

Abstract
The hepatitis C virus (HCV) non-structural (NS) 5A protein plays an essential role in replication of the viral RNA by the membrane-associated replication complex (RC). Recently, a putative NS5A inhibitor, BMS-790052, has the highest potency of any known anti-HCV compound in inhibiting HCV replication in vitro and showed a promising clinical effect in HCV-infected patients. The precise mechanism of action for this new class of potential anti-HCV therapeutics, however, is still unclear. In order to gain further insight into its mode of action, we sought to test the hypothesis that the antiviral effect of BMS-790052 might be mediated by interfering with the functional assembly of the HCV RC. We observed that BMS-790052 indeed altered the subcellular localization and biochemical fractionation of NS5A. Taken together, our data suggest that NS5A inhibitors such as BMS-790052 can suppress viral genome replication by altering the proper localization of NS5A into functional RCs.
Introduction
Hepatitis C virus (HCV) is an important human viral pathogen infecting more than 170 million people worldwide (Shepard, Finelli, and Alter, 2005). HCV infection is responsible for the development of chronic liver diseases such as liver cirrhosis and hepatocellular carcinoma (Alter et al., 1999; Di Bisceglie, 2000). Current standard of care for HCV infection using PEGylated interferon-α and ribavirin has significant toxicity and its efficacy is suboptimal for many patients (Liang et al., 2000; Zeuzem et al., 2000), emphasizing an urgent need to develop alternative anti-HCV therapeutics.
HCV is a positive strand RNA virus and the only member of the Hepacivirus genus of the Flaviviridae family. The HCV genome is composed of a ~9.6 kb long single-stranded RNA, which encodes a polyprotein of ~3000 amino acids. This viral polyprotein undergoes proteolytic cleavage by host and virally-encoded proteases to yield more than 10 different viral proteins (Grakoui et al., 1993a; Grakoui et al., 1993b). Among those viral proteins, structural viral proteins such as E1, E2, and core serve as components of the mature virus particle, whereas non-structural (NS) viral proteins such as NS3, NS4A, NS4B, NS5A, and NS5B serve as components of a functional replication complex (RC) that replicates the viral genome but are not packaged into mature virus particles (Blight, Kolykhalov, and Rice, 2000; Lohmann et al., 1999; Moradpour, Penin, and Rice, 2007).
HCV replicates its RNA genome in association with membranes that are derived in part from the endoplasmic reticulum (ER). Precisely how this RC is assembled and maintained, however, remains largely unknown. The NS5A protein is thought to play an essential role in the assembly of the viral RC although its exact molecular functions needed for this process are still poorly characterized (Hijikata et al., 1993; Moradpour et al., 1998). While enzymatic activities encoded in other HCV non-structural proteins such as NS3 (protease) and NS5B (RNA polymerase) (Manns et al., 2007) have enabled the development of anti-HCV therapeutics against those targets, the lack of an enzymatic activity described for NS5A, has made the latter a more challenging target against which to design specific anti-HCV drugs.
Recently, however, using a cell-based replicon screen a new class of anti-HCV compounds was identified that appear to target NS5A (Lemm et al., 2010). Surprisingly, one member of this class, BMS-790052, showed the highest in vitro potency of any known anti-HCV compound with a picomolar range of half-maximum effective concentration (EC50) against HCV replicons from various genotypes. In addition, this compound exhibited a very potent clinical effect on patients chronically infected with HCV in a phase I clinical trial (Gao et al., 2010). Analysis of mutations resistant to NS5A inhibitors identified the first 76 amino acids from NS5A as important determinants for a replicon’s susceptibility to NS5A inhibitors (Lemm et al., 2010). Although BMS-790052 and related compounds were reported to bind to NS5A from cell extracts, and be associated with decreased NS5A hyperphosphorylation, a correlate of genome replication, Gao et al., 2010; Lemm et al., 2010; Neddermann et al., 2004)), the apparent Kd for NS5A binding appears to be significantly different than the EC50 for inhibition of viral replication leaving BMS-790052’s precise mechanism of action unclear.
In this report, we sought to test the hypotheses that BMS-790052 might exert its powerful anti-HCV effect in part by disrupting the proper assembly of functional RCs, or by direct inhibition of RCs. We used morphologic and biochemical fractionation assays to show that BMS-790052 alters the subcellular localization of NS5A protein without affecting its expression level. We also determined that BMS-790052 has no activity in assays for either in vitro replication activity of pre-assembled RCs or NS5A self-dimerization. Taken together, our data suggest that NS5A inhibitors like BMS-790052 suppress viral genome replication by altering the subcellular localization of NS5A, thereby preventing the assembly of NS5A into functional RCs.
RESULTS
BMS-790052 blocks HCV genome replication
The NS5A inhibitors with a thiazolidinone core structure including BMS-858 and BMS-824 were originally identified by a cell-based high-throughput HCV/bovine viral diarrhea virus (BVDV) replicon screen (Lemm et al., 2010). They were further optimized by studying their structure and activity relationship to yield BMS-790052 (Fig. 1A). This compound is the most potent HCV replication inhibitor described to date, with reported in vitro EC50 values of 9 and 71 pM against HCV genotypes 1b and 2a, respectively. BMS-790052 also exhibited a high therapeutic index (CC50/EC50) (>100,000), demonstrating its high specificity against HCV and low toxicity to host cells (Gao et al., 2010).
Fig. 1. BMS-790052 blocks HCV genome replication.


(a) Structure of BMS-790052
(b) Effect of BMS-790052 on stable replication of a HCV replicon derived from genotype 1b. Huh7 cells harboring replicating subgenomic replicons derived from genotype 1b were treated with 0, 0.1, 1, 10, 100, or 1000 pM of BMS-790052 for 3 days. A total cell lysate from cells was prepared and an equal amount of each cell lysate was separated by SDS-PAGE. Levels of NS5A and β-actin protein expression were examined by western blot analysis using monoclonal anti-NS5A and anti-β-actin antibodies.
(c) Huh7.5 cells were infected with a vaccinia virus expressing a T7 RNA polymerase and then transfected with either Bart79I or Bartman (Con1) plasmid construct containing the NS viral proteins downstream of a T7 promoter. Transfected cells were incubated with 0.1 % of DMSO or 0.01, 0.1, 1, 10, or 100 nM of BMS-790052 for 9 hours. A total cell lysate was prepared and an equal amount of each cell lysate was separated by SDS-PAGE. Levels of NS5A protein expression were examined by western blot analysis using a monoclonal anti-NS5A antibody.
(d) All procedures were performed as in Fig. 1(c) except that levels of NS3 protein expression were examined by western blot analysis using a monoclonal anti-NS3 antibody.
