

Abstract
Parkinson’s disease is a neurodegenerative movement disorder characterized by loss of midbrain dopaminergic neurons leading to motor abnormalities and autonomic dysfunctions. Despite intensive research, the etiology of Parkinson’s disease remains poorly understood leaving us with no effective therapeutic options. However, the recent identification of genes linked to heritable forms of Parkinson’s disease has revolutionized research in the field and has begun to provide new clues to disease pathogenesis. Here we discuss these recent genetic advances and highlight their significance in our quest to better understand common underlying disease mechanisms that will help us identify innovative neuroprotective therapies for Parkinson’s disease.
Introduction
Parkinson’s disease is the second most common neurodegenerative disorder, affecting 1–2% of the population over the age of 65 [1]. It is a chronic movement disorder caused by relentless degeneration of specific neuronal populations in the brain, most notably the dopamine-producing neurons of the substantia nigra pars compacta region of the basal ganglia, which helps control voluntary movement. Patients generally suffer from the cardinal symptoms of slowness of movement, tremors while at rest, rigidity, and poor balance, and often also show autonomic, cognitive, and psychiatric disturbances. Parkinson’s disease is progressive and may last 10–20 (or more) years after diagnosis. It is typically partially treatable (mainly with dopamine-replacement therapy) for a few years after diagnosis, but this is generally followed by years of decline during which there is no effective therapy, eventually leading to premature death. Parkinson’s disease is usually a sporadic disorder with onset in later life and the causes of this disease are incompletely understood. For most of the twentieth century, genetic predisposition was thought to play a negligible role in the disease, but in the past 15 years the identification of distinct genetic loci responsible for (both the dominant and recessive) inherited forms of Parkinson’s disease has provided us with numerous clues to understanding its molecular causes. To date, about 16 Parkinson’s disease-related genetic loci (the PARK loci) and 11 genes associated with these PARK loci have been described (see Table 1). Among these genes, 5 have been studied extensively: α-synuclein (also known as SNCA), parkin (PARK2), PINK1 (PARK6), DJ-1 (PARK7), and LRRK2 (PARK8) [2]. Consequently, these will be the main focus of this review. Recent genome-wide association studies (GWAS) have found that having particular variants of these genes greatly increases the chance of developing Parkinson’s disease [3,4], suggesting perhaps that in the cases where the functions are known, the products of these genes are often proteins involved in the way brain cells cope with oxidative stress, mitochondrial dysfunction, and removal of misfolded proteins. A commonly held view is that Parkinson’s disease may result from environmental factors (such as toxins) damaging dopamine-producing neurons of the substantia nigra in an accumulative way in individuals who have defects in pathways dealing with oxidative stress, mitochondrial dysfunction, and either the ubiquitin proteasome system or the autophagy-lysosome pathway, which remove misfolded proteins (Figure 1). However, some of the implicated proteins have functions that do not necessarily fit into this model, making the scenario somewhat complicated. Here, we provide a brief overview of how genetic research and the ensuing molecular insights have led to substantial advances in our understanding of disease pathogenesis and in experimental approaches to study the disorder.
Table 1: Gene loci identified for Parkinson's disease and their probable functions.
