

Abstract
The S6K1 and S6K2 kinases are considered important mTOR signaling effectors, yet their contribution to tumorigenesis remains unclear. Aberrant mTOR activation is a frequent event in cancer, that commonly results from heterozygous loss of PTEN. Here, we show for the first time a differential protein expression between S6K1 and S6K2 in both mouse and human tissues. Additionally, the inactivation of S6k1 in the context of Pten heterozygosity (Pten+/-) suggests a differential requirement for this protein across multiple tissues. This tissue-specificity appears to be governed by the relative protein expression of S6k2. Accordingly, we find that deletion of S6k1 markedly impairs Pten+/- mediated adrenal tumorigenesis, specifically due to low expression of S6k2. Concomitant observation of low S6K2 levels in the human adrenal gland supports the development of S6K1-inhibitors for treatment of PTEN loss driven pheochromocytoma.
Introduction
Aberrant activation of the phosphatidylinositol-3 kinase (PI3K) pathway plays a key role in tumorigenesis through regulation of pivotal biological processes including proliferation, growth, survival and migration (1). A major regulator of this pathway is the tumor suppressor phosphatase and tensin homolog deleted on chromosome 10 (PTEN) gene. PTEN negatively regulates the PI3K signaling cascade and is now established as one of the most frequently mutated, deleted and silenced tumor suppressor genes in human cancer (2). We have previously established several faithful mouse models to elucidate the role of Pten in cancer [reviewed in (3)]. Mice engineered to have only one allele of Pten (Pten+/- mice) die prematurely as a result of an autoimmune disorder characterized by massive lympho-splenomegaly (4), and are extremely susceptible to developing a wide spectrum of epithelial tumors (5).
Downstream of the PI3K/PTEN pathway, the mammalian target of rapamycin (mTOR) complex 1 (mTORC1) has been demonstrated to be an essential effector in promoting cell proliferation and susceptibility to cancer development (reviewed in (6)). mTORC1 becomes highly active in the absence of PTEN and promotes cell growth through phosphorylation of important regulators of protein translation including the wellcharacterized ribosomal protein S6 kinase (S6K) and the eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) (reviewed in (7)). These two mTORC1 substrates represent two distinct signaling pathways downstream of mTORC1. Phosphorylation of 4EBP1 leads to its uncoupling from the eukaryotic initiation factor 4E (eIF4E), while activation of the S6K protein family results in the phosphorylation of additional downstream targets including the 40S ribosomal protein S6 (RpS6) (8), eIF4B (9), eEF2 kinase (10), and the tumor suppressor PDCD4 (11). Release of eIF4E from 4EBP1 and the phosphorylation of RpS6 result in the upregulation and activation of the translation machinery (7).
Downstream of mTOR, the role of the 4EBP1/eIF4E branch of the pathway in tumorigenesis has been extensively studied both in vivo and in vitro [(12), (13) and reviewed in (14)].
In contrast, the contribution of the S6K arm of the mTOR signaling cascade in oncogenesis has been less well investigated. The S6K family consists of two kinases, S6K1 and S6K2 (15-18). To date, both kinases are reported to be ubiquitously expressed, with mRNA expression detected in all mouse and human tissues examined (15-17). In addition, knockout mice for S6k1 or S6k2 alone demonstrate that there is redundancy between both genes, and it has also been suggested that inactivation of one may be compensated for through upregulation of the other (15, 19). This has made it difficult to properly understand the role of the S6K signaling arm in cancer. However, overexpression of both S6K1 and S6K2 have been reported in breast cancer (20), while a recent report showed that only S6k1 is required for insulinoma formation induced by expression of constitutively active Akt1 in the mouse pancreas (21). Thus, the tissue-specific requirement of both S6K1 and S6K2 in oncogenesis remains an open question, and one that is very relevant for the therapeutic targeting of this arm of the pathway.
Here we show that S6k1 genetic deletion can suppress phenotypes mediated by Pten haploinsufficiency in an exquisite tissue-specific fashion. This tissue-specificity appears to be dictated by the abundance of S6K2 protein levels. Indeed, we report for the first time that, in contrast to S6K1, S6K2 protein levels vary profoundly amongst different tissues.
