

Abstract
A series of computational methods were used to study how cytochrome P450 2A6 (CYP2A6) interacts with (S)-(−)-nicotine, demonstrating that the dominant molecular species of (S)-(−)-nicotine in CYP2A6 active site exists in the free base state (with two conformations, SRt and SRc), despite of the fact that the protonated state is dominant for the free ligand in solution. The computational results reveal that the dominant pathway of nicotine metabolism in CYP2A6 is through nicotine free base oxidation. Further, first-principles quantum mechanical/molecular mechanical free energy (QM/MM-FE) calculations were carried out to uncover the detailed reaction pathways for the CYP2A6-catalyzed nicotine 5′-hydroxlylation reaction. In the determined CYP2A6-(S)-(−)-nicotine binding structures, the oxygen of Compound I (Cpd I) can abstract a hydrogen from either the trans-5′- or the cis-5′-position of (S)-(−)-nicotine. CYP2A6-catalyzed (S)-(−)-nicotine 5′-hydroxylation consists of two reaction steps, i.e. the hydrogen transfer from the 5′-position of (S)-(−)-nicotine to the oxygen of Cpd I (the H-transfer step), followed by the recombination of the (S)-(−)-nicotine moiety with the iron-bound hydroxyl group to generate the 5′-hydroxynicotine product (the O-rebound step). The H-transfer step is rate-determining. The 5′-hydroxylation proceeds mainly with the stereoselective loss of the trans-5′-hydrogen, i.e. the 5′-hydrogen trans to the pyridine ring. The calculated overall stereoselectivity of ~97% favoring the trans-5′-hydroxylation is close to the observed stereoselectivity of 89~94%. This is the first time to demonstrate that a CYP substrate exists dominantly in one protonation state (cationic species) in solution, but uses its less-favorable protonation state (neutral free base) to perform the enzymatic reaction.
Introduction
Tobacco use kills more than 5 million people worldwide each year, and this number is expected to grow.1 Recent surveys2 in the United States show that cigarette smoking kills an estimated 440,000 Americans each year--more than alcohol, illegal drug use, homicide, suicide, car accidents, and AIDS combined. Cigarette smoking accounts for 90% of all cases of lung cancer, and about 38,000 deaths per year can be attributed to secondhand smoke. In face of the negative health consequences of cigarette smoking, most smokers have realized that tobacco is health-eroding and expressed a desire to stop or reduce smoking. However, more than 85% of these smokers get relapsed in a week. Nicotine, an alkaloid found in tobacco leaves, is the primary compound responsible for tobacco dependence, although cigarette smoke contains many other harmful chemicals, including tar, carbon monoxide, acetaldehyde, and nitrosamines etc.3–6
Nicotine activates reward pathway–the brain circuitry that regulates feelings of pleasure. Nicotine-dependent individuals regulate their smoking to maintain nicotine concentration in their blood and brain.7,8 In humans, the principal pathway of nicotine metabolism is 5′-hydroxylation by the hepatic enzyme cytochrome P450 2A6 (CYP2A6).9,10 The metabolite 5′-hydroxynicotine, which exists in equilibrium with Δ1′(5′)-iminium ion, is further oxidized to pharmacologically less active cotinine by cytosolic aldehyde oxidase.11–13 A deficiency in CYP2A6-mediated metabolism of nicotine may permit longer exposure to nicotine and, in turn, may decrease the number of cigarettes a person needs to smoke to obtain their desired blood nicotine concentration.14–16 Hence, inhibition of CYP2A6 could be used as a potential therapeutic strategy for smoking cessation and tobacco-use reduction.17
It has been reported that inhibition of CYP2A6 could cause the decrease in smoking.17 For this purpose, a number of compounds, such as tranylcypromine and methoxsalen, have been identified as inhibitors of CYP2A6.18,19 However, these compounds inhibit both the target CYP2A6 and some other cytochrome P450 enzymes and, thus, have unexpected side effects.20,21 To date, no satisfactory CYP2A6 inhibitor has been used in practical treatment of tobacco dependence.22–24 Potent and selective CYP2A6 inhibitors are highly desirable for therapeutic treatment that aims to reduce the metabolism of nicotine.
For rational design of novel inhibitors of CYP2A6, it is important to understand the detailed mechanism concerning how the enzyme catalyzes nicotine metabolism. A detailed understanding of the mechanism of CYP2A6-catalyzed nicotine metabolism could provide a valuable mechanistic base for rational design of a possible stable analogue of the rate-determining transition state as a novel type of CYP2A6 inhibitor.
Nicotine metabolism catalyzed by cytochrome P450 has attracted considerable attention for several decades.4,9,25–31 The CYP2A6-catalyzed hydroxylation of nicotine (1 in Scheme 1) at the prochiral 5′-position to form the Δ1′(5′)-iminium ion, 2, is the initial step in the biotransformation of nicotine to cotinine, which is the principal metabolite of nicotine in humans. Structurally, the two methylene hydrogen atoms at the 5′-position of nicotine molecule are diastereotopic, i.e. the 5′-hydrogen in trans position to the pyridine ring (trans-5′-hydrogen) and the 5′-hydrogen in cis position to the pyridine ring (cis-5′-hydrogen). This leads to two possible choices for hydrogen abstraction along the pathway of 5′-hydroxylation reaction, one is that the reaction may proceed to selectively abstract the trans-5′-hydrogen, and the other is to selectively abstract the cis-5′-hydrogen of nicotine. Are these two choices equally possible? If not, which one is dominant? Why is one choice dominant over the other? In pursuing the answers to these questions, extensive experimental studies have been performed. The metabolically generated iminium ion species 2 has been trapped and analyzed as the corresponding diastereomeric 5′-cyano adducts (5) by the inclusion of cyanide ion in the reaction mixture.9,10,26,27,32,33 An early study10 using human liver microsomal preparations assessed the stereochemical course of this transformation using the selectively deuterated trans- and cis-5′-monodeutero disatereomers 3 and 4, respectively, as substrate (Scheme 1). The deuterium content of the resulting mixture of iminium species 2a and 2b, as assessed by the GC/EIMS analysis of the corresponding cyano adducts 5a and 5b, established that Δ1′(5′)-iminium ion was formed after the stereoselective loss of the trans-5′-hydrogen. The percentage of stereoselectivity was found to be ~90% and ~89% from 3 and 4, resepectively, and was found to be independent of isotopic substitution. A later work by Carlson et al.33 using human liver microsomal preparations led to the similar result, i.e. ~94% of the 5′-cyanonicotine isomers obtained from 3 was protium enriched, whereas ~93% of the 5′-cyanonicotine isomers obtained from 4 was deuterium enriched. All these stereochemical investigations demostrated that CYP2A6-catalyzed hydroxylation of nicotine proceeded with a highly stereoselective loss of the trans-5′-hydrogen at the azaheterocyclic ring of the nicotine molecule, but the molecular mechanism for such selective loss of trans-5′-hydrogen remains intriguing and unsolved.
Scheme 1.

CYP2A6-catalyzed biotransformation of monodeutero substituted nicotines, 3 and 4, to the corresponding Δ1′(5′)-iminium ion species, 2, and their cyano adducts, 5.
