

Abstract
Systemic lupus erythematosus (SLE) is a severe multi-system autoimmune disease which results from both genetic predisposition and environmental factors. Many lines of investigation support interferon alpha (IFN-α) as a causal agent in human lupus, and high levels of serum IFN-α are a heritable risk factor for SLE. Interferon regulatory factors (IRFs) are a family of transcription factors involved in host defense, which can induce transcription of IFN-α and other immune response genes following activation. In SLE, circulating immune complexes which contain nucleic acid are prevalent. These complexes are recognized by endosomal Toll-like receptors, resulting in activation of downstream IRF proteins. Genetic variants in the IRF5 and IRF7 genes have been associated with SLE susceptibility, and these same variants are associated with increased serum IFN-α in SLE patients. The increase in serum IFN-α related to IRF5 and 7 genotypes is observed only in patients with particular antibody specificities. This suggests that chronic stimulation of the endosomal Toll-like receptors by autoantibody immune complexes is required for IRF SLE-risk variants to cause elevation of circulating IFN-α and subsequent risk of SLE. Recently, genetic variation in the IRF8 gene has been associated with SLE and multiple sclerosis, and studies support an impact of IRF8 genotype on the IFN-α pathway. In summary, the SLE-associated polymorphisms in the IRF family of proteins appear to be gain-of-function variants, and understanding the impact of these variants upon the IFN-α pathway in vivo may guide therapeutic strategies directed at the Toll-like receptor/IRF/IFN-α pathway in SLE.
Keywords: Interferon Alpha, Genetics, Systemic Lupus Erythematosus, Interferon Regulatory Factor, Autoantibodies, Autoimmunity
Introduction
Since the 1970’s, high levels of type I interferon have been observed in serum of SLE patients (1). Type I interferons include interferon alpha (IFN-α) and interferon beta (IFN-β), and both of these molecules signal through the same type I interferon receptor (2). IFN-α normally functions in viral defense, and forms a bridge between the innate and adaptive immune systems (3). In this way, IFN-α is also important in setting thresholds for self-reactivity and autoimmunity. In recent microarray studies comparing SLE patients to healthy controls, over-expression of IFN-α-induced genes was one of the most dominant findings in the peripheral blood mononuclear cells of SLE patients (4, 5). While the finding of overactive IFN-α pathway signaling in SLE patients does not allow us to infer whether IFN-α is causal or a secondary reactive finding, a number of lines of evidence suggest that this relationship between IFN-α and SLE is primary and causal.
Strikingly, some individuals who have received recombinant human IFN-α injections to treat chronic viral infections or malignancy have developed de novo SLE (6, 7). IFN-α-induced SLE frequently resolves with discontinuation of IFN-α therapy (7). This human experience provides some “proof-of-principal” that IFN-α can break tolerance (8), and that this IFN-α-induced tolerance break in some individuals results in the very specific autoimmune phenotype of SLE.
Additionally, abnormally high levels of IFN-α are present in healthy first degree relatives of SLE patients as compared to healthy unrelated subjects (9, 10), suggesting that high serum IFN-α is an inherited risk factor for SLE. High levels of IFN-α were not observed in spouses of SLE patients (Niewold TB et al, unpublished data), suggesting that genetic and not environmental influences are the cause of this familial clustering. A number of genetic variants have now been associated with increased IFN-α in SLE (11–15), outlining some of the genetic architecture of this SLE-associated trait. The high IFN-α trait is common across SLE patients of all ancestral backgrounds (16), however the particular genetic variants which underlie this SLE-associated trait sometimes differ between ancestral backgrounds (11, 17–20).
A number of genetic polymorphisms in genes directly within the IFN-α pathway have been associated with susceptibility to SLE, including IRF5 and IRF7 (21, 22). Consistent with the above data, these SLE-associated polymorphisms in IRF5 and IRF7 are gain-of-function variants, which are associated with higher serum IFN-α than the non-risk variants of the gene (19, 23–25). As their name would suggest, interferon regulatory factors (IRFs) play a crucial role in the induction of IFN-α, and thus occupy an essential position in this pathogenic mechanism in SLE. In this review we will synthesize some recent work examining the unique role that IRFs play in human lupus pathogenesis.