Before embarking on our mechanism of action studies for BMS-790052, we sought to confirm its specificity and potency against HCV genome replication. First, we examined its effect on transient HCV replication. Huh7 cells transiently transfected with luciferase-linked subgenomic replicon RNA (Blight, Kolykhalov, and Rice, 2000; Elazar et al., 2003; Tscherne et al., 2006) were treated with increasing doses of BMS-790052. As expected, BMS-790052 displayed a very potent inhibitory activity against transient HCV replication with an EC50 value of 1 pM against genotype 1b replicon and 18 pM against genotype 2a J6/JFH replicon, respectively (Supplementary Fig. 1). Assessment of antiviral potency in the stable genotype 1b subgenomic replicon (Bart79I (Elazar et al., 2003)) was based on the detection of NS5A protein content, and yielded an EC50 of 15 pM (Fig. 1B), a value comparable to the prior reported EC50 in the stable Con1 genotype 1b HCV replicon system (Gao et al., 2010), In contrast to Con1, the Bart79I replicon variant contains an adaptive mutation in the NS5A coding sequence, which impairs hyperphosphorylation of NS5A (Blight, Kolykhalov, and Rice, 2000). While treatment of Con1 replicon cells with BMS-790052 resulted in a reduced concentration of the intracellular hyperphosphorylated form of NS5A as previously reported (Lemm et al., 2010), this effect could not be observed in Bart79I replicon cells (Fig. 1C); yet BMS-790052 remained a highly potent inhibitor of HCV replication in these cells, suggesting that inhibition of hyperphosphorylation may not be a sole mechanism of action for NS5A inhibitors. Taken together, these data confirmed that BMS-790052 is a potent inhibitor of both transient as well as stable HCV genome replication.
BMS-790052 alters the subcellular localization of NS5A
Having confirmed the extraordinary potency of BMS-790052 against HCV replication, we next sought to study its mechanism of action. Since NS5A plays an essential role in the assembly of the viral RC, we hypothesized that BMS-790052 disrupts NS5A function(s) important for this assembly process. To test this hypothesis, we used a vaccinia virus-based HCV replicase assembly system to express and assemble NS proteins into RCs in the presence or absence of BMS-790052. In this system, the HCV non-structural proteins NS2 to NS5B are constitutively expressed via a T7 polymerase promoter, with a vaccinia virus recombinant that provides a source of T7 RNA polymerase. This allows the study of HCV RC assembly without interference from HCV replication inhibition.
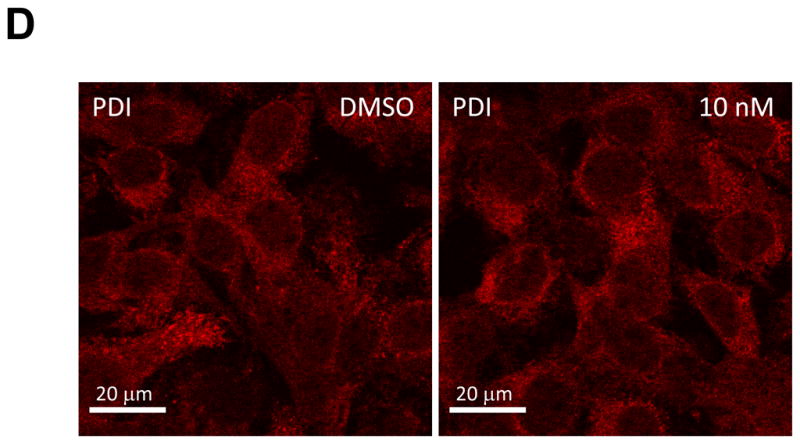
First, we examined the effect of BMS-790052 on the subcellular localization pattern of NS5A proteins using immunofluorescence analysis. As shown in Fig. 2Ai, when NS5A protein was expressed in the context of the HCV NS proteins, it was localized in large perinuclear and cytosolic foci (Fig. 2Ai). However, in cells treated with BMS-790052, NS5A protein showed a diffuse localization pattern (Fig. 2Aii and 2Aiii). Other NS proteins, however, were minimally affected. NS5B protein was localized in large foci in the absence of compound (Fig. 2Bi), and its localization pattern did not change even in the presence of BMS-790052 (Fig. 2Bii and 2Biii). The localization pattern of NS3 protein, which is less punctate than that of NS5A and NS5B proteins, was minimally affected by treatment of BMS-790052 (Fig. 2C). In addition, the subcellular localization of the ER marker protein, PDI, did not show any significant change upon treatment with BMS-790052 (Fig. 2D). We also observed co-localization of NS5A protein, when expressed exogenously in the context of the HCV polyprotein by using the vaccinia virus-based HCV replicase assembly system, with NS5A protein expressed endogenously in HCV replicon cells (Supplementary Fig. 2). The subcellular localization of NS5A expressed from the vaccinia virus-based HCV replicase assembly system was therefore similar to that observed in cells harboring HCV replicons.
Fig. 2. BMS-790052 alters the subcellular localization of NS viral proteins.



(a) Immunofluorescence analysis of NS5A. Huh7.5 cells were infected with a vaccinia virus expressing a T7 RNA polymerase and then transfected with a plasmid (Bart79I) expressing the HCV NS viral proteins downstream of a T7 promoter. Transfected cells were incubated with 0.1 % of DMSO (i), 100 pM (ii), or 1 nM of BMS-790052 (iii) for 9 hours. Cells were fixed, permeabilized, and stained with a monoclonal anti-NS5A mouse antibody. Anti-mouse Alexa 488-conjugated secondary antibody was used to visualize the NS5A protein in green. White scale bar represents 10 μm. The image on the right panel shows 4-fold enlarged view of the area marked with a white rectangle on the left panel, highlighting the differences in distribution patterns of NS5A.
(b) Immunofluorescence analysis of NS5B. Procedure was performed as described in Fig. 2a except that cells were stained with a polycolonal anti-NS5B rabbit antibody. Anti-rabbit Alexa 488-conjugated secondary antibody was used to visualize the NS5B protein in green.
(c) Immunofluorescence analysis of NS3. Procedure was performed as described in Fig. 2a except that cells were stained with a monoclonal anti-NS3 mouse antibody. Anti-mouse Alexa 488-conjugated secondary antibody was used to visualize the NS3 protein in green
(d) Immunofluorescence analysis of PDI. Procedure was performed as described in Fig. 2a except that cells were stained with a polyclonal anti-PDI rabbit antibody. Anti-rabbit Alexa 594-conjugated secondary antibody was used to visualize the PDI protein in red.
