpCAM staining and HCC grade showed an inverse correlation between EpCAM staining and HCC in grade 1 (χ2 = 10.2257; P < 0.005) and grade 2 (χ2 = 5.3831; P < 0.025), but not in grades 3 (χ2 = 1.3; P > 0.05) and 4 (χ2 = 1.06; P > 0.3). There was no correlation between β-catenin staining and tumor grade. Experiments with a larger number of samples should be conducted to address these relationships more thoroughly.
HBx up-regulates miR-181a
MicroRNA(s) are known to contribute importantly to the pathogenesis of many tumor types (33). When miRNA microarray analysis was performed on HepG2X and HepG2CAT cells, HBx was associated with up-regulation of miR-181a (2-fold) and miR-181b (1.5-fold) (data not shown). RT-PCR was performed to validate the microarray data in 20 paired HCC/nontumor cases (Suppl. Table 2). When these tissues were evaluated for HBx by staining and miR-181a by real-time RT/PCR, 11 of 15 HBx positive non-tumor liver samples had elevated miR-181a (73%) (P < 0.01) (Fig. 4). Among the corresponding tumors, 5 were HBx positive, but only one had higher levels of miR-181a compared to non-tumor. These results suggest that miR-181a is an epigenetic target of HBx both in cell lines and in liver. This suggests that EpCAM may be up-regulated by HBx targeting both β-catenin and miR-181.
Fig. 4.
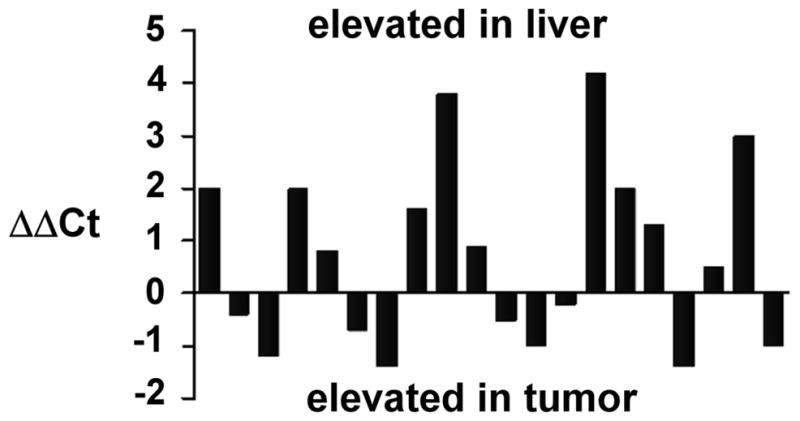
Expression of miR-181a in tumor (HCC) and non-tumor liver. Negative values mean that the miRNA was expressed more in tumor compared to nontumor from the same patient. Positive values mean the miRNA was expressed more in non-tumor compared to tumor.
Phenotypic characteristics of HBx-associated “stemness”
HepG2X and HepG2CAT cells were tested for their ability to form spheres in 3D culture systems (clonogenic potential), to induce tumors in nude mice, and promote cell migration. It has been shown that stem/progenitor cells and CSCs can form spheroids in vitro suggesting the presence of self-renewing cells (34, 35). In this work, HBx-expressing cells formed spheroids by day 3 after inoculation. About 4-fold more spheroids were derived from HepG2X compared to HepG2CAT cells after 16 days in culture. Some of these spheroids exceeded 100μm in diameter (Fig. 5A, C). In contrast, HepG2CAT cells formed mostly disorganized clusters that became adherent, with less floating spheroids (Fig. 5B). Primary spheroids enzymatically dissociated to single cells gave rise to secondary spheroids. When anchorage independent growth was evaluated after 28 days of culture, HepG2X cells formed 81± 20.5 colonies, while HepG2CAT cells produced 37 ± 4.9 colonies (P < 0.02) (Fig. 5D). Cell migration was then examined by Matrigel invasion assay and H&E staining after 24 hrs. About 3.5-fold more HepG2X than HepG2CAT cells migrated through Matrigel basement membrane matrix (P < 0.001) (Fig. 5E). In tumorigenicity studies, tumor onset was 30 days post-injection for HepG2X cells compared to 46 days for HepG2CAT cells (P < 0.01). HepG2X also yielded larger tumors (1.6 ± 0.3 cm3) than HepG2CAT cells (0.7 ± 0.4 cm3) (P < 0.01). Thus, HepG2X cells showed more pronounced phenotypes consistent with stem cell behavior compared to HepG2CAT control cells. Hence, the phenotypic characteristics presented above were consistent with the induction of “stemness” markers by HBx.