Toll-like Receptor System of IFN-α Generation in SLE
Toll-like receptors (TLRs) are pattern recognition receptors which are capable of responding to a number of microbe-associated molecular patterns. Ligation of TLRs in cells of the innate immune system results in cell activation and secretion of cytokines and chemokines, in an effort to activate the immune system against pathogens. TLR3, TLR7, and TLR9 physiologically respond to viral RNA and DNA, and these TLRs are located in the endosomal compartment within the cell (26). Activation of these endosomal TLRs results in the production of IFN-α, which normally functions in viral defense. In SLE, autoantibodies are present which can bind either double-stranded native DNA (dsDNA antibodies) or small nuclear RNA-binding proteins such as Ro, La, Sm, and RNP. Immune complexes formed by these autoantibodies contain DNA or RNA respectively. Self-derived nucleic acid contained within these immune complexes is delivered in an abnormal way to the endosomal TLRs after they are taken up by cells via Fc receptors. This abnormal delivery may then pathologically activate normal anti-viral immunity, amplifying an autoimmune response. Experiments examining SLE-associated immune complexes in vitro provide strong support for this model, demonstrating that these immune complexes can stimulate cells via the endosomal TLR system resulting in IFN-α production (27, 28) (Figure 1).
Figure 1.
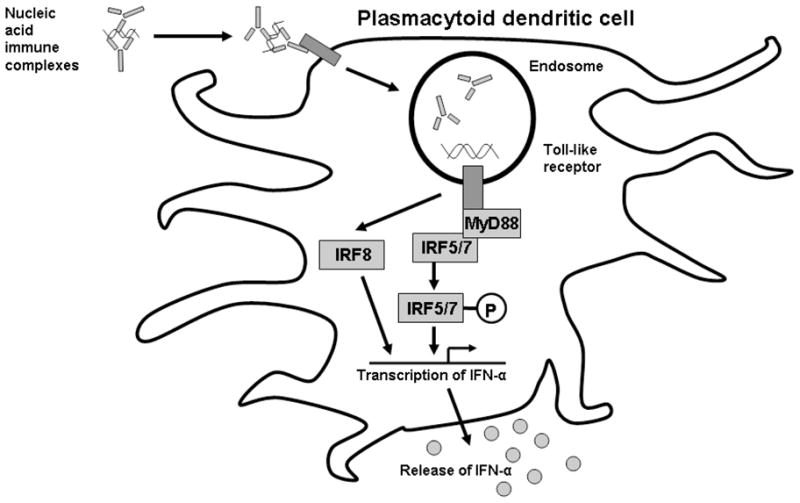
Proposed relationship between SLE immune complexes, endosomal TLRs, and downstream IRF proteins with respect to IFN-α production in SLE patients. A plasmacytoid dendritic cell is shown, as these cells are thought to be the major IFN-α producing cells.
In support of this model, autoantibody traits provide the strongest association we have observed between serum IFN-α and clinical features in SLE, and this association extends to SLE patients of all ancestral backgrounds (9, 16). While this clinical association is strong and in vitro models suggest a causal relationship between autoantibodies and IFN-α via the endosomal TLR system, the presence of these autoantibodies is not completely predictive of high IFN-α in patients in vivo (29). This suggests that other host factors influence the relationship between autoantibodies and serum IFN-α in humans. An interplay between SLE-associated autoantibodies and high- vs. low-functioning genetic variants in the TLR system is an attractive hypothesis to explain some of the variability in the relationship between autoantibodies and serum IFN-α in SLE patients in vivo.
Interferon Regulatory Factors
There are nine members of the IRF family in humans, numbered from IRF1 to IRF9 (30, 31). This transcription factor family shares a helix-turn-helix DNA binding motif, and IRFs are classically involved in type I interferon signaling (31). In addition to roles within the type I interferon pathway, IRFs have also been implicated in immune cell development and tumor suppression (30, 32, 33). In particular, IRF4 has been implicated in lymphoid malignancies by a number of different groups (34). Strikingly, genetic variations in three of the nine IRFs have been linked to SLE susceptibility, supporting a major role for this family of proteins in SLE pathogenesis.