Materials and methods
S6k1 and Pten mice
S6k1-/- and Pten+/- mice were previously generated (15, 22) and were crossed through several generations of breeding to ensure normalization of the background strains. All mouse work was performed in accordance with our IACUC approved protocol. For genotyping, tail DNA was subjected to PCR following the protocols previously described (22) (15).
Histopathology and immunoistochemistry
Mice were autopsied, tissue were extracted and fixed in 10% neutral-buffered formalin (Sigma) overnight, subsequently washed once with PBS, transferred into 50% ethanol and stored in 70% ethanol. After that, the tissues were embedded in paraffin, sectioned and stained with hematoxylin and eosin (H&E) in accordance with standard procedures. Sections were stained with the following antibodies: B220 (BD Pharmingen); CD3 (Dako); synaptophysin (Dako); phospho-S6 (S235/S236) (Cell Signaling Technology); Ki-67 (Novacastra); S6K1 (Cell Signaling Technology, 49D7 Rabbit mAb).
Quantitative RT-PCR
Quantitative RT-PCR. Total RNA was prepared from mice tissues using the Trizol method (Invitrogen). cDNA was obtained with iScriptTM cDNA Synthesis Kit (Bio-Rad). Taqman probes were obtained from Applied Biosystems (Foster City, California). Amplifications were run in a 7900 Real-Time PCR System (Applied Biosystems). Each value was adjusted by using Hprt1 levels as reference.
Western blot analysis on murine tissues
Cell lysates were prepared with RIPA buffer [1 × PBS, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail (Roche)] and cleaned by centrifugation. The following antibodies were used for western blot analysis: GAPDH (14C10 Cell Signaling Technology); S6K1 (Cell Signaling Technology, 49D7 Rabbit mAb); S6K2 [p70 S6 kinase β (C-19) Santa Cruz Biotechnology]; phospho-S6 (S235/S236) (Cell Signaling Technology); total S6 (Cell Signaling Technology); Pten (Cell Signaling Technology).
Human samples analysis
Human tissue lysates (Leinco Technologies) were probed with S6K1 (Cell Signaling Technology, 49D7 Rabbit mAb); S6K2 (Bethyl Laboratories); GAPDH (14C10 Cell Signaling Technology). Human sections were analyzed by IHC with S6K2 antibody (Bethyl Laboratories).
Original H & E sections from sixteen post-diagnostic cases of human pheochromocytoma were reviewed to verify the histological diagnosis. S6K1 immunostaining (Cell Signaling Technology, 49D7 Rabbit mAb) was evaluated independently by two pathologists (ML and GF) and scored as negative (0), weak (1), moderate (2) or strong (3). All human sections were collected following Institutional Review Board approval at the Brigham and Women's Hospital.
Phylogenetic analysis
Multiple alignments of amino acid sequences were performed with ClustalW (23). Amino acid sequences for human (Homo sapiens): RPS6Kbeta1 (NP_003152.1), RPS6Kbeta2 (NP_003943.2); cow (Bos taurus): RPS6Kbeta1 (NP_991385.1), RPS6Kbeta2 (XP_582478.4); rat (Rattus norvegicus): RPS6Kbeta1 (NP_114191.1), RPS6Kbeta2 (NP_001010962); mouse (Mus musculus): RPS6Kbeta1 (NP_001107806), RPS6Kbeta2 (NP_067460.1); rabbit (O cuniculus): RPS6Kbeta1 (NP_001095160); chicken (Gallus gallus): RPS6K (NP_001025892.1); zebrafish (Danio rerio): RPS6K (NP_998241.1); frog (Xenopus laevis): RPS6Kb-1 (NP_001080935).