Fig. 5.

Representative images of spheroids derived from (A) HepG2X cells, (B) HepG2CAT cells, and (C) relative numbers of spheroids for each culture. Data are shown as mean ± SD. D. Soft agar assay. (E) Migration through Matrigel basement membrane. All experiments were performed in triplicate.
Treatment of cells with Trichostatin A (TSA)
The finding, that HBx binds to histone deacetylase 1 (HDAC1) (8) and that HDAC epigenetically regulates the expression E-cadherin (8, 24), suggests that inhibition of HDAC1 would reverse the effects of HBx on E-cadherin. To test this hypothesis, HepG2X and HepG2CAT cells were treated with the HDAC1 inhibitor, TSA, and the levels of E-cadherin evaluated over time. TSA treatment resulted in a time dependent increase in H3 acetylation by 20-fold (Fig. 6). This was accompanied by increased levels of E-cadherin in HepG2X cells (7-fold), decreased levels of wild type β-catenin (5-fold) and EpCAM (9.5-fold) with little change in HepG2CAT cells. These results show that once HBx repressed E-cadherin expression is reversed by TSA, alterations in β-catenin and EpCAM are also observed, suggesting these events are linked.
Fig. 6.

Western blots showing levels of acetyl-H3, E-cadherin, β-catenin and EpCAM after the treatment of HepG2X and HepG2CAT cells with TSA. The blot is representative of three independent experiments.
Discussion
In the present study, the pluripotent stem cell transcription factors Oct-4, Nanog, and Klf-4, as well as the “stemness”-associated markers EpCAM and β-catenin, were up-regulated in HBx expressing cells (Fig. 1). These observations were validated in tumor and adjacent nontumor liver from HBV infected patients (Figs. 2–3). HBx expressing cells also demonstrated enhanced colony-formation capability and sphere-forming activity in vitro (Fig. 5), and were more tumorigenic in vivo. miRNA array analysis showed that miR-181, which targets EpCAM (16), was up-regulated in HepG2X cells, and this was validated in vivo (Fig. 4). The tight epidemiologic association between chronic HBV infection and the development of HCC (36), combined with evidence that HBx and CSCs contribute importantly to tumor development, suggest that HBV may promote hepatocarcinogenesis, in part, by triggering “stemness.” This may be mediated by HBx associated transcriptional trans-regulation and altered epigenetic regulation of host gene expression (8, 37) as well as activation of cellular kinases that alter signal transduction (5, 36). Together data suggest that HBx promoted characteristics of CSCs in liver cells.
Although chronic infection often extends for decades, it is proposed that HBx promotes “stemness” most readily in liver just prior to the appearance of HCC. This is because the levels of HBx expression in the liver increase with the length of time a carrier is infected, with the highest levels of HBx expression seen in the cirrhotic liver (2, 38). As chronic infection proceeds, virus replication decreases, and HBx expression from integrated templates increases (2). During this process, HBx impacts upon the host pathways and gene targets mentioned above. In the chronically infected liver, HBx expressed from integrated templates are often truncated at their C-terminal regions. Prior work has shown that these mutants share properties with full-length HBx (4, 39), suggesting that they may also promote the appearance of “stemness,” although further work with a series of mutants will need to be conducted to address this point conclusively. However, the observations that HBx promotes “stemness” may help to understand the promiscuous and pleiotrophic properties of HBx, since once “stemness” factors are activated, cell fate reprogramming can occur, even without the sustained expression of these markers. In this context, it is interesting that the strongest and most prevalent HBx staining was found in cirrhotic livers, and relatively little or none in the majority of HCC nodules (2, 38). This is consistent with the observation that HCC arises most often from cirrhotic livers, and with the notion that the HBx promotion of “stemness” in this pathological setting, is central to early stage tumor development.
The contribution of HBx to the development of “stemness” is supported by observations that Oct-4, Nanog, Klf-4 and c-myc are up-regulated in HepG2X compared to HepG2CAT cells (Fig. 1), and Oct-4 and Nanog were detectable in several HCC nodules (Fig. 2). Independent evidence showed strongly positive clusters of Oct-4 positive cells in HCC (40). Importantly, CSCs comprise a minor population of cells that re-establish the phenotypic heterogeneity in the primary tumor and exhibit self-renewing capability on serial passaging (9). Oct-4, Sox-2, Nanog and their binding partners act as key regulators of pluripotency in early mammalian development (41). These “stemness” genes are re-expressed in cancer cells, and the reactivation of these factors contributes to tumorigenesis in somatic tissues (10). For example, Oct-4 and Nanog expression was observed in seminoma, retinoblastoma, oral squamous cell carcinoma, bone sarcoma, breast and a colon cancer cell lines compared to their normal counterparts (42). It is proposed that some of these same pathways are turned on by HBx in hepatocarcinogenesis.
HBx contributes to the pathogenesis of HCC by several pathways, resulting in the sustained activation of EpCAM. Prior studies have shown that EpCAM is present on subsets of normal epithelia (bile duct epithelium), numerous tissue stem and progenitor cells (fetal hepatoblasts and hepatic stem cells) (12), and most carcinomas including HCC (43, 44). Although normal adult hepatocytes do not express EpCAM (12, 44), it is striking that HBV infected hepatocytes do (Fig. 2C), implying that HBx confers “stemness” properties on at least some infected cells. The significant co-staining between HBx and EpCAM in the nontumor liver is consistent with this hypothesis. EpCAM acts as a mitogenic signal transducer in vitro and in vivo via nuclear translocation of the EpICD, the latter of which was also observed in a few cases here (Fig. 2B). Importantly, the scaffolding protein, FHL2, which is a co-activator of β-catenin, bridges EpCAM with β-catenin and Lef-1, thus promoting interactions of EpCAM with DNA (13). Involvement of TCF/Lef-1, a major regulator of c-myc and cyclin E expression (45), may explain the ability of EpCAM to rapidly up-regulate their expression, thereby inducing cell proliferation. Importantly, EpCAM also has the ability to sustain “stemness” through EpICD binding to Oct-4, Nanog, Sox-2, and Klf-4 promoter regions, promoting the expression of these genes in human embryonic stem cells (14). The up-regulated expression of EpCAM by HBx is consistent with possibly similar events occurring in HCC.
Several studies have shown that HBx up-regulates and stabilizes β-catenin (Fig. 1) (20, 30). β-catenin transcriptionally up-regulates EpCAM via the presence of two Tcf binding elements in the EpCAM promoter (15). Nuclear accumulation of β-catenin induced EpCAM gene expression in cultured human hepatocytes and HCC cell lines (15). In tumor xenograft transplantation models, only EpCAM-positive cells efficiently initiated the development of invasive tumors, even after serial transplantation, demonstrating that EpCAM-positive cells display CSC-like characteristics (43). Thus, the HBx up-regulation of β-catenin (Fig. 1), and β-catenin mediated induction of EpCAM, may be critical in initiating and maintaining CSCs growth.
E-cadherin, which is important for the maintenance of cell polarity and the structural integrity in tissue, is down-regulated by HBx (24). Abnormalities in the expression and cellular distribution of E-cadherin are frequently associated with de-differentiation and invasiveness in a variety of human malignancies, including HCC (23). HBx has been found to repress E-cadherin expression at the transcriptional level through hypermethylation of the E-cadherin promoter (24), and possibly by alterations in HDAC function following complex formation with HBx (8). The latter is supported by evidence that TSA, an inhibitor of HDAC1, relieves the suppression of E-cadherin by HBx (Fig. 6). Independent evidence has shown that the inhibition of HDAC by TSA could induce hyperacetylation of histones and restore the expression of E-cadherin (46). HBx mediated repression of E-cadherin releases β-catenin from its role in cell adhesion. Prior work has shown that HBx induced adherens junction disruption is src kinase-dependent, and resulted in the accumulation of cytoplasmic β-catenin (25, 47). In the nucleus,β-catenin binds Tcf/Lef factor and acts as a transcriptional activator of growth-regulatory genes including those involved in self-renewal of stem cells. Studies have revealed that Tcf3 factor co-occupies almost all promoter regions occupied by stem cell specific transcription factors, including Oct-4 and Nanog, and this suggests the mechanism how β-catenin regulates the expression of “stemness” genes (17, 18). Hence, the finding that HBx constitutively activated wild type β-catenin in up to 80% of HCCs (20) underscores the potentially close relationship between HBx, activated wild type β-catenin, and stem cell renewal.
