

Abstract
A screening of conformationally constrained aromatic amino acids as base cores for the preparation of new NK1 receptor antagonists resulted in the discovery of three new NK1 receptor antagonists, 19 [Ac-Aba-Gly-NH-3′,5′-(CF3)2-Bn], 20 [Ac-Aba-Gly-NMe-3′,5′-(CF3)2-Bn] and 23 [Ac-Tic-NMe-3′,5′-(CF3)2-Bn], which were able to counteract the agonist effect of substance P, the endogenous ligand of NK1R. The most active NK1 antagonist of the series, 20 [Ac-Aba-Gly-NMe-3′,5′-(CF3)2-Bn], was then used in the design of a novel, potent chimeric opioid agonist-NK1 receptor antagonist, 35 [Dmt-D-Arg-Aba-Gly-NMe-3′,5′-(CF3)2-Bn], which combines the N-terminus of the established Dmt1-DALDA agonist opioid pharmacophore (H-Dmt-D-Arg-Phe-Lys-NH2) and 20, the NK1R ligand. The opioid component of the chimeric compound 35, i.e. Dmt-D-Arg-Aba-Gly-NH2 36, also proved to be an extremely potent and balanced μ- and δ opioid receptor agonist with subnanomolar binding and in vitro functional activity.
Keywords: NK1 receptor antagonists, opioids, multitarget drug design, designed multiple ligands
Introduction
Substance P (SP, Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met-NH2), an undecapeptide neurotransmitter or neuromodulator, is a member of the tachykinin family of peptides. These peptides mediate their biological functions through binding to three neurokinin G protein-coupled receptors, NK1, NK2 and NK3, which have as endogenous ligands the peptides SP, neurokinin A and neurokinin B, respectively.1 The NK1 receptor is expressed both in the CNS and in peripheral tissues. The observation that the release of SP is linked to the transmission of pain and inflammatory responses related to noxious stimuli,2 makes SP receptor antagonists (NK1R antagonists) potential therapeutic agents for pathologies such as postoperative pain, migraine,3 arthritis,4 asthma, nausea and emesis.1,5,6
Small peptidomimetic NK1R antagonists have been developed and often possess a bis-aromatic motif, typically consisting of a substituted (-OMe or -bis-CF3) phenyl group next to another aromatic ring that is coupled to a central scaffold (e.g. 1–4, Figure 1).7–9 The relative disposition of the aromatics in such ligands was investigated in a large set of SP antagonists and resulted in a hypothesis for the receptor-bound conformation of NK1R antagonists.9,10 In most cases, the two aromatic groups, crucial for high receptor affinity, are in proximity of each other and are proposed to adopt either a parallel face-to-face9 or a perpendicular “T” or edge-on “L” arrangement. 10
Figure 1.

Structures of reported peptidomimetic NK1R antagonists
Biologically, prolonged pain states lead to neuroplastic changes in the CNS that modify/increase the release of certain neurotransmitters, such as the pronociceptive neurotransmitter SP, and increase the expression of the corresponding receptors for these released pain-enhancing ligands.11–13 The current treatment of chronic pain cannot counteract these changes and hence the currently used analgesics are not effective in these pathological conditions. For this reason, an approach was taken to design bifunctional ligands that act as agonists at opioid receptors and as antagonists at NK1 receptors.11,16,17,18 The concept for this type of designed multiple ligand (DML)19 was originally developed by Lipkowski and coworkers.18,20–22 Antagonism at the NK1 receptors blocks the signals induced by SP, the native ligand for NK1R, and would potentially inhibit the increased secretion of the peptide hormone SP and the enhanced expression of NK1 receptors upon sustained opioid administration or in prolonged pain states, while the opioid subunit of these DMLs, on the other hand, would be responsible for the activation of μ-opioid (MOR) and δ-opioid (DOR) receptors to produce the analgesic effect. Moreover, the administration of morphine to NK1 knockout mice did not show the morphine-related rewarding properties.23
In general, the advantages of multitarget agents include enhanced efficacy, relative to drugs that use the “one active agent, one target” approach by modulating multiple targets simultaneously and lower risk of drug-drug interaction which can potentially cause unexpected side-effects and/or toxicity. Because poor safety and a lack in efficacy are the main causes of failure of compounds in clinical trials, the approach of multitarget drug discovery is a research area of increasing interest in drug discovery programs.24 Other advantages of a single ligand with multiple biological activities over a cocktail of individual drugs are: i) easy administration, ii) single biodistribution, iii) simple pharmacokinetic profile, and iv) higher concentrations at the synaptic cleft, which may lead to significant synergies in potency and efficacy.24
Hruby and Lipkowski synthesized three lead chimeras, 6 (TY 027, [H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-3′,5′-Bn(CF3)2]), 7 (H-Tyr-D-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-3′,5′-Bn(CF3)2) and 8 (H-Tyr D-Ala-Gly-Phe-NH-NH-Trp-Cbz) (Figure 2), that contain an enkephalin-based opioid pharmacophore at the N-terminus and a NK1 receptor-binding subunit at the C-terminus.11,16,22,24–27 Gratifyingly, the dual opioid-NK1R activity resulted in both enhanced antinociception in acute pain models and prevention of opioid-induced tolerance in chronic trials for the peptidic bifunctional ligand 6 and its ester analog 7.16,25 Compounds 6 and 7 were shown to potently attenuate neuropathic pain without producing opioid-induced tolerance and hence they seem to validate the concept or approach of using bifunctional opioid agonist-NK1 antagonist chimeras to tackle this type of pathology. 28
Figure 2.

Examples of reported bifunctional opioid-NK1 ligands 6, 7 and 8.
Previously, we have suggested the 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one 9 (Aba) and amino indoloazepinone 10 (Aia) (Figure 3) to represent privileged templates. The appropriate derivatization of the central core scaffolds 9 and 10 allowed to obtain (selective) ligands for several G protein-coupled receptors. Examples include: opioid GPCRs,29,30 bradykinin B231 and somatostatin receptors.32,33
Figure 3.

General structures of the 4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one 9 (Aba), amino indoloazepinone 10 (Aia), tetrahydroisoquinoline 11 (Tic) and tetrahydro-β-carboline 12 (Tcc) scaffolds.
In this work we present the 3′,5′-bistrifluoromethyl benzyl derivatization of the conformationally constrained amino acids Aba 9 and Aia 10 and, in addition, of the tetrahydroisoquinoline-3-carboxylic acid (Tic) 11 tetrahydro-β-carboline-3-carboxylic acid (Tcc) 12 templates for the development of new NK1R antagonists. The 1-phenyl-benzazepine core 9 (R1 = Ph) was chosen as an alternative for the benzodiazepine core in 4 (Figure 1),14 whereas the indoloazepine core 10 could be considered to lead to a constrained analogue of the potent antagonist 5.15
The biological evaluation of all newly prepared compounds for NK1R antagonism, led to the identification of 3 potent antagonists. This discovery allowed us to integrate the novel NK1R pharmacophore into the design of a compact peptidomimetic bifunctional opioid-NK1R ligand containing an opioid agonist component derived from the potent μ opioid agonist [Dmt1]DALDA (H-Dmt-D-Arg-Phe-Lys-NH2; Dmt = 2′,6′-tyrosine).34 Here, we present the design rationale, synthesis and in vitro biological evaluation of both NK1R ligands and a dual opioid-NK1R ligand.
Results and Discussion
Synthesis
Using an earlier reported methodology the Phth-(1S/R)-(4S)-amino-1,2,4,5-tetrahydro-2-benzazepin-3-ones stereoisomers 13 and 14 were obtained.35
N-methyl-3′,5′-bistrifluoromethyl benzylamine was prepared by N-methylation of the corresponding, commercially available, benzyl chloride.36 Coupling of this secondary amine, or the primary 3′,5′-bistrifluoromethyl benzylamine, with TBTU as a coupling reagent in the presence of Et3N gave the N-protected Phth-(1R)-4-amino-(4S/R)-benzazepinone intermediates. Removal of the phthaloyl protective group via hydrazinolysis was completed in refluxing EtOH and followed by a standard acetylation using acetic acid anhydride and DIPEA as a base. The final compounds 15 to 17 were purified by preparative RP-HPLC followed by lyophilization (purity ≥ 95%).