Results
S6k1 deletion impacts Pten heterozygous driven phenotypes in a tissue-specific manner
To explore the contribution of the S6K1 arm of mTORC1 signaling to the phenotype dictated by Pten heterozygosity, we crossed Pten+/- mice with Rps6kb1-null (S6k1-/-) mice. In particular, we have focused on 4 specific genotypes for our study: wild-type (wt), S6k1-/-, Pten+/-, and S6k1-/-;Pten+/- mice. Initially, these cohorts were evaluated for long-term survival. Surprisingly, genetic deletion of S6k1 had no effect on the lifespan of Pten+/- mice (Fig. 1A). We have previously reported that Pten+/- mice die from an autoimmune disorder, characterized by a severe lymphadenopathy affecting mainly the submandibular, axillary and inguinal lymph nodes (4). Thus, we examined the S6k1-/-;Pten+/- mice to determine whether this was also the case for this population of mice. Indeed, the lymph nodes from Pten+/- and S6k1-/-;Pten+/- mice were found to be indistinguishable in composition, with lymph nodes from both genotypes exhibiting an expansion of B- and T-lymphocytes as characterized by immunohistochemical staining (IHC) with the B-lymphocyte specific marker B220 and the T-lymphocyte marker CD3 (Fig. 1B). Furthermore, lymph nodes from Pten+/- and S6k1-/-;Pten+/- mice showed no significant differences in positive staining for the cell proliferation marker Ki-67, (Fig. 1C, upper panel). Thus, these data suggest that S6k1 deletion does not impact the increased lymph node proliferation triggered by Pten heterozygous loss. Additionally, staining of S6k1-/-;Pten+/- lymph nodes for phosphorylation of the S6K1 downstream target RpS6 (phospho-S6) showed only a slight reduction in phosphorylation as compared with lymph nodes from Pten+/- mice (Fig. 1C, lower panel). This observation was further confirmed by western blot analysis and its quantification (Supplementary Fig. 1A).
Figure 1. Impact of S6k1 genetic deletion on lymph nodes proliferation and pheochromocytoma formation triggered by heterozygous loss of Pten.
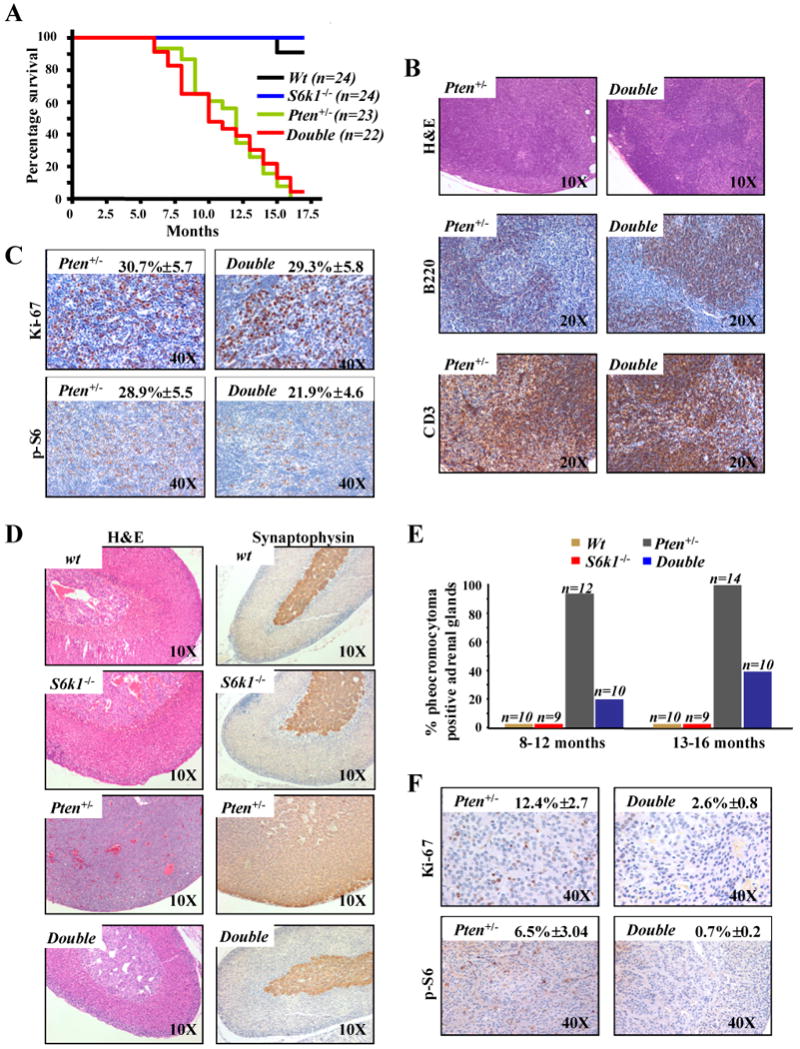
A, Kaplan-Meier plot for overall survival shows no difference between S61k-/-;Pten+/- (double) and Pten+/- mice. B, sections of lymph nodes from 12 month old S6k1-/-;Pten+/- and Pten+/- mice stained with: haematoxylin and eosin (H&E) (upper panel); anti-B220, a specific marker of B-lymphocytes (middle panel); anti-CD3, a specific marker of T-lymphocytes (lower panel). C, sections of lymph nodes from 12 month old S6k1-/-;Pten+/- and Pten+/- mice stained with: Ki-67 (upper panel); anti-phospho-S6 (lower panel). Quantifications of Ki-67 and phospho-S6 positive cells are indicated. D, sections of adrenal glands from 10 month old wt, S6k1-/-, Pten+/- and S6k1-/-;Pten+/- mice stained with: H&E (left panel) and anti-synaptophysin, a specific marker of the medulla of the adrenal gland (right panel). E, cumulative incidence of pheochromocytoma over time in wt, S6k1-/-, S6k1-/-;Pten+/- and Pten+/- mice. F, sections of adrenal gland from 10 month old S6k1-/-;Pten+/- and Pten+/- mice stained with: Ki-67 (upper panel); anti-phospho-S6 (lower panel). Quantifications of Ki-67 and phospho-S6 positive cells are indicated.
Taken together, our data suggest that S6k1 deletion has little impact on the proliferative advantage conferred by Pten heterozygous loss, consistent with the comparable survival rates between Pten+/- and S6k1-/-;Pten+/- cohorts of mice. Furthermore, the modest decrease in phospho-S6 staining and equivalent proliferation in the lymph nodes suggests redundancy between S6k1 and S6k2, as previously reported for the S6k1 deficient mice (15).
In order to further characterize the S6k1-/-;Pten+/- mice we examined the impact of S6k1 genetic deletion in tumorigenesis driven by Pten heterozygous loss. Pten+/- mice are highly prone to develop spontaneous epithelial tumors in a variety of tissues (5). However, some of the tumors driven by Pten heterozygosity occur after a long latency and at incomplete penetrance. Thus, we focused on pheochromocytoma as it is the most penetrant phenotype at earlier time points in our genetic background (5). As we have previously reported Pten+/- mice develop a marked expansion and proliferation of chromaffin cells in the medulla of the adrenal gland. This expansion is typical of pheochromocytoma, with 100% of Pten+/- mice showing this phenotype by 9 months of age (5). Strikingly, the S6k1-/-;Pten+/- mice showed a dramatic reduction in the proliferation of the chromaffin cells as compared with Pten+/- mice (Fig. 1D, left panel). This is confirmed by staining the adrenal glands with both synaptophysin and chromogranin A, specific markers of the adrenal medulla (Fig. 1D, right panel and Supplementary Fig. 1B respectively). Pathological analysis of the S6k1-/-;Pten+/- mice at 8-12 months of age revealed a 20% incidence of pheochromocytoma when compared to complete penetrance in age-matched Pten+/- mice (Fig. 1E). At 13-15 months of age the S6k1-/-;Pten+/- still showed a markedly lower penetrance of the tumor phenotype compared to Pten+/- mice (40% in S6k1-/-;Pten+/- versus 100% in Pten+/-) (Fig. 1E). We also identified a profound suppression of proliferation as characterized by Ki-67 staining in the adrenal medullas of the S6k1-/-;Pten+/- versus those of the Pten+/- mice (Fig. 1F, upper panel). In addition, deletion of S6k1 in the Pten+/- background resulted in a notable reduction in phoshpo-S6 in these adrenal glands (Fig. 1F, lower panel; and Fig. 2C), highlighting the relevance of the S6k1 arm of mTORC1 signaling in driving proliferation triggered by Pten heterozygous loss in this tissue. Thus, the marked impairment in Pten+/- driven pheochromocytoma indicates a lack of redundancy between S6k1 an S6k2 in this tissue and, in conjunction with the contrasting results obtained in the lymph nodes, clearly demonstrate that the genetic deletion of S6k1 has a differential impact on specific phenotypes and in a tissue-specific fashion.