| Locus | Gene | Chromosome | Inheritance | Probable function |
|---|---|---|---|---|
| PARK1 & PARK4 | ɑ-synuclein | 4q21 | Dominant | Presynaptic protein, Lewy body, lipid and vesicle dynamics |
| PARK2 | parkin | 6q25.2-27 | Recessive | Ubiquitin E3 ligase, mitophagy |
| PARK3 | Unknown | 2p13 | Dominant | Unknown |
| PARK5 | UCHL1 | 4p14 | Dominant | Ubiquitin C-terminal hydrolase |
| PARK6 | PINK1 | 1p35-36 | Recessive | Mitochondrial kinase |
| PARK7 | DJ-1 | 1p36 | Recessive | Oxidative stress |
| PARK8 | LRRK2 | 12p11.2 | Dominant | Kinase signaling, cytoskeletal dynamics, protein translation |
| PARK9 | ATP13A2 | 1p36 | Recessive | Unknown |
| PARK10 | Unknown | 1p32 | Dominant | Unknown |
| PARK11 | GIGYF2 | 2p37 | Dominant | IGF-1 signaling |
| PARK12 | Unknown | Xq21-q25 | X-linked | Unknown |
| PARK13 | Omi/HtrA2 | 2p13 | Unknown | Mitochondrial serine protease |
| PARK14 | PLA2G6 | 22q13 | Recessive | Phospholipase enzyme |
| PARK15 | FBXO7 | 22q11 | Recessive | Ubiquitin E3 ligase |
| PARK16 | Unknown | 1q32 | Unknown | Unknown |
ATP13A2, ATPase type 13A2; FBXO7, F-box protein 7; GIGYF2, GRB10 interacting GYF protein 2; HtrA2, HtrA serine peptidase 2 (also known as Omi); IGF-1, insulin-like growth factor 1; LRRK2, leucine-rich repeat kinase 2; PINK1, PTEN-induced putative kinase 1; PLA2G6, phospholipase A2, group VI (cytosolic, calcium-independent); UCHL1, ubiquitin carboxyl-terminal esterase L1. Adapted from Hum Mol Genet [68], © 2007.
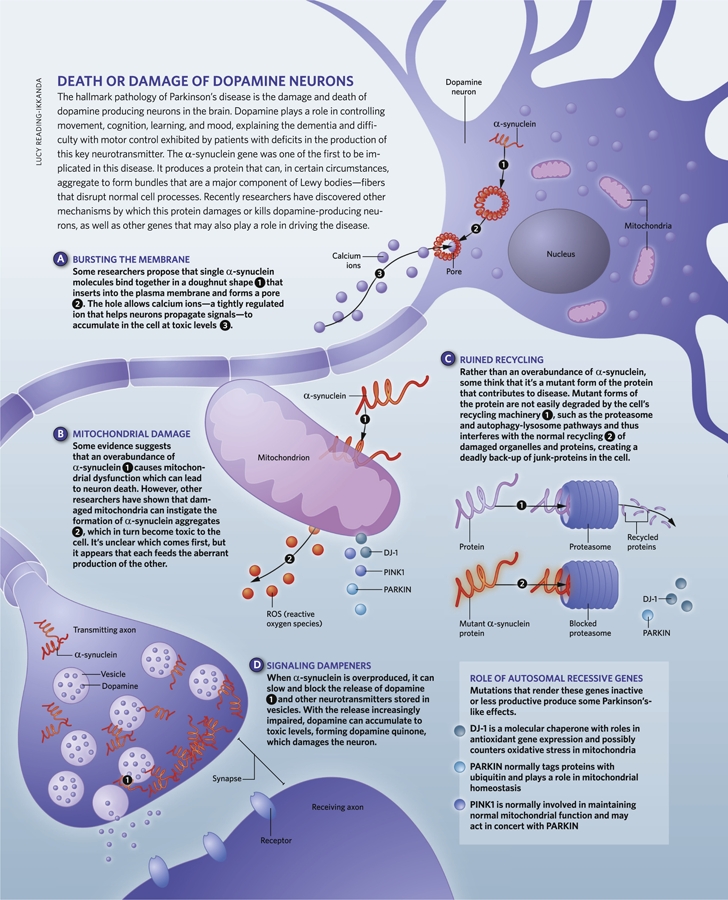
Figure 1. Underlying mechanism of dopaminergic neurodegeneration in Parkinson’s disease.