A Y93H mutation within NS5A was identified to be one of the major mutations conferring a 20 to 1000-fold resistance to BMS-790052 and related compounds, depending on which HCV genotype was assayed (Conte et al., 2009; Fridell et al., 2010; Gao et al., 2010). Therefore, we hypothesized that if the BMS-790052-induced alteration in the subcellular localization of NS proteins is required for its inhibition of HCV genome replication, the resistance variant (Y93H) should reverse this alteration at comparable concentrations of compound. To test this hypothesis, the Y93H mutation was introduced into the vaccinia virus-based HCV replicase assembly system and the effect of BMS-790052 on NS5A protein subcellular localization was examined. BMS-790052-resistant mutant NS5A protein (NS5A-Y93H) was localized into large foci, similar to wild-type NS5A protein, when expressed in the context of HCV NS proteins in the absence of compound (Fig 3Ai). Consistent with the above hypothesis, NS5A-Y93H protein was resistant to the BMS-790052 effect and remained localized in the large foci without showing diffused localization patterns in the presence of BMS-790052 at up to 1 nM (Fig. 3Aii and 3Aiii). These data suggest that the Y93H mutation in NS5A confers resistance to BMS-790052 by preventing the drug’s ability to alter the subcellular localization of NS5A protein. Just as the resistance to replication inhibition conferred by the Y93H mutation can be overcome with higher concentrations of BMS-790052, diffuse localization patterns of NS5A protein started to be observed with the replicon containing the NS5A Y93H resistance mutation when the concentration of BMS-790052 was increased higher than 100 nM (Supplementary Fig. 3A).
Fig. 3. Reversal of BMS-790052-induced alterations on subcellular localization of NS proteins expressed from a replicon containing the BMS-790052-resistant mutation (Y93H) in NS5A.


(a) Immunofluorescence analysis of NS5A. Procedure was performed as described in Fig. 2a except that Bart79I plasmid, containing the BMS-790052-resistant mutation Y93H (NS5A) was used to express NS viral proteins.
(b) Immunofluorescence analysis of NS5A. Procedure was performed as described in Fig. 2a except that the plasmid used was pEF6-NS5A, which expresses NS5A by itself.
Next, we sought to test whether the BMS-790052 induced alterations in the subcellular localization of NS proteins occurred when the proteins were expressed individually, as opposed to in the context of the HCV polyprotein. Interestingly, when NS5A was expressed in isolation, most of the NS5A proteins were diffusely localized throughout the cytoplasm (Fig. 3Bi), and this dispersed localization pattern was not affected by BMS-790052 (Fig. 3Bii). This suggests that the BMS-790052-induced alteration in subcellular localization of NS5A protein only takes place when NS5A protein is co-expressed with other NS proteins in the context of the viral polyprotein.
The BMS-790052-induced alteration in subcellular localization of NS5A protein is independent of NS protein expression levels
The observed BMS-790052-induced alteration in subcellular localization of NS5A protein might be the result of drug-induced changes in expression levels of NS proteins. To address this possibility, we performed western blot analyses to examine the expression levels of both NS5A and NS3 proteins in the presence of increasing concentrations of BMS-790052 in the vaccinia virus-based HCV replicase assembly system. As shown in Figs. 1C and 1D, BMS-790052 does not significantly affect the level of NS5A or NS3 expression, at concentrations up to 100 nM. This suggests that the BMS-790052-induced alterations in the subcellular localization of NS5A protein were not caused by reduced expression levels of NS proteins.
BMS-790052 alters the subcellular fractionation patterns of NS5A protein
The observation of BMS-790052-induced alterations in the subcellular localization of NS5A protein implies changes in the molecular interactions of NS5A protein with other viral proteins or host factors in the presence of BMS-790052. We therefore examined whether BMS-790052 alters the biochemical subfractionation patterns of NS proteins including NS5A and NS3. For this purpose, a whole cell lysate from the vaccinia virus-based HCV replicase assembly system was prepared in the absence or presence of 10 nM of BMS-790052. Cell lysates were fractionated on idioxanol density gradients (Vogelmann and Nelson, 2007) and the amounts of NS5A and NS3 proteins within the various gradient fractions were determined by western blot analysis. As shown in Fig. 4A, most of the NS5A protein was found to be concentrated in middle fractions (numbers 10 through 15), which correspond to intermediate density fractions. In the presence of BMS-790052, however, the distribution of NS5A was significantly shifted from the middle to the top, low density, fractions (numbers 1 through 8) (Fig. 4B). In contrast, the density gradient profiles of NS3 protein, as well as control cell-cell adhesion protein E-cadherin, were not significantly affected by treatment with 10 nM of BMS-790052. Thus there is morphologic and biochemical evidence that BMS-790052 induces a specific alteration in the subcellular distribution of NS5A protein and the resulting effect on the assembly of the HCV RCs may underlie BMS-790052’s mode of suppressing HCV genome replication.
Fig. 4. BMS-790052 alters the subcellular fractionation patterns of NS proteins.

Huh7.5 cells were infected with a vaccinia virus expressing a T7 RNA polymerase and then transfected with a plasmid (Bart79I) containing the NS viral proteins downstream of a T7 promoter. Transfected cells were incubated with 0.1 % of DMSO (A) or 10 nM of BMS-790052 (B) for 9 hours. Membrane-enriched cell lysates were prepared and fractionated on idioxanol density gradients. Distribution patterns of NS5A, NS3, and E-cadherin proteins were examined by western blot analysis using anti-NS5A, anti-NS3, and anti-E-cadherin antibodies.
BMS-790052 affects neither the in vitro replicase activity of pre-assembled RCs nor the self-dimerization of NS5A proteins
If a potential mechanism of action for BMS-790052 involves inhibition of proper assembly of a new viral RC by altering the localization of NS5A, we would predict that already assembled RCs might be insensitive to the effects of the drug. To test this hypothesis, the in vitro replicase activity (RNA synthesis activity) of membrane-associated RCs (Ma et al., 2005) was determined in the presence or absence of BMS-790052 using RCs isolated from Huh7 cells harboring a full-length J6/JFH (genotype 2a) replicon. As shown in Fig. 5A, in the absence of drug, robust in vitro RNA synthesis activity was detected. 10 μM of a chain-terminating nucleotide analog 3′-deoxycytidine triphosphate (3′-dCTP) completely inhibited HCV replicase activity, similar to previously reported data (Ma et al., 2005). In contrast, BMS-790052 failed to inhibit HCV replicase activity even at micromolar concentrations. This suggests that BMS-790052 is not able to affect pre-assembled RCs.
Fig. 5. BMS-790052 affects neither the in vitro replicase activity of pre-assembled RCs nor the self-dimerization of NS5A proteins.