In a previous study, QM reaction coordinate calculations were preformed on a simplified model system including nicotine and the active site (Compound I, Cpd I in brief) of cytochrome P450.34 According to the QM results, the metabolism of nicotine is progressed dominantly through the 5′-hydroxylation pathway, rather than the N-demethylation pathway. However, the stereoselectivity (trans-5′-hydroxylation vs cis-5′-hydroxylation) of the 5′-hydroxylation pathway observed in experimental studies has not been elaborated theoretically. In addition, the QM calculations on the simplified model system could not account for the actual protein environment of CYP2A6, although the bulky polarity and the N-H···S hydrogen bonding were used as a mimic. In order to understand the molecular mechanism about the 5′-hydroxylation pathway for the CYP2A6-catalyzed nicotine metabolism, and to reveal the structural/energetic determinants for the experimentally observed stereoselectivity of the catalytic reaction, we performed molecular modeling on the nicotine binding mode at the catalytic site of CYP2A6 enzyme and first-principles quantum mechanical/molecular mechanical-free energy (QM/MM-FE) calculations35–38 on the CYP2A6-catalyzed 5′-hydroxylation reaction. In the present study, we accounted for the actual protein environment of CYP2A6 for nicotine metabolism for the first time. First of all, the nicotine binding mode at the catalytic site of CYP2A6 was modeled by molecular docking, molecular dynamics (MD) simulations, and binding free energy calculations. For the CYP2A6-nicotine binding modeling, six possible molecular species of nicotine accounting for various free bases and their protonation states of (S)-(−)-nicotine39 (as shown in Table 1) were considered. In the QM/MM-FE calculations, first-principles QM/MM reaction coordinate calculations were followed by free energy perturbation (FEP) calculations in order to more reasonably account for the dynamic effects of the protein environment on the free energy barriers for the catalytic reaction processes. Our calculations demonstrated that CYP2A6-catalyzed 5′-hydroxylation of (S)-(−)-nicotine proceeds mainly with the stereoselective loss of the trans-5′-hydrogen.
Table 1.
Various molecular species for (S)-(−)-nicotine.
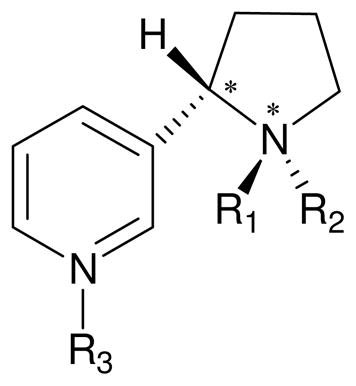 | ||||||
|---|---|---|---|---|---|---|
| Name | Config. | R1 | R2 | R3 | Charge (e) | pKa (ref.3) |
| SR | (S,R) | CH3 | lone pair | lone pair | 0 | |
| SRH | (S,R) | CH3 | H | lone pair | 1 | 8.0 |
| SRHH | (S,R) | CH3 | H | H | 2 | 3.0 |
| SS | (S,S) | lone pair | CH3 | lone pair | 0 | |
| SSH | (S,S) | H | CH3 | lone pair | 1 | |
| SSHH | (S,S) | H | CH3 | H | 2 | |
Computational Methods
First-principles electronic structure calculations
The geometries of all the (S)-(−)-nicotine species involved in this study were fully optimized by employing density functional theory (DFT) using Becke’s three-parameter hybrid exchange functional and the Lee-Yang-Parr correlation functional (B3LYP)40–42 with the 6-31+G* basis set. Then the optimized geometries were used to carry out second-order Møller-Plesset (MP2) single-point energy calculations with the 6-31+G* basis set. All these electronic structure calculations in the gas phase were performed by using the Gaussian 03 program.43
Self-consistent reaction field (SCRF) calculations were performed to calculate solvent shifts of the Gibbs free energies by using the geometries optimized at the B3LYP/6-31+G* level in the gas phase. The SCRF procedure used in the present study is our recently developed surface and volume polarization for electrostatic interactions (SVPE) model44–46 implemented in the Gaussian 03 program. The SVPE model is also known as the fully polarizable continuum model (FPCM),47–51 because it fully accounts for both surface and volume polarization effects in the SCRF calculation. Since the solute cavity surface is defined as a solute electron charge isodensity contour determined self-consistently during the SVPE iteration process, the SVPE results (converged to the exact solution of the Poisson equation with a given numerical tolerance) depend only on the value of the contour for a given dielectric constant under a given QM calculation level.44 This single parameter value has been calibrated to be 0.001 au on the basis of an extensive calibration study.45
Molecular docking and molecular dynamics simulations
In order to construct a reasonable binding structure for CYP2A6 binding with each molecular species of (S)-(−)-nicotine, molecular docking and MD simulations were performed. The A chain in the crystal structure of human microsomal CYP2A6 in complex with methoxsalen (PDB entry of 1Z11 at 2.05 Å resolution)52 was selected as the initial structure of CYP2A6. The first 29 residues at the N-terminal and the last 2 residues at the C-terminal were omitted as these residues did not show any regular secondary structure in the crystal. After all the ionizable residues of CYP2A6 were set to the standard protonated or deprotonated states, the hydrogen atoms were added by using the X-leap module of Amber 8 program.53
The molecular mechanical force field parameters for Cpd I were developed based on our first principles electronic structure calculations. The geometry of Cpd I was optimized by using the Gaussian 03 program43 with the B3LYP functional40–42 and the 6-31G* basis set. The atomic charges for Cpd I, and for various molecular species of (S)-(−)-nicotine, were the restrained electrostatic potential (RESP) charges determined by fitting with the standard RESP procedure implemented in the Antechamber module of the Amber 8 program. Bonded and nonbonded parameters for Cpd I were derived from the default libraries of Amber 8 program.53
In molecular mechanics (MM) minimizations and MD simulations, the AMBER ff03 force field54 was used for the protein and the general AMBER force field (gaff)55 was used for ligands. The whole CYP2A6 molecule including Cpd I and the water molecules in the original X-ray crystal structure was neutralized by adding chloride counterions and was solvated in a rectangular box of TIP3P56 water molecules, with a minimal distance of 10 Å from the protein to the box boundary. After the whole system was set up, a series of energy minimizations were carried out by using the Sander module of Amber 8 program.53 First, the atomic position of water molecules was minimized for 20,000 steps. Then, an additional 20,000 steps of minimization on the side chains of CYP2A6 along with water molecules was performed. Finally, the entire system was energy-minimized and a convergence criterion of 0.001 kcal/(mol·Å) was achieved. Based on the energy-minimized CYP2A6 structure, each of the considered species of (S)-(−)-nicotine (as listed in Table 1) was docked into the active site of CYP2A6 by using the AutoDock 4.0 program.57 During the docking process, the Lamarkian genetic algorithm (LGA) was applied to the conformational search for the protein-ligand binding structure. Among a series of docking parameters, the grid size was set to be 60 × 60 × 60 and the grid space was the default value of 0.375 Å. The poses with the lowest free energies of binding were selected as the best binding modes.