Genetic breakthroughs
The first big breakthrough came in studying the inherited autosomal-dominant form of Parkinson’s disease, when mutations of the α-synuclein gene locus were reported in several families with a history of Parkinson’s disease. Both point mutations (three point mutations) and multiplications [5-7], in addition to triplications [8] and duplications [9,10] in the α-synuclein gene cause Parkinson’s disease. Those with a family history of triplications tend to be younger at disease onset compared to those with duplications, and the fact that even one copy of the faulty gene is sufficient to cause the disease suggests that the increased levels of α-synuclein are in some way toxic [8,11]. Although our knowledge about the normal function of α-synuclein is limited (we know that it modulates synaptic plasticity and regulates neurotransmitter release), recent studies elucidating the structural properties of this protein have shed considerable light on its pathogenic involvement in Parkinson’s disease. Both pathogenic mutations and elevated concentrations will give α-synuclein a propensity to develop a β-sheet structure which readily polymerizes into oligomers and higher order aggregates such as fibrils [12]. α-synuclein aggregation was worsened by various types of post-translational modifications such as Ser-129 phosphorylation, calpain-mediated cleavage, O-glycosylation, tyrosine nitration, methionine oxidation, and C-terminal truncation [2], although the mechanisms are not clear. Insoluble α-synuclein fibrils are a key component of Lewy bodies (cytoplasmic bodies containing aggregated proteins), which are the pathogenic hallmark of Parkinson’s disease. There is some controversy as to whether Lewy bodies are a cause or a consequence of Parkinson’s disease, with some evidence suggesting that they play a protective role by sequestering toxic α-synuclein oligomers [13]. However, emerging evidence from in-vitro studies [14] and animal models [15-18] suggests that both the oligomer and fibrillar forms of α-synuclein aggregates are toxic to neurons. However, the precise roles of α-synuclein aggregation in mediating cell death in Parkinson’s disease remain elusive, although there is no shortage of hypotheses. One suggestion is that α-synuclein oligomers may alter plasma membrane stability or permeability by forming membrane pores that increase intracellular Ca2+ to toxic levels [20] (Figure 1). Another suggestion is that because α-synuclein is found at mitochondrial membranes of dopaminergic neurons [21], its overexpression may induce mitochondrial dysfunction (by inhibiting complex I), and since mitochondria are a major source of reactive oxygen species, this would result in increased oxidative stress, leading to neurodegeneration [22,23] (Figure 1). Yet another potential mechanism of α-synuclein toxicity is suggested by the observation that increased or mutant α-synuclein expression in synaptic vesicles interferes with synaptic transmission by causing the accumulation of docked vesicles at the presynaptic membrane [24], reducing the recycling vesicle pool [25] and thereby increasing cytosolic dopamine to toxic levels [26] (Figure 1). Finally, there is the suggestion that as mutant species of α-synuclein are poor substrates for proteasomal degradation, they inhibit proteolysis [27], block lysosomal function [28] (Figure 1) and chaperone-mediated autophagy [29], and disrupt endoplasmic reticulum-Golgi trafficking [30,31], causing toxicity. Whatever the exact mechanism(s) involved, it is clear that α-synuclein aggregates exert toxic effects on several important cellular functions necessary for survival of dopaminergic neurons.
The second gene mutation identified as causing the autosomal-dominant form of Parkinson’s disease is that of LRRK2. LRRK2 mutations cause early-onset Parkinson’s disease in families from diverse ethnic backgrounds, with a clinical profile identical to sporadic late-onset Parkinson’s disease. Leucine-rich repeat kinase 2 (LRRK2) is a large multidomain protein [32], but mutations associated with Parkinson’s disease are concentrated in the central catalytic regions of the GTPase and kinase domains, and most of them result in increased kinase activity in vitro [32]. Increased kinase activity is known to promote cell death in Parkinson’s disease, but the mechanisms are not yet known. LRRK2 undergoes autophosphorylation and also phosphorylates a number of protein substrates, so it is significant that the toxicity of disease-causing mutant forms of LRRK2 is related to their enhanced kinase activity and GTP binding [32-34]. Unsurprisingly, a great deal of effort has been focused on identifying the normal function of LRRK2, including studying its phosphosubstrates, binding partners, and regulators of its kinase and GTPase activities, in order to determine how it causes disease. LRRK2 is thought to play a role in neuronal outgrowth [35,36], protein translation by phosphorylation of the translation inhibitor eukaryotic initiation factor 4E (eIF4E) [37,38], and in cytoskeletal dynamics via phosphorylation of moesin, which anchors the cytoskeleton to the plasma membrane [39]. Mice that harbor LRRK2 mutations develop the cardinal abnormalities of Parkinson’s disease in the nigrostriatal system (one of the major dopaminergic pathways conveying impulses from the substantia nigra), such as stimulated dopamine neurotransmission or behavioral deficits [40]; others show progressive age-dependent motor deficits that can lead to immobility but are responsive to the dopaminergic drugs L-DOPA and apomorphine, and develop axonal spheroids in striatal and cortical projections composed of phosphorylated tau despite an absence of nigrostriatal dopaminergic neurodegeneration [41]. Strikingly, there is an additive effect of expressing mutant forms of LRRK2 and α-synuclein in mice, whereas deficiency of LRRK2 limits the toxic effects of mutant α-synuclein [42], suggesting that the two proteins interact. Until recently, it was unclear whether the neurological features in mice with mutant LRRK2 result from altered kinase activity. Recent studies elegantly demonstrated that viral-mediated overexpression of G2019S mutant LRRK2 in nigrostriatal dopaminergic neurons (those located in the substantia nigra pars compacta and striatum) resulted in marked neurotoxicity caused by LRRK2 kinase activity. By contrast, overexpression of normal LRRK2 or a “kinase-dead” version of the enzyme did not have this adverse effect [43]. What’s more, the mutant-LRRK2-mediated nigrostriatal dopaminergic neurotoxicity was blocked by selective LRRK2 kinase inhibitors GW5074 and indirubin-3′-monoxime [43]. This is encouraging as it suggests that LRRK2 kinase inhibitors could become a new treatment for Parkinson’s disease.
There have also been advances in understanding from studying the recessive form of the disease. There is compelling evidence suggesting that mutations causing a loss of function in three genes, parkin, PINK1, and DJ-1, underlie the autosomal-recessive Parkinson’s disease (and also a few cases of sporadic Parkinson’s disease) [44]. Recent studies have demonstrated that products of all three genes preserve mitochondrial function and protect against reactive oxygen species. Patients homozygous for loss-of-function parkin mutations or having compound heterozygous parkin mutations account for about 50% of all familial early-onset cases of Parkinson’s disease, with point mutations being the most frequent genetic lesion and deletions, duplications, and exonic rearrangements also contributing to disease [45]. Mutations in PINK1, the second most common autosomal-recessive mutation (following parkin) contribute to between 1% and 7% of early-onset Parkinson’s disease [46], whereas mutations in DJ-1 are a rare cause of Parkinson’s disease [47]. There has inevitably been much study of what the protein products of these genes do. Parkin tags protein lysine residues with ubiquitin, either targeting them for destruction via the 26S proteasome (by adding polyubiquitin chains via lysine K48) or influencing other signaling pathways such as DNA repair, endocytosis, transcriptional regulation, and protein trafficking (by ubiquitination via lysine K48 or K63) [48]. Disease-causing mutations in parkin lead to a loss of this E3 ubiquitin ligase activity [48], but although numerous substrates for parkin have been identified to date, no consensus has emerged on which of these may (if not ubiquitinated) lead to neurodegeneration in Parkinson’s disease (Figure 1).
Several PINK1 mutations are also thought to lead to a loss of function [49]. PTEN-induced putative kinase 1 (PINK1) is found in the mitochondrial intermembrane space and membranes of the mitochondria [50], with its serine/threonine kinase domain facing the cytosol [51], suggesting that its substrates may reside here. Several studies point towards both parkin and PINK1 having a prominent role in preserving mitochondrial function (Figure 1). This can be seen vividly in parkin or PINK1-mutant/deficient flies, which exhibit abnormal mitochondria together with enhanced sensitivity to oxidative stress, apoptotic muscle degeneration, and significant loss of a subset of dopaminergic neurons [52-54]. Furthermore, mice lacking parkin and PINK1 exhibit nigrostriatal deficits (without degeneration) and mitochondrial dysfunction (indicated by reduced activity of multiple respiratory chain complexes) [55,56]. Interestingly, increased expression of parkin improved dysfunctions in flies lacking PINK1, but increased expression of PINK1 had no effect on dysfunctions in flies lacking parkin [54,57]. This neatly puts parkin and PINK1 in a common pathway, with PINK1 functioning upstream from parkin. This is also consistent with recent studies where PINK1 was shown to act upstream of parkin to regulate degradation of damaged mitochondria by a process known as mitophagy [58,59]. Together these data provide compelling evidence by which both parkin and PINK1 play a crucial role in mitochondrial quality control for Parkinson’s disease. These further reinforce the similarities between sporadic and familial forms of the disease, which both implicate mitochondrial dysfunction as a common pathogenic mechanism.