(a) Effects of BMS-790052 on an in-vitro replicase activity from pre-assembled RCs from genotype 2a. 10 μM of 3′-dCTP, 0.1 % of DMSO, 1 or 10 μM of BMS-790052 was incubated with a crude membrane fraction isolated from Huh7.5 cells harboring replicating J6/JFH full-length replicons derived from genotype 2a. Newly replicated HCV RNAs were separated on an agarose gel and exposed to a phosphorimager.
(b) Effects of BMS-790052 on the dimerization of NS5A proteins. Equal amounts of pAct-NS5A, pBind-NS5A and pG5-Luc were transfected into Huh7.5 cells. Transfected cells were incubated with 0.1 % of DMSO or 10 nM of BMS-790052. Firefly luciferase assay was performed to measure levels of interaction between two NS5A fusion proteins. pAct and pBind are negative control vectors and pAct-Myo and pBind-ID are positive control vectors.
High resolution structural studies of NS5A protein reveal that the protein forms a dimer via contacts near the N-terminus, and the Y93H resistance mutation lies at the interface between two NS5A proteins (Tellinghuisen, Marcotrigiano, and Rice, 2005). We therefore hypothesized that BMS-790052 might inhibit RC assembly and HCV replication by interfering with self-dimerization of NS5A proteins. To test this hypothesis, we utilized a mammalian two-hybrid system. Sequences encoding a VP16 transactivation domain and a Gal4 DNA-binding domain were each individually fused to the N-terminus of NS5A so as to express VP16-NS5A and Gal4DBD-NS5A fusion proteins, respectively. Interactions of these two molecules were measured by a luciferase reporter driven by a promoter containing five Gal4 binding sites (Fearon et al., 1992). As shown in Fig. 5B, a robust interaction between the two NS5A fusion proteins was detected in DMSO-treated control cells. Their level of interaction was comparable to that of two positive control proteins, MyoD (myogenic regulatory protein) and ID (negative regulator of myogenic differentiation) (Weintraub et al., 1991). However, the NS5A-NS5A interaction was not affected by treatment with 10 nM of BMS-790052. This suggests that BMS-790052 may exert its effects of inhibiting functional RC assemblies by means other than interfering with NS5A dimerization.
Discussion
In this study, we confirmed the extraordinary potency of the putative NS5A inhibitor, BMS-790052 against HCV RNA genome replication. In order to gain further insight into its mechanism of action, we investigated the effect of BMS-790052 on the function and on the assembly of functional HCV RCs. To allow the investigation of inhibitors on RC assembly without interference from HCV replication inhibition, we used a vaccinia virus-based HCV replicase assembly system, which expresses T7 RNA polymerase and HCV non-structural proteins NS2 to NS5B under the control of the T7 RNA polymerase promoter. Using this system, we found that NS5A protein localizes to large foci in Huh-7 cells. BMS-790052 altered the subcellular localization of RC-associated NS5A protein, as assessed both morphologically by immunofluorescence and biochemically by subcellular fractionation. Interestingly, this occurred only when NS5A protein was co-expressed with other NS proteins in the context of the HCV polyprotein, suggesting that interactions between HCV NS proteins that contribute to RC formation are a prerequisite for the formation of large foci and for inhibition of HCV replication by BMS-790052. These data suggest that the NS5A inhibitor BMS-790052 exerts its potent anti-HCV replication activity by targeting NS5A function(s) or interactions that are essential for the formation of functional RCs.
While an in vitro EC50 value of BMS-790052 against genotype 1b replicon transient HCV replication was determined to be around 1 pM, 15 pM of BMS-790052 was required to reduce stable HCV replication in half (Fig. 1B). This difference in relative potency may be explained by the presence of pre-assembled RCs in the stable replicon, but not in the transient replicon system. As BMS-790052 only inhibits the formation of new RCs, a complete inhibition of HCV replication may depend on RCs turnover in stable replicon-harboring cells.
With regards to RCs stability, we were able to observe reduction in the amount of NS5A protein close to undetectable levels when stably HCV replicating cells were treated with 1 nM of BMS-790052 for 3 days (Fig. 1B). Similarly, administering NS3 protease inhibitor to HCV replicon cells was shown to eliminate most of NS3 protein within 3 days (Pause et al., 2003). In addition, it has been shown that the half life of NS5A protein in HCV replicon cells is around 16 hours as measured by pulse-chase experiments (Pietschmann et al., 2001). In spite of their different mechanisms of action, both NS5A inhibitors and NS3 protease inhibitors appear to act primarily on the new assembly of functional viral RCs without affecting the pre-existing viral RCs. Unlike NS3 protease inhibitors, however, which block the synthesis of new viral proteins through inhibition of polyprotein processing, the NS5A inhibitor does not directly affect the expression of viral proteins (Fig. 1C and 1D).
BMS-790052 altered the subcellular localization of NS5A protein from large foci to a diffuse localization pattern (Fig. 2A and 2B). The dose response of this effect with regards to compound concentration was different in the HCV replicon system and in the T7-RNA polymerase based HCV RC assembly system. In the replicon system, BMS-790052 inhibited HCV replication at low pM concentrations, while in the T7-polymerase driven RC assembly system, a BMS-790052 concentration of 100 pM and above was required to induced the phenotypic effect of subcellular localization change. The difference in effective BMS-790052 concentration between the transient replicon and the stable replicon was 5 to 15-fold, and between the stable replicon and the T7 RNA polymerase system was approximately 17-fold. We found that the T7-RNA polymerase-based HCV RC assembly system produces 3 to 4-fold higher levels of viral NS proteins compared with those found in HCV replicon cells due to its continuous production of HCV RNAs by a robust T7 RNA polymerase activity (Data not shown). Therefore, relatively higher concentrations of BMS-790052 might be needed in order to achieve a similar level of inhibition in the T7 RNA polymerase-based viral RC assembly system as compared to the replicon system. The difference in phenotypic sensitivity between the transient and the stable replicon systems may be due to the presence of pre-assembled RCs in the latter, which are not affected by BMS-790052.
In regards to its mode of action, reduction of the NS5A hyperphosphorylation by BMS-790052 was reported to be correlated with its anti-HCV replication activity (Lemm et al., 2010). We also were able to see a reduction of hyperphosphorylated NS5A by half upon treatment of 100 pM of BMS-790052 in the subgenomic Con1 replicon system (Fig. 1C). However, replication of the Bart79I HCV 1b replicon, which carries the adaptive mutation S232I in NS5A that significantly decreases the level of hyperphosphorylated NS5A (Blight, Kolykhalov, and Rice, 2000) was similarly sensitive to inhibition with BMS-790052 as was the Con1 replicon.
This indicates that inhibition of hyperphosphorylation may not be a sole mechanism of action for NS5A inhibitors. The subcellular localization of NS5A when expressed by itself is found to be diffuse in the cytoplasm of Huh-7 cells (Fig. 3B), indicating that other NS and/or host proteins are involved in targeting NS5A to large foci. We also observed that exogenously expressed NS5A protein in isolation driven by a CMV promoter does not colocalize with NS5A protein that is endogenously expressed from HCV replicons and incorporated into viral RCs (data not shown). When NS5A protein is expressed in the context of the HCV polyprotein by using the T7 RNA polymerase virus-based RC assembly system, however, such exogenously expressed NS5A colocalized with endogenous, HCV replicon-expressed NS5A (Supplementary Fig. 2). This suggests that expression and assembly of NS proteins by the T7 RNA polymerase-based RC assembly system is accurately mimicking the subcellular localization patterns of NS protein in HCV replicon cells.
The results of biochemical subcellular fractionation assays showed that BMS-790052 treatment of Huh-7 cells expressing HCV NS proteins induced the redistribution of NS5A from middle density fractions to high density fractions (Fig. 4A and 4B). Treatment with BMS-790052 was therefore associated with both morphologic and biochemical alterations in the subcellular distribution of NS5A, as observed by immunofluorescence analysis and subcellular fractionation assays, respectively, consistent with a change in NS5A protein interactions with viral or host factors.
Mutations that confer resistance to BMS-790052 have been mapped to residues 28–32 and 93 of NS5A (Fridell et al., 2010; Lemm et al., 2010), suggesting that BMS-790052 may disrupt an unknown function of the NS5A N-terminus, that is required for interaction with other NS proteins or host factors to allow localization in the large foci. Amino acids 1–30 of NS5A serve as a membrane anchor, and likely contribute to localizing NS5A to an ER-derived membrane, where the HCV RC is likely formed (Brass et al., 2002; Elazar et al., 2003; Moradpour et al., 1998). Although we found no evidence that BMS-790052 can prevent NS5A dimerization, the compound may affect the NS5A dimer conformation without preventing dimer formation or disrupt the interaction of NS5A with as yet unidentified host and/or viral proteins that are important for building RC assemblies. Therefore, the identification of NS5A-binding host and viral proteins which may be affected by NS5A inhibitors like BMS-790052 could provide important insights into understanding RC assembly and the detailed mechanism of action for BMS-790052 and related inhibitors.
Taken together, we found that BMS-790052, a representative of an exciting new class of putative NS5A inhibitors, induces alterations in the subcellular localization of NS5A protein from large foci to a diffuse cytoplasmic localization. The point mutant NS5A-Y93H protein confers resistance to the BMS-790052 induced effect on subcellular localization of NS5A. We propose therefore that BMS-790052 prevents the incorporation of NS5A into functional HCV RCs, while pre-formed RCs are not affected by BMS-790052. Together with other direct-acting antivirals, such as NS3 protease inhibitors, NS5B polymerase inhibitors, and NS4B inhibitors, NS5A inhibitors may provide a valuable component of future combination therapy against HCV infection. Therefore, a better understanding of the mechanism of action of this new class of anti-NS5A therapeutics will help develop new anti-HCV strategies and better understand the essential role of NS5A in the assembly process of viral RCs
Materials and methods
Synthesis of BMS-790052
BMS-790052 was synthesized by Roche according to the procedures described in Bachand, C. et al. Hepatitis C virus NS5A inhibitors, world patent WO-2008021927 (2008)
Plasmids
Bartman is a high-efficiency bicistronic subgenomic replicon of HCV derived from an HCV genotype 1b Con1 sequence that harbors the neomycin phosphotransferase gene in the first cistron and the HCV nonstructural proteins in the second cistron under the translational control of an EMCV IRES (Blight, Kolykhalov, and Rice, 2000). Bart79I is a modification of Bartman that contains an adaptive mutation (S2204I) in NS5A, which increases replication efficiency (Blight, Kolykhalov, and Rice, 2000). Bart79I-luc is a modification of the the Bart79I subgenomic replicon wherein the neomycinphosphotransferase gene of the first cistron has been replaced by a firefly luciferase reporter gene from pGL3 (Promega). Construction of Bart79I-luc was described elsewhere (Bryson et al., 2010) and construction of pEF6-NS5A, which expresses NS5A under the transcriptional control of the elongation factor 6 promoter, was also described elsewhere (Sklan et al., 2007). To introduce an BMS-790052-resistant mutation (Y93H) in NS5A into Bart79I, the nucleotide sequence TAC that encodes for tyrosine at amino acid position 93 of NS5A (at nucleotide position 4009 of Bart79I) was changed to CAC (encoding for histidine) using following two primers FW-NS5A-Y93H, 5′-CATTCCCCATTAACGCGCACACCACGGGCCCCTGCAC-3′, and RV-NS5A-Y93H, 5′-GTGCAGGGGCCCGTGGTGTGCGCGTTAATGGGGAATG-3′ through the use of Quick-Change™ XL site-directed mutagenesis kit (Stratagene) as described by the manufacturer. FL-J6/JFH-5′C19Rluc2AUbi is a monocistronic, full-length HCV genome that expresses Renilla luciferase and was derived from the previously described infectious genotype 2a HCV genome J6/JFH1 (Lindenbach et al., 2005). pAct and pBind are vectors to express fusion proteins with a VP16 transactivation domain and a Gal4 DNA-binding domain, respectively. pG5-Luc is a reporter plasmid containing five Gal4 DNA-binding sites. They are commercially available checkmate vectors (Promega) to perform a mammalian two-hybrid experiment. pAct-Myo and pBind-ID are positive control vectors provided with the kit. pAct-NS5A and pBind-NS5A were constructed by cloning the NS5A DNA fragment amplified by PCR from Bart79I into pAct and pBind plasmids by using XbaI and NotI sites, respectively.
Cell culture
Huh7.5 cell line of the human hepatoma origin were cultured in monolayers as described (Blight, McKeating, and Rice, 2002; Sklan et al., 2007), with media consisting of DMEM (Mediatech) supplemented with 1 % l-glutamine (Mediatech), 1 % penicillin, 1 % streptomycin (Mediatech), and 10 % fetal bovine serum (Omega Scientific). The establishment of Bart-HA cells (Huh7 cells harboring a subgenomic 1b replicon with an HA tag inserted in frame into the C-terminal segment of NS5A) was described elsewhere (Sklan et al., 2007).
