

Abstract
Stress triggers psychiatric conditions including depressive and anxiety disorders. The mechanisms by which stress produces persistent changes in behavior are not fully understood. Here we show in rats that stress (footshock) activates the transcription factor cAMP response element binding protein (CREB) within the nucleus accumbens shell (NAS), a brain area involved in encoding reward and aversion. To examine the behavioral significance of altered CREB function in the NAS, we used viral vectors to elevate or disrupt CREB in this region. Elevated CREB produced increases in intracranial self-stimulation thresholds, a depressive-like sign reflecting anhedonia (decreased sensitivity to reward), whereas disruption of CREB function by expression of a dominant-negative CREB had the opposite effect. To determine whether neuroadaptations that produce anhedonia subsequently affect vulnerability to stress-induced behavioral adaptations, we subjected rats with altered CREB function in the NAS to fear conditioning. Although neither elevation nor disruption of CREB function altered the development of conditioned fear, elevation of CREB impaired extinction of conditioned fear. To mimic downstream effects of CREB activation on expression of the opioid peptide dynorphin, we microinjected the κ-opioid receptor (KOR) agonist U50,488 directly into the NAS. KOR stimulation produced anhedonia but had no effect on expression or extinction of conditioned fear. These findings demonstrate that activation of CREB in the NAS produces multiple behavioral signs (anhedonia, impaired extinction) characteristic of experience-dependent psychiatric conditions such as posttraumatic stress disorder. Although CREB activation is a common trigger, expression of these individual signs appears to involve divergent downstream mechanisms.
Introduction
Stress is implicated in the etiology of depression (Keller et al., 2007) and anxiety disorders including posttraumatic stress disorder (PTSD) (Kessler et al., 1995; Keane et al., 2006). The mechanisms by which stress produces signs of these conditions are not understood. Studies of stress-related neuroadaptive responses may identify overlap in the pathophysiology of these frequently comorbid disorders, enabling improved diagnosis and treatments that target specific symptoms.
Anhedonia (reduced ability to experience pleasure or reward) is a sign of both depression and PTSD (American Psychiatric Association, 2000). One brain area known to play a crucial role in reward and motivation is the nucleus accumbens (NAc) (Nestler and Carlezon, 2006; Wise, 2008; Carlezon and Thomas, 2009). Pliakas et al. (2001) showed that forced swim stress activates (via phosphorylation) the transcription factor cAMP response element binding protein (CREB) in the NAc shell (NAS), and that this neuroadaptive response has functional effects on behavior. Specifically, elevating CREB expression to mimic activation in the NAS increases immobility behavior in the forced swim test, a putative depressive-like sign, whereas disruption of CREB in the NAS has antidepressant-like effects. Elevated expression of CREB in the NAS also alters responsiveness to cocaine in place conditioning tests (Carlezon et al., 1998; Pliakas et al., 2001), reducing the ability of high doses to establish place preferences. Whereas this effect may reflect anhedonia, it could also reflect increases in the aversive or anxiogenic effects of cocaine or cocaine withdrawal (Pliakas et al., 2001; Neumaier et al., 2002), generalized reductions in sensitivity to motivationally relevant stimuli (numbing), or alterations in learning and memory processes required for expression of conditioned reward. Models that quantify sensitivity to other rewards, or that reflect other core signs of depressive and anxiety disorders in humans, may help to clarify how elevated CREB function affects sensitivity to rewarding and aversive stimuli.
The present studies were designed to identify the behavioral significance of stress-induced elevation of CREB function within the NAS. We used intracranial self-stimulation (ICSS) to provide real-time measurements of the effects of altered CREB function on the rewarding impact of lateral hypothalamic brain stimulation (Carlezon and Chartoff, 2007). We also extended our studies to fear conditioning to determine whether other depressive- or PTSD-like behaviors are produced, such as vulnerability to fear or failure to extinguish fear (Myers and Davis, 2007). Finally, we examined the role of κ-opioid receptor (KOR) systems in the effects of CREB on reward and fear. CREB regulates dynorphin, an endogenous agonist at KORs, in striatum and NAS (Cole et al., 1995; Carlezon et al., 1998). Systemic administration of KOR agonists causes depressive-like effects in humans (Pfeiffer et al., 1986) and rats (Bals-Kubik et al., 1989; Mague et al., 2003; Todtenkopf et al., 2004), whereas KOR antagonists have antidepressant- (Mague et al., 2003; Beardsley et al., 2005) and anxiolytic-like effects (Knoll et al., 2007). We used microinjections of a selective KOR agonist [trans-(±)-3,4-dichloro-N-methyl-N[2-(pyrrolidinyl) cyclohexyl] benzeneacetamide methanesulfonate (U50,488)] to determine whether the behavioral effects of elevating CREB function in the NAS are mimicked by local KOR stimulation.
Materials and Methods
Rats.
A total of 144 male Sprague Dawley rats (300–350 g; Charles River Laboratories) were used. Rats were housed in pairs except those in the ICSS studies, which were housed singly after electrode implantation. Rats were maintained on a 12 h light/dark cycle (lights on 7:00 A.M. to 7:00 P.M.) with access to food and water except during testing. Experiments were conducted in accordance with the 1996 National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Viral vectors.
cDNAs for CREB, dominant-negative CREB (mCREB), and LacZ (encoding β-galactosidase) were inserted into the herpes simplex virus (HSV) amplicon HSV-PrpUC and packaged into virus using the helper 5dl1.2, as described previously (Carlezon et al., 1998, 2000; Pliakas et al., 2001; Paine et al., 2009). Virus was purified on a sucrose gradient, pelleted, and resuspended in 10% sucrose. The titer of the vector stocks was ∼4.0 × 107 infectious units per milliliter. For each study, aliquots from the same batches of the viral vectors were used. We have shown previously that transgene expression caused by these vectors is maximal 3–4 d after treatment and minimal by day 10 (Carlezon et al., 1998; Pliakas et al., 2001; Paine et al., 2009). We have also shown using these vectors that elevated CREB produces functional increases in the expression of CREB-regulated target genes (e.g., prodynorphin), whereas mCREB decreases expression of CREB-regulated target genes (Carlezon et al., 1998).
Drug solutions.
U50,488 and cocaine hydrochloride were purchased from Sigma-Aldrich and dissolved in triple-distilled water (vehicle). Doses (in micrograms) are expressed as the salt form of the drug per microliter. Rats received bilateral microinjections of 0.5 μl per side for a total of volume of 1.0 μl.
Protein (Western) immunoblotting.