The binding complexes of CYP2A6 with various species of (S)-(−)-nicotine obtained from the molecular docking were used as the starting structures for MD simulations. The MD simulations were performed by using the Sander module of Amber 8 program.53 Each aforementioned starting structure was neutralized by adding chloride counterions and was solvated in a rectangular box of TIP3P56 water molecules, with a minimal distance of 10 Å from the protein to the box boundary. The solvated system was subject to energy minimization in a similar way as that used to minimize the CYP2A6 itself. Then, each of the solvated CYP2A6-ligand complexes was gradually heated from 10 K to 298.15 K with a constraint force constant on the docked ligand (all atoms of ligand). The constraint force constant was gradually decreased and finally removed for the production MD simulations at 298.15 K. The production MD at 298.15 K was kept running 1.0 ns or longer to obtain a stable MD trajectory. For example, with SRH, the constraint force constant is 100 kcal/mol for 400 ps, 80 kcal/mol for 200 ps, 50 kcal/mol for 200 ps, and 20 kcal/mol for 200 ps, before the fully relaxed MD simulation for 1.5 ns, all at 298.15 K. During the MD simulations, a 10 Å non-bonded interaction cutoff was used and the non-bonded list was updated every 500 steps. The SHAKE algorithm58 was employed to constrain all covalent bonds involving hydrogen atoms, enabling the use of a 2-fs time step to numerically integrate the equations of motion. The particle mesh Ewald (PME) method was applied to treat long-range electrostatic interactions. Periodic boundary condition was used in the NTP ensemble at T = 298.15 K with Berendsen temperature coupling and at P = 1 atm with anisotropic molecule-based scaling.
Binding free energy calculations
The binding free energy (ΔGbind) for CYP2A6 with each molecular species of (S)-(−)-nicotine was calculated by using the molecular mechanics-Poisson-Boltzmann surface area (MM-PBSA) method,59 following the production MD simulations at 298.15 K. During the MD simulations, the atomic charges used for the ligand were the RESP charges calculated at the HF/6-31G* level (the same QM level used to determine the RESP charges used in the standard AMBER force field for proteins) without any scaling. In the MM-PBSA method, the free energy of the ligand binding with CYP2A6, ΔGbind, is calculated from the difference between the free energy of the CYP2A6-ligand complex (Gcomplex) and the sum of the free energies of the unbound CYP2A6 (GCYP2A6) and ligand (Gligand) as Eq. 1:
| (1) |
ΔGbind was evaluated as a sum of the changes in the MM gas-phase binding energy (ΔEMM), solvation free energy (ΔGsolv), and entropy contribution (−TΔS).
| (2) |
| (3) |
| (4) |
| (5) |
The MM binding energies were calculated with the Sander module of Amber 8 program.53 Electrostatic solvation free energy (ΔGPB) was calculated by the finite-difference solution to the PB equation implemented in the Delphi program.60,61 The MSMS program62 was used to calculate the solvent-accessible surface area (SASA) for the estimation of the nonpolar solvation energy (ΔGnp) using Eq. 5 with parameters γ = 0.00542 kcal/Å2 and β = 0.92 kcal/mol.
The entropy contribution, −TΔS, to the binding free energy was calculated using a local program (a standalone program) developed in our own laboratory. The computational procedure used to evaluate the −TΔS was the same as that described in our recent publication.63 As we described previously, the entropy contribution is divided into two parts: solvation entropy (ΔSsolv) and conformational entropy (ΔSconf):
| (6) |
The solvation entropy is gained by solvent water molecules on being displaced from the active site by the ligand during binding,64 and was calculated by using the parameters established previously.63 The contribution to the binding free energy from the conformational entropy change is proportional to the number (ΔNrot) of the lost rotatable bonds during the binding:
| (7) |
in which w is the scaling factor to be calibrated. Thus we have:
| (8) |
Although all of the other parameters used in our MM-PBSA calculations are the standard ones reported in literature or the default ones of the Amber 8 program. The adjustable parameter, w, was calibrated by fitting the ΔGbind value calculated for the most favorable binding mode to the corresponding ΔGbind value derived from experimentally measured KM values. There were multiple KM values reported in different experimental studies,65–70 and the averaged value was 37.6 μM. This averaged value of KM was used in the process of calibrating the value of adjustable w in Eq. 8. The experimentally derived ΔGbind was −6.04 kcal/mol. According to the reported pKa value of 8.0 for (S)-(−)-nicotine,3 the molar ratio of SRH to SR in solution should be ~4:1 at pH 7.4. By taking all these into account, the parameter w was calibrated to be 0.6434 kcal/mol, and this value was used in our MM-PBSA calculations of ΔGbind for all of the MD-simulated binding structures of CYP2A6 with (S)-(−)-nicotine.
The final binding free energy for each CYP2A6-ligand complex was taken as the average of the ΔGbind values calculated for the equally-distributed 100 snapshots of the stable MD trajectory for each binding structure.
Minimum-energy path determination
For each reaction pathway (trans- and cis-5′-hydroxlylation), a snapshot which was close to the average structure simulated was extracted from the stable trajectory as the starting structure. Specifically, we tracked the key distances between the oxygen of Cpd I and the (trans and cis) 5′-hydrogen atoms of nicotine, i.e. distances D1 and D2 in Figure 2 below. Within a stable MD trajectory, we obtained an average D1 value and average D2 value. We chose the snapshot with D1 and D2 values being closest to the corresponding D1 and D2 values. Prior to QM/MM calculations, each selected starting structure was energy-minimized for 1,000 steps by using the Sander module of Amber 8 program. As the outer water molecules were far away from the molecular surface of the CYP2A6-(S)-(−)-nicotine complex structure, the role of these outer water molecules was only to solvate inner water molecules nearby the molecular surface of CYP2A6-(S)-(−)-nicotine complex. In order to save computing resource and decrease the size of our QM/MM calculations, the water molecules beyond 40 Å of the iron atom were ignored. The final QM/MM system contained 909 water molecules with a total number of 10,377 atoms for the trans-5′-system, and the cis-5′-system had 1,039 water molecules with totally 10,767 atoms. As illustrated in Figures 4A and 5A, the substrate, the porphyrin-iron-oxygen complex, and the side chain of Cys439 were defined as QM atoms. The QM/MM interface has been described by a pseudobond approach.35–38 The pseudobond first-principles QM/MM approach used in the present study has been demonstrated to be a powerful tool in simulating a variety of enzymes,71–78 and some theoretical predictions71,73 have been confirmed by experimental studies.79–81
Figure 2.

Plots of the C-C-N-C dihedral (associated with numbers in pink) versus the simulation time.
Figure 4.

(A) Division of the QM/MM system. Atoms in blue are treated by the QM method. The boundary carbon atom (colored in red) is treated with the improved pseudobond parameters. All other atoms belong to the MM subsystem. (B) to (F) Optimized geometries of key configurations of CYP2A6-catalyzed trans-5′-hydroxylation of (S)-(−)-nicotine. The geometries were optimized at the QM/MM (B3LYP/B1:AMBER) level. Nicotine and Cpd I are shown in ball-and-stick style, the rest of CYP2A6 is displayed in cartoon style. Values outside the parentheses are for the quartet state, while the values in parentheses are for the doublet state. RC: reactant complex; TS: transition state; IM: intermediate; PC: product complex. Distances are in angstrom (Å) and angles are in degrees.
Figure 5.