These compounds were structurally analogous to investigated benzodiazepines of type 4 (Figure 1) that possessed the desired NK1R antagonism.14 In this benzodiazepine series, as described by Armour et al., the role and necessity of the third aromatic moiety was questioned and this motivated us to make the compounds without the 1-phenyl substitution (Scheme 2). As mentioned in the introduction, numerous examples of NK1R antagonists containing only two aromatics are published (selected examples: 1 to 3 and 5 in Figure 1).
Scheme 2.

Synthesis of Aba and Tic analogues 19, 20, 22 and 23.
Reaction conditions: a) 1.1 eq. 3′,5′-bistrifluoromethyl benzylamine or N-methyl-3′,5′-bistrifluoromethyl benzylamine, 1.1 eq. TBTU, 3 eq. TEA, CH2Cl2, r.t. 2h; b) TFA/CH2Cl2/anisole 49:49:2, r.t. 3h; c) 5 eq. Ac2O, TEA, EtOH, r.t., 3h.
We started from the dipeptide mimetic Boc-(S)-Aba-Gly-OH 1837 which was again coupled to both N-methyl- and 3′-5′-bistrifluoromethyl benzylamine. Removal of the Boc group by classic acidolysis (TFA/CH2Cl2/anisole 49:49:2) and acetylation, followed by HPLC purification, gave aminobenzazepinones 19 and 20. The identical pathway starting from commercial Boc-(S)-Tic-OH 21 yielded tetrahydroisoquinoline analogues 22 and 23.
The seven- and six-membered Trp-based constrained Aia and Tcc analogues were prepared from the Boc-2′-formyl-(S/R)-Trp-OH 24a/b32 and Boc-(S/R)-Tcc-OH 25a/b,38 respectively (Scheme 3). Reductive amination with methyl glycinate and sodium cyanoborohydride was immediately followed by EDC-mediated lactamization. The latter reaction was slow and needed 48h to give complete conversion to 26a/b. Saponification with LiOH and standard coupling using TBTU gave 27a/b. The final acetylated structures 28a/b were obtained using the procedure for the Aba and Tic ligands, after acidolysis and treatment with acetic anhydride.
Scheme 3.

Synthesis of Aia and Tcc analogues 28a/b, 30 and 32a/b.
Reaction conditions: a) 1.05 eq. H-GlyOMe.HCl, 20 wt% MgSO4, 2.5 eq. NaBH3CN, CH2Cl2, pH 6, r.t., 4h; b) 1.5 eq. EDC.HCl, 2.5 eq. pyridine, acetonitrile:water 1:1, r.t., 2 days; c) 4 eq. 1M aq. LiOH, MeOH, r.t., 1.5h; d) 1.1 eq. N-methyl-3′,5′-bistrifluoromethyl benzylamine, 1.1 eq. TBTU, 3 eq. NEt3, r.t., 2h; e) 5% H2O in TFA:acetonitrile 4:1, r.t., 1h; f) acetonitrile:H2O 1:1, NEt3, pH 6, 5 eq. Ac2O, r.t., 2h; g) 1.05 eq. 3′,5′-bistrifluoromethyl benzylamine, 20 wt% MgSO4, 2.5 eq. NaBH3CN, CH2Cl2, pH 6, r.t., 4h.
A constrained analogue of 5 (Ac-Trp-O-3′,5′-(CF3)2Bn,39 Figure 1) was synthesized starting from 24a via reductive alkylation with 3′,5′-bistrifluoromethyl benzylamine and carbodiimide–induced cyclization, to afford indoloazepinone 29. Standard Boc deprotection and acetylation gave 30. Moreover, both (S)- and (R)-Tcc derivatized structures 32a and 32b were obtained after a coupling/deprotection/acetylation sequence similar to that used for the Tic-analogues 22 and 23. Finally, the chimeric opioid-NK1R DML 35 (Scheme 4) was prepared after identification of the most potent NK1R antagonist, proven to be compound 20 (see section Biological evaluation). For the synthesis of the double pharmacophore-containing peptidomimetic structure we combined an adapted (constrained) version of Dmt1-[DALDA] (Dmt-D-Arg-Phe-Lys-NH2) with 20. As such, the N-terminal fragment Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH was first prepared using a Fmoc solid phase peptide synthesis strategy on 2-chlorotrityl resin with DIC as a coupling reagent and HOBt as an additive (Scheme 4). Boc-Dmt-OH was coupled as the final residue, so that the N-terminal Boc group would be cleaved simultaneously with the acid labile Pbf side-chain protective group. First, the protected sequence Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH 33 was removed from the 2-Cl-trityl resin under mild acidic conditions (1% TFA in CH2Cl2 for 30 min). Introduction of the C-terminal pharmacophoric unit, the N-methyl-3′,5′-bistrifluoromethyl benzylamide, was performed by a coupling the secondary amine with 33 by means of BOP in the presence of diisopropylethylamine in dichloromethane over 3h at room temperature. Final cleavage of the protection groups in 34 was achieved by treatment with TFA/CH2Cl2 49/49 (2h at r.t.) and anisole (2%) to scavenge the released carbocations during deprotection. The final compound 35 was purified by preparative HPLC (≥95% purity ).
Scheme 4.

Synthesis of bifunctional chimera 35.
Reaction conditions: a) Fmoc-Aba-Gly-OH, diisopropylethylamine, CH2Cl2, r.t., overnight; b) 20% piperidine/DMF (5min+15min); c) Coupling of Fmoc-D-Arg(Pbf)-OH and DIC/HOBt in DMF r.t., 2h; d) Coupling of Boc-Dmt-OH and DIC/HOBt in DMF r.t., 2h; e) 1% TFA in CH2Cl2 for 30 min; f) Coupling of N-methyl-3′,5′-bistrifluoromethyl benzylamine with BOP/DIPEA in DMF, r.t., 3h; g) TFA/CH2Cl2/anisole 49:49:2, r.t., 2h.
The peptidic sequence for verification of the opioid activity of the N-terminal opioid component, H-Dmt-D-Arg-Aba-Gly-NH2 36, was prepared by standard SPPS using Nα-Fmoc chemistry on Rink resin with DIC/HOBt as the coupling mixture (not shown). After cleavage from the solid support, the solvent was removed in vacuo, the resulting crude mixture was washed with chilled Et2O, and the precipitate purified via preparative HPLC.
Biological Evaluation and Structure-Activity Relationships
The functional activity of the potential NK1 receptor ligands was evaluated for NK1R agonism and antagonism using an assay measuring the agonist-induced calcium-dependent aequorine-luminescence of cells expressing NK1 receptors.40
None of the derivatized 1-phenyl-Aba- (Scheme 1), Aba- and Tic- (Scheme 2), and Aia- and Tcc-analogues (Scheme 3) showed any significant agonism up to a concentration of 10−4M (not shown). This is in contrast to SP which gave the expected NK1R activation and served as the positive agonist control compound. Next, the targeted neurokinin 1 receptor antagonism of all compounds was evaluated. Three compounds (19, 20 and 23) were able to counteract the effect of SP, as proven by the rightward shift of the dose-response curve of SP in the functional receptor assay (see supporting information for graphical data).
Scheme 1.

Synthesis of 1-phenyl Aba analogues 15 to 17.
Reaction conditions: a) 1.1 eq. 3′,5′-bistrifluoromethyl benzylamine or N-methyl-3′,5′-bistrifluoromethyl benzylamine, 1.1 eq. TBTU, 3 eq. TEA, CH2Cl2, r.t. 2h; b) 2 eq. NH2NH2.H2O, EtOH, reflux, 1.5h; c) 5 eq. Ac2O, TEA, EtOH, r.t., 3h.
These results demonstrate that both Tic and Aba can serve as a core scaffold for the development of NK1R antagonists and that for the Aba-type central scaffolds, the 1-phenyl substituent hinders antagonism at N1KR for both (4S)- and (4R) configurations (15–17). In contrast, the constrained tryptophan analogues 28a/b, 30 and 32a/b, obtained by derivatization of both 24a/b and 25a/b, did not show any antagonist properties. This indicates that these types of conformational constraints are not mimicking the bioactive conformation of 5 or that the change of the ester function in 5 into an amide function is not tolerated. These structural features are however also present in the most potent NK1R antagonist in the series, analogue 20. The preferred conformations of 20 was studied by molecular modeling using Macromodel.54 The three lowest energy conformations which differ by only 4.72 kJ/mol, have orientations of the aromatic rings corresponding to the stacked (Figure 4A), the perpendicular T- (Figure 4B) or L-orientations (Figure 4C).9,10 The distances between the centroids of the aromatic rings (4.25, 5.33 and 5.49 Å, respectively) are within the range proposed for 3′,5′-bistrifluoromethyl benzyl-containing NK1R antagonists.10 This suggests that 20 is able to orient its aromatic rings for the most efficient interaction with the NK1R. In the indole analogue of 20, compound 28a, similar conformations were found. However, the pyrrole ring in 28a shifts the annulated benzene ring to a further distance from the 3′,5′-bistrifluoromethyl benzene ring than in 20. This might account for the difference in NK1R antagonist potency.