Parkinson’s disease-associated mutations in the third gene, DJ-1, produce an inactive molecule that can’t form a dimer, or lead to no expression at all [60,61]. DJ-1 is a molecular chaperone with a variety of functions and is found in the cytosol, the mitochondrial matrix, and mitochondrial intermembrane space [62]. In cellular models, it regulates oxidation-reduction-dependent signaling pathways and acts as a regulator of antioxidant gene expression [63], and gene deletion studies show that it counters oxidative stress in mitochondria [64]. Recent studies indicate that DJ-1 deficiency is associated with apoptosis, perturbed mitochondrial dynamics, and autophagic dysregulation [65-67], linking it with functions mediated by both parkin and PINK1 (Figure 1). Future studies focused on the interplay between PINK1, parkin, and DJ-1 will hopefully greatly advance the understanding of how mutations in these genes cause Parkinson’s disease and what the common underlying mechanisms are.
Future directions
The identification and characterization of familial Parkinson’s disease-linked genes has sparked an extremely fruitful line of research, delineating molecular pathways that are involved in the pathogenesis of Parkinson’s disease. Although the genetic mutations in known genes account for only a limited fraction of the heritable forms of Parkinson’s disease, the use of high-throughput exome and genome sequencing in the future are likely to identify additional rare variants that will further expand our knowledge of pathogenic disease mechanisms. So far, the proteins that have been linked to Parkinson’s disease by genetic studies have roles in lipid and vesicle dynamics (α-synuclein), the ubiquitin-proteasome system (parkin), cytoskeletal dynamics, protein translation, abnormal kinase function (LRRK2), oxidative stress, and mitochondrial dysfunction (DJ-1, PINK1, parkin). Evidently, these disparate functions must overlap as they lead to the dysfunction and death of dopaminergic neurons characteristic of Parkinson’s disease. As discussed above these molecular pathways and functions are not only relevant for the rare familial variants of Parkinson’s disease, but also to the more common sporadic version of the disease.
However, while we have made great strides in understanding, the relationships between these functions are not direct and the connections between them are not immediately apparent. Therefore, the major focus in the future should be to identify common underlying mechanisms by which familial Parkinson’s disease-linked genes affect dopaminergic neuronal survival, to hopefully provide the basis for new and tractable targets for new drugs to prevent and treat Parkinson’s disease.
Acknowledgments
BT is supported by National Institutes of Health grants NS060885 and NS062165 and a grant from the Michael J. Fox Foundation for Parkinson’s Research. MFB is supported by National Institutes of Health grant ES017295, and grants from the Parkinson’s Disease Foundation, Department of Defense, and Michael J. Fox Foundation for Parkinson’s Research.
Abbreviations
- GWAS
genome-wide association studies
- LRRK2
leucine-rich repeat kinase 2
- PINK1
PTEN-induced putative kinase 1
Competing interests
The authors declare that they have no competing interests.