(A) Division of the QM/MM system. Atoms in blue are treated by the QM method. The boundary carbon atom (colored in red) is treated with the improved pseudobond parameters. All other atoms belong to the MM subsystem. (B) to (F) Optimized geometries of key configurations for CYP2A6-catalyzed cis-5′-hydroxylation of (S)-(−)-nicotine. The geometries were optimized at the QM/MM (B3LYP/B1:AMBER) level. Nicotine and Cpd I are shown in ball-and-stick style, the rest of CYP2A6 is displayed in cartoon style. Values outside the parentheses are for the quartet state, while the values in parentheses for the doublet state. RC: reactant complex; TS: transition state; IM: intermediate; PC: product complex. Distances are in angstrom (Å) and angles are in degrees.
With a reaction coordinate driving method and an interactive energy minimization procedure,36 the enzymatic reaction path was determined by the pseudobond QM/MM calculations, in which the QM calculations were performed with the B3LYP functional40–42 by using a modified version of the Gaussian 03 program43 and the MM calculations were performed by using a modified version of the AMBER 8 program.53 Normal mode analyses were performed to characterize the reactant, transition state, intermediate, and final product. During the geometry optimization, the basis set for the QM part was a mixed basis set, involving 6-31G* for iron and the six atoms coordinated to iron, and 6-31G basis set for all other atoms, which is denoted by B1 in brief. In addition, the optimized geometries were used to carry out the QM/MM single-point energy calculations with a larger basis set, denoted by B2, which describes iron by the Wachters+f basis set82,83 and the rest of the atoms by the 6-31+G* basis set. No cutoff for nonbonded interactions was used in the QM/MM calculations. For the QM subsystem, the SCF convergence criterion for geometry optimizations was set to 10−6. For the MM subsystem, the geometry optimization convergence criterion was that the rmsd of energy gradient is less than 0.1 kcal/(mol·Å). Then atoms within 20 Å of the iron atom were allowed to move while all the other atoms outside this range were frozen in all of the QM/MM calculations, resulting in 2,893 movable atoms in the QM/MM calculations for the trans-5′-system and 3,007 movable atoms in the QM/MM calculations for the cis-5′-system.
Free energy perturbation
After the minimum-energy path was determined by the QM/MM calculations, the free energy changes associated with the QM-MM interactions were determined by using the FEP method.36 In FEP calculations, sampling of the MM subsystem was carried out with the QM subsystem frozen at different geometries along the reaction path. The point charges on the frozen QM atoms used in the FEP calculation are those determined by fitting the electrostatic potential (ESP) in the QM part of the QM/MM calculation. The total free energy difference between the transition state and the reactant was obtained from the following formula:
| (9) |
where ΔFqm/mm(RC→TS) is the free energy change associated with the QM-MM interaction, ΔEqm(RC→TS) refers to the QM energy difference between the two QM subsystems, and is the change in contribution from the QM subsystem fluctuation to the free energy difference.84 The FEP calculations enabled us to more reasonably determine relative free energy changes due to the QM-MM interaction. Technically, the final (relative) free energy determined by the QM/MM-FE calculations is the QM part of the QM/MM energy (excluding the Coulombic interaction energy between the point charges of the MM atoms and the ESP charges of the QM atoms) plus the relative free energy change determined by the FEP calculations. In FEP calculations, the time step used was 2 fs, and bond lengths involving hydrogen atoms were constrained. In sampling of the MM subsystem by MD simulations, the temperature was maintained at 298.15 K. Each FEP calculation consisted of 50 ps of equilibration and 300 ps of production sampling.
The first-principles electronic structure calculations, MD simulations, and QM/MM-FE calculations were performed on a supercomputer (e.g. IBM X-series Cluster with 340 nodes or 1,360 processors) at the Center for Computational Sciences, University of Kentucky. The other modeling and computations were carried out on SGI Fuel workstations in our own laboratory.
Results and Discussion
Binding mode of CYP2A6 with (S)-(−)-nicotine
As observed in the CYP2A6 structure, the active site of CYP2A6 is highly hydrophobic, and Asn297 is the only polar residue involved in ligand binding. The aromatic cage at the binding site is composed of several aromatic residues including Phe107, Phe111, Phe118, Phe209, and Phe480. Depicted in Figure 1 are the representative structures of CYP2A6-ligand complexes obtained from our MD simulations. As shown in Figure 1, each of the (S)-(−)-nicotine species fits well in the narrow active-site cavity of CYP2A6. The pyridine ring of (S)-(−)-nicotine packs closely with the aromatic side chains of residues at the aromatic cage, while the azaheterocyclic ring sits just above Cpd I in the active site of CYP2A6. As seen in Table 1, SR and SS of (S)-(−)-nicotine are two different molecular species of the free base (neutral species). But these two neutral species can exchange very rapidly in solution because the free energy barriers for the exchange are very low.39 We examined CYP2A6 binding with both the SR and SS species and found that CYP2A6 can bind only with SR. During the MD simulation on the complex of CYP2A6 binding with the SS species, the SS structure of (S)-(−)-nicotine gradually changed to the SR structure. The C-C-N-C dihedral (associated with numbers in pink in Figure 2) is about 140° in the SS species and about −130° in the SR species. As seen in Figure 2, the SS species changed to SR in the heating process of MD simulation. Thus, the SS species of (S)-(−)-nicotine actually became the SR species to bind with CYP2A6 in the same way as SR.
Figure 1.

Representative CYP2A6-(S)-(−)-nicotine binding structures derived from the trajectory of MD simulations. Cpd I of CYP2A6 is shown in stick style, and residues within 5 Å around the ligand are shown in lines. (S)-(−)-nicotine is displayed in ball-and-stick style. Possible hydrogen bonds between CYP2A6 and (S)-(−)-nicotine are represented by dashed lines with the distances (Å) labeled. A: CYP2A6-SRt; B: CYP2A6-SRc; C: CYP2A6-SRH; D: CYP2A6-SSH; E: CYP2A6-SRHH; and F: CYP2A6-SSHH.
Further, we noted that the SR species can take two slightly different conformations (i.e. SRt and SRc) in its binding with CYP2A6. In both SRt and SRc, the planes of the pyridine and azaheterocyclic rings are nearly vertical to each other; SRt becomes SRc when the azaheterocyclic ring rotates for about 180° as one can see in Figure 1A and 1B. In the SRt conformation, the nitrogen atom on the pyridine ring of (S)-(−)-nicotine forms a stable hydrogen bond with the amine group of Asn297 side chain during the MD simulation, and the average N···H distance is ~2.3 Å as shown in Figure 1A. The pyridine ring of SRt is surrounded by residues Phe107, Phe111, and Phe118 through face-to-edge aromatic interactions.
In the mode of CYP2A6 binding with SRc conformation of (S)-(−)-nicotine (see Figure 1B), placement of the cis-5′-hydrogen in the immediate vicinity of the oxygen of Cpd I allows for reaction at the experimentally determined cis-5′-site.10,33 As noted in Figure 1B, SRc interacts with CYP2A6 in a way similar to that of the CYP2A6-SRt binding, including the aforementioned hydrogen bonding interactions and the face-to-edge aromatic interactions between the pyridine ring of (S)-(−)-nicotine and the surrounding residues.