Footshock was delivered in the same apparatus used for fear-potentiated startle (FPS) (see below section on FPS) (Knoll et al., 2007). Rats were habituated to handling and confinement in customized startle cages (19 × 9 × 14 cm) with steel-rod floor bars for 30 min/d on the 2 d before the experiment. On the day of the experiment, rats were placed in the startle cages, and after a 5 min acclimation period, some (n = 9) received five 0.6 mA scrambled footshock (3 min mean interstimulus interval) delivered through the floor bars by an external shock generator (Med Associates). Control rats (n = 8) did not receive footshock. Ten minutes after the completion of this session, rats were killed by rapid decapitation; brains were removed and snap frozen in 2-methylbutane chilled with dry ice. This time point was chosen to enable qualitative comparisons with previous work showing increased CREB phosphorylation in the NAS 10 min after forced swim stress (Pliakas et al., 2001). Brains were sectioned on a cryostat to the level of the striatum. To obtain bilateral punches of the NAS, a 1.0 mm sample corer was used to first remove each nucleus accumbens core (NAC, centered on the anterior commissure); a 2.0 mm sample corer was then used to obtain each NAS in a second, crescent-shaped punch made immediately medial to the NAC. Brain sections (40 μm) in the area of the dissection were retained for each rat. Tissue was sonicated in buffer containing 1× phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich) in 1% SDS and diluted to 2 μg of protein per microliter. Samples were diluted in lithium dodecyl sulfate sample buffer (Invitrogen) heated at 70°C for 10 min and loaded (20 μg of protein) onto NuPAGE Novex 4–12% Bis-Tris gels (Invitrogen). After electrophoretic transfer, the polyvinylidene membranes were blocked in 5% milk and then incubated in phospho-CREB (P-CREB) antibody (1:1000; lot 4; Cell Signaling Technology) overnight at 4°C. After incubation in secondary antibody (HRP-conjugated goat anti-mouse IgG, 1:5000; Vector Laboratories) for 1 h, immunoreactivity was visualized with chemiluminescence (PerkinElmer) using a Kodak Image Station 4000 mm (Carestream). Membranes were then stripped in 62.5 mm Tris, 2% SDS, and 100 mm 2-mercaptoethanol, pH 6.8, at 50°C, blocked, and reprobed with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:20,000; Sigma-Aldrich) to normalize the results (Espinosa et al., 2008; Green et al., 2008). Blots were quantified by image analysis (Carestream), and data were expressed as the ratio of P-CREB/GAPDH in rats exposed to footshock and those not exposed to footshock. Group differences in P-CREB were analyzed using a Student's t test.
ICSS.
Forty-five rats were anesthetized with an intraperitoneal injection of pentobarbital (65 mg/kg) and given subcutaneous atropine sulfate (0.25 mg/kg) to reduce bronchial secretions. Monopolar stainless-steel electrodes (0.250 mm diameter; Plastics One) were implanted in the right medial forebrain bundle (MFB) at the level of the lateral hypothalamus at a 10° angle (final electrode tip coordinates, 3.0 mm posterior to bregma, 1.6 mm lateral from midsaggital suture, 7.6 mm below dura) (Paxinos and Watson, 1997). The electrodes were coated with polyimide insulation except at the flattened tip. Simultaneously, rats were implanted with bilateral guide cannulae (20 gauge; Plastics One) aimed at the NAS (final injection site coordinates, 1.2 mm anterior to bregma, 0.8 mm lateral from midsaggital suture, 6.9 mm below dura). For rats in the gene transfer studies, obturators were flush with the bottom of the guide cannulae (because viral vector expression is improved when infused into intact tissue) (Carlezon et al., 2000), whereas for rats in the drug studies, obturators extended 1.0 mm beyond the bottom of the guide cannulae. Skull screws (one of which served as the ground) and the electrode and cannula were secured to the skull with dental acrylic.
After 1 week recovery, the rats were trained on a continuous reinforcement schedule (FR1) to respond to brain stimulation as described previously (Todtenkopf et al., 2006). Briefly, each lever press earned a 0.5 s train of square-wave cathodal pulses (0.1 ms pulse duration) at a frequency of 141 Hz. The stimulation current (100–300 μA) was adjusted gradually to the lowest value that would sustain a reliable rate of responding (at least 40 rewards per minute). Once an appropriate current was found for each rat, it was held constant throughout the remainder of the experiment. Each rat was then adapted to brief tests (1 min trials) with each of a descending series of 15 stimulation frequencies. For each frequency tested, there was an initial 5 s “priming” phase during which noncontingent stimulation was given followed by a 50 s test phase during which the number of responses was counted. After the test phase, there was a 5 s time-out period during which no stimulation was available. The stimulation frequency was then lowered by ∼10% (0.05 log10 units), and another trial was started. After responding had been evaluated at each of the 15 frequencies (a “pass”), the procedure was repeated such that each rat was given four passes per day (60 min of training). To characterize the functions relating response strength to reward magnitude, a least-squares line of best fit was plotted across the frequencies that sustained responding at 20, 30, 40, 50, and 60% of the maximum rate using customized analysis software. ICSS threshold was defined as the frequency at which the line intersected the x-axis (theta 0) (Miliaressis et al., 1986); theoretically, this represents the point at which the stimulation becomes rewarding. Testing began when mean daily ICSS thresholds varied by <15% over five consecutive training sessions.
ICSS: effects of CREB vectors.
Thirty-three of the rats received microinjections of viral vectors using methods described previously (Todtenkopf et al., 2006). Immediately after the final training session, rats were lightly anesthetized with isoflurane and placed in a stereotaxic apparatus. The needle of a 26 gauge Hamilton syringe modified with a metallic tab so that it would extend 1.0 mm beyond the bottom of the guide cannulae was lowered into place; rats then received bilateral (2.0 μl/side) microinjections of HSV-CREB (n = 9), HSV-mCREB (n = 11), or HSV-LacZ (n = 13) into the NAS over 10 min, and the syringes were left in place for an additional 5 min to allow diffusion. One of the rats that received HSV-LacZ was not tested further, and killed 4 d later to provide a representative depiction of HSV-mediated gene transfer within the NAS (Fig. 2), using previously described methods (Todtenkopf et al., 2006). The remaining 32 rats were tested for ICSS beginning 24 h after the microinjections in 15 min sessions on 8 consecutive days. The mean threshold for each day was calculated and compared to the baseline threshold, which was defined as the mean of the three training sessions that immediately preceded gene transfer. Treatment effects on ICSS thresholds or maximal response rates over the 8 d test period were evaluated with separate two-way ANOVAs (treatment by day) with repeated measures, and significant effects were analyzed further using post hoc Fisher's protected t tests.
Figure 2.

Expression of β-galactosidase in the NAS 4 d after HSV-mediated gene transfer into the NAS. Brain is costained for calbindin. The number (in millimeters) at the bottom left depicts distance from bregma. Scale bar, 500 μm.
ICSS: effects of the KOR agonist U50.
Twelve of the rats were used for the drug microinjection studies. On each test day, two rate-frequency functions (passes) obtained immediately before drug treatment were averaged to determine baseline (threshold and maximal response rates) parameters. Microinjections were then administered using a syringe pump and Hamilton syringes connected to polyethylene tubing, which was fitted to injectors extending 1.0 mm beyond the end of the cannulae. All rats received infusions at a rate of 0.2 μl/min. The injectors were left in place for an additional 2.5 min to allow for drug diffusion, and then the rats were tested for 30 min (two passes). The order of drug treatment was the same for all animals (vehicle, 1.0 μg of U50, 3.0 μg of U50, and 20 μg of cocaine). There was a minimum of 3 d of restabilized responding between microinjections. Effects of U50 on ICSS thresholds or maximal response rates were evaluated with separate one-way ANOVAs (treatment) with repeated measures, and significant effects were analyzed further using post hoc Fisher's protected t tests. Effects of cocaine on ICSS thresholds and maximum rates were analyzed with Student's t tests (correlated groups).
FPS.
A total of 82 rats were used in experiments involving fear conditioning; all were implanted with bilateral NAS guide cannulae as described above and given 1 week for recovery. FPS studies were conducted in the startle cages described above using established methods (Knoll et al., 2007). Each cage was mounted on a load cell within a fan-ventilated 64 × 40 × 60 cm sound-attenuating cabinet; temperature was monitored constantly and maintained at ∼20°C. Changes in load cell voltage caused by cage movement were amplified, digitized, and recorded using a computer and corresponding hardware interface card. Startle amplitude was proportional to the amount of cage movement and was defined as the largest peak-to-peak voltage that occurred during the first 200 ms after the onset of the startle stimulus. Constant wide-band background noise (60 dB, 10–20 kHz) and 50 ms white noise startle stimuli (1–32 kHz, 1 ms rise and decay) were produced by an audio generator and delivered using loudspeakers positioned 4 cm behind the startle cage. An 8 W (300 lumen) fluorescent lamp located 6 cm from the cage was used as the conditioned stimulus. The unconditioned stimulus was a scrambled footshock (0.6 mA; 500 ms) delivered through the floor of each cage by an external electric stimulator, identical to that used for the immunoblotting studies described above. The calibration, presentation, and sequencing of all stimuli were controlled by a computer and customized software.
FPS: effects of CREB vectors.
Sixty of the rats received microinjections of viral vectors. The first experiment examined the effects of altering CREB function before training on the development and expression of FPS and extinction. In these studies, rats received bilateral infusions of HSV-CREB (n = 9), HSV-mCREB (n = 10), HSV-LacZ (n = 7), or 10% sucrose vehicle (n = 9) aimed at the NAS as described above on experimental day 1. [Since there were no differences in behavior between sucrose vehicle and HSV-LacZ, sucrose was used for subsequent studies to minimize the use of viral vectors (Fig. 5).] After a recovery period of two days, rats were placed in startle cages on experimental day 4 and allowed a 5 min acclimation period before a conditioning session, during which they received 10 light–shock pairings (3 s light coterminating with a footshock). The mean interval between stimulus presentations was 3 min, with a range of 2–4 min. Testing occurred on the following day (experimental day 5): rats were returned to startle cages and allowed 5 min acclimation before the presentation of nine habituating startle stimuli (three stimuli at 95, 100, and 105 dB; 30 s interstimulus interval). Rats then received 18 startle stimuli (six each at 95, 100, and 105 dB). Nine of the startle stimuli were preceded by a 3 s presentation of the light. Across trials, the presentation of sound intensities and the light stimulus were made in a semirandom order. Conditioned fear was defined as the difference in startle amplitude elicited in the presence or absence of the light (percentage FPS = [(startle in presence of light − startle in dark)/startle in dark] × 100).
Figure 5.

Effect of intra-NAS gene transfer on behavior in the FPS test. A, When gene transfer was performed before fear conditioning, there were no group differences in FPS (mean ± SEM) during test 1, but elevated CREB caused persistent increases in FPS that were resistant to extinction. B, When gene transfer was performed after fear conditioning, elevated CREB produced similar effects. *p < 0.05; **p < 0.01, Fisher's protected t tests.
On the day after FPS testing (experimental day 6), rats underwent extinction training. During extinction training sessions, rats were returned to startle cages and were exposed to 60 presentations of the light (3 s duration, 30 s interstimulus interval). Rats then received a second test for FPS on experimental day 7. Two days later, rats were given a second extinction training session (experimental day 9) and a third test for FPS the following day (experimental day 10).
The second experiment examined the effects of altering CREB function after training on the development and expression of FPS and extinction. These studies were designed to use the same order of training and testing as in the first experiment, but CREB function was manipulated after FPS had been established. Rats were conditioned with 10 light–shock pairings (experimental day 1) and tested for FPS the following day (experimental day 2). Rats were assigned to three groups with matching levels of FPS and the next day (experimental day 3) given intra-NAS infusions of HSV-CREB (n = 7), HSV-mCREB (n = 10), or sucrose vehicle (n = 8). After 2 d recovery, rats received extinction training (experimental day 6) and were given a second test for FPS the following day (experimental day 7). Two days later, rats were given a second extinction training session (experimental day 9), and a third test for FPS the following day (experimental day 10).
FPS data from each experiment were analyzed using two-way ANOVAs (treatment by day) with repeated measures, and significant effects were analyzed further using post hoc Fisher's protected t tests.
FPS: effects of the KOR agonist U50.
Twenty-two rats were used to determine whether microinjections of U50 into the NAS were sufficient to mimic the effect of elevated CREB. Since we found evidence (Fig. 5) that rats treated with HSV-CREB had elevated startle regardless of whether they were treated with the vector before or after training, these studies focused on whether elevated KOR function during testing or during extinction training would increase startle. Training and testing protocols were similar to those used for the viral vector studies. All rats received 10 light–shock pairings on experimental day 1 and were tested for FPS on experimental day 2. On the basis of this test, rats were separated into groups with equivalent FPS. Experimental day 3 involved extinction training, as described above. On this day, a subgroup of rats received intra-NAS microinjections of U50 (1.0 μg; n = 7) or vehicle (n = 7) immediately preceding extinction training, and subsequent FPS testing on experimental day 4. The remaining rats were given extinction training on experimental day 3, but then received intra-NAS microinjections of U50 (1.0 μg; n = 4) or vehicle (n = 4) immediately preceding FPS testing on experimental day 4. For each subgroup, differences in FPS on day 4 were analyzed using Student's t tests.
Histology.
Immediately after the final test, rats were overdosed with pentobarbital (130 mg/kg, i.p.) and perfused with 0.9% saline followed by 4% paraformaldehyde as described previously (Todtenkopf et al., 2006). Brains were kept overnight in 20% glycerol before slicing (40 μm). Electrode and injection placements were verified in histological analyses (Carlezon et al., 1998) by an observer unaware of the treatment conditions.
Results
Effect of footshock stress on CREB activity in the NAS
Footshock stress increased expression of PCREB in the NAS (t(15) = 2.28; p < 0.05) (Fig. 1A). Effects were localized to the NAS by first dissecting the NAC (Fig. 1B) and then obtaining the NAS in a second, crescent-shaped punch (Fig. 1C). These data indicate that the type of footshock used in fear-conditioning tests (Knoll et al., 2007) increases CREB activity in the NAS.
Figure 1.
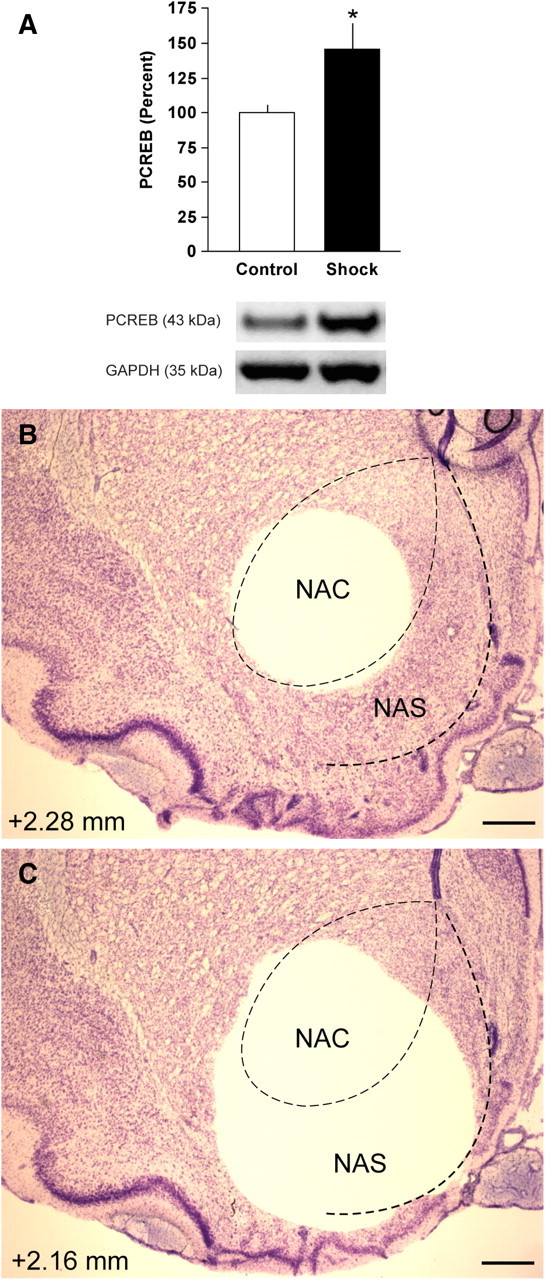
Effect of footshock on P-CREB in the NAS. A, Western immunoblot of P-CREB in the NAS after exposure to footshock. Control rats were treated identically in all respects except they did not receive footshock. Exposure to footshock significantly elevated P-CREB in the NAS. GAPDH was used to normalize protein loading in each lane; data are expressed as the mean ± SEM of the P-CREB/GAPDH ratio. *p < 0.05, Student's t test. B, C, Effects were localized to the NAS by first dissecting the NAC (B) and then obtaining the NAS in a second, crescent-shaped punch (C). The number (in millimeters) at the bottom left depicts distance from bregma. Scale bar, 500 μm.
Effect of altered CREB function on ICSS
Viral vectors expressing CREB, mCREB, or β-galactosidase (the protein product of LacZ) (Fig. 2) were microinjected into the NAS to examine how alterations in CREB function might affect the function of brain reward systems, as reflected by changes in the rewarding impact of lateral hypothalamic brain stimulation. Over the 8 d period of testing, ICSS thresholds depended on an interaction of treatment and day (F(14,203) = 2.46; p < 0.01). Whereas HSV-LacZ had no effect (Fig. 3A,C), elevated expression of CREB increased ICSS thresholds (Fig. 3A); this is reflected by parallel rightward shifts in rate-frequency functions caused by reduced effectiveness of stimulation frequencies that sustained responding before gene transfer (i.e., baseline) (Fig. 3D). ICSS thresholds were significantly higher in rats treated with HSV-CREB than in rats treated with HSV-LacZ on day 2 (p < 0.01, Fisher's protected t tests), day 3 (p < 0.01), day 4 (p < 0.05), day 5 (p < 0.01), and day 7 (p < 0.05) (Fig. 3A). In contrast, disruption of CREB function by expression of mCREB decreased ICSS thresholds (Fig. 3A); this is reflected by parallel leftward shifts in rate-frequency functions caused by increased effectiveness of stimulation frequencies that failed to sustain responding before gene transfer (Fig. 3E). ICSS thresholds were significantly lower in rats treated with HSV-mCREB than in rats treated with HSV-LacZ on day 2 (p < 0.05, Fisher's protected t tests), day 3 (p < 0.05), day 4 (p < 0.05), day 5 (p < 0.01), and day 6 (p < 0.05) (Fig. 3A). None of the viral vector treatments affected response rates during the test period [F(2,29) = 0.19; not significant (n.s.)] (Fig. 3B), although there was a main effect of days (F(7,203) = 3.36; p < 0.01). Simple main effect tests revealed that relative to day 1, response rates were significantly higher on days 2–7 (p values < 0.01). ICSS electrodes were localized to the MFB at the level of the lateral hypothalamus (Fig. 4), and the placements were indistinguishable from those depicted previously (Todtenkopf et al., 2004, 2006; Mague et al., 2005; Tomasiewicz et al., 2008; DiNieri et al., 2009). These data indicate that elevated CREB expression in the NAS reduces the rewarding impact of MFB stimulation.
Figure 3.

Effect of intra-NAS gene transfer on behavior in the ICSS test over the 8 d test period. A, Elevated CREB increased the mean (± SEM) ICSS thresholds, whereas expression of mCREB decreased thresholds relative to rats treated with HSV-LacZ. B, None of the treatments differentially affected maximal response rates, although a main effect of day revealed that response rates were generally lower the first day after gene transfer. Data are expressed as percentage pre-gene-transfer baselines. The transient time course of the behavioral effects is qualitatively similar to the transient time course of HSV-mediated transgene expression, which is maximal on days 3–4 and waning by days 7–8 (Carlezon et al., 2000). *p < 0.05; **p < 0.01, Fisher's protected t tests. C–E, Comparisons between responding during pre-gene-transfer baseline and responding on day 3. Elevated expression of β-galactosidase in the NAS had no effects on ICSS, but elevated CREB expression caused rightward shifts in rate-frequency functions, indicating that higher stimulation frequencies were necessary to motivate responding. In contrast, expression mCREB caused leftward shifts, indicating that lower stimulation frequencies were necessary to motivate responding. Data are from representative rats.
Figure 4.

ICSS electrode track in a Nissl-stained section from a representative rat. The number (in millimeters) at the bottom left depicts distance from bregma. Scale bar, 500 μm. fx, Fornix; mtt, mammilothalamic tract; ot, optic tract; VMH, ventromedial nucleus of the hypothalamus.
Effect of altered CREB function on FPS
Considering that elevated CREB function in the NAS produced a depressive-like sign (anhedonia) in the ICSS test, we extended our studies to the FPS test to determine whether it produces other behavioral signs that are characteristic of stress-related disorders. To examine whether nonspecific increases in transgene expression affect startle, rats that received intra-NAS microinjections of HSV-LacZ were compared to those that received intra-NAS microinjections of vehicle (10% sucrose). Because the responses in these groups were virtually identical (main effect of treatment, F(1,14) = 0.0002, n.s.; treatment by day interaction, F(2,28) = 0.15, n.s.) (data not shown), they were consolidated into a single group (control). When the viral vectors were used to manipulate CREB function before training (Fig. 5A), startle responses depended on treatment (F(2,32) = 5.76; p < 0.01) and test number (F(2,64) = 18.8; p < 0.01), although the interaction of these factors did not reach statistical significance (F(4,64) = 2.23; p = 0.07). Simple main effects tests on treatment revealed that there were no differences among groups on day 1 (F(2,32) = 1.26, n.s.), but that there was an effect during test 2 (F(2,32) = 7.32; p < 0.01) because of elevated startle in the group that received HSV-CREB (p < 0.01). Similarly, there was an effect during test 3 (F(2,32) = 3.80; p < 0.05) because of elevated startle in the group that received HSV-CREB (p < 0.05). Simple main effects tests on test number revealed effects in the group treated with HSV-CREB (F(2,30) = 20.1; p < 0.01), HSV-mCREB (F(2,30) = 12.3; p < 0.01), and control (F(2,30) = 17.8; p < 0.01). Post hoc analysis revealed that, compared to test 1, startle responsiveness was lower during test 2 and test 3 in the HSV-mCREB and control groups (p values < 0.01), but only on test 3 in the HSV-CREB group (p < 0.01). These findings suggest impaired extinction of FPS in the HSV-CREB group.
To address the possibility that the rats treated with HSV-CREB before training had stronger consolidation of FPS rather than extinction deficits per se, a second set of rats received gene transfer after training. When the viral vectors were used to manipulate CREB function after training (Fig. 5B), startle responses depended on treatment (F(2,22) = 4.94; p < 0.05) and test number (F(2,44) = 7.65; p < 0.01), although the interaction of these factors did not reach statistical significance (F(4,44) = 2.08; p = 0.10). Simple main effects tests on treatment revealed that there were no differences among groups on test 1 (F(2,22) = 0.26, n.s.), but that there was an effect during test 2 (F(2,22) = 3.34; p < 0.05) because of elevated startle in the group that received HSV-CREB (p < 0.05). Similarly, there was an effect on test 3 (F(2,22) = 4.68; p < 0.05) because of elevated startle in the group that received HSV-CREB (p < 0.05). Simple main effects tests on test number revealed no effects in the HSV-CREB group (F(2,18) = 0.08, n.s.), but significant effects in HSV-mCREB (F(2,18) = 10.6; p < 0.01) and control (F(2,18) = 17.8). Post hoc analysis revealed that, compared to test 1, startle responsiveness was lower in the HSV-mCREB group during test 2 (p < 0.05) and test 3 (p < 0.01). Similarly, startle responsiveness was lower in the control group during test 2 and test 3 (p values < 0.01). These findings are also consistent with impaired extinction in the HSV-CREB group.
Effect of KOR stimulation on ICSS
Previous work has shown that CREB regulates dynorphin (Douglass et al., 1994; Cole et al., 1995; Simpson and McGinty, 1995) and that HSV-CREB causes elevations in dynorphin gene expression (Carlezon et al., 1998). Elevated dynorphin tone in the NAS would be expected to increase stimulation of KORs located on the terminals of ventral tegmental area inputs (Svingos et al., 1999), decreasing dopamine (DA) release and producing anhedonia (Todtenkopf et al., 2004; Carlezon et al., 2006; Tomasiewicz et al., 2008; DiNieri et al., 2009; Carlezon and Thomas, 2009). We used microinjections of U50 to determine whether the anhedonia produced in the ICSS test by elevated expression of CREB in the NAS is recapitulated by KOR stimulation localized to this brain region (Fig. 6), using microinjections of vehicle as a control. ICSS thresholds depended on treatment (F(2,22) = 3.46; p < 0.05) (Fig. 7A). Whereas microinjections of vehicle had no effect (Fig. 7A,C), microinjections of 1.0 μg of U50 increased ICSS thresholds (p < 0.05) (Fig. 7A,D), as reflected by parallel rightward shifts in rate-frequency functions caused by reduced effectiveness of stimulation frequencies that sustained responding during baseline. A higher dose of U50 (3.0 μg) produced smaller, nonsignificant effects (Fig. 7A). After the U50 studies, we microinjected cocaine (20 μg) into the NAS as a positive control. This treatment decreased ICSS thresholds (t(11) = 3.86; p < 0.01) (Fig. 7A,E), as reflected by parallel leftward shifts in rate-frequency functions caused by increased effectiveness of stimulation frequencies that failed to sustain responding during baseline. Response rates were not affected by U50 (F(2,22) = 2.80, n.s.) or cocaine (t(11) = 1.41, n.s.) (Fig. 7B). ICSS electrode placements were indistinguishable from those in the viral vector studies (Fig. 4). These data indicate that the consequences of elevated CREB expression in the NAS on reward function are mimicked by KOR stimulation.
Figure 6.
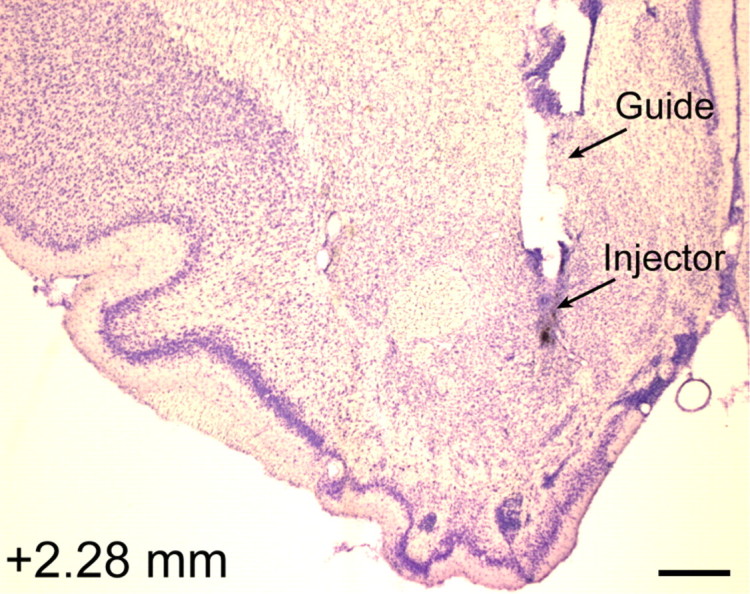
Histology section from a representative rat depicting the location of the guide cannula and injector used to deliver drug microinjections for ICSS and FPS studies. The number (in millimeters) at the bottom left depicts distance from bregma. Scale bar, 500 μm.
Figure 7.

Effect of intra-NAS drug treatments on behavior in the ICSS test. A, Microinjections of 1.0 μg of U50 increased the mean (± SEM) ICSS thresholds, although a higher dosage (3.0 μg) was less effective. In the same rats, subsequent microinjections of 20 μg of cocaine decreased ICSS thresholds. *p < 0.05, Fisher's protected t test; ⋀p < 0.05, Student's t test (correlated groups). B, None of the treatments differentially affected maximal response rates. Data are from 30 min tests and are expressed as percentage daily pretreatment baselines. C–E, Comparisons between responding during pretreatment baseline and responding after treatment. Microinjections of vehicle had no effects on ICSS, but 1.0 μg of U50 caused rightward shifts in rate-frequency functions, indicating that higher stimulation frequencies were necessary to motivate responding. In contrast, 20 μg of cocaine caused leftward shifts, indicating that lower stimulation frequencies were necessary to motivate responding. Data are from representative rats.
Effect of KOR stimulation on FPS
Because the anhedonia produced in the ICSS test by elevated expression of CREB in the NAS was recapitulated by intra-NAS microinjections of U50 (1.0 μg), we examined whether this treatment would also affect startle. After fear conditioning training and a single test session, rats were matched into two groups with equivalent FPS (control, 117.5 ± 12.3; U50, 113.9 ± 20.1). Rats were further subdivided into equivalent groups that received either U50 (or vehicle) immediately before FPS testing or extinction training. In rats that received treatment immediately before FPS testing, there were no differences between rats treated with intra-NAS microinjections of U50 or vehicle (t(6) = 0.45, n.s.) (Fig. 8). Likewise, there were no differences in FPS in rats that had been treated with intra-NAS microinjections of U50 or vehicle during an extinction training session (t(12) = 0.25, n.s.) (Fig. 8). Together these data indicate that the consequences of elevated CREB expression in the NAS on fear are not recapitulated by KOR stimulation.
Figure 8.

Effect of intra-NAS microinjections of κ agonist on behavior in the FPS test. U50 (1.0 μg) had no effect on FPS (mean ± SEM) when administered immediately before an FPS test session or immediately before an extinction training session that occurred the previous day.
Discussion
Footshock activated the transcription factor CREB in the NAS of rats, and in turn CREB activation in this region triggered depressive- and anxiety-like behaviors. Viral vector-mediated elevation of CREB expression within the NAS, which increases CREB-mediated transcription (Carlezon et al., 1998; Sakai et al., 2002), reduced the effectiveness of brain stimulation that sustained robust ICSS before gene transfer. A qualitatively similar effect is caused by treatments that cause depressive behaviors in humans and laboratory animals (Pfeiffer et al., 1986; Markou et al., 1992; Todtenkopf et al., 2004), suggesting that it reflects decreases in the rewarding impact of the stimulation (anhedonia). Elevated CREB expression also induced persistent increases in FPS despite extinction training. Impaired extinction of conditioned fear was not caused merely by elevated CREB function during FPS training, because it was also evident when CREB function was elevated after training. These findings demonstrate that CREB activation within the NAS produces multiple behavioral signs (anhedonia, impaired extinction) characteristic of experience-dependent psychiatric conditions such as PTSD. Microinjections of the KOR agonist U50 directly into the NAS produced effects on ICSS that were indistinguishable from those of elevated CREB, suggesting that CREB-mediated increases in dynorphin expression (Carlezon et al., 1998) and subsequent increases in KOR stimulation are sufficient to produce anhedonia. However, microinjections of U50 failed to elevate startle when given during FPS testing or extinction training. These findings raise the possibility that although CREB activation is a common trigger for anhedonia and impaired extinction, expression of these individual behaviors involves divergent downstream mechanisms.
Stress activation of the NAS: consequences for behavior
The finding that footshock activates CREB in the NAS extends evidence that stress activates this region. For example, stress increases Fos immunoreactivity in the NAS (Barrot et al., 1999; Bruijnzeel et al., 1999; Nikulina et al., 2004). Forced swim stress activates CREB in the NAS (Pliakas et al., 2001), and various types of stressors activate CRE-mediated gene transcription in this area (Barrot et al., 2002). These new data are important because different types of stressors—indeed, even different types of shock—can produce different effects on motivated behavior (Shalev et al., 2000). To ensure relevance to behavior, we administered footshock using the same procedures we use for fear conditioning (Knoll et al., 2007).
ICSS is a highly trained behavior that is sensitive to treatments that induce rewarding or depressive states in humans, and it enables real-time analysis of treatment effects (Carlezon and Chartoff, 2007). Previous work has shown that elevated CREB in the NAS reduces cocaine- and morphine-induced conditioned place preferences (Carlezon et al., 1998; Pliakas et al., 2001; Barrot et al., 2002), effects interpreted as decreased sensitivity to reward (anhedonia). Place conditioning depends on the ability to learn and remember associations between drug and environment (Bardo and Bevins, 2000), and thus these previous studies reflect changes in conditioned (rather than real-time) effects. Increased CREB function has recently been implicated in reduced motivation to self-administer cocaine, although that work focused on striatal regions dorsal to the NAS (Hollander et al., 2010). The finding that elevated CREB in the NAS reduces sensitivity to rewarding brain stimulation mitigates concerns that previous findings with place conditioning reflect nonspecific effects on learning and memory processes, and extends applicability to a behavior not motivated by addictive drugs. These ICSS data also mitigate concerns that CREB effects were caused by alterations in drug time course or the severity of drug withdrawal, each of which can affect place conditioning (Pliakas et al., 2001; Neumaier et al., 2002). The effects of HSV-CREB on brain stimulation reward followed the time course of this vector, peaking 3–4 d after gene transfer and waning by days 7–8 (Carlezon et al., 2000). As such, elevated CREB expression in the NAS causes effects on ICSS that are qualitatively similar to those of footshock (Kamata et al., 1986). The CREB effects are also similar to those caused by elevated GluR1 in the NAS (Todtenkopf et al., 2006). These effects may be related: elevated GluR1 expression favors formation of calcium-permeable AMPA receptors (Hollmann et al., 1991), stimulation of which could trigger calcium-dependent CREB activation (Dash et al., 1991; Sheng et al., 1991).
The fact that elevated CREB in the NAS produced anhedonia in the ICSS test enabled tests of the hypothesis that this depressive state increases vulnerability to fear and anxiety-related behaviors. There is some evidence that mesolimbic DA is critical for long-term fear memories (Pezze and Feldon, 2004). As one example, fear conditioning is absent in mice lacking the ability to synthesize DA, but intact when DA input to the nucleus accumbens is restored (Fadok et al., 2010). Our data raise the possibility that activation of D1-receptor-linked intracellular signaling pathways that culminate in CREB activation (Carlezon et al., 2005) plays an especially important role in long-term fear expression. Although CREB elevation before training produced nominal differences in FPS, effects were small and variable. The observation that CREB elevation after FPS training produces similar effects may indicate decreases in the impact of extinction training, which effectively reduced FPS in the control groups. Although this effect depends on phosphorylation at Ser 133, since it was not seen with HSV-mCREB, its physiological basis is not known. Elevated CREB function in NAS medium spiny neurons increases their excitability (Dong et al., 2006); this effect might alter gating of information flow or occlude physiological processes required for extinction, which reflects new learning (Myers and Davis, 2007). Regardless, our data indicate that the NAS is embedded within brain circuits that regulate fear and provide new evidence that CREB in this region is preferentially involved in the persistence of learning-dependent fear- and anxiety-related behaviors.
Divergent CREB-regulated mechanisms
Microinjections of U50 (1.0 μg) into the NAS mimicked the anhedonia-inducing effect of elevated CREB function. The depressive-like effects of intra-NAS U50 are likely mediated by stimulation of KORs located on the terminals of mesolimbic DA inputs (Svingos et al., 1999), which decreases DA release (Carlezon et al., 2006). This finding strengthens the relationship among stress, CREB activation, KOR stimulation, and depressive behavior. The observation that 3.0 μg of U50 failed to cause larger effects might be attributable to transient internalization of KOR receptors at this concentration (Li et al., 1999; Reyes et al., 2010). The ability of microinjections of cocaine, which were given as the final treatment, to decrease ICSS thresholds suggests that the lack of effect was not attributable to tissue damage caused by repeated injections. In contrast, microinjections of U50 failed to mimic the persistent increases in FPS produced by elevated CREB, regardless of whether they were given during startle testing or extinction training. Although higher doses of U50 might produce changes in startle, such changes would not be accompanied by anhedonia, and thus would not reproduce the effects of elevated CREB function. These data suggest that the anhedonia- and anxiety-producing effects of elevated CREB are mediated by different target genes. CREB also regulates expression of proteins such as corticotropin-releasing factor and brain-derived neurotrophic factor (Carlezon et al., 2005), the intra-NAS actions of which have been implicated in expression of anxiety-related behaviors (Berton et al., 2006). Although our work provides evidence that KOR stimulation in the NAS does not influence expression or extinction of FPS, identifying the factor(s) involved will require systematic studies of many CREB-regulated proteins.
Our data suggest that elevated CREB function in the NAS does not lead to numbing-like reductions in sensitivity to all stimuli (Barrot et al., 2003). Whereas CREB decreased sensitivity to reward, it increased responsiveness to cues previously associated with an aversive stimulus (footshock), although extinction training was required to reveal this effect. The ability of stress to produce CREB-mediated elevations of KOR function may likewise contribute to increased sensitivity to cues that trigger drug-seeking behaviors (McLaughlin et al., 2003). However, the ability of elevated CREB to reduce the effectiveness of extinction training might be considered a numbing-like effect. The mechanisms by which disruption of CREB function (via HSV-mCREB) increased reward in the ICSS test but did not affect extinction in the FPS test are not understood. Although we used parameters that produced strong FPS, it is possible that mCREB effects might have been detectable with different training parameters. More intense shock produces inverted U-shaped functions and paradoxical reductions in FPS behavior that reflect a shift to passive coping (Walker et al., 1997), complicating interpretation of experiments involving stronger stimulus parameters. Importantly, expression of mCREB in the basolateral amygdala also did not affect fear conditioning (Josselyn et al., 2001). One possibility is that CREB function in NAS cells not infected by the viral vectors offsets mCREB effects.
Conclusions
This work provides new insights on how CREB might contribute to the development and expression of neuropsychiatric disorders. In particular, it provides a molecular mechanism that might help to explain comorbidity of depression and PTSD. The diversity in molecular targets of CREB illustrates the difficulty in developing new treatments that ameliorate the full spectrum of signs of depressive and anxiety disorders and raises the possibility that, to the extent possible, it may be easier to prevent rather than reverse the symptoms of stress-related disorders (Knoll and Carlezon, 2010).
Footnotes
This work was supported by National Institute of Mental Health Grant MH063266 to W.A.C.
References
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. Ed 4. Washington, DC: American Psychiatric Association; 2000. [Google Scholar]
- Bals-Kubik R, Herz A, Shippenberg TS. Evidence that the aversive effects of opioid antagonists and kappa-agonists are centrally mediated. Psychopharmacology. 1989;98:203–206. doi: 10.1007/BF00444692. [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology. 2000;153:31–43. doi: 10.1007/s002130000569. [DOI] [PubMed] [Google Scholar]
- Barrot M, Marinelli M, Abrous DN, Rougé-Pont F, Le Moal M, Piazza PV. Functional heterogeneity in dopamine release and in the expression of Fos-like proteins within the rat striatal complex. Eur J Neurosci. 1999;11:1155–1166. doi: 10.1046/j.1460-9568.1999.00525.x. [DOI] [PubMed] [Google Scholar]
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci U S A. 2002;99:11435–11440. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beardsley PM, Howard JL, Shelton KL, Carroll FI. Differential effects of the novel kappa opioid receptor antagonist, JDTic, on reinstatement of cocaine-seeking induced by footshock stressors vs cocaine primes and its antidepressant-like effects in rats. Psychopharmacology. 2005;183:118–126. doi: 10.1007/s00213-005-0167-4. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bruijnzeel AW, Stam R, Compaan JC, Croiset G, Akkermans LM, Olivier B, Wiegant VM. Long-term sensitization of Fos-responsivity in the rat central nervous system after a single stressful experience. Brain Res. 1999;819:15–22. doi: 10.1016/s0006-8993(98)01350-x. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Chartoff EH. Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation. Nat Protoc. 2007;2:2987–2995. doi: 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thomas M. Biological substrates of reward and aversion: a nucleus accumbens activity hypothesis. Neuropharmacology. 2009;56(Suppl 1):122–132. doi: 10.1016/j.neuropharm.2008.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Nestler EJ, Neve RL. Viral-mediated gene transfer as a tool for neuropsychiatric research. Crit Rev Neurobiol. 2000;14:47–68. doi: 10.1080/08913810008443546. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Duman RS, Nestler EJ. The many faces of CREB. Trends Neurosci. 2005;28:436–445. doi: 10.1016/j.tins.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Carlezon WA, Jr, Beguin C, DiNieri J, Baumann MH, Richards M, Todtenkopf MS, Rothman RB, Ma Z, Lee DY-L, Cohen BM. Depressive-like effects of the kappa-opioid receptor agonist Salvinorin A on behavior and neurochemistry in rats. J Pharmacol Exp Ther. 2006;314:440–447. doi: 10.1124/jpet.105.092304. [DOI] [PubMed] [Google Scholar]
- Cole RL, Konradi C, Douglass J, Hyman SE. Neuronal adaptation to amphetamine and dopamine: molecular mechanisms of prodynorphin gene regulation in rat striatum. Neuron. 1995;14:813–823. doi: 10.1016/0896-6273(95)90225-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiNieri JA, Nemeth C, Parsegian A, Carle T, Gurevich VV, Gurevich E, Neve RL, Nestler EJ, Carlezon WA., Jr Altered sensitivity to rewarding and aversive drugs in mice with inducible disruption of CREB function within nucleus accumbens. J Neurosci. 2009;29:1855–1859. doi: 10.1523/JNEUROSCI.5104-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neuroscience. 2006;9:475–477. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Douglass J, McKinzie AA, Pollock KM. Identification of multiple DNA elements regulating basal and protein kinase A-induced transcriptional expression of the rat prodynorphin gene. Mol Endocrinol. 1994;8:333–344. doi: 10.1210/mend.8.3.8015551. [DOI] [PubMed] [Google Scholar]
- Espinosa VP, Liu Y, Ferrini M, Anghel A, Nie Y, Tripathi PV, Porche R, Jansen E, Stuart RC, Nillni EA, Lutfy K, Friedman TC. Differential regulation of prohormone convertase 1/3, prohormone convertase 2 and lated cyclic-AMP-response element binding protein by short-term and long-term morphine treatment: implications for understanding the “switch” to opiate addiction. Neuroscience. 2008;156:788–799. doi: 10.1016/j.neuroscience.2008.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok JP, Darvas M, Dickerson TM, Palmiter RD. Long-term memory for pavlovian fear conditioning requires dopamine in the nucleus accumbens and basolateral amygdala. PLoS One. 2010;5:e12751. doi: 10.1371/journal.pone.0012751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green TA, Alibhai IN, Roybal CN, Winstanley CA, Theobald DE, Birnbaum SG, Graham AR, Unterberg S, Graham DL, Vialou V, Bass CE, Terwilliger EF, Bardo MT, Nestler EJ. Environmental enrichment produces a behavioral phenotype mediated by low cyclic adenosine monophosphate response element binding (CREB) activity in the nucleus accumbens. Biol Psychiatry. 2008;67:28–35. doi: 10.1016/j.biopsych.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Im HI, Amelio A, Kocerha J, Bali P, Lu Q, Willoughby D, Conkright M, Kenny PJ. Striatal microRNA controls cocaine intake through regulation of CREB signaling. Nature. 2010;466:197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA-gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi CJ, Carlezon WA, Jr, Neve RL, Nestler EJ, Davis M. Long-term memory is facilitated by cAMP response-element binding protein overexpression in the amygdala. J Neurosci. 2001;21:2404–2412. doi: 10.1523/JNEUROSCI.21-07-02404.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata K, Yoshida S, Kameyama T. Antagonism of footshock stress-induced inhibition of intracranial self-stimulation by naloxone or methamphetamine. Brain Res. 1986;371:197–200. doi: 10.1016/0006-8993(86)90830-9. [DOI] [PubMed] [Google Scholar]
- Keane TM, Marshall AD, Taft CT. Posttraumatic stress disorder: etiology, epidemiology, and treatment outcome. Annu Rev Clin Psychol. 2006;2:161–197. doi: 10.1146/annurev.clinpsy.2.022305.095305. [DOI] [PubMed] [Google Scholar]
- Keller MC, Neale MC, Kendler KS. Association of different adverse life events with distinct patterns of depressive symptoms. Am J Psychiatry. 2007;164:1521–1529. doi: 10.1176/appi.ajp.2007.06091564. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Arch Gen Psychiatry. 1995;52:1048–1060. doi: 10.1001/archpsyc.1995.03950240066012. [DOI] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Meloni EG, Thomas JB, Carroll FI, Carlezon WA., Jr Anxiolytic-like effects of kappa-opioid receptor antagonists in behavioral models of unlearned and learned fear in rats. J Pharmacol Exp Ther. 2007;323:838–845. doi: 10.1124/jpet.107.127415. [DOI] [PubMed] [Google Scholar]
- Li JG, Luo LY, Krupnick JG, Benovic JL, Liu-Chen LY. U50,488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J Biol Chem. 1999;274:12087–12094. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Mague SD, Andersen SL, Carlezon WA., Jr Early developmental exposure to methylphenidate reduces cocaine-induced potentiation of brain stimulation reward in rats. Biol Psychiatry. 2005;57:120–125. doi: 10.1016/j.biopsych.2004.10.037. [DOI] [PubMed] [Google Scholar]
- Markou A, Hauger RL, Koob GF. Desmethylimipramine attenuates cocaine withdrawal in rats. Psychopharmacology. 1992;109:305–314. doi: 10.1007/BF02245878. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miliaressis E, Rompre PP, Laviolette P, Philippe L, Coulombe D. The curve-shift paradigm in self-stimulation. Physiol Behav. 1986;37:85–91. doi: 10.1016/0031-9384(86)90388-4. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M. Mechanisms of fear extinction. Mol Psychiatry. 2007;12:120–150. doi: 10.1038/sj.mp.4001939. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59:1151–1159. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Neumaier JF, Vincow E, Arvanitogiannis A, Wise RA, Carlezon WA., Jr Elevated expression of 5HT1B receptor within the mesolimbic system sensitizes rats to cocaine. J Neurosci. 2002;22:10856–10863. doi: 10.1523/JNEUROSCI.22-24-10856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikulina EM, Covington HE, 3rd, Ganschow L, Hammer RP, Jr, Miczek KA. Long-term behavioral and neuronal cross-sensitization to amphetamine induced by repeated brief social defeat stress: Fos in the ventral tegmental area and amygdala. Neuroscience. 2004;123:857–865. doi: 10.1016/j.neuroscience.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Paine TA, Neve RL, Carlezon WA., Jr Attention deficits and hyperactivity following inhibition of cAMP-dependent protein kinase (PKA) within the medial prefrontal cortex of rats. Neuropsychopharmacology. 2009;34:2143–2155. doi: 10.1038/npp.2009.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 3. Orlando, FL: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Pezze MA, Feldon J. Mesolimbic dopaminergic pathways in fear conditioning. Prog Neurobiol. 2004;74:301–320. doi: 10.1016/j.pneurobio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Pfeiffer A, Brantl V, Herz A, Emrich HM. Psychotomimesis mediated by kappa opiate receptors. Science. 1986;233:774–776. doi: 10.1126/science.3016896. [DOI] [PubMed] [Google Scholar]
- Pliakas AM, Carlson R, Neve RL, Konradi C, Nestler EJ, Carlezon WA., Jr Altered responsiveness to cocaine and increased immobility in the forced swim test associated with elevated cAMP response element binding protein expression in nucleus accumbens. J Neurosci. 2001;21:7397–7403. doi: 10.1523/JNEUROSCI.21-18-07397.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Chavkin C, Van Bockstaele EJ. Agonist-induced internalization of kappa-opioid receptors in noradrenergic neurons of the rat locus coeruleus. J Chem Neuroanat. 2010;40:301–309. doi: 10.1016/j.jchemneu.2010.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Thome J, Newton SS, Chen J, Kelz MB, Steffen C, Nestler EJ, Duman RS. Inducible and brain region-specific CREB transgenic mice. Mol Pharmacol. 2002;61:1453–1464. doi: 10.1124/mol.61.6.1453. [DOI] [PubMed] [Google Scholar]
- Shalev U, Highfield D, Yap J, Shaham Y. Stress and relapse to drug seeking in rats: studies on the generality of the effect. Psychopharmacology. 2000;150:337–346. doi: 10.1007/s002130000441. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca(2+)-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Simpson JN, McGinty JF. Forskolin induces preproenkephalin and preprodynorphin mRNA in rat striatum as demonstrated by in situ hybridization histochemistry. Synapse. 1995;19:151–159. doi: 10.1002/syn.890190302. [DOI] [PubMed] [Google Scholar]
- Svingos AL, Colago EE, Pickel VM. Cellular sites for dynorphin activation of kappa-opioid receptors in the rat nucleus accumbens shell. J Neurosci. 1999;19:1804–1813. doi: 10.1523/JNEUROSCI.19-05-01804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA., Jr Effects of kappa opioid ligands on intracranial self-stimulation in rats. Psychopharmacology. 2004;172:463–470. doi: 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- Todtenkopf MS, Parsegian A, Neve RL, Carlezon WA., Jr Brain reward regulated by glutamate receptor subunits in the nucleus accumbens shell. J Neurosci. 2006;26:11665–11669. doi: 10.1523/JNEUROSCI.3070-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA., Jr The kappa-opioid agonist U69,593 blocks cocaine-induced enhancement of brain stimulation reward. Biol Psychiatry. 2008;64:982–988. doi: 10.1016/j.biopsych.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DL, Cassella JV, Lee Y, De Lima TC, Davis M. Opposing roles of the amygdala and dorsolateral periaqueductal gray in fear-potentiated startle. Neurosci Biobehav Rev. 1997;21:743–753. doi: 10.1016/s0149-7634(96)00061-9. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine and reward: the anhedonia hypothesis 30 years on. Neurotoxicol Res. 2008;14:169–183. doi: 10.1007/BF03033808. [DOI] [PMC free article] [PubMed] [Google Scholar]