Figure 2. S6K1 and S6K2 proteins are differentially expressed in mouse tissues.
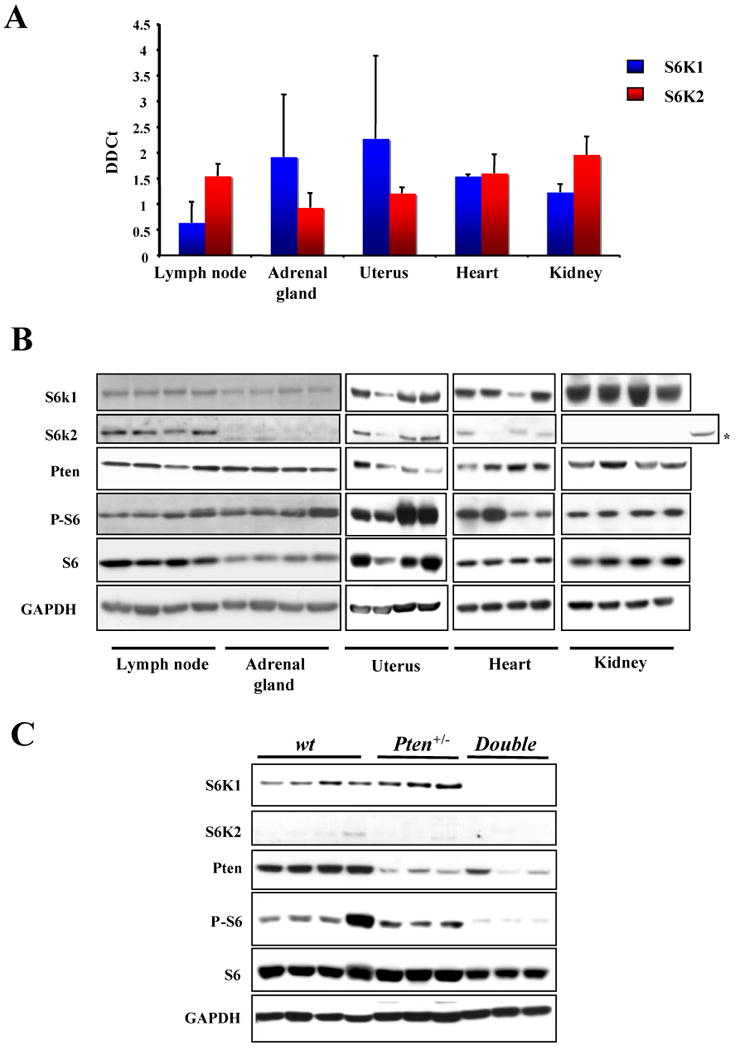
A, TaqMan RT-PCR analysis of the RNA from a panel of tissues from wt mice with S6k1 and S6k2 specific probes. Error bars show S.D. from three independent experiments. B, western blot analysis from the same tissues showed in A. The asterisk indicates S6k2 found in the lymph node loaded as a control C, western blot analysis from the adrenal gland of three different 13 month old mice of the indicated genotypes.
Interestingly, an analysis of the effect of S6k1 inactivation on uterine tumor formation in Pten+/- female mice, a less penetrant phenotype in this background (5), leads to a mildly reduced penetrance when compared with Pten+/- females alone (supplementary Fig. 2). However, the reduced penetrance is not as striking as that observed for the adrenal glands. These data suggest that there may be incomplete redundancy between S6k1 and S6k2 in this tissue which is in contrast to the complete redundancy observed with the lymph node phenotype of S6k1-/-;Pten+/- mice, and again support a tissue specific role for the S6k1 and S6k2 proteins.