Concerning CYP2A6 binding with other (protonated) molecular species, the overall orientation of the ligand in the binding structures is similar to that of SRt and SRc in the CYP2A6-SRt and CYP2A6-SRc binding structures. The primary difference exists in the hydrogen bonding with the enzyme, as depicted in Figure 1. For example, as shown in Figure 1C, the N···H-N hydrogen bond (with a N···H distance of ~2.5 Å) between the nitrogen atom on the pyridine ring of SRH and the amine group of Asn297 side chain is not as strong as that in the CYP2A6-SRt or CYP2A6-SRc binding structure (with a N···H distance of ~2.3 Å). In the CYP2A6-SSH binding structure (Figure 1D), the above-mentioned N···H-N hydrogen bond with Asn297 does not exist. Instead, the proton on the azaheterocyclic nitrogen of SSH interacts with the oxygen of Cpd I through an N-H···O hydrogen bond with a H···O distance of ~2.1 Å. In the CYP2A6-SRHH binding structure, an N-H···O hydrogen bond exists between the protonated nitrogen on the pyridine ring of SRHH and the backbone oxygen of Asn297 (with a H···O distance of ~2.2 Å as shown in Figure 1E). Meanwhile, there is an N-H···O hydrogen bond (with a H···O distance of ~1.9 Å, see Figure 1E) between the protonated nitrogen on its azaheterocyclic ring and the oxygen of Cpd I. The CYP2A6-SSHH binding structure involves two intermolecular hydrogen bonds. One is the N-H···O hydrogen bond between the protonated nitrogen on the pyridine ring of SSHH and the carbonyl oxygen of Asn297 side chain (Figure 1F), with a H···O distance of ~2.2 Å. The other is N-H···O hydrogen bond between the protonated azaheterocyclic nitrogen of SSHH and the oxygen of Cpd I, with a H···O distance of ~1.8 Å.
Among the computationally determined six binding structures depicted in Figure 1, the CYP2A6-SRt, CYP2A6-SRc, and CYP2A6-SRH binding structures may be considered suitable for the experimentally observed enzymatic 5′-hydroxylation reaction of (S)-(−)-nicotine. This is because in these binding structures, one of the 5′-hydrogen atoms on the azaheterocyclic ring of SRt, SRc, or SRH is orientated pointing to the oxygen of Cpd I. Specifically, in the CYP2A6-SRt and CYP2A6-SRH binding structures (see Figure 1A and 1C), the trans-5′-hydrogen on the azaheterocyclic ring of (S)-(−)-nicotine is positioned to allow for the hydroxylation reaction. In the CYP2A6-SRc binding structure (Figure 1B), the cis-5′-hydrogen on the azaheterocyclic ring of (S)-(−)-nicotine is positioned to allow for the hydroxylation reaction. The determined CYP2A6-SRt, CYP2A6-SRc, and CYP2A6-SRH binding structures are qualitatively consistent with reported experimental data indicating that hydroxylation occurs at both the trans-5′-hydrogen and cis-5′-hydrogen on the azaheterocyclic ring of (S)-(−)-nicotine although the stereoselectivity for the trans-5′-hydrogen was commonly observed.10,33 In the CYP2A6-SSH, CYP2A6-SRHH, and CYP2A6-SSHH binding structures, the protonated nitrogen on the azaheterocyclic ring of (S)-(−)-nicotine forms a favorable hydrogen bond with the oxygen of Cpd I. Thus, the 5′-hydrogen atoms on the azaheterocyclic ring of SSH, SRHH, or SSHH cannot reach the oxygen of Cpd I such that the 5′-hydroxylation reaction cannot occur with these binding structures. Hence, the CYP2A6-SSH, CYP2A6-SRHH, and CYP2A6-SSHH binding structures may be excluded from the 5′-hydroxylation reaction pathway.
Summarized in Table 2 are the binding free energies calculated by using the MM-PBSA approach for CYP2A6 binding with (S)-(−)-nicotine in all of the binding structures. As shown in Table 2, the van der Waals interactions contributed mostly to the calculated binding free energies for CYP2A6 binding with SRt, SRc, SSH, and SRH of (S)-(−)-nicotine. This may suggest that the major driving force for the binding of the enzyme with these molecular species of (S)-(−)-nicotine is the van der Waals interactions, and this is in line with the fact that the active site of CYP2A6 is mainly composed of hydrophobic residues. Although the electrostatic interactions between the SRHH or SSHH species of (S)-(−)-nicotine and the CYP2A6 contributed largely to the enzyme-ligand binding, such favorable contribution was counteracted by the unfavorable desolvation. As listed in Table 2, the binding free energies for both the CYP2A6-SRt complex and CYP2A6-SRc binding structures are low enough and comparable with these ΔGbind values for the CYP2A6-SRHH and CYP2A6-SSHH binding structures.
Table 2.
Calculated binding free energies (ΔGbind, in kcal/mol) for CYP2A6 binding with (S)-(−)-nicotine in all of the binding structures, and in comparison with the ΔGbind value derived from experimental data.
| Ligand | ΔEele | ΔEvdw | ΔEMM | ΔGsolv | −TΔS | ΔGbind | Expt. ΔGbinda | Distributionb |
|---|---|---|---|---|---|---|---|---|
| SRt | −7.89 | −28.39 | −36.28 | 21.4 | 8.02 | −6.86 | −6.86 | 0.95 |
| SRc | −5.96 | −30.77 | −36.73 | 20.93 | 10.37 | −5.42 | 4.41×10−2 | |
| SRH | 8.33 | −29.97 | −21.64 | 10.38 | 9.21 | −2.04 | 1.11×10−3 | |
| SSH | −1.73 | −29.41 | −31.14 | 16.95 | 9.47 | −4.73 | 2.69×10−4 | |
| SRHH | −60.76 | −27.48 | −88.24 | 72.6 | 9.53 | −6.11 | 4.25×10−5 | |
| SSHH | −62.14 | −27.64 | −89.78 | 72.8 | 10.96 | −6.02 | 9.43×10−8 |
Distribution of binding structures in solution
In solution, the different diastereomeric species SR and SS can exchange to each other through the pyramidal inversion of the azaheterocyclic nitrogen. The conformations SRt and SRc can also exchange to each other when the azaheterocyclic ring rotates for about 180°. The distribution of different diastereomeric species or conformations of (S)-(−)-nicotine (L1 and L2) in solution can be evaluated using the Boltzmann distribution as
| (10) |
in which ΔG(L2→L1) is the free energy change from L2 to L1. The species SR, SRH, and SRHH can reach a thermodynamic equilibrium through the protonation/deprotonation processes. In the same manner, the thermodynamic equilibrium can also exist between SS, SSH, and SSHH. The distribution of different protonation states of (S)-(−)-nicotine (L) in solution can be evaluated as
| (11) |
in which LH is the protonated form of L.
As multiple molecular species or conformations of (S)-(−)-nicotine can exist in solution, they can also exist at the CYP2A6 binding site. The thermodynamic distribution of each CYP2A6-(S)-(−)-nicotine binding structure is determined by both the distribution of the molecular species (or conformations) of the free ligand in solution and the relative binding free energies associated with the binding structures. Considering a receptor-ligand binding structure (RL) formed from the CYP2A6 enzyme (R) and a specific molecular species or conformation (L) of (S)-(−)-nicotine, we have
| (12) |
| (13) |
in which ΔGbind is the binding free energy between R and L, and Kd is the dissociation constant of the binding complex RL. The calculated distribution of the CYP2A6-(S)-(−)-nicotine binding structures in solution are listed in the last column of Table 2.
According to the experimental pKa of (S)-(−)-nicotine (i.e. 8.0),3 the primary molecular species in solution under physiologic condition (pH 7.4) should be SRH. However, combining the distribution of the free ligands in solution and the binding free energies of the binding structures, the dominant binding structure should be the CYP2A6-SRt complex (~95.4%), while the CYP2A6-SRc binding structure should also have a significant distribution (~4.4%), as seen in Table 2. Combining the structural features (Figure 1) and the results of binding free energy calculations (Table 2), both the CYP2A6-SRt and CYP2A6-SRc binding structures were selected for the QM/MM reaction coordinate calculations (see below for the results).
It is interesting to note that the CYP2A6-SRt and CYP2A6-SRc binding structures are similar to that of coumarin, methoxsalen, and some other inhibitors complexed with CYP2A6 determined by Yano et al. using crystallography,52,85 involving a hydrogen bond with the side chain of Asn297 and through face-to-edge aromatic interactions with the phenylanalines around the pyridine ring of (S)-(−)-nicotine. The oxygen of Cpd I acts to hold the azaheterocyclic ring with either the trans-5′- (Figure 1A) or the cis-5′-hydrogen (Figure 1B) close to the porphyrin-iron- oxygen species. The distances from the oxygen of Cpd I for each hydrogen atom (trans-5′-hydrogen or cis-5′-hydrogen) are shown in Figure 3. In the CYP2A6-SRt complex, the trans-5′- hydrogen is closer to the oxygen of Cpd I than the cis-5′-hydrogen. The average distance between the trans-5′-hydrogen to the oxygen is ~3.1 Å, while the average distance for the cis-5′- hydrogen is ~4.8 Å. Likewise, in the CYP2A6-SRc complex, the cis-5′-hydrogen is pointed toward the oxygen of Cpd I. The average distances from the cis-5′-hydrogen and trans-5′- hydrogen to the oxygen of Cpd I is ~2.5 and ~3.8 Å, respectively. From each of these complexes, the oxygen of Cpd I can abstract a hydrogen atom from the 5′-position of (S)-(−)-nicotine.
Figure 3.

Plots of the key internuclear distances versus simulation time for (a) SRt and (b) SRc binding with CYP2A6. D1 refers to the distance between the trans-5′-hydrogen and the oxygen of Cpd I, D2 refers to the distance between the cis-5′-hydrogen and the oxygen of Cpd I.
Fundamental reaction pathways
Molecular docking and MD simulations led to dynamically stable CYP2A6-SRt and CYP2A6-SRc binding complexes. These stable complexes were chosen as the initial structures to perform QM/MM reaction coordinate calculations. Since the active species of CYP2A6, Cpd I, involves two spin states (i.e. the high-spin quartet state and the low-spin doublet state),86 we carried out the QM/MM calculations on both the quartet and doublet states to explore the possible reaction pathways associated with the trans-5′-hydroxylation and cis-5′-hydroxylation. Throughout this report, the superscript 4/2 represents the quartet/doublet spin state of the reaction system.
The 5′-hydroxylation reaction involves the breaking of the C-H bond, the formation of the H-O bond, the formation of the C-O bond, and the breaking of the O-Fe bond. The changes of the distance (RC-H) between the C and H atoms, the distance (RH-O) between the H and O atoms, the distance (RC-O) between the C and O atoms, and the distance (RO-Fe) between the O and Fe atoms reflect the nature of the chemical reaction process. Thus, the distances RC-H, RH-O, RC-O, and RO-Fe were chosen to establish the reaction coordinate as RC-H − RH-O − RC-O + RO-Fe for CYP2A6-catalyzed 5′-hydroxylation of (S)-(−)-nicotine. Our QM/MM reaction-coordinate calculations at the B3LYP/B1:AMBER level reveals that the CYP2A6-catalyzed 5′-hydroxylation of (S)-(−)-nicotine consists of two steps. The first step is the hydrogen transfer from the 5′-position of (S)-(−)-nicotine to the oxygen of Cpd I (the H-transfer step). The second step is the recombination of the (S)-(−)-nicotine moiety with the iron-bound hydroxyl group to generate the 5′-hydroxynicotine product (the O-rebound step). The optimized geometries of the reactant complex (RC), transition state (TS), intermediate (IM), and final product complex (PC) are depicted in Figures 4 and 5.
Free energy barriers
On the basis of the QM/MM-optimized geometries at the B3LYP/B1:AMBER level, we carried out QM/MM single-point energy calculation at the B3LYP/B2:AMBER level on each geometry point along the minimum-energy path of CYP2A6-catalyzed 5′-hydroxlylation of (S)-(−)-nicotine. For each geometry, the ESP charges determined in the QM part of the QM/MM single-point energy calculation were used in subsequent FEP simulations for estimating the free energy changes along the reaction path. Depicted in Figure 6 are the free energy profiles determined by the QM/MM-FE calculations.
Figure 6.

Free energy profile determined by the QM/MM-FE calculations at the B3LYP/B2:AMBER level. Values are relative free energies including zero-point and thermal corrections for the QM subsystem.
Figure 6 shows the free energy profiles obtained from the QM/MM-FE calculations on the trans-5′-hydroxylation and cis-5′-hydroxylation. For the trans-5′-hydroxylation, the reaction proceeds in two steps on both the quartet (Figure 6A) and doublet states (Figure 6B), i.e. the H-transfer and the O-rebound steps. The free energy barrier with thermal corrections for the QM subsystem is 16.9/14.1 kcal/mol on the quartet/doublet state for the H-transfer step, and the free energy barrier for the O-rebound step is 0.1/1.1 kcal/mol. With this low rebound barrier, the intermediate can easily go through the transition state and down to the product complex. For cis-5′-hydroxylation (Figure 6C and 6D), the reaction process is also a stepwise one with an intermediate, and there is a low barrier of 0.3/0.9 kcal/mol for the rebound step. The free energy barrier with thermal corrections for the QM subsystem is 16.5/14.4 kcal/mol for the H-transfer step.
As seen in Figure 6, for each reaction pathway and each spin state, the free energy barrier, that is, the energy change from RC to TSH, calculated for the H-transfer step is much higher than that of the O-rebound step. Thus, the hydrogen-transfer step is rate-determining during the 5′-hydroxylation reaction. The difficulties of reaching conclusions about rate-determining steps in enzymatic reactions have been addressed by Northrop.87–89 The complication of P450 catalysis86,90,91 makes it more difficult to determine which step is rate-determining in the whole catalytic cycle, more than one steps may have a strong influence on the catalytic constant kcat.91 Nevertheless, the free energy barriers calculated here can be used to predict the relative reaction rates of the two reaction pathways, i.e. the stereoselectivity of nicotine 5′-hydroxylation.
Distribution of products associated with the stereoselectivity
Experimental investigations revealed that CYP2A6-catalyzed 5′-hydroxylation of nicotine proceeds with a highly stereoselective loss of the trans-5′-hydrogen at the azaheterocyclic ring (89%~94%).10,33 The small percentage of product obtained by the hydroxylation of the cis-5′-hydrogen can be explained through the existence of a second, less favorable, binding mode in which the conformation of (S)-(−)-nicotine in its interaction with the enzyme would be oriented such that the cis-5′-hydrogen is positioned for reaction. The lack of an observable isotope effect33 could be attributed to the slow exchange between the two binding modes (CYP2A6-SRt and CYP2A6-SRc) relative to the forward rate of the catalytic reaction. Generally speaking, the experimentally measured stereoselectivity10,33 of 89%~94% is the phenomenological stereoselectivity which reflects the contributions from both the relative distribution of CYP2A6-SRt and CYP2A6-SRc binding structures in solution and the relative free energy barriers for the trans-5′-hydroxylation and cis-5′-hydroxylation reactions.
The lowest free energy barriers calculated for tran-5′-hydroxylation and cis-5′-hydroxylation are 14.1 and 14.4 kcal/mol, respectively. Thus, the QM/MM-FE free energy barriers alone indicate that loss of the trans-5′-hydrogen is competitive with loss of the cis-5′-hydrogen. However, the stereoselectivity of CYP2A6-catalyzed (S)-(−)-nicotine 5′-hydroxylation, i.e. the product distribution of trans-5′-hydroxynicotine and cis-5′-hydroxynicotine, is determined not only by the relative free energy barriers of the two reaction pathways, but also by the distribution of the CYP2A6-SRt and SYP2A6-SRc binding structures in solution. The overall product distribution can be evaluated as
| (14) |
in which ΔGbind is the binding free energy (see Table 1 for the specific values), ΔG(SRc→SRt) (= ~1.4 kcal/mol) is the free energy change from SRc to SRt. ΔGtrans (= 14.1 kcal/mol) and ΔGcis (= 14.4 kcal/mol) are the free energy barriers calculated for tran-5′-hydroxylation and cis-5′- hydroxylation, respectively. Thus, the calculations in the present study predict a product distribution of ~36:1 for trans-5′-hydroxynicotine and cis-5′-hydroxynicotine. The finally calculated overall stereoselectivity for trans-5′-hydroxylation over cis-5′-hydroxylation of (S)-(−)-nicotine catalyzed by CYP2A6 is ~97%, which is close to the experimental observed stereoselectivity of 89~94%.10,33
As mentioned above, the QM/MM-FE free energy barriers alone predict a competition of trans-5′-hydroxylation and cis-5′-hydroxylation since the difference between the two lowest barriers is only 0.3 kcal/mol. Insight into the origin of the preference of trans-5′-hydroxynicotine in the product distribution can be gained from the distribution of CYP2A6-SRt and CYP2A6-SRc binding structures in solution. The slight preference of SRt over SRc free ligand in solution and the higher binding affinity of SRt with CYP2A6 compared to SRc binding with the enzyme lead to the predominance of the CYP2A6-SRt binding structure in solution and, therefore, the stereoselectivity of the (S)-(−)-nicotine 5′-hydroxylation reaction.
Conclusion
Molecular docking, molecular dynamics simulations, and binding free energy calculations, in combination with first-principles electronic structure calculations accounting for solvent effects, carried out in this study have demonstrated how (S)-(−)-nicotine species bind at the active site of CYP2A6. It has been demonstrated that the dominant molecular species of (S)-(−)-nicotine in the active site of the enzyme exists in the neutral state (the free base), despite of the fact that the dominant molecular species of the free (S)-(−)-nicotine in aqueous solution is the protonated state (cation). In other words, the present study demonstrates for the first time that a CYP substrate exists dominantly in one protonation state (cationic species) in solution, but uses its less-favorable protonation state (neutral free base) to perform the enzymatic reaction. This novel mechanistic insight suggests that future studies on the possible mechanisms for a CYP-catalyzed (or other enzymatic) reaction should account for not only the most favorable protonation state, but also the less-favorable protonation state, of substrates in solution.
For the determined enzyme-(S)-(−)-nicotine binding structures, the only polar residue, i.e. Asn297, in the active site provides a hydrogen bond donor to the pyridine nitrogen of (S)-(−)-nicotine. The phenylanalines around the pyridine ring contact with (S)-(−)-nicotine through face-to-edge aromatic interactions. The oxygen of Cpd I acts to hold the azaheterocyclic ring with either the trans-5′- or the cis-5′-hydrogen close to the porphyrin-iron-oxygen species.
The computational results reveal that the dominant pathway of nicotine metabolism in CYP2A6 is the CYP2A6-catalyzed oxidation of the nicotine free base (neutral state), rather than the protonated nicotine. The detailed reaction pathway of CYP2A6-catalyzed 5′-hydroxylation of (S)-(−)-nicotine has been studied further by using the first-principles QM/MM-FE approach. (S)-(−)-nicotine 5′-hydroxylation catalyzed by CYP2A6 involves two steps. The first step is the hydrogen transfer from the 5′-position of (S)-(−)-nicotine to the oxygen of Cpd I (the H-transfer step). The second step is the recombination of the (S)-(−)-nicotine moiety with the iron-bound hydroxyl group to generate the 5′-hydroxynicotine product (the O-rebound step). Reaction step one where the oxygen of Cpd I abstracts a hydrogen from the 5′-position of (S)-(−)-nicotine is found to be rate-determining during the 5′-hydroxylation reaction.
It has been demonstrated that CYP2A6-catalyzed (S)-(−)-nicotine 5′-hydroxylation proceeds mainly with the stereoselective loss of the trans-5′-hydrogen, that is, the 5′-hydrogen atom trans to the pyridine ring. The calculated overall stereoselectivity of ~97% favoring the trans-5′-hydroxynicotine is close to the experimentally observed stereoselectivity of 89~94%.
Concerning the origin of the preference of trans-5′-hydroxynicotine in the product distribution, computational results and analysis reveal that the slight preference of SRt over SRc in solution and the higher binding affinity of SRt with CYP2A6 compared to SRc binding with the enzyme lead to the predominance of the CYP2A6-SRt binding structure and, thus, the stereoselectivity of the (S)-(−)-nicotine 5′-hydroxylation reaction.
The computationally determined binding modes of (S)-(−)-nicotine at the active site of CYP2A6, the detailed mechanism of CYP2A6-catalyzed 5′-hydroxylation, the structures of hydrogen-transfer transition states, and the insights into the stereoselectivity of the 5′-hydroxylation reaction described in the present study provide a solid base for rational design of selective CYP2A6 inhibitors (e.g. substrate analogs with a similar binding mode of (S)-(−)-nicotine with CYP2A6, or stable analogs of the transition-state structures or intermediates) and should facilitate the future design of potent and selective compounds in smoking cessation studies and reduce tobacco dependence.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (grants R01DA013930 and R01DA025100 to C.-G. Zhan), NKBRSF (grant 2007CB815202 to K. Han), and NSFC (grants 20721004 and 20833008 to K Han). D. Li worked in C.-G. Zhan’s laboratory at the University of Kentucky as an exchange graduate student from Dalian Institute of Chemical Physics, Chinese Academy of Sciences and she is in the Ph.D. grogram of the Graduate School of the Chinese Academy of Sciences. The authors also acknowledge the Center for Computational Sciences at University of Kentucky for supercomputing time.
Footnotes
Supporting Information Available: More information about the energetic results and the detailed reaction pathways; complete citations of refs. 43 and 53. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO Report on the Global Tobacco Epidemic, 2009. World Health Organization (WHO); 2009. [Google Scholar]
- 2.NIDA Research Report Series. National Institute on Drug Abuse; 2009. [Google Scholar]
- 3.Gorrod JW, Jacob P III, editors. Analytical Determination of Nicotine and Related Compounds and their Metabolites. Elsevier Science; Amsterdam: 1999. [Google Scholar]
- 4.Tricker AR. Toxicology. 2003;183:151. doi: 10.1016/s0300-483x(02)00513-9. [DOI] [PubMed] [Google Scholar]
- 5.Tutka P, Mosiewicz J, Wielosz M. Pharmacol Rep. 2005;57:143. [PubMed] [Google Scholar]
- 6.Benowitz NL. Annu Rev Pharmacol Toxicol. 2009;49:57. doi: 10.1146/annurev.pharmtox.48.113006.094742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell MAH, Feyerabend C. Drug Metab Rev. 1978;8:29. doi: 10.3109/03602537808993776. [DOI] [PubMed] [Google Scholar]
- 8.Henningfield JE, Goldberg SR. Pharmacol Biochem Behav. 1988;30:221. doi: 10.1016/0091-3057(88)90449-2. [DOI] [PubMed] [Google Scholar]
- 9.Murphy PJ. J Biol Chem. 1973;248:2796. [PubMed] [Google Scholar]
- 10.Peterson LA, Trevor A, Castagnoli N. J Med Chem. 1987;30:249. doi: 10.1021/jm00385a004. [DOI] [PubMed] [Google Scholar]
- 11.Brandänge S, Lindblom L. Biochem Biophys Res Commun. 1979;91:991. doi: 10.1016/0006-291x(79)91977-6. [DOI] [PubMed] [Google Scholar]
- 12.Gorrod JW, Hibberd AR. Eur J Drug Metab Pharmacokinet. 1982;7:293. doi: 10.1007/BF03189632. [DOI] [PubMed] [Google Scholar]
- 13.Obach RS. Drug Metab Dispos. 2004;32:89. doi: 10.1124/dmd.32.1.89. [DOI] [PubMed] [Google Scholar]
- 14.Tyndale RF, Sellers EM. Drug Metab Dispos. 2001;29:548. [PubMed] [Google Scholar]
- 15.Pianezza ML, Sellers EM, Tyndale RF. Nature. 1998;393:750. doi: 10.1038/31623. [DOI] [PubMed] [Google Scholar]
- 16.Malaiyandi V, Sellers EM, Tyndale RF. Clin Pharmacol Ther. 2005;77:145. doi: 10.1016/j.clpt.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 17.Sellers EM, Kaplan HL, Tyndale RF. Clin Pharmacol Ther. 2000;68:35. doi: 10.1067/mcp.2000.107651. [DOI] [PubMed] [Google Scholar]
- 18.Sellers EM, Ramamoorthy Y, Zeman MV, Djordjevic MV, Tyndale RF. Nicotine Tobacco Res. 2003;5:891. doi: 10.1080/14622200310001615231. [DOI] [PubMed] [Google Scholar]
- 19.Zhang W, Kilicarslan T, Tyndale RF, Sellers EM. Drug Metab Dispos. 2001;29:897. [PubMed] [Google Scholar]
- 20.Koenigs LL, Trager WF. Biochemistry (Mosc) 1998;37:13184. doi: 10.1021/bi981198r. [DOI] [PubMed] [Google Scholar]
- 21.Tantcheva-Poor I, Servera-Llaneras M, Scharffetter-Kochanek K, Fuhr U. Br J Dermatol. 2001;144:1127. doi: 10.1046/j.1365-2133.2001.04233.x. [DOI] [PubMed] [Google Scholar]
- 22.Nides M. Am J Med. 2008;121:S20. doi: 10.1016/j.amjmed.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Reus VI, Smith BJ. Int J Clin Pract. 2008;62:1753. doi: 10.1111/j.1742-1241.2008.01885.x. [DOI] [PubMed] [Google Scholar]
- 24.Buchhalter AR, Fant RV, Henningfield JE. Drugs. 2008;68:1067. doi: 10.2165/00003495-200868080-00005. [DOI] [PubMed] [Google Scholar]
- 25.Hucker HB, Gillette JR, Brodie BB. J Pharmacol Exp Ther. 1960;129:94. [PubMed] [Google Scholar]
- 26.Nguyen T-L, Gruenke LD, Castagnoli N. J Med Chem. 1976;19:1168. doi: 10.1021/jm00231a018. [DOI] [PubMed] [Google Scholar]
- 27.Peterson LA, Castagnoli N. J Med Chem. 1988;31:637. doi: 10.1021/jm00398a024. [DOI] [PubMed] [Google Scholar]
- 28.Kyerematen GA, Vesell ES. Drug Metab Rev. 1991;23:3. doi: 10.3109/03602539109029754. [DOI] [PubMed] [Google Scholar]
- 29.Neurath GB. Clin Investig. 1994;72:190. doi: 10.1007/BF00189309. [DOI] [PubMed] [Google Scholar]
- 30.Hukkanen J, Jacob P, Benowitz NL. Pharmacol Rev. 2005;57:79. doi: 10.1124/pr.57.1.3. [DOI] [PubMed] [Google Scholar]
- 31.Hecht SS, Hochalter JB, Villalta PW, Murphy SE. Proc Natl Acad Sci U S A. 2000;97:12493. doi: 10.1073/pnas.220207697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen T-L, Gruenke LD, Castagnoli N. J Med Chem. 1979;22:259. doi: 10.1021/jm00189a008. [DOI] [PubMed] [Google Scholar]
- 33.Carlson TJ, Jones JP, Peterson L, Castagnoli N, Iyer KR, Trager WF. Drug Metab Dispos. 1995;23:749. [PubMed] [Google Scholar]
- 34.Li D, Wang Y, Han K, Zhan C-G. J Phys Chem B. 2010;114:9023. doi: 10.1021/jp102225e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Lee T-S, Yang W. J Chem Phys. 1999;110:46. [Google Scholar]
- 36.Zhang Y, Liu H, Yang W. J Chem Phys. 2000;112:3483. [Google Scholar]
- 37.Zhang Y. J Chem Phys. 2005;122:024114. doi: 10.1063/1.1834899. [DOI] [PubMed] [Google Scholar]
- 38.Zhang Y. Theor Chem Acc. 2006;116:43. [Google Scholar]
- 39.Huang XQ, Zheng F, Crooks PA, Dwoskin LP, Zhan CG. J Am Chem Soc. 2005;127:14401. doi: 10.1021/ja052681+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 41.Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]
- 42.Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623. [Google Scholar]
- 43.Frisch MJ, et al. revision C02 ed. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 44.Zhan CG, Bentley J, Chipman DM. J Chem Phys. 1998;108:177. [Google Scholar]
- 45.Zhan C-G, Chipman DM. J Chem Phys. 1998;109:10543. [Google Scholar]
- 46.Zhan CG, Chipman DM. J Chem Phys. 1999;110:1611. [Google Scholar]
- 47.Zhan CG, de Souza ON, Rittenhouse R, Ornstein RL. J Am Chem Soc. 1999;121:7279. [Google Scholar]
- 48.Zhan C-G, Zheng F. J Am Chem Soc. 2001;123:2835. doi: 10.1021/ja005529a. [DOI] [PubMed] [Google Scholar]
- 49.Zhan C-G, