

Summary
Methylated CpG Binding Protein 2 (MeCP2) is a nuclear protein named for its ability to selectively recognize methylated DNA. Much attention has been focused on understanding MeCP2 structure and function in the context of its role in Rett syndrome, a severe neurodevelopmental disorder that afflicts one in 10,000–15,000 girls. Early studies suggested a connection between DNA methylation, MeCP2, and establishment of a repressive chromatin structure at specific gene promoters. However, it is now recognized that MeCP2 can both activate and repress specific genes depending on the context. Likewise, in the cell, MeCP2 is bound to unmethylated DNA and chromatin in addition to methylated DNA. Thus, to understand the molecular basis of MeCP2 functionality, it is necessary to unravel the complex interrelationships between MeCP2 binding to unmethylated and methylated regions of the genome. MeCP2 is unusual and interesting in that it is an intrinsically disordered protein, that is, much of its primary sequence fails to fold into secondary structure and yet is functional. The unique structure of MeCP2 is the subject of the first section of this article. We then discuss recent investigations of the in vitro binding of MeCP2 to unmethylated and methylated DNA, and the potential ramifications of this work for in vivo function. We close by focusing on mechanistic studies indicating that the binding of MeCP2 to chromatin results in compaction into local (secondary) and global (tertiary) higher order structures. MeCP2 also competes with histone H1 for nucleosomal binding sites. The recent finding that MeCP2 is found at near stoichiometric levels with nucleosomes in neuronal cells underscores the multiple modes of engagement of MeCP2 with the genome, which include the cooperative tracking of methylation density.
Keywords: chromatin architectural protein, genome, intrinsically disordered, natively unstructured, allosteric, DNA methylation, histones, nucleosomes, nucleosomal arrays, DNA-dependent dimerization
Introduction
Methylated CpG Binding Protein 2 (MeCP2), named for its ability to selectively recognize methylated DNA, is a chromosomal protein particularly abundant in neuronal cells (1). While the initial investigations of MeCP2 focused on its methylated DNA binding properties, recently the protein has received a great deal of attention because of its involvement in Rett syndrome (RTT), an X chromosome-linked neurodevelopmental disorder that afflicts one in 10,000–15,000 girls (2). Hundreds of different mutations in MeCP2 have been identified that are associated with the RTT phenotype (http://www.rettsyndrome.org/mutation-databases.html). Hence, there presently is intense interest in the structure/function relationships of normal MeCP2 and the mutations that result in disease.
Historically, the ability of MeCP2 to bind methylated DNA has been interpreted in the context of transcriptional repression. This is based on the long-standing correlation between DNA methylation and repression of gene expression (3). For example, MeCP2 initially was proposed to mediate transcriptional repression by binding to the co-repressor, Sin3A, and recruiting histone deacetylase (HDAC) to methylated promoters, creating a hypo-acetylated locally repressive chromatin environment (4). However, subsequent studies have revealed additional roles of MeCP2, including higher order compaction of unmethylated chromatin (5), large scale chromatin looping (6), and RNA splicing (7). More recently, the concept of MeCP2 as a multifunctional protein has been further emphasized by genome-wide studies showing that MeCP2 both represses and activates genes, and that MeCP2 binds to both methylated and unmethylated regions of the genome in vivo (8, 9). Thus, while MeCP2 may in some cases function as a repressor at specific methylated promoters, there seem to be other MeCP2-mediated genomic functions as well, some of which do not involve DNA methylation or transcriptional repression. Given this background, the intent of this article is to critically review the recent literature relating to MeCP2 as a DNA and chromatin binding protein. We first discuss the recent insight into the unusual protein chemistry of MeCP2. We then examine the relationship between methylated DNA binding and MeCP2 action. We conclude by highlighting advances in our understanding of MeCP2 as a protein that binds to unmethylated DNA and chromatin, properties of MeCP2 that until recently, have received less attention.
MeCP2—The Protein
Humans have two isoforms of MeCP2 produced by alternative splicing of a short segment at the extreme N-terminus of the protein. The e2 isoform is the most widely studied and will be referred to as MeCP2 from here on. Human MeCP2 has 486 residues and a mass of ∼53 kDa. Sedimentation equilibrium experiments indicate that, when free in solution, MeCP2 behaves as a monomer over a nearly 1000-fold concentration range (10). Sedimentation velocity experiments yield a sedimentation coefficient of 2.2 S (10, 11), independent of salt concentration (10). From the mass and the sedimentation coefficient, the frictional coefficient (f/fo) for the MeCP2 monomer is calculated to be 2.4 (a sphere has a frictional ratio of 1.0). This is a very high, anomalous value for a ∼53 kDa protein, and indicates that MeCP2 has random coil-like hydrodynamic properties in solution. Consistent with the hydrodynamic behavior of the protein, circular dichroism (CD) revealed that MeCP2 is 60–65% unstructured (10, 12). Together, the biochemical data indicate that MeCP2 falls into the category of an “intrinsically disordered” protein (13), in which all or part of the primary sequence fails to fold into classical secondary structure elements such as alpha helix or beta sheet. Computational algorithms (e.g., FoldIndex, PONDR) also predict that MeCP2 is extensively disordered (10, 14) (Fig. 1). Given the large degree of disorder in the protein, MeCP2 is almost certainly quasi-stable in solution and in equilibrium between multiple conformations, and this conformational plasticity is likely related to the multifunctionality of MeCP2 in vivo. Clearly, one of the keys to understand the action of MeCP2 in health and disease will be to determine why so much of this protein lacks traditional folded structure yet is functional and subject to deleterious mutations.
Figure 1.
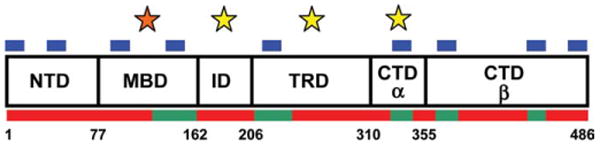
Domain organization and properties of human MeCP2. Shown are the six MeCP2 domains defined based on trypsin digestion (10). Listed at the bottom are the residue numbers at the domain junctions. Red bars along the bottom are regions predicted by PONDR-VLXT to be disordered, while green bars are regions of predicted order (14). Blue bars along the top are sites of predicted MoRFs (15). The domains with yellow stars above them bind unmethylated DNA with high affinity in vitro (14). The orange star above the MBD highlights that this domain binds methylated DNA with high affinity and unmethylated DNA with lower affinity (14, 16).
Two domains of MeCP2, the methyl DNA binding domain (MBD; residues 78–163) and the transcriptional regulation domain (TRD; residues 207–310), have been identified based on function. The MBD was defined as the minimum continuous stretch of residues of MeCP2 necessary to selectively recognize methylated DNA (17), whereas the TRD encompasses the smallest polypeptide needed to repress transcription in transient transfection reporter assays (18). Limited protease digestion yields information about the domain organization of an intact protein based on structure. Despite its extensively disordered nature, digestion of MeCP2 with trypsin reproducibly produces a characteristic pattern of six limiting peptides (10). N-terminal sequencing of the peptides revealed the linear domain organization shown in Fig. 1 (10). Importantly, two of the peptides correspond to the known functional domains, the MBD and TRD. There is no general agreement on domain nomenclature other than for the MBD and TRD. Thus, the N-terminal domain (NTD), the intervening domain (ID), and the C-terminal domain alpha and beta (CTD-α, -β) have been named based on their location relative to the MBD and TRD. Unfortunately, this has tended to create a very “MBD/TRD”-centric view of MeCP2. However, we emphasize that missense and nonsense mutations associated with RTT are found throughout the entire MeCP2 primary sequence, indicating that each of the six domains identified biochemically are in some way essential for the normal function(s) of the intact full length protein.
Because it is so disordered, gaining a structural understanding of full length MeCP2 presents a major technical challenge. CD analysis indicates that all the individually isolated MeCP2 domains are 60–80% unstructured, with the exception of the MBD, which is ∼60% ordered (12, 14). The structure of the isolated MBD alone has been solved by NMR (19) and in complex with methylated DNA by X-ray crystallography (20). The isolated MBD is comprised of a 4-stranded beta sheet, one alpha-helix, and several long stretches of disordered residues. Importantly, in addition to recognizing methylated DNA, the MBD also appears to make up the structural hub of the protein. Biophysical studies revealed that the R133C, F155S, and T158M RTT mutations all decrease the stability of the MBD relative to wild type. There is a single Trp residue in MeCP2 that can be used to report on the solvent accessibility and stability of the MBD within the native protein. This residue is found at position 104 in the MBD and is protected from solvent in the native state. Truncation peptides lacking combinations of the NTD, TRD, and CTD all showed increased solvent accessibility of Trp104 relative to full length MeCP2 (12), indicating that there is inter-domain coupling originating from the MBD in the intact protein. Inter-domain interactions have also been observed in cis (14). A careful examination of the location of the RTT mutations indicates that there are number of “hotspots” that often are found at or very near domain junctions, for example, MBD/ID, TRD/CTD, further suggesting the potential functional importance of inter-domain coupling.
DNA Binding
MeCP2 Avidly Binds Both Methylated and Unmethylated DNA
Although MeCP2 was isolated on the basis of its preferential binding to methylated DNA (21, 22), it soon became clear that, like many DNA-binding proteins, it also binds non-specifically to unmethylated DNA (5, 17, 23, 24). MeCP2 is unusual, however, in that its affinity for methylated DNA is only ∼3-fold greater than for unmethylated DNA (23, 25), whereas for the sequence-specific Lac repressor, the affinity for binding to the specific versus non-specific sequence is ∼104-fold higher (26). The small difference between methylation specific and non-specific binding affinity is reflected in chromatin immunoprecipitation-microarray studies demonstrating that MeCP2 binds to both unmethylated and methylated promoters in cultured neuronal cell lines (8). Although at first sight the recent report that MeCP2 binding efficiently “tracks” methyl-CpG density in the genome (1), appears to contradict the small difference in affinity, as discussed below, the density of methylated CpG sites has a powerful influence on binding affinity, and may play a key role in controlling MeCP2 occupancy.
Sedimentation equilibrium studies of MeCP2-DNA interactions provide quantitative insight into the factors that may modulate the dynamic nature of MeCP2 binding. Sedimentation equilibrium analyses confirm the ∼3 fold higher binding affinity of MeCP2 for a DNA segment carrying a single symmetrically methylated CpG, compared to unmethylated DNA of identical sequence (16). In contrast to most sequence-specific DNA-binding proteins which have few high affinity sites in the genome, MeCP2 has a large number of relatively high(er) affinity sites in a typical mammalian cell in which ∼3% of all cytosines are methylated (27). Thus for MeCP2, the moderately higher affinity for methylated DNA, coupled with the high frequency of encounters with methylated CpGs in vivo is likely to lead to more efficient methylation selectivity than expected from the small ∼3-fold difference in binding affinity discussed above.
The methylated CpG specific binding capacity of MeCP2 is mediated entirely by the core MBD domain. X-ray crystallo-graphic analysis reveals that the methyl specificity is conferred by hydrophilic interactions between the MBD and water molecules creating a hydration shell around the 5′methyl-cytosine (20). An important electrostatic component to binding is also indicated by the finding that higher salt concentrations are needed to elute densely methylated DNA from bound MBD than DNA with low methylation density (28). Interestingly, the NTD, which does not bind DNA, considerably improves the DNA binding affinity of the MBD, possibly through allosteric coupling (14). This further supports the concept that the MBD constitutes an interaction hub of MeCP2 through its structural coupling with the NTD, ID, and TRD domains (14). These conformational couplings, which are likely to modulate specific and nonspecific DNA binding by MeCP2, are perturbed by MBD-located RTT-causing mutations.
MeCP2 Possesses Multiple Non-specific DNA Binding Sites
Gel mobility shift and fluorescence anisotropy studies of isolated MeCP2 domains have revealed that the protein contains multiple non-specific binding sites for double stranded unmethylated DNA (Fig. 1). The MBD is intriguing because it can bind both unmethylated and methylated DNA with high affinity (see above). A DNA binding domain has also been identified in the ID (14) that, when mutated, reduces in vivo association with chromatin (29). The TRD fragment also possesses a non-specific DNA binding site (10, 14). Finally, there is a distinct non-specific binding site for unmethylated DNA in the CTDα (14). It is well established that non-specific DNA binding allows a protein to bind weakly to any site, and subsequently migrate along the DNA, thus greatly reducing the effective search time for a specific binding site (30). Non-specific binding also facilitates specific site binding through intrastrand and interstrand hopping involving repeated association and dissociation (30) as well as “intersegmental transfer” (30). Thus, the unusual non-specific, unmethylated DNA binding properties of MeCP2 may facilitate the ability of the protein to track methylated DNA in vivo.
MeCP2 Binds Cooperatively to DNA
Binding site saturation analysis reveals that one molecule of MeCP2 occupies 11-bp DNA (16), with the MBD protecting ∼6 bp (20). Although MeCP2 is strictly monomeric in solution (10), it binds cooperatively to DNA long enough to accommodate at least two molecules of MeCP2 (16), indicating it oligomerizes in conjunction with DNA binding. Sedimentation equilibrium analysis of the binding of MeCP2 to DNA substrates of differing length and sequence further reveals that binding affinity increases with DNA length and with the density of methylated CpG-(A/T)≥4 motifs. The DNA-dependent cooperative oligomerization of MeCP2 monomers is also strongly enhanced by increase in methylation density (16), which may account for the in vivo correlation between MeCP2 occupancy of nucleosomes and methylation density (1). MeCP2 undergoes marked structural transitions upon DNA binding (14), and it is possible that it is these structural changes that promote cooperative MeCP2–MeCP2 interactions. In this case, DNA would serve the dual role of substrate and allosteric modulator (31). In this respect, it is noteworthy that MeCP2 is predicted to contain an unusually high number of Molecular Recognition Features (MoRFs) (14) (Fig. 1). MoRFs are intrinsically disordered protein regions that likely constitute combinatorial interaction sites for binding partners by assuming different specific secondary structures upon binding to different surfaces (15). A cooperative association between MeCP2 monomers would result in local enrichment of MoRFs, potentially facilitating the local recruitment of multiple regulatory proteins at densely methylated control elements. Interestingly, cooperative binding is entirely abolished in the RTT causing C-terminal truncation mutant R294X (16), suggesting that the CTD, which harbors four predicted MoRFs, is essential for mediating DNA-dependent MeCP2–MeCP2 interactions.
MeCP2 Induces DNA Bridging and Looping
Serial analysis of gene expression suggests that genes undergoing coordinated expression are often spatially arranged in common chromatin domains (32). A common mechanism of coordinated control of genes is through looping of neighboring genes whereby their regulatory regions are brought together through homo- or hetero-association between regulatory proteins bound to these sites (33, 34). In addition to control of gene expression, DNA looping has been suggested to account for binding site tracking by proteins through large step sizes (≥400 bp) involving a mechanism termed “intersegmental transfer.” In this scenario, protein movement occurs through a transient loop formation, where the protein bound to two distant DNA sites exists as a short-lived intermediate between transfers from one site to another (30). In the case of MeCP2, such a mechanism is favored by its multiple independent DNA binding sites, and indeed, EM and AFM images of MeCP2-DNA interactions provide direct confirmation that single MeCP2 molecules can bring together two distant sites on a strand of DNA, creating a loop (5, 16, 24). An in vivo role for MeCP2 in induction and maintenance of large-scale repressive chromatin has been suggested for the DLX5 and DLX6 loci (6), although the general role of MeCP2 in the imprinting of these loci has been challenged (35). Periodic binding of MeCP2 in intergenic regions has also been suggested to create a looped organization of imprinted loci (8).
Nucleosome and Chromatin Binding
Chromatin is the genetic material of eukaryotes. The first level of chromatin organization is the nucleosome, in which 147 bp of chromosomal DNA is wrapped around an octamer of core histone proteins. At the next level, core histone octamers are spaced at 20–60 bp intervals along a DNA molecule to form chromatin. The free DNA that connects adjacent nucleosomal subunits in chromatin is referred to as linker DNA. Acting through both intrinsic and protein-mediated mechanisms, chromatin is packaged into highly condensed chromosomal fibers. In vitro studies have shown that MeCP2 is a chromatin architectural protein that condenses unmethylated or methylated chromatin fibers into highly compact and regular folded structures and “bridges” individual fibers together into supramolecular assemblies (5, 24). Importantly, given that the stoichiometry of MeCP2 in neuronal cells is very high (∼0.5 MeCP2/nucleosome), local and global chromatin compaction are likely to be important cellular functions of MeCP2.
Multiple Modes of MeCP2 Engagement with Nucleosomal Substrates
At the molecular level, we propose that the concerted engagement of MeCP2 with multiple different DNA/chromatin binding sites leads to fiber condensation. There are four potential binding targets for MeCP2 in chromatin, the most likely being the free linker DNA that connects adjacent nucleosomes. A second potential target is the curved and distorted DNA wrapped around each nucleosome. A third possible binding site is the highly contoured protein surface of the nucleosome. Finally, MeCP2 may interact with one or more of the solvent exposed core histone N-terminal tail domains. Even though nucleosome bound MeCP2 has been shown to be in close proximity to histone H3 (36), no direct interactions between MeCP2 and core histones have been documented, and it is not clear whether MeCP2-histone interactions are required to induce chromatin condensation.
Multiple modes of interaction are involved in the binding of MeCP2 to nucleosomes and chromatin. The early studies of Wolffe and coworkers (37) compared the interactions of Xenopus laevis MeCP2 with nucleosome cores and mononucleosomes with extranucleosomal linker DNA, where the nucleosomal DNA was either unmethylated or methylated. Based on nuclease footprinting, they found that MeCP2 could interact with methylated nucleosomal DNA at the nucleosome dyad axis and near the nucleosome core boundary, and with free linker DNA. Further, they reported that MeCP2 preferentially bound to nucleosomes containing linker DNA and protected the linker DNA from nuclease digestion. We have also observed asymmetric MeCP2 binding to nucleosomes and found that MeCP2 protects 11 bp of linker DNA from micrococcal nuclease digestion when bound to chromatin (36). At the chromatin fiber level, binding of MeCP2 to nucleosomal arrays promotes nucleosome–nucleosome clustering and DNA–nucleosome–DNA interactions as judged by electron microscopy (5, 36).
Domain Function During Chromatin Condensation
Several lines of evidence suggest a functional connection between the CTD and chromatin dynamics. The R168X truncation mutant (which is essentially the NTD-MBD fragment), is able to bind chromatin but not condense it into folded secondary chromatin structures (5). Recently, it was observed that the isolated CTD-β could bind to chromatin but not to linear dsDNA, suggesting that chromatin-specific binding sites are present in this domain (14). Consistent with this result, the TRD-CTD fragment can compact arrays of nucleosomes to the same extent as the full-length protein, while other domains and domain pairs cannot (14). Thus, the MeCP2 CTD clearly has properties that are essential for mediating chromatin folding.
The observation that the TRD–CTD fragment is able to effectively condense chromatin into folded structures suggests that MeCP2 is organized into two functional halves. The NTD-MBD-ID serves to direct MeCP2 to methylated GpGs given an appropriate cellular context. The MBD also acts as a nucleosome-binding domain if the nucleosomal DNA is methylated. The second half of the protein comprises the TRD–CTD, and it is this domain combination that is responsible for the nucleosome–nucleosome clustering that leads to chromatin folding. Based on in vitro gel shift studies, non-specific DNA binding is a component of both functional halves of the protein (10, 14).
MeCP2 and Histone H1 Compete for Nucleosomal Binding Sites
Linker histones are the most abundant chromatin architectural proteins in the cells of most metazoans. Linker histone H1 and MeCP2 both induce marked condensation of nucleosomal arrays in vitro and exhibit similar in vivo dynamics (29, 38). Furthermore, with short oligonucleosomes where the fundamental changes in geometry induced by MeCP2 are not obscured by secondary interactions, the underlying zigzag array architecture is remarkably similar to that induced by H1 (16, 39, 40). The binding dynamics of MeCP2 and H1 to nucleosomes and reconstituted nucleosomal arrays reveal strong competition between these two proteins for nucleosomal binding sites (16, 18). Further, an intimate relationship between H1 and MeCP2 expression and function is suggested by the recent observation that H1 undergoes a two-fold elevation in expression level in cells devoid of MeCP2 (1). Moreover, a fluorescence recovery after photobleaching (FRAP) based study of the binding kinetics of MeCP2 and H1 has revealed that the chromatin binding dynamics of MeCP2 is influenced by the concentration of linker histone H1 in the nucleus and vice versa (16). Interestingly, H1, which competes efficiently with nonspecifically bound MeCP2 but not with the tightly bound fraction of MeCP2, shifts the MeCP2 binding equilibrium towards specific binding by increasing the abundance of free MeCP2 and lowering the abundance of available non-specific sites in the nucleus (16). This suggests that in cells in which the expression level of MeCP2 is significantly lower than H1, MeCP2 is likely to have more local, gene specific functions than global functions. In contrast, in cells expressing equivalent or near equivalent levels of H1 and MeCP2, MeCP2 is likely to have both gene specific and global functions related to chromatin binding.
MeCP2 Distribution and Dynamics In Vivo
In the nucleus, MeCP2 shows strong heterochromatin localization that is particularly prominent in mouse fibroblast cells, where highly repetitive pericentromeric heterochromatin (PCH) from several chromosomes associates to form distinct foci. PCH typically has a high density of methylated CpGs leading to a strong focal distribution of MeCP2 which depends both on the integrity of the MBD domain, and the high density of CpG methylation. Thus, in mutants that disrupt the MBD (18) and in cells depleted of methylated DNA by 5-Aza-dC treatment, the localization of MeCP2 in PCH is reduced or abolished (16). Further, loss of methylation increases the FRAP kinetics in PCH foci (16). Note that the MBD alone cannot induce short or long-range chromatin condensation (14). Thus, the in vivo localization data support a model in which methylated DNA-binding activity and chromatin compaction reside in different functional units of the protein.
Conclusions
Since the discovery in 1999 that MeCP2 mutations are the primary cause of Rett syndrome (41), much attention has been focused on the role of MeCP2 in neurodevelopment with the ultimate aim of finding an effective treatment. As part of this effort, it is important to understand the nature of the protein itself, the details of its interactions with methylated and unmethylated DNA and chromatin, and the “downstream” events that occur subsequent to MeCP2 binding. The effect of MeCP2 is likely to be modulated by the tissue and cell specific distribution of methylated and unmethylated CpG units that clearly constitute an important control mechanism for gene expression (42). However, the evidence that MeCP2 is a “multifunctional” protein (43) suggests that it mediates many downstream events depending on the local context.
An important future goal will be to generate a better understanding of the structural and mechanistic roles of the non-MBD domains of MeCP2, which when mutated or truncated, inhibit MeCP2 function and cause disease. Similarly, although it is clear that several domains of MeCP2 acquire structure upon DNA binding, the molecular mechanisms involved remain to be determined.
The multiple independent DNA binding sites of MeCP2 promote DNA looping in vitro, and a similar role in vivo, where binding could promote or maintain functional chromatin loops is an attractive possibility (6). However, more evidence that MeCP2-mediated looping is widespread is needed (35). Here, the rapid advances in chromatin conformation capture (i.e., 3C) strategies (44), should allow a much more definitive understanding of the role of MeCP2 in large-scale chromatin domain organization.
The stereospecific binding of chromatin compacting proteins such as MeCP2 to densely methylated CpG dinucleotides might constitute an initial stage in the compaction of a large-scale chromatin domain, in turn increasing the local density of binding sites for MeCP2 and other chromatin architectural proteins. Such self-organization could lead to the formation of a dynamic interactive network involving MeCP2 and its binding partners. The correlated changes in the level of MeCP2 and H1 in neurons (1) and their competitive chromatin binding dynamics (16) suggests that a dynamic interactive network of this nature may contribute to the regulation by MeCP2 of neuronal chromatin plasticity (45).
Acknowledgments
This work was supported in part by NIH GM066834 (to J.C.H.) and by the Rett Syndrome Foundation and NIH GM070897 (to C.L.W.).
References
- 1.Skene PJ, Illingworth RS, Webb S, Kerr ARW, James KD, Turner DJ, Andrews R, Bird AP. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bienvenu T, Chelly J. Molecular genetics of Rett syndrome: when DNA methylation goes unrecognized. Nat Rev Genet. 2006;7:415–426. doi: 10.1038/nrg1878. [DOI] [PubMed] [Google Scholar]
- 3.Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. doi: 10.1038/321209a0. [DOI] [PubMed] [Google Scholar]
- 4.Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386–389. doi: 10.1038/30764. [DOI] [PubMed] [Google Scholar]
- 5.Georgel PT, Horowitz-Scherer RA, Adkins N, Woodcock CL, Wade PA, Hansen JC. Chromatin compaction by human MeCP2. Assembly of novel secondary chromatin structures in the absence of DNA methylation. J Biol Chem. 2003;278:32181–32188. doi: 10.1074/jbc.M305308200. [DOI] [PubMed] [Google Scholar]
- 6.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 7.Young JI, Hong EP, Castle JC, Crespo-Barreto J, Bowman AB, Rose MF, Kang D, Richman R, Johnson JM, Berget S. Regulation of RNA splicing by the methylation-dependent transcriptional repressor methyl-CpG binding protein 2. Proc Natl Acad Sci USA. 2005;102:17551–17558. doi: 10.1073/pnas.0507856102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yasui DH, Peddada S, Bieda MC, Vallero RO, Hogart A, Nagarajan RP, Thatcher KN, Farnham PJ, LaSalle JM. Integrated epigenomic analyses of neuronal MeCP2 reveal a role for long-range interaction with active genes. Proc Natl Acad Sci. 2007;104:19416–19421. doi: 10.1073/pnas.0707442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chahrour M, Jung SY, Shaw C, Zhou X, Wong STC, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320:1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Adams VH, McBryant SJ, Wade PA, Woodcock CL, Hansen JC. Intrinsic disorder and autonomous domain function in the multifunctional nuclear protein, MeCP2. J Biol Chem. 2007;282:15057–15064. doi: 10.1074/jbc.M700855200. [DOI] [PubMed] [Google Scholar]
- 11.Klose RJ, Bird AP. MeCP2 behaves as an elongated monomer that does not stably associate with the Sin3a chromatin remodeling complex. J Biol Chem. 2004;279:46490–46496. doi: 10.1074/jbc.M408284200. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, Woodcock CL. Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem. 2008;283:20523–20534. doi: 10.1074/jbc.M803021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Uversky VN, Oldfield CJ, Dunker AK. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu Rev Biophys. 2008;37:215–246. doi: 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh RP, Nikitina T, Horowitz-Scherer RA, Gierasch LM, Uversky VN, Hite K, Hansen JC, Woodcock CL. Unique physical properties and interactions of the domains of methylated DNA binding protein 2. Biochemistry. 2010;49:4395–4410. doi: 10.1021/bi9019753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohan A, Oldfield CJ, Radivojac P, Vacic V, Cortese MS, Dunker AK, Uversky VN. Analysis of molecular recognition features (MoRFs) J Mol Biol. 2006;362:1043–1059. doi: 10.1016/j.jmb.2006.07.087. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol. 2010;30:4656–4670. doi: 10.1128/MCB.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nan X, Meehan RR, Bird A. Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res. 1993;21:4886–4892. doi: 10.1093/nar/21.21.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 19.Wakefield RID, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, Barlow PN. The solution structure of the domain from MeCP2 that binds to methylated DNA1. J Mol Biol. 1999;291:1055–1065. doi: 10.1006/jmbi.1999.3023. [DOI] [PubMed] [Google Scholar]
- 20.Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, Walkinshaw MD. MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell. 2008;29:525–531. doi: 10.1016/j.molcel.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 21.Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell. 1992;69:905–914. doi: 10.1016/0092-8674(92)90610-o. [DOI] [PubMed] [Google Scholar]
- 22.Meehan RR, Lewis JD, Bird AP. Characterization of MeCP2, a vertebrate DNA binding protein with affinity for methylated DNA. Nucleic Acids Res. 1992;20:5085–5092. doi: 10.1093/nar/20.19.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765–1774. doi: 10.1093/nar/gkg249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikitina T, Shi X, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Woodcock CL. Multiple modes of interaction between the methylated DNA binding protein MeCP2 and chromatin. Mol Cell Biol. 2007;27:864–877. doi: 10.1128/MCB.01593-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishibashi T, Thambirajah AA, Ausió J. MeCP2 preferentially binds to methylated linker DNA in the absence of the terminal tail of histone H3 and independently of histone acetylation. FEBS Lett. 2008;582:1157–1162. doi: 10.1016/j.febslet.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Revzin A, Von Hippel PH. Direct measurement of association constants for the binding of Escherichia coli lac repressor to non-operator DNA. Biochemistry. 1977;16:4769–4776. doi: 10.1021/bi00641a002. [DOI] [PubMed] [Google Scholar]
- 27.Nafee TM, Farrell WE, Carroll WD, Fryer AA, Ismail KMK. Epigenetic control of fetal gene expression. BJOG. 2008;115:158–168. doi: 10.1111/j.1471-0528.2007.01528.x. [DOI] [PubMed] [Google Scholar]
- 28.Brinkman AB, Simmer F, Ma K, Kaan A, Zhu J, Stunnenberg HG. Whole-genome DNA methylation profiling using MethylCap-seq. Methods. 2010 doi: 10.1016/j.ymeth.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 29.Kumar A, Kamboj S, Malone BM, Kudo S, Twiss JL, Czymmek KJ, LaSalle JM, Schanen NC. Analysis of protein domains and Rett syndrome mutations indicate that multiple regions influence chromatin-binding dynamics of the chromatin-associated protein MECP2 in vivo. J Cell Sci. 2008;121:1128–1137. doi: 10.1242/jcs.016865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Halford SE, Marko JF. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004;32:3040–3052. doi: 10.1093/nar/gkh624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lefstin JA, Yamamoto KR. Allosteric effects of DNA on transcriptional regulators. Nature. 1998;392:885–888. doi: 10.1038/31860. [DOI] [PubMed] [Google Scholar]
- 32.Caron JM, Vega LR, Fleming J, Bishop R, Solomon F. Single site alpha-tubulin mutation affects astral microtubules and nuclear positioning during anaphase in Saccharomyces cerevisiae: possible role for palmitoylation of alpha-tubulin. Mol Biol Cell. 2001;12:2672–2687. doi: 10.1091/mbc.12.9.2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bulger M, Groudine M. Looping versus linking: toward a model for long-distance gene activation. Genes Dev. 1999;13:2465–2477. doi: 10.1101/gad.13.19.2465. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Fu XD. DNA interaction networks: an information highway for regulated gene expression in the 3-dimentional space of the nucleus. Cell Res. 2009;19:1316–1319. doi: 10.1038/cr.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schüle B, Li HH, Fisch-Kohl C, Purmann C, Francke U. DLX5 and DLX6 expression is biallelic and not modulated by MeCP2 deficiency. Am J Hum Genet. 2007;81:492–506. doi: 10.1086/520063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikitina T, Ghosh RP, Horowitz-Scherer RA, Hansen JC, Grigoryev SA, Woodcock CL. MeCP2-chromatin interactions include the formation of chromatosome-like structures and are altered in mutations causing Rett syndrome. J Biol Chem. 2007;282:28237–28245. doi: 10.1074/jbc.M704304200. [DOI] [PubMed] [Google Scholar]
- 37.Chandler SP, Guschin D, Landsberger N, Wolffe AP. The Methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry. 1999;38:7008–7018. doi: 10.1021/bi990224y. [DOI] [PubMed] [Google Scholar]
- 38.Misteli T, Gunjan A, Hock R, Bustin M, Brown DT. Dynamic binding of histone H1 to chromatin in living cells. Nature. 2000;408:877–881. doi: 10.1038/35048610. [DOI] [PubMed] [Google Scholar]
- 39.Hamiche A, Schultz P, Ramakrishnan V, Oudet P, Prunell A. Linker histone-dependent DNA structure in linear mononucleo-somes. J Mol Biol. 1996;257:30–42. doi: 10.1006/jmbi.1996.0144. [DOI] [PubMed] [Google Scholar]
- 40.Bednar J, Horowitz RA, Grigoryev SA, Carruthers LM, Hansen JC, Koster AJ, Woodcock CL. Nucleosomes, linker DNA, and linker histone form a unique structural motif that directs the higher-order folding and compaction of chromatin. Proc Natl Acad Sci USA. 1998;95:14173–14178. doi: 10.1073/pnas.95.24.14173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 42.Illingworth RS, Bird AP. CpG islands—“A rough guide. FEBS Lett. 2009;583:1713–1720. doi: 10.1016/j.febslet.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 43.Hite KC, Adams VH, Hansen JC. Recent advances in MeCP2 structure and function. Biochem Cell Biol. 2009;87:219–227. doi: 10.1139/o08-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dulac C. Brain function and chromatin plasticity. Nature. 2010;465:728–735. doi: 10.1038/nature09231. [DOI] [PMC free article] [PubMed] [Google Scholar]