Immunofluorescence microscopy
Huh7.5 cells were grown on coverslips to 70 % confluency. Coverslips were rinsed in phosphate-buffered saline (PBS) three times. After a vaccinia virus infection and a transfection, cells were fixed at room temperature for 15 min in 4 % paraformaldehyde, permeabilized in 0.1 % Triton-X in PBS for 5 min, rinsed three times in PBS, and blocked with PBS with 2 % fetal bovine serum (FBS). Anti-NS5A (1:1000, 6F3, Virostat, Portland, ME), anti-NS3 (1:1000, 6D7, Virostat, Portland, ME), anti-NS5B (1:200, house-made by Roche), or anti-PDI antibodies (1:500, SPA-890, Enzo Life Sciences, Plymouth Meeting, PA) were applied, and the mixture was incubated for 2 hours. After three washes in PBS, coverslips were incubated with Alexa 488-conjugated anti-mouse or Alexa 594-conjugated anti-rabbit IgG secondary antibodies (Invitrogen, Carlsbad, CA) for 1 hour. Following three washes with PBS, coverslips were mounted onto slides using Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA) and sealed. Fluorescent signals were examined and captured by LSM 510 Carl Zeiss confocal laser scanning microscope.
Transient replication assays
Transient replication assays were performed as previously described (Cho et al., 2010). Briefly, Huh7.5 cells were trypsinized and resuspended at 1.5×107 cells per ml in PBS buffer. 5 μg of in vitro transcribed Bart79I-luc or FL-J6/JFH-5′C19Rluc2AUbi RNAs were mixed with 400 μl of Huh7.5 cells in a 2-mm-gap cuvette (BTX) and immediately pulsed (0.68 kV (Bart79I-luc) and 0.82 kV (FL-J6/JFH-5′C19Rluc2AUbi) and, five 99 ms pulses) with a BTX-830 electroporator. Pulsed cells were immediately diluted into 20 ml of pre-warmed growth medium and seeded in 96-well plates (17000–20000 cells per well). At 6 hours after electroporation, different concentrations of BMS-790052 were added to the cells. After 72 hours of treatment, cells were incubated for 2 hours at 37°C in the presence of 10 % Alamar Blue reagent (TREK Diagnostic Systems) to assess cytotoxicity. Plates were then scanned and fluorescence was detected by using a FLEXstation II 384 (Molecular Devices, Sunnyvale, California). Viral RNA replication was determined using firefly (Bart79I-luc) or renilla (FL-J6/JFH-5′C19Rluc2AUbi) luciferase assays (Promega).
Stable replication assays
The establishment of Huh7.5 cells stably maintaining a Bart79I subgenomic replicon in the presence of G418 selection was described elsewhere (Cho et al., 2010). Briefly, in vitro-transcribed Bart79I RNAs were electroporated into Huh7.5 cells. The electroporated cells were supplemented with G418 to a final concentration of 750 μg/ml. This selection medium was replaced every 3 days for 3 weeks. Different concentrations of BMS-790052 were added to the G418-resistant Huh7.5 cells for 3 days.
Infection and transfection expression
Infection and transfection was performed as previously described (Elazar et al., 2003). Briefly, a vaccinia virus that expresses the T7 RNA polymerase was used to infect Huh7.5 cells at a multiplicity of infection (MOI) of 10. After 45 min of incubation at 37°C, the cells were washed twice with Opti-MEM (Invitrogen) and subjected to transfection with Bart79I plasmids containing the T7 promoter. The cells were supplemented with growth media containing DMSO or different concentrations of BMS-790052 and incubated for 9 hours at 37°C followed by western blot and immunofluorescence analyses.
Western blot analysis
Whole-cell extracts were prepared in RIPA buffer (150 mM NaCl, 50 mM Tris/HCl (pH 8.0), 5 mM EDTA, 0.5 mM dithiothreitol, 100 mM sodium fluoride, 200 mM sodium orthovanadate, 1 % NP-40, 0.5 % sodium deoxycholate, 0.1 % SDS, and 1 mM PMSF) containing a cocktail of protease inhibitors (Complete, Mini; Roche Diagnostic at final concentration of 1 tablet per 10 ml RIPA buffer) and quantitated by the Bradford assay (Bio-Rad). Equal amounts of protein were electrophoresed on an SDS-polyacrylamide gel, subsequently transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford), and probed with a mouse anti-NS5A or anti-NS3 monoclonal antibodies (1:1000, 6F3 for NS5A and 1:1000, 6D7 for NS3, Virostat, Portland, ME). Proteins were visualized via enhanced chemiluminescence (GE healthcare).
Subcellular fractionation assays
Subcellular fractionation assays using an idioxanol gradient were performed as described previously (Vogelmann and Nelson, 2007). Briefly, transfected cells were washed three times with ice-cold Ringers solution (10 mM HEPES-NaOH, pH 7.4, 154 mM NaCl, 7.2 mM KCl, 1.8 mM CaCl2). Then, cells were cross-linked by treatment of 200 μg/ml of Dithiobis (succinimidylpropionate, Pierce Biotechnology Inc.) for 20 min. A cross-linking reaction was stopped by rinsing with a quenching buffer (120 mM NaCl, 10 mM Tris-HCl, pH 7.4, 50 mM NH4Cl). Total cell lysates were prepared in Homogenization buffer (20 mM HEPES KOH, pH7.2, 90 mM K-acetate, 2 mM Mg-acetate, 25 mM sucrose, 300 nM Pefabloc and a protease inhibitor cocktail Mini (Roche)) by using a ball-bearing homogenizer with 0.3747′ ball and spun down at 930g for 10min at 4C to pellet nuclei and unbroken cells. Remaining supernatant was loaded on top of 10, 20, and 30 % of idioxanol gradients and was ultra-centrifuged at 350,000g for 3 hrs 5 min using Beckman Vti65.1rotor. After a centrifugation, 0.5 ml of a protein-idioxanol mixture was retrieved from top to bottom to make total 24 fractions. Total proteins were precipitated in each fraction by using methanol and chloroform for western blot analysis.