The three IRF family members in which genetic variation has been linked to SLE susceptibility are IRF5, IRF7, and IRF8 (22, 35, 36). IRF5 and IRF7 interact with the MyD88 adaptor protein downstream of Toll-like receptor signaling, and are phosphorylated and activated following Toll-like receptor engagement (30). IRF8 has not been shown to directly interact with MyD88, but does seem to play a role in the Toll-like receptor pathway as dendritic cells lacking IRF8 do not produce inflammatory cytokines in response to Toll-like receptor 9 ligand (37). In addition to direct roles inducing IFN-α and IFN-induced genes downstream of Toll-like receptor activation, the SLE-associated IRFs may also play a role in tumor suppression and immune cell development (33, 38).
IRF5 in SLE
IRF5 was an ideal candidate gene to study for an impact upon serum IFN-α levels in SLE patients. IRF5 can induce transcription of IFN-α mRNA (39), and strong genetic associations between IRF5 gene variants and SLE were discovered long before large-scale GWAS screening studies (21). A robust body of work supports genetic variation in the IRF5 gene as a risk factor for human SLE across multiple global populations (21, 35, 40–44). In Europeans, a risk haplotype containing multiple functional genetic elements has been defined, including a promoter insertion deletion, a splice site variation, a 30 base pair in-frame insertion/deletion, and an alternate polyadenlyation site in the 3’ UTR region (35, 45, 46). Due to high linkage-disequilibrium between these variants, it is not currently clear which functional element or minimum combination of elements is required for SLE susceptibility. We have shown that this IRF5 SLE-risk haplotype is associated with increased serum IFN-α in SLE patients (23), and subsequent studies have supported this concept by showing that SLE-associated IRF5 variants are associated with increased activation of the IFN-α pathway (24, 25).
Interestingly, in our study the association between IRF5 SLE-risk genotype and increased serum IFN-α was completely dependent upon autoantibodies (23). Thus, in SLE patients who lacked anti-dsDNA and anti-RNA-binding protein autoantibodies, there was no relationship between IRF5 and serum IFN-α levels. These data were striking, and suggest that the autoantibodies represent a chronic stimulus to the endosomal TLR system which is required for the IRF5 SLE-risk haplotype to result in dysregulation of circulating IFN-α levels (Figure 2). Thus, these data support a “gene + antibody = high IFN-α” model, which we would explore further in the context of IRF7 genetic variants in SLE patients. It is also possible that IRF5 SLE-risk genotype may predispose to the production of autoantibodies in the first place, as IRF5 deficient lupus model mice show a major defect in autoantibody generation (47). If this is the case in human SLE, then a feed-forward model would be supported, in which IRF5 genotype predisposes to autoantibody formation, and then subsequently also predisposes to greater IFN-α production in the setting of those same autoantibodies.
Figure 2.
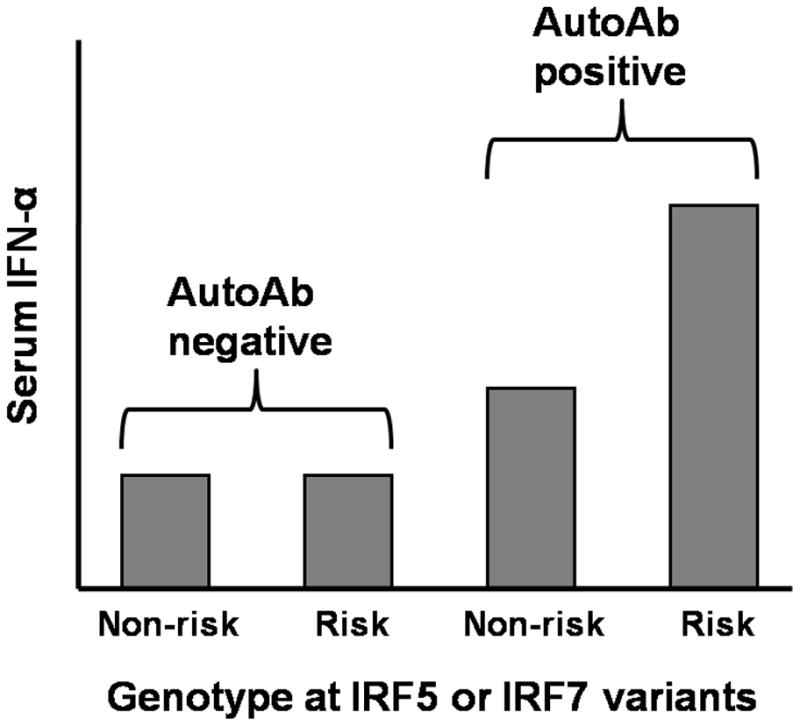
Representation of the interaction between autoantibodies and IRF5 and IRF7 genotype upon serum IFN-α levels in SLE patients. Bars on the graph represent median serum IFN-α levels in a group of SLE patients defined by the labels on the graph. “Risk” refers to subjects who carry IRF5 or IRF7 polymorphisms associated with SLE susceptibility in case-control studies, and “non-risk” refers to subjects who lack these variants.