Differential expression of S6k1 and S6k2 proteins in mouse tissues
In order to understand the molecular basis underlying the tissue specific differential response to deletion of S6k1, we decided to analyze in depth the expression status of both S6k1 and S6k2 across a panel of tissues. Previously published data have reported the mRNA for S6k1 and S6k2 to be ubiquitously expressed in all tissues analyzed (15-17). Indeed, using TaqMan real-time PCR assays we confirmed the expression of mRNA for both genes in tissues obtained from wt mice, indicating that they are actively transcribed in all organs tested (Fig. 2A). Surprisingly however, western blot analysis revealed a differential expression of the two proteins, with S6k2 levels found to vary profoundly (Fig. 2B and Supplementary Fig. 3). For example, comparing the amount of S6k2 protein between the lymph node and the adrenal gland shows strong differences in expression levels, while in contrast the S6k1 protein levels are comparable (Fig. 2B, left panel). In addition, it should be noted that S6k2 showed clear protein expression in the uterus. This data suggests the existence of a post-transcriptional mechanism that fine-tunes the levels of the S6k2 protein in a tissue specific manner. Additionally, we carried out western analysis on adrenal glands from wt, Pten+/- and S6k1-/-;Pten+/- mice and observed that S6k2 protein levels did not increase in the S6k1-/-;Pten+/- glands (Fig. 2C). This finding indicates that S6k2 does not compensate for the loss of S6k1 expression in this tissue.
Taken together, these data offer an explanation why S6k1 deletion has a differential impact on the phenotypes dictated by Pten heterozygosity. The minimal effect of S6k1 loss on uterine tumor formation and the lack of an effect on lymph node proliferation may be accounted for through compensation and redundancy with S6k2, while S6k1 deletion has a pronounced effect on the pheochromocytoma incidence due to low protein expression of S6k2 in the adrenal gland.
Analysis of S6K1 and S6K2 protein expression in human tissues
Given the surprising finding that S6k2 protein levels were differentially expressed in the mouse, we sought to uncover if this was also the case for the human adrenal gland and kidney, the two mouse tissues that showed dramatically reduced S6k2 expression. It has been previously reported that both S6K1 and S6K2 are ubiquitously expressed in humans at the mRNA level (16). However, as we showed in the mouse, western blot analysis of human adrenal gland revealed dramatically reduced S6K2 protein expression (Fig. 3A, left panel). In contrast, the human kidney tissue showed a relatively high S6K2 protein level, unlike our findings in mouse. This result was further confirmed by IHC (Fig. 3A, right panel). From these data it appears that not only can S6k2 protein expression vary dramatically between different tissues, but also the tissue specific pattern of expression can change between organisms. While S6k2 protein levels were markedly different amongst the human tissues tested, we again found S6K1 protein expression to be more consistent (Fig. 3A). This finding further highlights the key role of S6K1 in signaling downstream mTORC1 in the human adrenal gland, as is the case for the mouse.
Figure 3. S6K1 and S6K2 proteins expression in human tissues.