In vitro replicase assay
Cytoplasmic membrane fractions were prepared from Huh7.5 derived cell line containing an autonomously replicating HCV subgenomic replicon (J6/JFH for 2a genotype) as follows. Cell rupture and cytoplasmic membrane fraction isolation were performed as previously described (Takeda et al., 1986). In vitro replicase assay was performed as previously described (Ma et al., 2005). Briefly, the standard replicase assay reactions contained 10 μl cytoplasmic membrane fraction, 50 mM HEPES, pH 7.5, 10 mM KCl, 10 mM DTT, 5 mM MgCl2, 20 μg/ml actinomycin D, 1 mM ATP, GTP, and UTP, 30 μCi [α-32P]-CTP (3000 Ci/mmol, 10 mCi/ml), 1 U/Al SUPERase. In (Ambion), 10 mM creatine phosphate, and 200 μg/ml creatine phosphokinase in a final volume of 25 μl. BMS-790052 was added to make a final concentration of 1 μM and 10 μM, respectively. 10 μM of a chain-terminating nucleotide analog, 3′-deoxycytidine triphosphate (3′-dCTP) (Sigma Aldrich) was also added as a positive control. The reaction mixtures were incubated at 30°C for 120 min and stopped by the addition of 50 mM EDTA and 0.5 % SDS. The RNA products were recovered by phenol–chloroform extraction and ethanol precipitation following removal of proteins by proteinase K digestion. The RNA products were denatured in glyoxal sample loading buffer and resolved in a 1 % agarose gel using 10X RNA running buffer (Ambion). An agarose gel was dried using a GelAir drying system (Bio-Rad) and exposed to a phosphorimager for 4 hours.
Mammalian two-hybrid experiment
Equal amounts of pAct-NS5A, pBind-NS5A, and pG5-Luc were transfected into Huh7.5 cells by using a lipofectamine 2000 transfection reagent (Invitrogen) as described by the manufacturer. Transfected cells were plated onto a 96 well plate and supplemented with DMSO or 10 nM of BMS-790052. At 2 days after incubation, firefly and renilla luciferase activities were measured by using a dual glow luciferase kit (Promega). The renilla luciferase activity was used to normalize transfection efficiency. The firefly luciferase activity from pAct-Myo and pBind-ID cotransfection was defined as 1 to plot the firefly luciferase activity from pAct-NS5A and pBind-NS5A cotransfection.
Supplementary Material
(a) Effect of BMS-790052 on transient replication of a HCV replicon derived from genotype 1b. Huh7.5 cells were electroporated with 5 μg of in vitro-transcribed luciferase-linked Bart79I RNAs. Electroporated cells were treated with 0, 0.01, 0.1, 1, 10, 100, 1000, and 5000 pM of BMS-790052 for 3 days. Luciferase activities were measured at 3 days after electroporation. Replication is reported as a percentage of the non-treated control. Each data point is the mean of four replicates, and error bars indicate standard deviations.
(b) Effect of BMS-790052 on transient replication of a HCV replicon derived from genotype 2a. Huh7.5 cells were electroporated with 5 μg of in vitro-transcribed luciferase-linked J6/JFH1 RNAs. Electroporated cells were treated with 0, 0.1, 0.5, 1, 5, 10, 25, 50, 100, and 1000 pM of BMS-790052 for 3 days. Replication is reported the same way as in Fig. 1b.
Huh7.5 cells stably maintaining Bart79I-HA replicons were infected with a vaccinia virus expressing a T7 RNA polymerase and then transfected with a Bart79I plasmid expressing the HCV NS viral proteins downstream of a T7 promoter. NS5A proteins expressed exogenously from transfected Bart79I plasmids were visualized in green by using a monoclonal mouse anti-NS5A antibody followed by anti-mouse Alexa 488-conjugated secondary antibody. NS5A proteins expressed endogenously from Bart79I-HA replicons (Sklan et al., 2007) were visualized in red by using a polyclonal rabbit anti-HA antibody followed by anti-rabbit Alexa 594-conjugated secondary antibody. (Note that the inserted HA epitope destroys the binding activity of the employed anti-NS5A monoclonal antibody).
(a) Immunofluorescence analysis of NS5A. Procedure was performed as described in Fig. 2a except that a Bart79I plasmid containing the BMS-790052-resistant mutation (Y93H) in NS5A was used to express NS viral proteins in the presence of 200 nM of BMS-790052.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alter MJ, Kruszon-Moran D, Nainan OV, McQuillan GM, Gao F, Moyer LA, Kaslow RA, Margolis HS. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N Engl J Med. 1999;341(8):556–62. doi: 10.1056/NEJM199908193410802. [DOI] [PubMed] [Google Scholar]
- Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290(5498):1972–4. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- Blight KJ, McKeating JA, Rice CM. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J Virol. 2002;76(24):13001–14. doi: 10.1128/JVI.76.24.13001-13014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brass V, Bieck E, Montserret R, Wolk B, Hellings JA, Blum HE, Penin F, Moradpour D. An amino-terminal amphipathic alpha-helix mediates membrane association of the hepatitis C virus nonstructural protein 5A. J Biol Chem. 2002;277(10):8130–9. doi: 10.1074/jbc.M111289200. [DOI] [PubMed] [Google Scholar]
- Bryson PD, Cho NJ, Einav S, Lee C, Tai V, Bechtel J, Sivaraja M, Roberts C, Schmitz U, Glenn JS. A small molecule inhibits HCV replication and alters NS4B’s subcellular distribution. Antiviral Res. 2010;87(1):1–8. doi: 10.1016/j.antiviral.2010.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NJ, Dvory-Sobol H, Lee C, Cho SJ, Bryson P, Masek M, Elazar M, Frank CW, Glenn JS. Identification of a class of HCV inhibitors directed against the nonstructural protein NS4B. Sci Transl Med. 2010;2(15):15ra6. doi: 10.1126/scitranslmed.3000331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31:1014–1018. doi: 10.1053/he.2000.5762. [DOI] [PubMed] [Google Scholar]
- Elazar M, Cheong KH, Liu P, Greenberg HB, Rice CM, Glenn JS. Amphipathic helix-dependent localization of NS5A mediates hepatitis C virus RNA replication. J Virol. 2003;77(10):6055–61. doi: 10.1128/JVI.77.10.6055-6061.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon ER, Finkel T, Gillison ML, Kennedy SP, Casella JF, Tomaselli GF, Morrow JS, Van Dang C. Karyoplasmic interaction selection strategy: a general strategy to detect protein-protein interactions in mammalian cells. Proc Natl Acad Sci U S A. 1992;89(17):7958–62. doi: 10.1073/pnas.89.17.7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridell RA, Qiu D, Wang C, Valera L, Gao M. Resistance Analysis of the HCV NS5A Inhibitor, BMS-790052, in the In Vitro Replicon System. Antimicrob Agents Chemother. 2010 doi: 10.1128/AAC.00556-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Nettles RE, Belema M, Snyder LB, Nguyen VN, Fridell RA, Serrano-Wu MH, Langley DR, Sun JH, O’Boyle DR, 2nd, Lemm JA, Wang C, Knipe JO, Chien C, Colonno RJ, Grasela DM, Meanwell NA, Hamann LG. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature. 2010;465(7294):96–100. doi: 10.1038/nature08960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM. A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci U S A. 1993a;90(22):10583–7. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grakoui A, Wychowski C, Lin C, Feinstone SM, Rice CM. Expression and identification of hepatitis C virus polyprotein cleavage products. J Virol. 1993b;67(3):1385–95. doi: 10.1128/jvi.67.3.1385-1395.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata M, Mizushima H, Tanji Y, Komoda Y, Hirowatari Y, Akagi T, Kato N, Kimura K, Shimotohno K. Proteolytic processing and membrane association of putative nonstructural proteins of hepatitis C virus. Proc Natl Acad Sci U S A. 1993;90(22):10773–7. doi: 10.1073/pnas.90.22.10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm JA, O’Boyle D, 2nd, Liu M, Nower PT, Colonno R, Deshpande MS, Snyder LB, Martin SW, St Laurent DR, Serrano-Wu MH, Romine JL, Meanwell NA, Gao M. Identification of hepatitis C virus NS5A inhibitors. J Virol. 2010;84(1):482–91. doi: 10.1128/JVI.01360-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132(4):296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. Complete replication of hepatitis C virus in cell culture. Science. 2005;309(5734):623–6. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science. 1999;285(5424):110–3. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- Ma H, Leveque V, De Witte A, Li W, Hendricks T, Clausen SM, Cammack N, Klumpp K. Inhibition of native hepatitis C virus replicase by nucleotide and non-nucleoside inhibitors. Virology. 2005;332(1):8–15. doi: 10.1016/j.virol.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Manns MP, Foster GR, Rockstroh JK, Zeuzem S, Zoulim F, Houghton M. The way forward in HCV treatment--finding the right path. Nat Rev Drug Discov. 2007;6(12):991–1000. doi: 10.1038/nrd2411. [DOI] [PubMed] [Google Scholar]
- Moradpour D, Kary P, Rice CM, Blum HE. Continuous human cell lines inducibly expressing hepatitis C virus structural and nonstructural proteins. Hepatology. 1998;28(1):192–201. doi: 10.1002/hep.510280125. [DOI] [PubMed] [Google Scholar]
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5(6):453–63. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- Neddermann P, Quintavalle M, Di Pietro C, Clementi A, Cerretani M, Altamura S, Bartholomew L, De Francesco R. Reduction of hepatitis C virus NS5A hyperphosphorylation by selective inhibition of cellular kinases activates viral RNA replication in cell culture. J Virol. 2004;78(23):13306–14. doi: 10.1128/JVI.78.23.13306-13314.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pause A, Kukolj G, Bailey M, Brault M, Do F, Halmos T, Lagace L, Maurice R, Marquis M, McKercher G, Pellerin C, Pilote L, Thibeault D, Lamarre D. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J Biol Chem. 2003;278(22):20374–80. doi: 10.1074/jbc.M210785200. [DOI] [PubMed] [Google Scholar]
- Pietschmann T, Lohmann V, Rutter G, Kurpanek K, Bartenschlager R. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J Virol. 2001;75(3):1252–64. doi: 10.1128/JVI.75.3.1252-1264.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis. 2005;5(9):558–67. doi: 10.1016/S1473-3099(05)70216-4. [DOI] [PubMed] [Google Scholar]
- Sklan EH, Staschke K, Oakes TM, Elazar M, Winters M, Aroeti B, Danieli T, Glenn JS. A Rab-GAP TBC domain protein binds hepatitis C virus NS5A and mediates viral replication. J Virol. 2007;81(20):11096–105. doi: 10.1128/JVI.01249-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, Kuhn RJ, Yang CF, Takegami T, Wimmer E. Initiation of poliovirus plus-strand RNA synthesis in a membrane complex of infected HeLa cells. J Virol. 1986;60(1):43–53. doi: 10.1128/jvi.60.1.43-53.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellinghuisen TL, Marcotrigiano J, Rice CM. Structure of the zinc-binding domain of an essential component of the hepatitis C virus replicase. Nature. 2005;435(7040):374–9. doi: 10.1038/nature03580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol. 2006;80(4):1734–41. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelmann R, Nelson WJ. Separation of cell-cell adhesion complexes by differential centrifugation. Methods Mol Biol. 2007;370:11–22. doi: 10.1007/978-1-59745-353-0_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub H, Davis R, Tapscott S, Thayer M, Krause M, Benezra R, Blackwell TK, Turner D, Rupp R, Hollenberg S, et al. The myoD gene family: nodal point during specification of the muscle cell lineage. Science. 1991;251(4995):761–6. doi: 10.1126/science.1846704. [DOI] [PubMed] [Google Scholar]
- Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai MY, Gane E, O’Grady J, Reichen J, Diago M, Lin A, Hoffman J, Brunda MJ. Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med. 2000;343(23):1666–72. doi: 10.1056/NEJM200012073432301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Effect of BMS-790052 on transient replication of a HCV replicon derived from genotype 1b. Huh7.5 cells were electroporated with 5 μg of in vitro-transcribed luciferase-linked Bart79I RNAs. Electroporated cells were treated with 0, 0.01, 0.1, 1, 10, 100, 1000, and 5000 pM of BMS-790052 for 3 days. Luciferase activities were measured at 3 days after electroporation. Replication is reported as a percentage of the non-treated control. Each data point is the mean of four replicates, and error bars indicate standard deviations.
(b) Effect of BMS-790052 on transient replication of a HCV replicon derived from genotype 2a. Huh7.5 cells were electroporated with 5 μg of in vitro-transcribed luciferase-linked J6/JFH1 RNAs. Electroporated cells were treated with 0, 0.1, 0.5, 1, 5, 10, 25, 50, 100, and 1000 pM of BMS-790052 for 3 days. Replication is reported the same way as in Fig. 1b.
Huh7.5 cells stably maintaining Bart79I-HA replicons were infected with a vaccinia virus expressing a T7 RNA polymerase and then transfected with a Bart79I plasmid expressing the HCV NS viral proteins downstream of a T7 promoter. NS5A proteins expressed exogenously from transfected Bart79I plasmids were visualized in green by using a monoclonal mouse anti-NS5A antibody followed by anti-mouse Alexa 488-conjugated secondary antibody. NS5A proteins expressed endogenously from Bart79I-HA replicons (Sklan et al., 2007) were visualized in red by using a polyclonal rabbit anti-HA antibody followed by anti-rabbit Alexa 594-conjugated secondary antibody. (Note that the inserted HA epitope destroys the binding activity of the employed anti-NS5A monoclonal antibody).
(a) Immunofluorescence analysis of NS5A. Procedure was performed as described in Fig. 2a except that a Bart79I plasmid containing the BMS-790052-resistant mutation (Y93H) in NS5A was used to express NS viral proteins in the presence of 200 nM of BMS-790052.