The electronic version of this article is the complete one and can be found at: http://f1000.com/reports/m/3/7
References
- 1.Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol. 2003;157:1015–22. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- 2.Thomas B. Parkinson’s disease: from molecular pathways in disease to therapeutic approaches. Antioxid Redox Signal. 2009;11:2077–82. doi: 10.1089/ars.2009.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Satake W, Nakabayashi Y, Mizuta I, Hirota Y, Ito C, Kubo M, Kawaguchi T, Tsunoda T, Watanabe M, Takeda A, Tomiyama H, Nakashima K, Hasegawa K, Obata F, Yoshikawa T, Kawakami H, Sakoda S, Yamamoto M, Hattori N, Murata M, Nakamura Y, Toda T. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated by X William Yang 01 Dec 2009
- 4.Simón-Sánchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Krüger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated by X William Yang 01 Dec 2009
- 5.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 6.Krüger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet. 1998;18:106–8. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 7.Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol. 2004;55:164–73. doi: 10.1002/ana.10795. [DOI] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated by Thomas Gasser 18 Feb 2004, John Hardy 22 Mar 2004, Albert La Spada 21 Apr 2004
- 8.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]; F1000 Factor 9Evaluated by Sue Malcolm 16 Dec 2003, Vicky Brandt 19 Dec 2003
- 9.Chartier-Harlin MC, Kachergus J, Roumier C, Mouroux V, Douay X, Lincoln S, Levecque C, Larvor L, Andrieux J, Hulihan M, Waucquier N, Defebvre L, Amouyel P, Farrer M, Destée A. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet. 2004;364:1167–9. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- 10.Ibáñez P, Bonnet AM, Débarges B, Lohmann E, Tison F, Pollak P, Agid Y, Dürr A, Brice A. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 2004;364:1169–71. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- 11.Winkler S, Hagenah J, Lincoln S, Heckman M, Haugarvoll K, Lohmann-Hedrich K, Kostic V, Farrer M, Klein C. alpha-Synuclein and Parkinson disease susceptibility. Neurology. 2007;69:1745–50. doi: 10.1212/01.wnl.0000275524.15125.f4. [DOI] [PubMed] [Google Scholar]
- 12.Lee VM, Trojanowski JQ. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: new targets for drug discovery. Neuron. 2006;52:33–8. doi: 10.1016/j.neuron.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka M, Kim YM, Lee G, Junn E, Iwatsubo T, Mouradian MM. Aggresomes formed by alpha-synuclein and synphilin-1 are cytoprotective. J Biol Chem. 2004;279:4625–31. doi: 10.1074/jbc.M310994200. [DOI] [PubMed] [Google Scholar]
- 14.Zhou W, Freed CR. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J Biol Chem. 2005;280:43150–8. doi: 10.1074/jbc.M507124200. [DOI] [PubMed] [Google Scholar]
- 15.Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–8. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- 16.Periquet M, Fulga T, Myllykangas L, Schlossmacher MG, Feany MB. Aggregated alpha-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci. 2007;27:3338–46. doi: 10.1523/JNEUROSCI.0285-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.St Martin JL, Klucken J, Outeiro TF, Nguyen P, Keller-McGandy C, Cantuti-Castelvetri I, Grammatopoulos TN, Standaert DG, Hyman BT, McLean PJ. Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem. 2007;100:1449–57. doi: 10.1111/j.1471-4159.2006.04310.x. [DOI] [PubMed] [Google Scholar]
- 18.Kirik D, Annett LE, Burger C, Muzyczka N, Mandel RJ, Björklund A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: a new primate model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2003;100:2884–9. doi: 10.1073/pnas.0536383100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim Biophys Acta. 2009;1792:651–63. doi: 10.1016/j.bbadis.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Furukawa K, Matsuzaki-Kobayashi M, Hasegawa T, Kikuchi A, Sugeno N, Itoyama Y, Wang Y, Yao PJ, Bushlin I, Takeda A. Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells. J Neurochem. 2006;97:1071–7. doi: 10.1111/j.1471-4159.2006.03803.x. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura K, Nemani VM, Wallender EK, Kaehlcke K, Ott M, Edwards RH. Optical reporters for the conformation of alpha-synuclein reveal a specific interaction with mitochondria. J Neurosci. 2008;28:12305–17. doi: 10.1523/JNEUROSCI.3088-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, Wong J, Takenouchi T, Hashimoto M, Masliah E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–10. doi: 10.1016/S0002-9440(10)64553-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin LJ, Pan Y, Price AC, Sterling W, Copeland NG, Jenkins NA, Price DL, Lee MK. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J Neurosci. 2006;26:41–50. doi: 10.1523/JNEUROSCI.4308-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci. 2006;26:11915–22. doi: 10.1523/JNEUROSCI.3821-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 9Evaluated by Jesse Hay 28 Jan 2010, Robert Burgoyne 29 Jan 2010
- 26.Lotharius J, Brundin P. Pathogenesis of Parkinson’s disease: dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci. 2002;3:932–42. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 27.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J Biol Chem. 2003;278:11753–9. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 28.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci. 2001;21:9549–60. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Daniel Klionsky 09 Sep 2004
- 30.Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science. 2006;313:324–8. doi: 10.1126/science.1129462. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 21Evaluated by Ted Dawson 30 Jun 2006, Adam Linstedt 31 Jul 2006, Nektarios Tavernarakis 01 Aug 2006, Alain Prochiantz 15 Aug 2006, Monte Gates 21 Aug 2006, Ruth Collins 14 Feb 2007, Jesse Hay 09 Mar 2007
- 31.Thayanidhi N, Helm JR, Nycz DC, Bentley M, Liang Y, Hay JC. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol Biol Cell. 2010;21:1850–63. doi: 10.1091/mbc.E09-09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West AB, Moore DJ, Choi C, Andrabi SA, Li X, Dikeman D, Biskup S, Zhang Z, Lim KL, Dawson VL, Dawson TM. Parkinson’s disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–32. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- 33.Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–41. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 34.Smith WW, Pei Z, Jiang H, Dawson VL, Dawson TM, Ross CA. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–3. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- 35.MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–93. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Sakaguchi-Nakashima A, Meir JY, Jin Y, Matsumoto K, Hisamoto N. LRK-1, a C. elegans PARK8-related kinase, regulates axonal-dendritic polarity of SV proteins. Curr Biol. 2007;17:592–8. doi: 10.1016/j.cub.2007.01.074. [DOI] [PubMed] [Google Scholar]
- 37.Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J. 2008;27:2432–43. doi: 10.1038/emboj.2008.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tain LS, Mortiboys H, Tao RN, Ziviani E, Bandmann O, Whitworth AJ. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat Neurosci. 2009;12:1129–35. doi: 10.1038/nn.2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaleel M, Nichols RJ, Deak M, Campbell DG, Gillardon F, Knebel A, Alessi DR. LRRK2 phosphorylates moesin at threonine-558: characterization of how Parkinson’s disease mutants affect kinase activity. Biochem J. 2007;405:307–17. doi: 10.1042/BJ20070209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106:14622–7. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated by Roger Barker 11 Mar 2010
- 41.Li Y, Liu W, Oo TF, Wang L, Tang Y, Jackson-Lewis V, Zhou C, Geghman K, Bogdanov M, Przedborski S, Beal MF, Burke RE, Li C. Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci. 2009;12:826–8. doi: 10.1038/nn.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Roger Barker 11 Mar 2010
- 42.Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson’s-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–27. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee BD, Shin JH, VanKampen J, Petrucelli L, West AB, Ko HS, Lee YI, Maguire-Zeiss KA, Bowers WJ, Federoff HJ, Dawson VL, Dawson TM. Inhibitors of leucine-rich repeat kinase-2 protect against models of Parkinson’s disease. Nat Med. 2010;16:998–1000. doi: 10.1038/nm.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by George Ricaurte 22 Oct 2010
- 44.Dodson MW, Guo M. Pink1, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson’s disease. Curr Opin Neurobiol. 2007;17:331–7. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 45.Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet. 2004;13(Suppl 1):R127–133. doi: 10.1093/hmg/ddh089. [DOI] [PubMed] [Google Scholar]
- 46.Gasser T. Mendelian forms of Parkinson’s disease. Biochim Biophys Acta. 2009;1792:587–96. doi: 10.1016/j.bbadis.2008.12.007. [DOI] [PubMed] [Google Scholar]
- 47.Heutink P. PINK-1 and DJ-1--new genes for autosomal recessive Parkinson’s disease. J Neural Transm Suppl. 2006;70:215–219. doi: 10.1007/978-3-211-45295-0_33. [DOI] [PubMed] [Google Scholar]
- 48.Dawson TM, Dawson VL. The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord. 2010;25(Suppl 1):S32–39. doi: 10.1002/mds.22798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–60. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]; F1000 Factor 10Evaluated by M Flint Beal 05 May 2004, Michele Ramsay 16 Aug 2004
- 50.Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–92. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- 51.Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A. 2008;105:12022–7. doi: 10.1073/pnas.0802814105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–83. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–94. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- 54.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]; F1000 Factor 10Evaluated by M Flint Beal 03 Aug 2006, Michael Ehlers 21 Nov 2006
- 55.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated byThomas Gasser 31 Mar 2004
- 56.Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–9. doi: 10.1073/pnas.0802076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]; F1000 Factor 12Evaluated by Monte Gates 30 May 2006, M Flint Beal 03 Aug 2006, Michael Ehlers 21 Nov 2006
- 58.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 9Evaluated by Kenneth Downing 09 Dec 2008, Robert Parton 16 Dec 2008
- 59.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A. 2010;107:378–83. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 6Evaluated by Massimo Zeviani 11 Feb 2010
- 60.Macedo MG, Anar B, Bronner IF, Cannella M, Squitieri F, Bonifati V, Hoogeveen A, Heutink P, Rizzu P. The DJ-1L166P mutant protein associated with early onset Parkinson’s disease is unstable and forms higher-order protein complexes. Hum Mol Genet. 2003;12:2807–16. doi: 10.1093/hmg/ddg304. [DOI] [PubMed] [Google Scholar]
- 61.Moore DJ, Dawson VL, Dawson TM. Genetics of Parkinson’s disease: what do mutations in DJ-1 tell us? Ann Neurol. 2003;54:281–2. doi: 10.1002/ana.10740. [DOI] [PubMed] [Google Scholar]
- 62.Cookson MR, Bandmann O. Parkinson’s disease: insights from pathways. Hum Mol Genet. 2010;19:R21–27. doi: 10.1093/hmg/ddq167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med. 2009;47:1354–61. doi: 10.1016/j.freeradbiomed.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 64.Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, Ko HS, Sasaki M, Ischiropoulos H, Przedborski S, Dawson TM, Dawson VL. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–12. doi: 10.1073/pnas.0703219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aleyasin H, Rousseaux MW, Marcogliese PC, Hewitt SJ, Irrcher I, Joselin AP, Parsanejad M, Kim RH, Rizzu P, Callaghan SM, Slack RS, Mak TW, Park DS. DJ-1 protects the nigrostriatal axis from the neurotoxin MPTP by modulation of the AKT pathway. Proc Natl Acad Sci U S A. 2010;107:3186–91. doi: 10.1073/pnas.0914876107. [DOI] [PMC free article] [PubMed] [Google Scholar]; F1000 Factor 8Evaluated by M Flint Beal 11 Feb 2010
- 66.Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M, Lutz AK, Rousseaux MW, Bevilacqua L, Jahani-Asl A, Callaghan S, MacLaurin JG, Winklhofer KF, Rizzu P, Rippstein P, Kim RH, Chen CX, Fon EA, Slack RS, Harper ME, McBride HM, Mak TW, Park DS. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010;19:3734–46. doi: 10.1093/hmg/ddq288. [DOI] [PubMed] [Google Scholar]
- 67.Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, Wolburg H, Gizatullina Z, Gellerich FN, Woitalla D, Riess O, Kahle PJ, Proikas-Cezanne T, Krüger R. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS One. 2010;5:e9367. doi: 10.1371/journal.pone.0009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thomas B, Beal MF. Parkinson’s disease. Hum Mol Genet. 2007;16(Suppl 2):R183–94. doi: 10.1093/hmg/ddm159. [DOI] [PubMed] [Google Scholar]