Figure 4.

The three lowest energy conformations of compound 20 showing a stacked orientation of the aromatics (A), a T-perpendicular orientation (B, 3.94 kJ/mol above A) and a L-perpendicular orientation
The calculated pA2 values (Table 1) allowed to rank the relative potency of the Aba- and Tic-derived structures 19, 20 and 23.41,42 These values indicate that a N-methylation at the level of the C-terminal amide bond resulted in improved antagonist activity [pA2(19) = 6.2 vs. pA2(20) = 8.4 and pA2(23) = 7.5] and that the lactam constraint of 20, in combination with the carboxymethyl chain of Gly, was favored over the isoquinoline constraint in 23.
Table 1.
Structures, functional activities and affinities of NK1-, opioid- and opioid-NK1 ligands
| Structure | Comp. | NK1R pA2a | hNK1R Ki (nM)b | GPI(μ) IC50 (nM) c | MVD(δ) IC50 (nM) c | MOR Ki (nM) c | DOR Ki (nM)c | KOR Ki (nM) c |
|---|---|---|---|---|---|---|---|---|
|
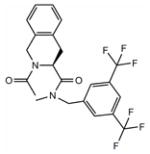
23 | 7.5 | 32±4 | / | / | / | / | / |
|
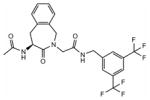
19 | 6.2 | 387±66 | / | / | / | / | / |
|
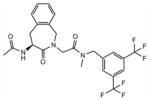
20 | 8.4 | 27±4 | / | / | / | / | / |
|
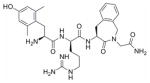
36 | / | / | 0.32 ± 0.04 | 0.42 ± 0.02 | 0.15 ± 0.02 | 0.60 ± 0.07 | 118 ± 12 |
|
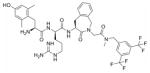
35 | 7.8 | 0.5±0.1 | 8.51 ± 0.62 | 43.3 ± 6.3 | 0.416 ± 0.012 | 10.4± 0.6 | 445 ± 81 |
The pA2 values were calculated using the Schild’s equation.41
Inhibitory constants (Ki) of NK1 receptor ligands, measured for the receptor prototype [3H]-SP in the presence of hNK1-CHO membranes. Results are means ± SEM of three independent experiments. Binding data were calculated using the nonlinear regression/one site competition fitting options of the GraphPad Prism Software.
Values represent means of 3–6 experiments ± SEM. The GPI functional assay is representative of MOR activation, whereas the MVD is a δ receptor-representative assay. Binding affinities of compounds for μ and δ opioid receptors were determined by displacing [3H]DAMGO and [3H]DSLET, respectively, from rat brain membrane binding sites and binding affinities for κ opioid receptors were measured by displacement of [3H]U69,593 from guinea pig brain membrane binding sites.
In a next step, the binding affinity (Ki) of the active compounds was determined on CHO membranes expressing the human NK1 receptor. These data show that the antagonist potency, indicated as pA2, of these compounds is in accordance with the receptor binding affinities and gives the relative rank order of 19 < 23 < 20. The moderate nanomolar binding affinity of 19 and lack of antagonism of 22 offers proof that, for compounds of type 19–20 and 22–23, the presence of a hydrogen bond donor at the C-terminal benzyl amide, is unfavorable for receptor binding and antagonist activity. Both 20 and 23 possess a tertiary amide linkage which results in increased hydrophobicity and eliminates the possibility for hydrogen bond formation at the amide bond site.
Earlier results have demonstrated that the use of the Aba scaffold in opioid peptides results in very potent agonists with high receptor affinity.30,43–45 Several examples were reported in which Aba serves as the third residue in synthetic peptidic opioid ligands (e.g. Tyr1-D-Ala2-Aba3-Gly4-NH2,39 Tyr1-D-Ala2-Aba3-Gly4-Tyr5-Pro6-Ser7-NH2.46 This conformational constraint yields a substantial enhancement in DOR affinity, while maintaining MOR binding and functional activity. In addition, the presence of a positive charge in the side chain of a D-Arg residue in position 2 is suggested to play a role in the excellent membrane transport properties of Dmt 1-D-Arg2-Phe3-Lys4-NH2.47,48 For these reasons, we prepared the opioid ‘control’ compound Dmt-D-Arg-Aba-Gly-NH2 36 via solid phase peptide synthesis using Fmoc chemistry with Rink amide resin as a solid support. As can be seen from Table 1 both the opioid receptor affinities and functional activities of 36 are extremely high and balanced for the μ- and δ-receptor, as indicated by subnanomolar values in all assays. On the other hand, this compound showed weak receptor binding affinity. These data also indicate that D-Arg in position 2 can be combined with the conformational constraint imposed by Aba3 to provide a potent and balanced μ/d opioid component for the DML.
The framework combination24 of potent ligands for the two separate targets resulted in multitarget ligand 35, which is a combination of the most potent NK1R antagonist of the series, Ac-Aba-Gly-NMe-3′,5′-(CF3)2Bn 20, and the very potent opioid ligand Dmt-D-Arg-Aba-Gly-NH2 36. Although a modest loss in NK1R antagonism was observed for 35 (pA 2 8.4 → 7.8), subnanomolar hNK1R binding (Ki 0.5 nM) was determined and proves that the presence of the opioid subunit even adds favorable features for efficient binding to this G protein-coupled receptor. As for the opioid in vitro characteristics, one can see that MOR binding is less disrupted, compared to DOR affinity, when both frameworks are combined. A three-fold loss in μ opioid receptor -but still subnanomolar- binding affinity was determined (Ki(μ) 0.15 → 0.416 nM), whereas a 17-fold loss in DOR affinity was observed (Ki(δ) 0.6 → 10.4 nM), relative to the opioid ‘control’ compound 36. As expected on the basis of the weak receptor binding affinity of 36, chimera 35 showed weak κ receptor binding affinity as well. The receptor binding affinities are in agreement with the results obtained in the in vitro functional GPI and MVD tissue bioassays. In the guinea pig ileum assay, reflecting mainly μ opioid receptor activation, a low nanomolar IC50 value of 8.51 nM indicates that, even though a potency loss is observed relative to 36, DML 35 still is a very potent agonist at the opioid μ receptor. The results of the MVD assay indicate that DOR agonist activity is still maintained but only with a two digit nanomolar IC50 value.
The creation of bifunctional ligands of the type of compound 35 in general is a difficult task because the connected pharmacophores can interfere with each other to change or reduce their binding and activity at the corresponding target receptors. Therefore the merged DML 35 is a very successful example of a dual ligand in which a large overlap of pharmacophores is present. The central Aba structure is part of both pharmacophores. In addition, our earlier research had shown that a benzyl amide moiety was tolerated in the shorter opioid ligand Dmt-Aba-Gly-NH- Bn for both MOR and DOR,30 although this subunit was shifted by one position (D-Arg in position 2 is inserted) in hybrid 35 described here.
Conclusions
Different constrained Phe and Trp amino acid mimics were used as central scaffolds in the design of new NK1R analogues. Unfortunately, all Trp-derivatives were inactive, both as agonists and antagonists, in the functional assay measuring the agonist-induced calcium-dependent aequorine- luminescence of cells expressing NK1 receptors. Gratifyingly, the derivatization of the Ac-Tic and Ac-Aba-Gly scaffolds as the 3′,5′-bistrifluoromethyl benzylamides resulted in moderate (structure 19) to good (structures 20 and 23) NK1R antagonists. The commonality of the Aba ring in both opioid and NK1R ligands, prompted us to combine both pharmacophores and produce successfully a compact bifunctional opioid-NK1R ligand (structure 35) that targets both the opioid and the neurokinin system. This study also identified opioid control compound 36, as an extremely potent and balanced μ- and δ opioid receptor agonist in vitro, which deserves further investigation.
Chimeric ligand 35 can be considered as a small DML with highly merged frameworks. In general medicinal chemists try to maximize the degree of overlap, and hence minimize molecular weight, in order to have a better chance at oral activity.24 Ongoing research consists of the structural fine-tuning of this new lead compound. More extensive in vitro and in vivo assays are scheduled to verify the membrane permeability of this dual opioid agonist-NK1 receptor antagonist through important biological barriers, such as the blood-brain barrier (BBB).
Experimental section
General
Thin layer chromatography (TLC) was performed on plastic sheet pre-coated with silica gel 60F254 (Merck, Darmstadt, Germany) using specified solvent systems. Mass Spectrometry (MS) was recorded on a Micromass Q-Tof Micro spectrometer using electrospray (ESP) ionization (positive or negative ion mode). Data collection was done with Masslynx software. Analytical RP-HPLC was performed using an Agilent 1100 Series system (Waldbronn, Germany) with a Supelco Discovery BIO Wide Pore® (Bellefonte, PA, USA) RP C-18 column (25 cm × 4.6 mm, 5 μm) using UV detection at 215 nm. The mobile phase (water/acetonitrile) contained 0.1% TFA. The gradient consisted of a 20 min run from 3 to 97% acetonitrile at a flow rate of 1mL/min. Preparative HPLC was performed on a Gilson apparatus and controlled with the software package Unipoint. The Reverse Phase C18-column (Discovery BIO Wide Pore 25 cm × 21.2 mm, 10 μm) was used under the same conditions as the analytical RP-HPLC, but with a flow rate of 20 mL min−1. A purity of more than 95% was determined for all compounds by analytical RP-HPLC using the conditions described above. 1H-NMR and 13C-NMR spectra were recorded at 250 MHz and 63 MHz, respectively, on a Bruker Avance 250 spectrometer or at 500 MHz and 125 MHz on a Bruker Avance II 500. Calibration was done with TMS (tetramethylsilane) or residual solvent signals as an internal standard. The solvent used is mentioned in all cases and the abbreviations used are: s (singlet), d (doublet), dd (double doublet), t (triplet), br s (broad singlet), m (multiplet).
Functional NK1R assay40
Cell line and cell culture conditions
The Chinese Hamster Ovary K1 (CHO-K1) cell line, stably expressing human NK1 receptor (hereafter referred to as CHO-NK1 cells), was transfected with an apoaequorin expression vector (pER2) using Fugene6 (Roche Applied Science). The cell line and expression vector were obtained from Euroscreen (Belgium). The CHO-NK1 cells were cultured in sterile DMEM/HAM’s F12 medium (Sigma) supplemented with 10% foetal bovine serum, 100 IU/ml penicillin, 100 μg/ml streptomycin and 400 μg/ml G 418 (Geneticin, Gibco), at 37°C with 5% CO2 and were trypsinized every 3 days.
Aequorin charging protocol
Transfected cells in the mid-log phase were detached by changing the growth medium for PBS buffer supplemented with 5mM EDTA (pH 8). The cells were spun down and incubated for 4h at a concentration of 5× 106 cells/ml in DMEM-F12 medium without phenol red (Gibco) supplemented with 0.1% BSA (BSA-medium) and 5 μM coelenterazine h (Molecular Probes). After coelenterazine loading, the cells were diluted 10-fold in the same medium and incubated for an additional period of 30 minutes. The cells were mildly shaken during the incubation periods.
Aequorin luminescence assay
A dilution series of peptide agonist (Substance P was purchased from Sigma) ranging from 10−11 to 10−4 M was distributed in a white 96-well plate. For investigating antagonism, the synthetic compounds were added to these wells to obtain the desired concentrations (ranging from 10−8 to 10−4 M). One negative control sample (BSA medium only) was included in each row of the 96-well plate. The plate was loaded in a “Multimode Reader Mithras, LB940” (Berthold). The wells were screened one by one and each measurement started at the moment of injection of 50μl of the coelenterazine-loaded cell suspension, containing 2.5× 104 cells. Light emission was measured every second for 30s after which 50μl of 10nM ATP solution (positive control) was injected. Each measurement was carried out in duplicate. Light emission was recorded for an additional period of 10s per well and the data were presented in Relative Light Units (RLU).
Data Analysis
Luminescence data (peak integration) were calculated using MikroWin 2000 software (Berthold), which was linked to the Microsoft Excel program. All statistical and curve-fitting analyses were performed using Prism 4.0 (GraphPad) software. Data are expressed in percentage (% RLU) of the maximal luminescence that was detected with 10−4 M SP (without antagonist). The competitive nature of antagonism was evaluated using the Schild plot method.41 All antagonists analyzed in this study provided linear regression plots and were considered competitive. The pA2 values were calculated using the Schild’s equation.42
hNK1/CHO cell membrane preparation and Radioligand binding assay
Recombinant hNK1/CHO cells were grown to confluency in 37 °C, 95% air and 5% CO2, humidified atmosphere, in a Forma Scientific (Thermo Forma, OH, USA) incubator in Ham’s F12 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin,100 μg/ml streptomycin and 500 μg/ml Geneticin. The confluent cell monolayers were then washed with Ca2+, Mg 2+-deficient phosphate-buffered saline (PD buffer) and harvested in the same buffer containing 0.02% EDTA. After centrifugation at 2700 rpm for 12 min, the cells were homogenized in ice-cold 10 mM Tris-HCl and 1 mM EDTA, pH 7.4 buffer. A crude membrane fraction was collected by centrifugation at 18,000 rpm for 12 min at 4°C and the pellet was suspended in 50mM Tris-Mg buffer and protein concentration of the membrane preparation was determined by using Bradford assay.
Six different concentrations of the test compound were each incubated, in duplicates, with 20 μg of membrane homogenate and 0.4 nM [3H] substance P (135 Ci/mmol, Perkin-Elmer, USA) in 1 mL final volume of assay buffer (50 mM Tris, pH 7.4, containing 5 mM MgCl2, 50 μg/mL bacitracin, 30 μM bestatin, 10 μ M captopril, and 100 μM phenylmethylsulfonylfluoride) Substance P at 10 μM was used to define the nonspecific binding. The samples were incubated in a shaking water bath at 25 °C for 20 min. The [3H] substance P concentration and the incubation time was selected based on the studies of Yamamoto et al.26 The reaction was terminated by rapid filtration through Whatman grade GF/B filter paper (Gaithersburg, MD, USA) presoaked in 1% polyethyleneimine, washed 4 times each with 2 mL of cold saline, and the filter bound radioactivity was determined by liquid scintillation counting (Beckman LS5000 TD). The media and chemicals listed above were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA) unless otherwise stated.
Data analysis
Analysis of data collected from three independent experiments performed in duplicates is done using GraphPad Prizm 4 software (GraphPad, San Diego, California). Log IC50 values for each test compound were determined from nonlinear regression. The inhibition constant (Ki) was calculated from the antilogarithmic IC50 value by the Cheng and Prusoff equation.49
Functional GPI and MVD assays
The guinea pig ileum (GPI)50 and mouse vas deferens (MVD)51 bioassays were carried out as described in detail elsewhere. 52,53 A dose-response curve was determined with [Leu5]enkephalin as standard for each ileum and vas preparation and IC50 values of the compounds being tested were normalized according to a published procedure.54
Opioid receptor binding assays
Opioid receptor binding studies were performed as described in detail elsewhere.52 Binding affinities for μ and δ opioid receptors were determined by displacing, respectively, [3H]DAMGO (Multiple Peptide Systems, San Diego, CA) and [3H]DSLET (Multiple Peptide Systems) from rat brain membrane binding sites, and κ opioid receptor binding affinities were measured by displacement of [3H]U69,593 (Amersham) from guinea pig brain membrane binding sites. Incubations were performed for 2 h at 0 °C with [3H]DAMGO, [3H]DSLET and [3H]U69,593 at respective concentrations of 0.72, 0.78 and 0.80 nM. IC50 values were determined form log-dose displacement curves, and Ki values were calculated from the IC50 values by means of the equation of Cheng and Prusoff,49 using values of 1.3, 2.6 and 2.9 nM for the dissociation constants of [3H]DAMGO, [ 3H]DSLET and [3 H]U69,593, respectively.
Molecular Modeling
The calculations were carried out using Macromodel 5.054 with Maestro 8.0 as a graphic interface. The MM3* force field55 was used for energy minimization in combination with the GB/SA solvation model of Still et al.,56 using MacroModel’s default parameters for an aqueous medium. Conformational searches were carried out using the Pure Low Mode Search.57 Structures were generated and minimized by means of the Polak-Ribière conjugate gradient method as implemented in MacroModel, using a gradient convergence criterion of 0.1 kJ/mol Å. The resulting conformations were again minimized to an energy convergence of 0.01 kJ/mol Å. Duplicate structures and those greater than 50kJ/mol above the global minimum were discarded. The remaining structures were clustered into families using Xcluster 1.7 (the aromatic carbons were used as comparison atoms). A RMSD value of 0.2 Å was used.
General synthetic procedures
Boc deprotection
Boc-protected amine (1.8 mmol) was dissolved in a mixture of TFA:water 95:5 (13 mL) and acetonitrile was added (6 mL). The reaction was stirred for 1 h and the mixture was evaporated. Crude TFA salts were used in the next reactions.
Formation of amide bonds
Carboxylic acid (54 mmol) was dissolved in dry CH2Cl2 (250 mL). NEt3 (22.5 mL, 162 mmol, 3 equiv) and TBTU (19.07 g, 59 mmol, 1.1 equiv) were added and the mixture was stirred at room temperature for 10 min. Amine TFA salt (59 mmol, 1.1 equiv) was added and the pH was kept at 8 by the addition of NEt3. The reaction was stirred for 1 h. The solution was extracted with HCl (1M, 3×80 mL), NaHCO3 (saturated, 3×80 mL) and brine (3×80 mL). The organic layer was dried over MgSO 4 and after filtration, the residue was evaporated. No further purification was necessary.
Acetylation
Amine TFA salt (0.89 mmol, 1 equiv) was dissolved in acetonitrile:water 1:1 (10 mL) and the pH was adjusted to 6 by the addition of NEt3. Ac2O (0.42 mL, 4.5 mmol, 5 equiv) was added in three portions while the pH was kept at 6. The reaction was stirred during 2 h at room temperature. The solution was evaporated and after the addition of EtOAc (25 mL), it was extracted with HCl (1M, 3×15 mL), NaHCO3 (saturated, 3×15 mL) and brine (3×15 mL). The organic layer was dried over MgSO 4 and after filtration, the solution was evaporated.
Saponification
Methyl ester (4.1 mmol) was dissolved in MeOH (96 mL) and aqueous LiOH (1M, 4 equiv, 16 mmol, 16.2 mL) was added. The reaction was monitored by TLC (silica, EtOAc) and after 1.5 h it was complete. MeOH was evaporated, and water (100 mL) and EtOAc (50 mL) were added. After extraction, the aqueous layer was acidified to pH4 and extracted with EtOAc (3× 50 mL). The organic layers were combined and washed with brine (3× 100 mL). The organic phase was dried over MgSO4 and after filtration, the mixture was evaporated.
Peptide synthesis
Peptide 36 was synthesized manually by Na-Fmoc solid phase methodology on Rink amide resin (0.189 mmol scale) using DIC and HOBt as the coupling reagents. A threefold excess of the building blocks (Fmoc-Aba-Gly-OH, Fmoc-D-Arg(Pbf)-OH and Fmoc-Dmt-OH) and activating agents was applied in the presence of a ninefold excess of diisopropylethylamine and dry DMF was used as a solvent. Fmoc-deprotections were carried out by treating the resin twice (5 min and 15 min) with 20% piperidine in DMF. Final cleavage of the peptide as well as the Pbf side chain protection group removal was accomplished by treatment with TFA/TES/water 90:5:5 for 90 minutes. The peptide was isolated and purified by RP-HPLC on a SUPELCO DiscoveryBIO wide pore preparative C18 column in 45% overall yield and was >95% pure as determined by analytical RP-HPLC. The structure of pure compound 36 was confirmed by high-resolution electrospray ionization (ESP) mass-spectrometry.
Hybrid 35 was prepared through a two-step approach. First, Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH was prepared on 2-chlorotrityl resin (0.15 mmol). Fmoc-Aba-Gly-OH (1.2 equiv) and DIPEA (5 equiv) in CH2Cl2 were added to the swollen solid support and the reaction mixture was shaken overnight. The resin was washed three times with DMF, and three times with CH2Cl2. Subsequently, Fmoc-D-Arg(Pbf)-OH and Boc-Dmt-OH were coupled as described above. The protected C-terminal free acid Boc-Dmt-D-Arg(Pbf)-Aba-Gly-OH 33 was obtained after treatment of the resin with 1% v/v TFA in CH2Cl2 for 30 min. The obtained crude peptide was obtained after removal of the solvent in vacuo. The crude peptide was pure enough to be used directly in the second step, the coupling step with N-methyl-3′,5′-bistrifluoromethyl benzylamine. This reaction was carried out on 33 (0.043mmol) in CH2Cl2 (10mL) after adding BOP (1.1 equiv) in the presence of DIPEA (2.5 equiv), and using CH2Cl2 as a solvent. After stirring the reaction mixture for 3h, an extraction with saturated sodium bicarbonate (5mL) was performed and the organic phase was dried over magnesium sulphate and the solvent was removed. After final treatment with TFA/CH2Cl2/anisole 49:49:2 for 2h, evaporation of the solvent under vacuum yielded crude compound 35 which was purified and characterized by preparative RP-HPLC and HRMS as mentioned above.
Compound characterization
Boc-(S)-Aba-Gly-NH-3′,5′-(CF3)2-Bn
Flash chromatography (EtOAc/Hexane 1:1) yielded the desired compound (white solid, 83%). HPLC (standard gradient): tRet 19.3 min; TLC Rf (EtOAc/hexane 1:1): 0.43; MS (ESP+) found m/z 560 [M + H]+, C26H27F6N3O4 requires 559.19, 1H NMR 250 MHz (CDCl3): δ 7.78 (s, 1H), 7.65 (s, 2H), 7.30–7.02 (m, 4H), 6.72 (br s, 1H), 5.73 (d, 1H, J= 7.2 Hz), 5.29 (m, 2H), 4.44 (t, 2H, J= 6.5 Hz), 4.28 (d, 1H, J= 15.8 Hz), 4.07 (d, 1H, J = 15.8 Hz), 3.45 (dd, 1H, J1= 17.2 Hz, J2= 5.0 Hz), 2.96 (dd, 1H, J1= 17.3 Hz, J2= 14.4 Hz), 1.45 (s, 9H). 13C NMR (CDCl3) δ 173.0, 168.8, 155.3, 140.6, 135.2, 132.6, 132.1, 131.7, 130.9, 128.8, 128.6, 127.8, 126.6, 121.5, 80.4, 53.6, 52.6, 50.4, 42.6, 36.9, 28.5
Ac-(S)-Aba-Gly-NH-3′,5′-(CF3)2-Bn 19
Preparative HPLC yielded the desired compound (white powder, 43%). HPLC (standard gradient): tRet 15.85 min; TLC Rf (EtOAc): 0.39; MS (ESP+) found m/z 502.14 [M + H]+, C23H21F6N3O3 requires 501.15, 1H NMR 250 MHz (CDCl3): δ 8.27 (br s, 1H), 7.78 (m, 3H), 7.37–6.99 (m, 5H), 5.41 (m, 1H), 5.29 (d, 1H, J=16.8 Hz), 4.53 (m, 3H), 4.00 (d, 1H, J= 16.8 Hz), 3.79 (d, 1H, J= 16.8 Hz), 3.45 (dd, 1H, J1= 17.7 Hz, J2= 5.5 Hz), 2.95 (dd, 1H, J1= 17.3 Hz, J2= 13.7 Hz), 2.04 (s, 3H). 13C NMR (CDCl3) δ 171.8, 169.0, 168.2, 141.2, 135.0, 132.4, 131.2, 130.6, 130.3, 128.2, 127.6, 127.3, 125.7, 120.3, 52.6, 50.9, 48.3, 41.6, 35.7, 22.6
Boc-(S)-Aba-Gly-NMe-3′,5′-(CF3)2-Bn
After flash chromatography (EtOAc/Hexane 1:1) the desired compound was obtained (white solid, 81%). HPLC (standard gradient): tRet 18.9 min; TLC Rf (EtOAc/hexane 1:1): 0.49; MS (ESP+) found m/z 574 [M + H]+, C27H29F6N3O4 requires 573.21, 1H NMR 250 MHz (CDCl3): δ 7.81 (s, 1H), 7.66 (s, 2H), 7.00–7.25 (m, 4H), 5.89 (d, 1H, J= 5.3 Hz), 5.34 (d, 1H, J= 15.9 Hz), 5.26 (m, 1H), 4.78 (s, 1H), 4.71 (s, 1H), 4.51 (d, 1H, J= 15.9 Hz), 4.00 (m, 2H), 3.51 (dd, 1H, J1= 16 Hz and J2= 4.4 Hz), 3.02 (m, 1H), 2.96 (s, 3H), 1.47 (s, 9H). 13C NMR (CDCl3) δ 173.1, 168.5, 155.2, 139.3, 135.7, 132.4, 132.7, 131.9, 130.9, 128.6, 128.1, 126.5, 126.2, 121.8, 79.9, 53.2, 51.0, 49.5, 49.3, 37.2, 34.7, 28.3
Ac-(S)-Aba-Gly-NMe-3′,5′-(CF3)2-Bn 20
Preparative HPLC yielded the desired compound 20 (white powder, 47%). HPLC (standard gradient): tRet 15.69 min; TLC Rf (EtOAc/hexane 1:1): 0.40; MS (ESP+) found m/z 516 [M + H]+, C24H23F6N3O3 requires 515.16, 1H NMR 250 MHz (CDCl3): δ 7.82 (s, 1H), 7.69 (d, 2H), 7.31–6.96 (m, 4H), 5.49 (m, 1H), 5.36 (d, 1H, J=17.2 Hz), 4.80 (d, 1H, J=7.2 Hz), 4.74 (d, 1H, J= 7.2 Hz), 4.51 (d, 1H, J=17.2 Hz,), 4.51 (d, 1H, J= 17.0 Hz), 3.99 (m, 2H), 3.58 (dd, 1H, J1= 17.6 Hz, J2= 5.6 Hz), 2.98 (s, 3H), 2.10 (s, 3H). 13C NMR (CDCl3) δ 172.2, 170.2, 168.2, 139.3, 135.5, 132.4, 132.5, 131.9, 131.0, 128.6, 128.3, 128.0, 126.3, 121.4, 53.5, 51.0, 48.7, 49.5, 36.3, 34.8, 23.1
Boc-Tic-NMe-3′,5′-(CF3)2-Bn
After flash chromatography (EtOAc/Hexane 1:1) the desired compound was obtained with a yield of 90% (white solid, 65 mg). HPLC (standard gradient): tRet 19.85 min; TLC Rf (EtOAc/hexane 1:1): 0.75; MS (ESP+) found m/z 517 [M + H]+, C25H26F6N2O3 requires 516.18, 1H NMR 250 MHz (CDCl3): δ(cis/trans) 7.80 (s, 1H), 7.68 (s, 2H), 7.07–7.25 (m, 4H), 5.31 (t, 1H, J = 5.5 Hz), 5.15 (t, 1H, J = 5.5 Hz), 4.80 (m, 2H), 4.53 (m, 2H), 3.14 (s, 3H), 3.05 (m, 2H), 1.50 and 1.37 (2s, 9H). 13C NMR (CDCl3) δ(cis/trans) 172.6, 155.2, 140.0, 134.4, 132.1, 131.8, 128.4, 127.8, 127.0, 126.7, 125.8, 124.3, 121.4, 80.7, 50.8, 50.7, 46.0, 45.0, 35.2, 34.9, 31.2, 29.8, 28.3
Ac-Tic-NMe-3′,5′-(CF3)2-Bn 23
Purification was performed by preparative HPLC (51%, white powder). HPLC (standard gradient): tRet 16.82 min; TLC Rf (EtOAc/hexane 1:1): 0.72; MS (ESP+) found m/z 459 [M + H]+, C22H20F6N2O2 requires 458.14, 1H NMR 250 MHz (CDCl3): δ(cis/trans mixture) 7.77 (s, 1H), 7.65 (s, 2H), 7.29–7.14 (m, 4H), 5.41 (pseudo t, 1H, J = 6.0 Hz), 4.80–4.63 (m, 4H), 3.18 (s, 3H), 3.12 (m, 2H), 2.26 (s, 3H). 13C NMR (CDCl3) δ(cis/trans mixture) 172.4, 170.6, 139.8, 133.7, 132.9, 132.7, 130.5, 127.9, 127.8, 127.0, 125.6, 121.5, 51.8, 50.4, 47.4, 35.6, 30.9, 21.8
H-Dmt-D-Arg-Aba-Gly-NH2 36
Preparative HPLC yielded the desired compound (white powder, 45%). HPLC (standard gradient): t Ret 8.73 min; TLC Rf (EBAW): 0.19; HRMS (ESP+) found m/z 581.3199 [M+H]+, C29H41N8O5 requires 581.3195.
H-Dmt-D-Arg-Aba-Gly-NMe-3′,5′-(CF3)2Bn 35
Preparative HPLC yielded the desired compound (white powder, 54%). HPLC (standard gradient): tRet 14.19 min; TLC Rf (EBAW): 0.33; HRMS (ESP+) found m/z 821.3580 [M+H]+, C39H47F6N8O5 requires 821.3574.
Supplementary Material
Acknowledgments
SB and DF are postdoctoral fellows of the Research Foundation – Flanders (F.W.O.), which also supported the work by grant G.0008.08. The Institute for Innovation through Science and Technology (I.W.T. Vlaanderen) granted KB a PhD scholarship. This work was also supported by grants from the MDEIE (to DT and PWS), NIH (DA-25196 to PWS) and CIHR (MOP-89716 to PWS). The authors gratefully thank Sofie Van Soest and Hans Peter Vandersmissen for technical support (NK1R aequorin assays) and Euroscreen (Belgium) for the kind gift of CHO-NK1 cells and apoaequorin expression construct. The authors gratefully acknowledge the Interuniversity Attraction Poles program (Belgian Science Policy Grant P6/14) for financial support (JVdB).
Abbreviations
- Aba
4-amino-1,2,4,5-tetrahydro-2-benzazepin-3-one
- Aia
4-amino-1,2,4,5-tetrahydro-indolo[2,3-c]azepin-3-one
- CHO cells
Chinese hamster ovary cells
- CNS
central nervous system
- DML
designed multiple ligand
- DOR
δ opioid receptor
- EBAW, ethyl acetate/n-butanol/acetic acid/water 1
1:1:1 (v/v)
- EDC
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide
- KOR
κ opioid receptor
- MOR
μ opioid receptor
- NK1R
neurokinin 1 receptor
- SP
substance P
- TBTU
O-(Benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium tetrafluoroborate
- Tcc
1,2,3,4-tetrahydro-β-carboline-3-carboxylic acid
- Tic
1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid
- Pbf
2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl
- Phth
phthaloyl
Footnotes
Supporting Information Available: Spectroscopic, analytical and graphical data on antagonism of SP-induced aequorin luminescence are included and is available free of charge via the internet at http://pubs.acs.org.
References
- 1.Longmore J, Hill RG, Hargreaves RJ. Neurokinin-Receptor Antagonists: Pharmacological Tools and Therapeutic Drugs. Can J Physiol Pharm. 1997;75:612–621. doi: 10.1139/cjpp-75-6-612. [DOI] [PubMed] [Google Scholar]
- 2.Maggi CA, Patacchini R, Rovero P, Giachetti A. Tachykinin Receptors and Tachykinin Receptor Antagonists. Autonomic Pharmacol. 1993;13:23–93. doi: 10.1111/j.1474-8673.1993.tb00396.x. [DOI] [PubMed] [Google Scholar]
- 3.Moskowitz MA. Neurogenic Versus Vascular Mechanisms of Sumatriptan And Ergot Alkaloids in Migraine. Trends Pharmacol Sci. 1992;13:307–311. doi: 10.1016/0165-6147(92)90097-p. [DOI] [PubMed] [Google Scholar]
- 4.Lotz M, Carson DA, Vaughan JH. Substance-P Activation of Rheumatoid Synoviocytes – Neural Pathway in Pathogenesis of Arthritis. Science. 1987;235:893–895. doi: 10.1126/science.2433770. [DOI] [PubMed] [Google Scholar]
- 5.Leroy V, Mauser P, Gao Z, Peet NP. Neurokinin Receptor Antagonists. Exp Opin Invest Drugs. 2000;9:735–746. doi: 10.1517/13543784.9.4.735. [DOI] [PubMed] [Google Scholar]
- 6.Tattersall FD, Rycroft W, Francis B, Pearce D, Merchant K, MacLeod AM, Ladduwahetty T, Keown L, Swain C, Baker R, Cascieri M, Ber E, MacIntyre DE, Hill RG, Hargreaves RJ. Tachykinin NK1 Receptor Antagonists Act Centrally to Inhibit Emesis Induced By The Chemotherapeutic Agent Cisplatin in Ferrets. Neuropharmacology. 1996;35:1121–1129. doi: 10.1016/s0028-3908(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 7.Young JR, Eid R, Turner C, DeVita RJ, Kurtz MM, Tsao KLC, Chicchi GG, Wheeldon A, Carlson E, Mills SG. Pyrrolidine-Carboxamides and Oxadiazoles As Potent hNK1 Antagonists. Bioorg Med Chem Lett. 2007;17:5310–5315. doi: 10.1016/j.bmcl.2007.08.028. [DOI] [PubMed] [Google Scholar]
- 8.Desai MC, Lefkowitz SL, Thadeio PF, Longo KP, Snider RM. Discovery of a Potent Substance P Antagonist: Recognition of the Key Molecular Determinant. J Med Chem. 1992;35:4911–4913. doi: 10.1021/jm00104a018. [DOI] [PubMed] [Google Scholar]
- 9.Lewis RT, Macleod AM, Merchant KJ, Kelleher F, Sanderson I, Herbert RH, Cascieri MA, Sadowski S, Ball RG, Hoogsteen K. Tryptophan-Derived NK1 Antagonists: Conformationally Constrained Heterocyclic Bioisosteres of The Ester Linkage. J Med Chem. 1995;38:923–933. doi: 10.1021/jm00006a011. [DOI] [PubMed] [Google Scholar]
- 10.Takeuchi Y, Berkley Shands EF, Beusen DD, Marshall GR. Derivation of a Three-Dimensional Pharmacophore Model of Substance P Antagonists Bound to the Neurokinin-1 Receptor. J Med Chem. 1998;41:3609–3623. doi: 10.1021/jm9700171. [DOI] [PubMed] [Google Scholar]
- 11.Yamamoto T, Nair P, Jacobsen NE, Kulkarni V, Davis P, Ma S, Navratilova E, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. Biological and Conformational Evaluation of Bifunctional Compounds for Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists Possessing Two Penicillamines. J Med Chem. 2010;53:5491–5501. doi: 10.1021/jm100157m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalso E. Improving Opioid Effectiveness: From Ideas to Evidence. Eur J Pain. 2005;9:131–135. doi: 10.1016/j.ejpain.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 13.King T, Gardell LR, Wang R, Vardanyen A, Ossipov MH, Malan TP, Jr, Vanderah TW, Hunt SP, Hruby VJ, Lai J, Porreca F. Role of NK-1 Neurotransmission in Opioid-induced Hyperalgesia. Pain. 2005;115:276–288. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armour DR, Aston NM, Morriss KML, Congreve MS, Hawcock AB, Marquess D, Mordaunt JE, Richards SA, Ward P. 1,4-Benzodiazepin-2-one Derived Neurokinin-1 Receptor Antagonists. Bioorg Med Chem Lett. 1997;7:2037–2042. [Google Scholar]
- 15.Yamamoto T, Nair P, Vagner J, Largent-Milnes T, Davis P, Ma S, Navratilova E, Moye S, Tumati S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. A Structure-Activity Relationship Study and Combinatorial Synthetic Approach of C-terminal Modified Bifunctional Peptides That Are δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J Med Chem. 2008;51:1369–1376. doi: 10.1021/jm070332f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamamoto T, Nair P, Ma SW, Davis P, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. The Biological Activity and Metabolic Stability of Peptidic Bifunctional Compounds That Are Opioid Receptor Agonists And Neurokinin 1 Receptor Antagonists With a Cysteine Moiety. Bioorg Med Chem. 2009;17:7337–7343. doi: 10.1016/j.bmc.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballet S, Pietsch M, Abell AD. Multiple Ligands in Opioid Research. Protein Pept Lett. 2008;15:668–682. doi: 10.2174/092986608785133672. [DOI] [PubMed] [Google Scholar]
- 18.Lipkowski AW. Cooperative reinforcement of opioid pharmacophores. Polish J Pharmacol Pharm. 1987;39:585–596. [PubMed] [Google Scholar]
- 19.Morphy R, Rancovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J Med Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 20.Misterek K, Maszczynska I, Dorociak A, Gumulka SW, Carr DB, Szyfelbein SK, Lipkowski AW. Spinal co-administration of peptide substance P antagonist potentiates antinociceptive effect of opioid peptide. Life Sci. 1994;54:939–944. doi: 10.1016/0024-3205(94)00494-3. [DOI] [PubMed] [Google Scholar]
- 21.Lipkowski AW, Carr DB, Misicka A, Misterek K. Biological activities of a peptide containing both casomorphin-like and substance P antagonist structural characteristics. In: Brantl V, Teschemacher H, editors. β-casomorphins and related peptides: Recent developments. VCH; Weinheim: 1994. pp. 113–118. [Google Scholar]
- 22.Bonney IM, Foran SE, Marchand JE, Lipkowski AW, Carr DB. Spinal antinociceptive effects of AA501, a novel chimeric peptide with opioid receptor agonist and tachykinin receptor antagonist moieties. Eur J Pharmacol. 2004;488:91–99. doi: 10.1016/j.ejphar.2004.02.023. [DOI] [PubMed] [Google Scholar]
- 23.Ripley TL, Gadd CA, De Filipe C, Hunt SP, Stephens DN. Lack of Self- Administration and Behavioural Sensitization to Morphine, But Not Cocaine, in Mice Lacking NK1 Receptors. Neuropharmacology. 2002;43:1258–1268. doi: 10.1016/s0028-3908(02)00295-2. [DOI] [PubMed] [Google Scholar]
- 24.Morphy R, Rancovic Z. Design of Multitarget Ligands. In: Rancovic Z, Morphy R, editors. Lead Generation Approaches in Drug Discovery. John Wiley & Sons, Inc; Hoboken: 2010. pp. 141–164. [Google Scholar]
- 25.Hruby VJ. Organic Chemistry and Biology: Chemical Biology Through the Eyes of Collaboration. J Org Chem. 2009;74:9245–9264. doi: 10.1021/jo901767e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamamoto T, Nair P, Vagner J, Davis P, Ma SW, Navratilova E, Moye S, Tumati S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. Design, Synthesis, And Biological Evaluation of Novel Bifunctional C-terminal-Modified Peptides for δ/μ Opioid Receptor Agonists and Neurokinin-1 Receptor Antagonists. J Med Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yamamoto T, Nair P, Jacobsen NE, Davis P, Ma SW, Navratilova E, Moye S, Lai J, Yamamura HI, Vanderah TW, Porreca F, Hruby VJ. The Importance of Micelle-Bound States for the Bioactivities of Bifunctional Peptide Derivatives for δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J Med Chem. 2008;51:6334–6347. doi: 10.1021/jm800389v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Largent-Milnes TM, Yamamoto T, Nair P, Moulton JW, Hruby VJ, Lai J, Porreca F, Vanderah TW. Spinal or Systemic TY005, A Peptide Opioid Agonist/Neurokinin 1 Receptor Antagonist, Attenuates Pain With Reduced Tolerance. Br J Pharmacol. 2010;161:986–1001. doi: 10.1111/j.1476-5381.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Van den Eynde I, Laus G, Schiller PW, Kosson P, Chung NN, Lipkowski AW, Tourwé D. A New Structural Motif for Mu Opioid Antagonists. J Med Chem. 2005;48:3644–3648. doi: 10.1021/jm0491795. [DOI] [PubMed] [Google Scholar]
- 30.Ballet S, Feytens D, De Wachter R, De Vlaeminck M, Marczak ED, Salvadori S, de Graaf C, Rognan D, Negri L, Lattanzi R, Lazarus LH, Tourwé D, Balboni G. Conformationally Constrained Opioid Ligands: The Dmt-Aba and Dmt-Aia versus Dmt-Tic Scaffold. Bioorg Med Chem Lett. 2009;19:433–437. doi: 10.1016/j.bmcl.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballet S, De Wachter R, Van Rompaey K, Tomboly Cs, Feytens D, Toth G, Quartara L, Cucchi P, Meini S, Tourwé D. Bradykinin Analogues Containing The 4- Amino-2-Benzazepin-3-one Scaffold at the C-Terminus. J Pept Sci. 2007;13:164–170. doi: 10.1002/psc.827. [DOI] [PubMed] [Google Scholar]
- 32.Feytens D, Cescato R, Reubi JC, Tourwé D. New sst(4/5)-selective Somatostatin Peptidomimetics Based on a Constrained Tryptophan Scaffold. J Med Chem. 2007;50:3397–3401. doi: 10.1021/jm070246f. [DOI] [PubMed] [Google Scholar]
- 33.Feytens D, De Vlaeminck M, Cescato R, Tourwé D, Reubi JC. Highly Potent 4-Amino-Indolo[2,3-c]Azepin-3-One-Containing Somatostatin Mimetics With a Range of sst Receptor Selectivities. J Med Chem. 2009;52:95–104. doi: 10.1021/jm801205x. [DOI] [PubMed] [Google Scholar]
- 34.Schiller PW, Nguyen TMD, Berezowska I, Dupuis S, Weltrowska G, Chung NN, Lemieux C. Synthesis and In Vitro Opioid Activity Profiles of DALDA Analogues. Eur J Med Chem. 2000;35:895–901. doi: 10.1016/s0223-5234(00)01171-5. [DOI] [PubMed] [Google Scholar]
- 35.Ballet S, Urbanczyk-Lipkowska Z, Tourwé D. Synthesis of Substituted 4-Amino-2-Benzazepin-3-Ones Via N-Acyliminium Ion Cyclizations. Synlett. 2005:2791–2795. [Google Scholar]
- 36.Mondini S, Dall Avo M, Guerrato A. WO2007107818(A2) Process for the Preparation of 3,5- Bistrifluoromethyl-N-Methylbenzylamine. 2008
- 37.Tourwé D, Verschueren K, Frycia A, Davis P, Porreca F, Hruby VJ, Toth G, Jaspers H, Verheyden P, Van Binst G. Conformational Restriction of Tyr and Phe Side Chains in Opioid Peptides: Information About Preferred and Bioactive Side-Chain Topology. Biopolymers. 1996;38:1–12. doi: 10.1002/(sici)1097-0282(199601)38:1<1::aid-bip1>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 38.Iterbeke K, Laus G, Verheyden P, Tourwé D. Lett Pept Sci. 1998;5:121. [Google Scholar]; Millet R, Goossens JF, Bertrand-Caumont K, Chavatte P, Houssin R, Hénichart JP. Lett Pept Sci. 1999;6:221. doi: 10.1002/psc.326. [DOI] [PubMed] [Google Scholar]
- 39.Cascieri MA, Macleod AM, Underwood D, Shiao LL, Ber E, Sadowski S, Yu H, Merchant KJ, Swain CJ, Strader CD, Fong TM. Characterization of the Interaction of N-Acyl-L-Tryptophan Benzyl Ester Neurokinin Antagonists With the Human Neurokinin-1 Receptor. J Biol Chem. 1994;269:6587–6591. [PubMed] [Google Scholar]
- 40.Janecka A, Poels J, Fichna J, Studzian K, Vanden Broeck J. Comparison of Antagonist Activity of Spantide Family at Human Neurokinin Receptors Measured by Aequorin Luminescence-Based Functional Calcium Assay. Regul Pept. 2005;131:23– 28. doi: 10.1016/j.regpep.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 41.Schild HO. pA2, A New Scale For The Measurement of Drug Antagonism. Br J Pharmacol. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arunlakshana O, Schild HO. Some Quantitative Uses of Drug Antagonists. Br J Pharmacol Chemother. 1959;14:45–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballet S, Frycia A, Piron J, Chung NN, Schiller PW, Kosson P, Lipkowski AW, Tourwé D. Synthesis and Biological Evaluation of Constrained Analogues of the Opioid Peptide H-Tyr-D-Ala-Phe-Gly-NH2 Using the 4-Amino-2-Benzazepin-3-One Scaffold. J Pept Res. 2005;66:222–230. doi: 10.1111/j.1399-3011.2005.00291.x. [DOI] [PubMed] [Google Scholar]
- 44.Ballet S, Misicka A, Kosson P, Lemieux C, Chung NN, Schiller PW, Lipkowski AW, Tourwé D. Blood-Brian Barrier Penetration By Two Dermorphin Tetrapeptide Analogues: Role of Lipophilicity vs. Structural Flexibility. J Med Chem. 2008;51:2571–2574. doi: 10.1021/jm701404s. [DOI] [PubMed] [Google Scholar]
- 45.Ballet S, Marczak ED, Feytens D, Salvadori S, Sasaki Y, Abell AD, Lazarus LH, Balboni G, Tourwé D. Novel Multiple Opioid Ligands Based on 4-Aminobenzazepinone (Aba), Azepinoindole (Aia) and Tetrahydroisoquinoline (Tic) Scaffolds. Bioorg Med Chem Lett. 2010;20:1610–1613. doi: 10.1016/j.bmcl.2010.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tourwé D, Verschueren K, Van Binst G, Davis P, Porreca F, Hruby VJ. Dermorphin Sequence With High Delta Affinity By Fixing the Phe Side-chain to Trans at Alfa-1. Bioorg Med Chem Lett. 1992;2:1305–1308. [Google Scholar]
- 47.Schiller PW. Bi- or Multifunctional Opioid Peptide Drugs. Life Sci. 2010;86:598–603. doi: 10.1016/j.lfs.2009.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao K, Luo G, Zhao GM, Schiller PW, Szeto HH. Transcellular Transport of a Highly Polar 3+ Net Charge Opioid Tetrapeptide. J Pharmacol Exp Ther. 2003;304:425–432. doi: 10.1124/jpet.102.040147. [DOI] [PubMed] [Google Scholar]
- 49.Cheng YC, Prusoff WH. Relationship Between the Inhibition Constant (Ki) and the Concentration of Inhibitor Which Causes 50 per cent (I50) of an Enzymatic Reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]; 46 Paton WDM. The Action of Morphine and Related Substances on Contraction and on Acetylcholine Output of Coaxially Stimulated Guinea-pig ileum. Br J Pharmacol Chemother. 1957;12:119–127. doi: 10.1111/j.1476-5381.1957.tb01373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henderson G, Hughes J, Kosterlitz HW. A New Example of a Morphine-Sensitive Neuro-Effector Junction: Adrenergic Transmission in the Mouse Vas Deferens. Br J Pharmacol. 1972;46:764–766. doi: 10.1111/j.1476-5381.1972.tb06901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schiller PW, Lipton A, Horrobin DF, Bodansky M. Unsulfated C-Terminal 7- Peptide of Cholecystokinin: A New Ligand of the Opiate Receptor. Biochem Biophys Res Commun. 1978;85:1332–1338. doi: 10.1016/0006-291x(78)91149-x. [DOI] [PubMed] [Google Scholar]
- 52.DiMaio J, Nguyen TMD, Lemieux C, Schiller PW. Synthesis and Pharmacological Characterization in Vitro of Cyclic Enkephalin Analogues: Effect of the Conformational Constraints on Opioid Receptor Selectivity. J Med Chem. 1982;25:1432–1438. doi: 10.1021/jm00354a008. [DOI] [PubMed] [Google Scholar]
- 53.Waterfield AA, Leslie FM, Lord JAH, Ling N, Kosterlitz HW. Opioid Activities of Frangments of β-Endorphin and Its Leucine5-Analogue. Comparison of the Binding Properties of Methionine- and Leucine-Enkephalin. Eur J Pharmacol. 1979;58:11–18. doi: 10.1016/0014-2999(79)90334-0. [DOI] [PubMed] [Google Scholar]
- 54.Mohamadi F, Richards NGJ, Guida WC, Liskamp R, Lipton M, Caufield C, Chang G, Hendrickson T, Still WC. Macromodel - An Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. J Comp Chem. 1990;11:440–467. [Google Scholar]
- 55.Allinger NL, Yuh YH, Lii JH. Molecular Mechanics - The MM3 Force-field for Hydrocarbons 1. J Am Chem Soc. 1989;111:8551–8566. [Google Scholar]
- 56.Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalytical Treatment of Solvation for Molecular Mechanics and Dynamics. J Am Chem Soc. 1990;112:6127–6129. [Google Scholar]
- 57.Kolossvary I, Guida WC. Low mode search. An efficient, automated computational method for conformational analysis: Application to Cyclic and Acyclic Alkanes and Cyclic Peptides. J Am Chem Soc. 1996;118:5011–5019. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.