Recent studies show that altered expression of specific miRNAs is involved in tumorigenesis (33). Microarray data from HBV associated HCC and nontumor liver tissues showed that highly conserved miR-181 family members were over-expressed in HSCs and CSCs and thus contributed to the maintenance of “stemness.” Forced miR-181 expression enriched EpCAM+ HCC cells with stem cell properties, whereas miR-181 blockage induced differentiation.16 Interestingly, data from this study showed elevated miR-181 expression in HepG2X cells compared with HepG2CAT cells, and in most HBx positive tumor and nontumor samples (Fig. 4), suggesting that miR-181 is an epigenetic target of HBx. Collectively, these data suggest that HBx may contribute to HCC by promoting the development of “stemness.”
Supplementary Material
Acknowledgments
Financial support. This work was supported by grants CA104025 and CA111427 awarded to Dr. Feitelson.
The authors would like to thank Drs. Yongwen Chen and Cheng-ying Yang from the Third Military Medical University for the paraffin blocks of tissue, and Dr. Eugeney Bichenkov for assistance with the figures.
Abbreviations
- HBx
hepatitis B x antigen
- HBV
hepatitis B virus
- CLD
chronic liver disease
- HCC
hepatocellular carcinoma
- CSCs
cancer stem cells
- MDR1
multi-drug resistance gene 1
- HSCs
hepatic stem cells
- EpCAM
epithelial cell adhesion molecule
- H & E
hematoxylin and eosin
- TSA
Trichostatin A
- HDAC1
histone deacetylase 1
Footnotes
Conflict of interest statement: none declared.
References
- 1.Marrero JA. Hepatocellular carcinoma. Current Opinion in Gastroenterology. 2006;22:248–253. doi: 10.1097/01.mog.0000218961.86182.8c. [DOI] [PubMed] [Google Scholar]
- 2.Feitelson MA, Duan LX. Hepatitis B virus X antigen in the pathogenesis of chronic infections and the development of hepatocellular carcinoma. Am J Pathol. 1997;150 (4):1141–1157. [PMC free article] [PubMed] [Google Scholar]
- 3.Matsubara K, Tokino T. Integration of HBV DNA and its implications for hepatocarcinogenesis. Mol Biol Med. 1990;7:243–260. [PubMed] [Google Scholar]
- 4.Takada S, Koike K. Trans-activation function of a 3′ truncated X gene-cell fusion product from integrated HBV DNA in chronic hepatitis tissues. Proc Natl Acad Sci USA. 1990;87:5628–5632. doi: 10.1073/pnas.87.15.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henkler F, Koshy R. Hepatitis B virus transcriptional activators: mechanisms and possible role in oncogenesis. J Viral Hepat. 1996;3:109–121. doi: 10.1111/j.1365-2893.1996.tb00001.x. [DOI] [PubMed] [Google Scholar]
- 6.Diamantis I, McGandy C, Chen T, Liaw YF, Gudat F, Bianchi Ll. Hepatitis B X-gene expression in hepatocellularar carcinoma. J Hepatol. 1992;15:400–403. doi: 10.1016/0168-8278(92)90077-3. [DOI] [PubMed] [Google Scholar]
- 7.Huang J, Kwong J, Sun ECY, Liang TJ. Proteasome complex as a potential cellular target of hepatitis B virus X protein. J Virol. 1996;70:5582–5591. doi: 10.1128/jvi.70.8.5582-5591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng DL, Zhang L, Cheng N, Xu X, Deng Q, Teng XM, et al. Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. Journal of Hepatology. 2009;50(2):377–387. doi: 10.1016/j.jhep.2008.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Marquardt JU, Thorgeirsson SS. Stem cells in hepatocarcinogenesis: evidence from genomic data. Semin Liver Dis. 2010;30(1):26–34. doi: 10.1055/s-0030-1247130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Monk M, Holding C. Human embryonic genes re-expressed in cancer cells. Oncogene. 2001;20:8085–8091. doi: 10.1038/sj.onc.1205088. [DOI] [PubMed] [Google Scholar]
- 11.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121:465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 12.Schmelzer E, Wauthier E, Reid LM. The phenotypes of pluripotent human hepatic progenitors. Stem Cells. 2006;24:1852–1858. doi: 10.1634/stemcells.2006-0036. [DOI] [PubMed] [Google Scholar]
- 13.Munz M, Baeuerle PA, Gires O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009;69(14):5627–5629. doi: 10.1158/0008-5472.CAN-09-0654. [DOI] [PubMed] [Google Scholar]
- 14.Lu TY, Lu RM, Liao MY, Yu J, Chung CH, Kao CF, et al. Epithelial cell adhesion molecule regulation is associated with the maintenance of the undifferentiated phenotype of human embryonic stem cells. J Biol Chem. 2010;285(12):8719–8732. doi: 10.1074/jbc.M109.077081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-β-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007;67:10831–10839. doi: 10.1158/0008-5472.CAN-07-0908. [DOI] [PubMed] [Google Scholar]
- 16.Ji J, Yamashita T, Budhu A, Forgues M, Jia HL, Li C, et al. Identification of microRNA-181 by genome-wide screening as a critical player in EpCAM-positive hepatic cancer stem cells. Hepatology. 2009;50(2):472–480. doi: 10.1002/hep.22989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takigawa Y, Brown AM. Wnt signaling in liver cancer. Curr Drug Targets. 2008;9(11):1013–1024. doi: 10.2174/138945008786786127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pereira L, Yi F, Merrill BJ. Repression of Nanog gene transcription by Tcf3 limits embryonic stem cell self-renewal. Mol Cell Biol. 2006;26(20):7479–7491. doi: 10.1128/MCB.00368-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Polakis P. The oncogenic activation of beta-catenin. Curr Opin Genet Dev. 1999;9(1):15–21. doi: 10.1016/s0959-437x(99)80003-3. [DOI] [PubMed] [Google Scholar]
- 20.Ding Q, Xia W, Liu JC, Yang JY, Lee DF, Xia J, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19(2):159–170. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39(3):305–318. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 22.Takeichi M. Cadherin cell adhesion receptors as a morphogenetic regulator. Science. 1991;251:1451–1455. doi: 10.1126/science.2006419. [DOI] [PubMed] [Google Scholar]
- 23.Osada T, Sakamoto M, Ino Y, Iwamatsu A, Matsuno Y, Muto T, et al. E-cadherin is involved in the intrahepatic metastasis of hepatocellular carcinoma. Hepatology. 1996;24:1460–1467. doi: 10.1053/jhep.1996.v24.pm0008938181. [DOI] [PubMed] [Google Scholar]
- 24.Liu J, Lian Z, Han S, Waye MM, Wang H, Wu MC, et al. Downregulation of E-cadherin by hepatitis B virus X antigen in hepatocellullar carcinoma. Oncogene. 2006;25(7):1008–1017. doi: 10.1038/sj.onc.1209138. [DOI] [PubMed] [Google Scholar]
- 25.Müller T, Choidas A, Reichmann E, Ullrich A. Phosphorylation and free pool of beta-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J Biol Chem. 1999;274(15):10173–10183. doi: 10.1074/jbc.274.15.10173. [DOI] [PubMed] [Google Scholar]
- 26.Lian Z, Pan J, Liu J, Zhang S, Zhu M, Arbuthnot P, et al. The translation initiation factor, hu-Sui1 may be a target of hepatitis B X antigen in hepatocarcinogenesis. Oncogene. 1999;18(9):1677–1587. doi: 10.1038/sj.onc.1202470. [DOI] [PubMed] [Google Scholar]
- 27.Feitelson MA, Millman I, Duncan GD, Blumberg BS. Presence of antibodies to the polymerase gene product(s) of hepatitis B and woodchuck hepatitis virus in natural and experimental infections. J of Med Virol. 1988;24:121–136. doi: 10.1002/jmv.1890240202. [DOI] [PubMed] [Google Scholar]
- 28.Carruba G, Cervello M, Miceli MD, Farruggio R, Notarbartolo M, et al. Truncated form of β-catenin and reduced expression of wild-type catenins feature HepG2 human liver cancer cells. Ann NY Acad Sci. 1999;886:212–216. doi: 10.1111/j.1749-6632.1999.tb09419.x. [DOI] [PubMed] [Google Scholar]
- 29.Lian Z, Liu J, Li L, Li X, Clayton M, Wu MC, et al. Enhanced cell survival of Hep3B cells by the hepatitis B x antigen effector, URG11, is associated with upregulation of beta-catenin. Hepatology. 2006;43(3):415–424. doi: 10.1002/hep.21053. [DOI] [PubMed] [Google Scholar]
- 30.Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/β catenin signaling in hepatoma cells. Hepatology. 2004;39:1683–1693. doi: 10.1002/hep.20245. [DOI] [PubMed] [Google Scholar]
- 31.Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 2011;300(2):162–172. doi: 10.1016/j.canlet.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 32.Wang W, London WT, Feitelson MA. HBxAg in HBV carrier patients with liver cancer. Cancer Res. 1991;51:4971–4977. [PubMed] [Google Scholar]
- 33.Agami R. microRNAs, RNA binding proteins and cancer. Eur J Clin Invest. 2010;40(4):370–374. doi: 10.1111/j.1365-2362.2010.02279.x. [DOI] [PubMed] [Google Scholar]
- 34.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 35.Barclay WW, Axanova LS, Chen W, Romero L, Maund SL, Soker S, et al. Characterization of adult prostatic progenitor/stem cells exhibiting self-renewal and multilineage differentiation. Stem Cells. 2008;26(3):600–610. doi: 10.1634/stemcells.2007-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feitelson MA. Hepatitis B virus in hepatocarcinogenesis. J Cell Physiol. 1999;181:188–202. doi: 10.1002/(SICI)1097-4652(199911)181:2<188::AID-JCP2>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Shon JK, Shon BH, Park IY, Lee SU, Fa L, Chang KY, et al. Hepatitis B virus-X protein recruits HDAC1 to repress IGF binding protein 3 transcription. Virus Res. 2009;139:14–21. doi: 10.1016/j.virusres.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 38.Diamantis ID, McGandy CE, Chen TJ, Liaw YF, Gudat F, Bianchi L. Hepatitis B X-gene expression in hepatocellular carcinoma. J Hepatol. 1992;15(3):400–403. doi: 10.1016/0168-8278(92)90077-3. [DOI] [PubMed] [Google Scholar]
- 39.Liu X, Wang L, Zhang S, Lin J, Zhang S, Feitelson MA, et al. Mutations in the C-terminus of the X protein of hepatitis B virus regulate Wnt-5a expression in hepatoma Huh7 cells: cDNA microarray and proteomic analyses. Carcinogenesis. 2008;29(6):1207–1214. doi: 10.1093/carcin/bgn111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci USA. 2008;105(7):2445–2450. doi: 10.1073/pnas.0705395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Niwa H. How is pluripotency determined and maintained? Development. 2007;134(4):635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- 42.Uche I, Ezeh UI, Turek PJ, Reijo RA, Amander T, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer. 2005;104(10):2255–2265. doi: 10.1002/cncr.21432. [DOI] [PubMed] [Google Scholar]
- 43.Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang HY, et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136(3):1012–1024. doi: 10.1053/j.gastro.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Breuhahn K, Baeuerle PA, Peters M, Prang N, Töx U, Köhne-Volland R, et al. Expression of epithelial cellular adhesion molecule (EpCAM) in chronic (necro-)inflammatory liver diseases and hepatocellular carcinoma. Hepatol Res. 2006;34(1):50–56. doi: 10.1016/j.hepres.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 45.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ou JN, Torrisani J, Unterberger A, Provençal N, Shikimi K, et al. HDAC inhibitor Trichostatin A induces global and gene-specific DNA demethylation in human cancer cell lines. Biochem Pharmacol. 2007;73(9):1297–1307. doi: 10.1016/j.bcp.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 47.Lara-Pezzi E, Roche S, Andrisani OM, Sanchez-Madrid F, Lopez-Cabrera M. The hepatitis B virus HBx protein induces adherens junction disruption in a src-dependent manner. Oncogene. 2001;20:3323–3331. doi: 10.1038/sj.onc.1204451. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.