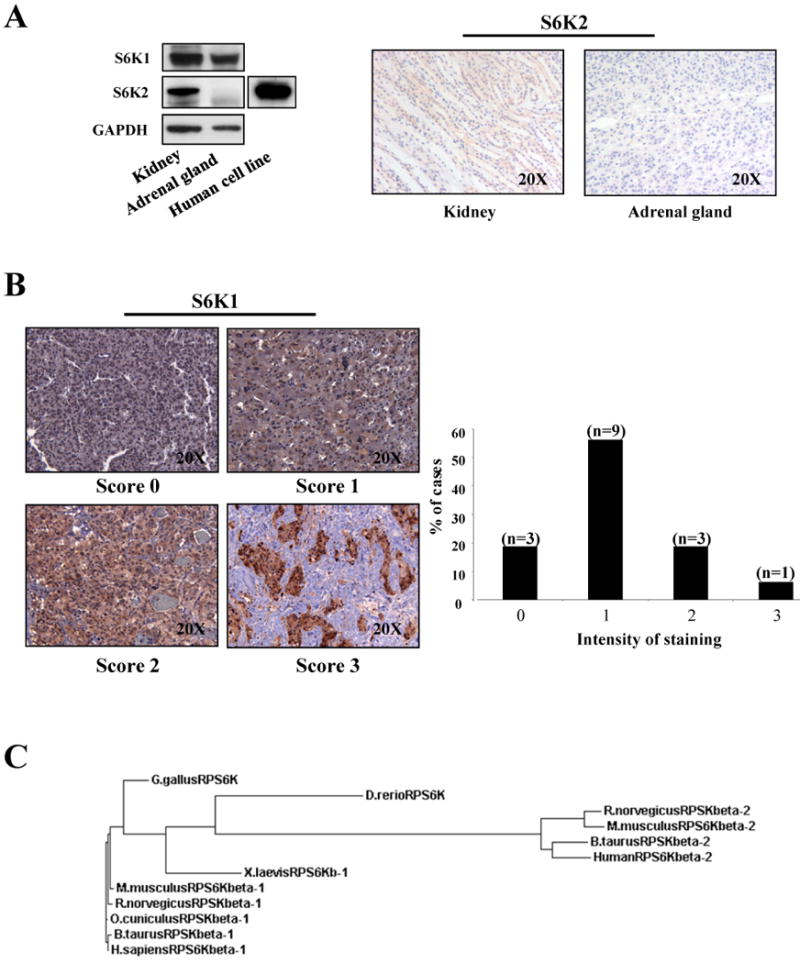
A, left panel: western blot analysis on human tissues lysates. As a control we have used a lysate from MCF7 human cell line. Right panel: IHC with anti-S6K2 on normal human tissues. B, staining with anti-S6K on 16 cases of human pheochromocytoma. S6K1 immunostaining was scored as negative (0), weak (1), moderate (2) or strong (3). C, phylogenetic tree indicates the relationship and evolutionary descent of various species based on protein sequence of S6K1 and S6K2. Specifically, RPSKbeta-1 refers to S6K1 protein and RPSKbeta-2 to S6K2.
Given that our data in mouse indicate a pivotal role for S6K1 in the proliferation of the adrenal medulla, and the fact that the human adrenal gland expresses mainly S6K1, we decided to analyze the protein expression status of S6K1 in human pheochromocytoma. We carried out IHC analysis on a number of human pheochromocytoma cases. Staining of S6K1 overexpression was quantified using a scale going from negative (0), weak (1), moderate (2) to strong (3) (Fig. 3B). The vast majority of pheochromocytoma cases expressed S6K1 at various levels (Fig. 3B), while the normal adjacent tissue scored as very weak (i.e. 0-1) for S6K1 expression (supplementary Fig. 4). Although these data set may be limited by a lack of additional information regarding the genetic determinants underlying these tumors, it is important to note that, 25% (4 out of 16) pheochromocytomas showed very high levels of S6K1 (Fig. 3B), suggesting that targeting of this kinase may represent a potential avenue for treatment of this tumor.
Discussion
Overall, our data are in agreement with a model whereby the response of Pten heterozygous phenotypes to the genetic deletion of S6k1 is tissue-specific. This specificity may be dictated by the relative protein expression of S6k2. Importantly, we have shown that S6K2 protein levels vary dramatically between different tissues, in humans as well as in mice, while S6K1 is less susceptible to such extreme differences in protein expression. A phylogenetic analysis demonstrates the fact that S6K1 is the ancestral kinase (Fig. 3C) and that S6K2 is only found in mammalian organisms (15). This suggests that S6K2 may have arisen at a later time and underscores our observation regarding the protein expression level of S6K1 relative to that for S6K2 across multiple tissues, highlighting the importance of S6K1 for therapeutic targeting.
Specifically, our data constitute the rationale to develop an S6K1-specific inhibitor to target tumors triggered by loss of PTEN in tissues having low expression of S6K2. The precise targeting of S6K1 in such tissues allows for inhibition of signaling downstream of mTORC1, while avoiding a general toxicity due to inhibition of both in normal tissues, as suggested by the perinatal lethality of the S6k1-/-;S6k2-/- mice. An excellent example for such an S6K1 targeted therapy is represented by the treatment of pheochromocytoma, as is clearly demonstrated through our genetic inactivation of S6k1 in the adrenal gland of Pten+/- mice and our concomitant human data.
Supplementary Material
Acknowledgments
We thank Leonardo Salmena and all the members of the Pandolfi laboratory for discussion and comments. We are grateful to Priyanka B. Patel and Kaitlyn Webster for their technical help with the histological work. This work is supported through MMHCC and NCI grants (5U01CA141464-02 and CA082328-12).
References
- 1.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–19. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 2.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–85. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 3.Nardella C, Carracedo A, Salmena L, Pandolfi PP. Faithfull Modeling of PTEN Loss Driven Diseases in the Mouse. Curr Top Microbiol Immunol. 347:135–68. doi: 10.1007/82_2010_62. [DOI] [PubMed] [Google Scholar]
- 4.Di Cristofano A, Kotsi P, Peng YF, Cordon-Cardo C, Elkon KB, Pandolfi PP. Impaired Fas response and autoimmunity in Pten+/- mice. Science. 1999;285:2122–5. doi: 10.1126/science.285.5436.2122. [DOI] [PubMed] [Google Scholar]
- 5.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–4. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 6.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471–84. doi: 10.1016/j.cell.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors. 2007;25:209–26. doi: 10.1080/08977190701779101. [DOI] [PubMed] [Google Scholar]
- 9.Mieulet V, Roceri M, Espeillac C, et al. S6 kinase inactivation impairs growth and translational target phosphorylation in muscle cells maintaining proper regulation of protein turnover. Am J Physiol Cell Physiol. 2007;293:C712–22. doi: 10.1152/ajpcell.00499.2006. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Li W, Williams M, Terada N, Alessi DR, Proud CG. Regulation of elongation factor 2 kinase by p90(RSK1) and p70 S6 kinase. Embo J. 2001;20:4370–9. doi: 10.1093/emboj/20.16.4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science. 2006;314:467–71. doi: 10.1126/science.1130276. [DOI] [PubMed] [Google Scholar]
- 12.Ruggero D, Montanaro L, Ma L, et al. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–6. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 13.Furic L, Rong L, Larsson O, et al. eIF4E phosphorylation promotes tumorigenesis and is associated with prostate cancer progression. Proc Natl Acad Sci U S A. 107:14134–9. doi: 10.1073/pnas.1005320107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sonenberg N. eIF4E, the mRNA cap-binding protein: from basic discovery to translational research. Biochem Cell Biol. 2008;86:178–83. doi: 10.1139/O08-034. [DOI] [PubMed] [Google Scholar]
- 15.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. Embo J. 1998;17:6649–59. doi: 10.1093/emboj/17.22.6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gout I, Minami T, Hara K, et al. Molecular cloning and characterization of a novel p70 S6 kinase, p70 S6 kinase beta containing a proline-rich region. J Biol Chem. 1998;273:30061–4. doi: 10.1074/jbc.273.46.30061. [DOI] [PubMed] [Google Scholar]
- 17.Koh H, Jee K, Lee B, et al. Cloning and characterization of a nuclear S6 kinase, S6 kinase-related kinase (SRK); a novel nuclear target of Akt. Oncogene. 1999;18:5115–9. doi: 10.1038/sj.onc.1202895. [DOI] [PubMed] [Google Scholar]
- 18.Lee-Fruman KK, Kuo CJ, Lippincott J, Terada N, Blenis J. Characterization of S6K2, a novel kinase homologous to S6K1. Oncogene. 1999;18:5108–14. doi: 10.1038/sj.onc.1202894. [DOI] [PubMed] [Google Scholar]
- 19.Pende M, Um SH, Mieulet V, et al. S6K1(-/-)/S6K2(-/-) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol Cell Biol. 2004;24:3112–24. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Filonenko VV, Tytarenko R, Azatjan SK, et al. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp Oncol. 2004;26:294–9. [PubMed] [Google Scholar]
- 21.Alliouachene S, Tuttle RL, Boumard S, et al. Constitutively active Akt1 expression in mouse pancreas requires S6 kinase 1 for insulinoma formation. J Clin Invest. 2008;118:3629–38. doi: 10.1172/JCI35237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–55. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.