IRF7 in SLE
IRF7 is another IRF family member which functions downstream of the endosomal TLRs which can induce transcription of IFN-α mRNA (48). A genome-wide association scan in European ancestry SLE patients detected a significant association near the IRF7 gene (22). To date, the causal functional genetic elements are not well described in the IRF7 locus, which also includes another gene named PHD and ring finger domains 1 (PHRF1). IRF7 seems like the more plausible candidate in this region given the potential biological relevance of the IRF family of genes in SLE pathogenesis, and upcoming fine-mapping and resquencing efforts will likely improve our ability to localize the genetic association with SLE in this locus.
We have recently explored the IRF7/PHRF1 locus in SLE patients, examining the relationship between genotype, autoantibodies, and serum IFN-α (19). We found a very similar model as that described above for IRF5, in which an impact of IRF7 genotype upon IFN-α was only observed in the presence of particular autoantibodies (19). Interestingly, the particular genetic variants and the particular associated autoantibodies differed somewhat between ancestral backgrounds, but the overall “gene + antibody = high IFN-α” model was conserved between different backgrounds. A direct genetic association between these particular autoantibodies and the respective IRF7 genotypes was also observed. In this case, SLE patients with the particular IRF7 genotypes were more likely to have the autoantibodies which cooperated with that genotype to result in higher serum IFN-α. It seems likely that this represents a gene-microenvironment interaction, in which the effect of the risk genotype is brought out by the microenvironmental stimulus. When IRF5 and IRF7 genotypes were considered jointly in the same patients, there was an additive effect observed between the risk genotypes of these two polymorphisms upon serum IFN-α in the autoantibody positive group, which was not present in the autoantibody negative group (19). The data above support the idea that this autoantibody-IRF7 interaction is important to disease susceptibility and pathogenesis in this subset of SLE patients, and provide a strategy for molecular stratification of the SLE population that will be useful in future studies of this locus in SLE patients.
IRF8 in SLE and Multiple Sclerosis
A recent genetic association study has strongly implicated a variant near the IRF8 gene in SLE susceptibility in Europeans (36). Interestingly, a different genetic variation in the IRF8 gene has been implicated in multiple sclerosis (49). Multiple sclerosis is treated effectively with recombinant human IFN-β (50), a type I interferon which signals through the same receptor as IFN-α (2). Paradoxically, the genetic variation in IRF8 which was associated with multiple sclerosis was also associated with increased type I IFN-induced gene expression in a human cell line, despite the therapeutic benefit of recombinant type I IFN in the treatment of the disease (49). We are currently performing studies of this locus in relation to serum IFN-α levels in SLE, as it is striking that different genetic variations in the same IRF gene are associated with two autoimmune syndromes which demonstrate an opposite relationship with type I IFN (causal in SLE, and therapeutic in multiple sclerosis). Similar to IRF7, the discovery of genetic associations between IRF8 variants in both SLE and multiple sclerosis are recent, and further fine-mapping and sequencing of this region will likely be required to understand the functional genetic variations underlying these associations. Given the association of the IRF8 locus with two disparate autoimmune diseases, we expect that further study of this locus will broadly impact our understanding of the ways in which IRFs can impact human autoimmunity.
Conclusions
A variety of study techniques including gene expression, familial cytokine analysis, and genetic association studies all strongly support the importance of over-activity in the IFN-α pathway as a causal factor in human SLE (3, 4, 9, 14, 51). IRFs are important mediators of IFN-α and IFN-α-induced gene expression, playing a critical role in viral defense downstream of endosomal TLR activation. These endosomal TLRs are pathogenically activated by SLE-associated autoantibody immune complexes (27), which likely provide a chronic endogenous stimulus to the IFN-α pathway in SLE patients. Detailed investigations of the SLE-associated genetic polymorphisms in IRF5 and IRF7 support the idea that these polymorphisms are gain-of-function in humans in vivo (19, 23), resulting in some of the IFN-α pathway activation observed in the disease. The impact of both of these polymorphisms upon serum IFN-α in SLE patients is dependent upon particular autoantibodies, providing evidence for a gene-microenvironment interaction (gene + autoantibody = high IFN-α, as shown in Figure 2).
It is not currently clear whether the observed gene-autoantibody interaction upon IFN-α is dependent upon other parts of the SLE phenotype. This is difficult to test in humans, as healthy subjects typically do not have SLE-associated autoantibodies. The genetic associations between gain-of-function variants in IRF genes and SLE susceptibility may also be relevant in SLE patients who lack the particular associated autoantibodies. It is possible that nucleic acid containing immune complexes represent one of many TLR stimuli which could interact pathologically with SLE-associated IRF variants. For example, Epstein-Barr virus infections have been implicated in SLE pathogenesis (52), and viral infection would provide a strong stimulus to the TLR/IRF system. In this case, a similar “gene + microenvironmental stimulus = high IFN-α” model may still apply, although potentially in a more self-limited manner than the consistent production of nucleic acid-containing immune complexes that is frequently observed in SLE patients.
Genetic studies support the relevance of IRF8 in both SLE and multiple sclerosis (36, 49). The type I IFN system is of critical importance in both of these conditions, although in multiple sclerosis type I IFN is therapeutic while it is thought to be pathogenic in SLE. These studies all support the importance of the IRF family of proteins in human SLE, and further work in human disease may provide improved methods for diagnosis, classification, and possibly novel therapeutic strategies. While the IRF proteins have not been directly targeted by therapeutics to date, agents directed at upstream targets such as endosomal TLRs (53) and downstream targets such as IFN-α (54) are in early stage clinical trials in SLE patients. We expect that an improved understanding of the role of IRFs in human SLE will inform these therapeutic efforts and help to enable personalized medicine in SLE.
Acknowledgments
Grant Support and Disclosures: R Salloum – none, TB Niewold - NIH/NIAID K08 Award AI083790, NIH P30 DK42086, NIAID Clinical Research Loan Repayment AI071651, CTSA Collaborative Pilot Grant from UL1 RR024999, Arthritis National Research Foundation Eng Tan Scholar Award, and grants from the France-Chicago Center, the Alliance for Lupus Research, and the Lupus Research Institute
The authors have no financial conflict of interest to declare. Funding sources include NIH/NIAID K08 Award AI083790, NIH P30 DK42086, NIAID Clinical Research Loan Repayment AI071651, CTSA Collaborative Pilot Grant from UL1 RR024999, Arthritis National Research Foundation Eng Tan Scholar Award, and grants from the France-Chicago Center, the Alliance for Lupus Research, and the Lupus Research Institute. No one beyond the authors listed provided any assistance in writing or editing the manuscript, and the funding sources played no part in the study design, interpretation of data, or decision to publish the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL. Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med. 1979;301(1):5–8. doi: 10.1056/NEJM197907053010102. [DOI] [PubMed] [Google Scholar]
- 2.Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- 3.Niewold TB, Clark DN, Salloum R, Poole BD. Interferon alpha in systemic lupus erythematosus. J Biomed Biotechnol. 2010;2010:948364. doi: 10.1155/2010/948364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crow MK, Kirou KA, Wohlgemuth J. Microarray analysis of interferon-regulated genes in SLE. Autoimmunity. 2003;36(8):481–90. doi: 10.1080/08916930310001625952. [DOI] [PubMed] [Google Scholar]
- 5.Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A. 2003;100(5):2610–5. doi: 10.1073/pnas.0337679100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronnblom LE, Alm GV, Oberg KE. Possible induction of systemic lupus erythematosus by interferon-alpha treatment in a patient with a malignant carcinoid tumour. J Intern Med. 1990;227(3):207–10. doi: 10.1111/j.1365-2796.1990.tb00144.x. [DOI] [PubMed] [Google Scholar]
- 7.Niewold TB, Swedler WI. Systemic lupus erythematosus arising during interferon-alpha therapy for cryoglobulinemic vasculitis associated with hepatitis C. Clin Rheumatol. 2005;24(2):178–81. doi: 10.1007/s10067-004-1024-2. [DOI] [PubMed] [Google Scholar]
- 8.Niewold TB. Interferon alpha-induced lupus: proof of principle. J Clin Rheumatol. 2008;14(3):131–2. doi: 10.1097/RHU.0b013e318177627d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niewold TB, Hua J, Lehman TJ, Harley JB, Crow MK. High serum IFN-alpha activity is a heritable risk factor for systemic lupus erythematosus. Genes Immun. 2007;8:492–502. doi: 10.1038/sj.gene.6364408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Niewold TB, Adler JE, Glenn SB, Lehman TJ, Harley JB, Crow MK. Age- and sex-related patterns of serum interferon-alpha activity in lupus families. Arthritis Rheum. 2008;58(7):2113–9. doi: 10.1002/art.23619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kariuki SN, Franek BS, Kumar AA, Arrington J, Mikolaitis RA, Utset TO, et al. Trait-stratified genome-wide association study identifies novel and diverse genetic associations with serologic and cytokine phenotypes in systemic lupus erythematosus. Arthritis Res Ther. 2010;12(4):R151. doi: 10.1186/ar3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kariuki SN, Crow MK, Niewold TB. The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 2008;58(9):2818–23. doi: 10.1002/art.23728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kariuki SN, Moore JG, Kirou KA, Crow MK, Utset TO, Niewold TB. Age- and gender-specific modulation of serum osteopontin and interferon-alpha by osteopontin genotype in systemic lupus erythematosus. Genes Immun. 2009;10(5):487–94. doi: 10.1038/gene.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kariuki SN, Niewold TB. Genetic regulation of serum cytokines in systemic lupus erythematosus. Transl Res. 2010;155(3):109–17. doi: 10.1016/j.trsl.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kariuki SN, Kirou KA, MacDermott EJ, Barillas-Arias L, Crow MK, Niewold TB. Cutting edge: autoimmune disease risk variant of STAT4 confers increased sensitivity to IFN-alpha in lupus patients in vivo. J Immunol. 2009;182(1):34–8. doi: 10.4049/jimmunol.182.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, et al. Network analysis of associations between serum interferon alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum. 2011 doi: 10.1002/art.30187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kariuki SN, Franek BS, Mikolaitis RA, Utset TO, Jolly M, Skol AD, et al. Promoter variant of PIK3C3 is associated with autoimmunity against Ro and Sm epitopes in African-American lupus patients. J Biomed Biotechnol. 2010;2010:826434. doi: 10.1155/2010/826434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lodolce JP, Kolodziej LE, Rhee L, Kariuki SN, Franek BS, McGreal NM, et al. African-derived genetic polymorphisms in TNFAIP3 mediate risk for autoimmunity. J Immunol. 2010;184(12):7001–9. doi: 10.4049/jimmunol.1000324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salloum R, Franek BS, Kariuki SN, Rhee L, Mikolaitis RA, Jolly M, et al. Genetic variation at the IRF7/PHRF1 locus is associated with autoantibody profile and serum interferon-alpha activity in lupus patients. Arthritis Rheum. 2010;62(2):553–61. doi: 10.1002/art.27182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pothlichet J, Niewold TB, Vitour D, Solhonne B, Chignard M, Crow MK, et al. A loss-of-function variant of the antiviral molecule MAVS is associated with a subset of systemic lupus patients. EMBO Mol Med. 2011 doi: 10.1002/emmm.201000120. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38(5):550–5. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 22.Harley JB, Alarcon-Riquelme ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 2008;40(2):204–10. doi: 10.1038/ng.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheum. 2008;58(8):2481–7. doi: 10.1002/art.23613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rullo OJ, Woo JM, Wu H, Hoftman AD, Maranian P, Brahn BA, et al. Association of IRF5 polymorphisms with activation of the interferon alpha pathway. Ann Rheum Dis. 2010;69(3):611–7. doi: 10.1136/ard.2009.118315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng D, Stone RC, Eloranta ML, Sangster-Guity N, Nordmark G, Sigurdsson S, et al. Genetic variants and disease-associated factors contribute to enhanced interferon regulatory factor 5 expression in blood cells of patients with systemic lupus erythematosus. Arthritis Rheum. 2010;62(2):562–73. doi: 10.1002/art.27223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang ZL. Important aspects of Toll-like receptors, ligands and their signaling pathways. Inflamm Res. 2010;59(10):791–808. doi: 10.1007/s00011-010-0208-2. [DOI] [PubMed] [Google Scholar]
- 27.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50(6):1861–72. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 28.Lovgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Ronnblom L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjogren's syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54(6):1917–27. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 29.Kariuki SN, Crow MK, Niewold TB. The PTPN22 C1858T polymorphism is associated with skewing of cytokine profiles toward high interferon-alpha activity and low tumor necrosis factor alpha levels in patients with lupus. Arthritis Rheum. 2008;58(9):2818–2823. doi: 10.1002/art.23728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paun A, Pitha PM. The IRF family, revisited. Biochimie. 2007;89(6–7):744–53. doi: 10.1016/j.biochi.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu Rev Immunol. 2008;26:535–84. doi: 10.1146/annurev.immunol.26.021607.090400. [DOI] [PubMed] [Google Scholar]
- 32.Battistini A. Interferon regulatory factors in hematopoietic cell differentiation and immune regulation. J Interferon Cytokine Res. 2009;29(12):765–80. doi: 10.1089/jir.2009.0030. [DOI] [PubMed] [Google Scholar]
- 33.Wang H, Morse HC., 3rd IRF8 regulates myeloid and B lymphoid lineage diversification. Immunol Res. 2009;43(1–3):109–17. doi: 10.1007/s12026-008-8055-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, et al. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–31. doi: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Graham RR, Kyogoku C, Sigurdsson S, Vlasova IA, Davies LR, Baechler EC, et al. Three functional variants of IFN regulatory factor 5 (IRF5) define risk and protective haplotypes for human lupus. Proc Natl Acad Sci U S A. 2007;104(16):6758–63. doi: 10.1073/pnas.0701266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gateva V, Sandling JK, Hom G, Taylor KE, Chung SA, Sun X, et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat Genet. 2009;41(11):1228–33. doi: 10.1038/ng.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsujimura H, Tamura T, Kong HJ, Nishiyama A, Ishii KJ, Klinman DM, et al. Toll-like receptor 9 signaling activates NF-kappaB through IFN regulatory factor-8/IFN consensus sequence binding protein in dendritic cells. J Immunol. 2004;172(11):6820–7. doi: 10.4049/jimmunol.172.11.6820. [DOI] [PubMed] [Google Scholar]
- 38.Barnes BJ, Kellum MJ, Pinder KE, Frisancho JA, Pitha PM. Interferon regulatory factor 5, a novel mediator of cell cycle arrest and cell death. Cancer Res. 2003;63(19):6424–31. [PubMed] [Google Scholar]
- 39.Barnes BJ, Moore PA, Pitha PM. Virus-specific activation of a novel interferon regulatory factor, IRF-5, results in the induction of distinct interferon alpha genes. J Biol Chem. 2001;276(26):23382–90. doi: 10.1074/jbc.M101216200. [DOI] [PubMed] [Google Scholar]
- 40.Reddy MV, Velazquez-Cruz R, Baca V, Lima G, Granados J, Orozco L, et al. Genetic association of IRF5 with SLE in Mexicans: higher frequency of the risk haplotype and its homozygozity than Europeans. Hum Genet. 2007;121(6):721–7. doi: 10.1007/s00439-007-0367-6. [DOI] [PubMed] [Google Scholar]
- 41.Shin HD, Kim I, Choi CB, Lee SO, Lee HW, Bae SC. Different genetic effects of interferon regulatory factor 5 (IRF5) polymorphisms on systemic lupus erythematosus in a Korean population. J Rheumatol. 2008;35(11):2148–51. doi: 10.3899/jrheum.080124. [DOI] [PubMed] [Google Scholar]
- 42.Kawasaki A, Kyogoku C, Ohashi J, Miyashita R, Hikami K, Kusaoi M, et al. Association of IRF5 polymorphisms with systemic lupus erythematosus in a Japanese population: support for a crucial role of intron 1 polymorphisms. Arthritis Rheum. 2008;58(3):826–34. doi: 10.1002/art.23216. [DOI] [PubMed] [Google Scholar]
- 43.Kelly JA, Kelley JM, Kaufman KM, Kilpatrick J, Bruner GR, Merrill JT, et al. Interferon regulatory factor-5 is genetically associated with systemic lupus erythematosus in African Americans. Genes Immun. 2008 doi: 10.1038/gene.2008.4. [DOI] [PubMed] [Google Scholar]
- 44.Siu HO, Yang W, Lau CS, Chan TM, Wong RW, Wong WH, et al. Association of a haplotype of IRF5 gene with systemic lupus erythematosus in Chinese. J Rheumatol. 2008;35(2):360–2. [PubMed] [Google Scholar]
- 45.Sigurdsson S, Goring HH, Kristjansdottir G, Milani L, Nordmark G, Sandling JK, et al. Comprehensive evaluation of the genetic variants of interferon regulatory factor 5 (IRF5) reveals a novel 5 bp length polymorphism as strong risk factor for systemic lupus erythematosus. Hum Mol Genet. 2008;17(6):872–81. doi: 10.1093/hmg/ddm359. [DOI] [PubMed] [Google Scholar]
- 46.Wen F, Ellingson SM, Kyogoku C, Peterson EJ, Gaffney PM. Exon 6 variants carried on systemic lupus erythematosus (SLE) risk haplotypes modulate IRF5 function. Autoimmunity. 2010 doi: 10.3109/08916934.2010.491842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richez C, Yasuda K, Bonegio RG, Watkins AA, Aprahamian T, Busto P, et al. IFN regulatory factor 5 is required for disease development in the FcgammaRIIB−/−Yaa and FcgammaRIIB−/− mouse models of systemic lupus erythematosus. J Immunol. 2010;184(2):796–806. doi: 10.4049/jimmunol.0901748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Pagano JS. Structure and function of IRF-7. J Interferon Cytokine Res. 2002;22(1):95–101. doi: 10.1089/107999002753452700. [DOI] [PubMed] [Google Scholar]
- 49.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41(7):776–82. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Javed A, Reder AT. Therapeutic role of beta-interferons in multiple sclerosis. Pharmacol Ther. 2006;110(1):35–56. doi: 10.1016/j.pharmthera.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 51.Weckerle CE, Niewold TB. The unexplained female predominance of systemic lupus erythematosus: clues from genetic and cytokine studies. Clin Rev Allergy Immunol. 2011;40(1):42–9. doi: 10.1007/s12016-009-8192-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Poole BD, Scofield RH, Harley JB, James JA. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity. 2006;39(1):63–70. doi: 10.1080/08916930500484849. [DOI] [PubMed] [Google Scholar]
- 53.Lenert P. Nucleic acid sensing receptors in systemic lupus erythematosus: development of novel DNA- and/or RNA-like analogues for treating lupus. Clin Exp Immunol. 2010;161(2):208–22. doi: 10.1111/j.1365-2249.2010.04176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, et al. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum. 2009;60(6):1785–96. doi: 10.1002/art.24557. [DOI] [PubMed] [Google Scholar]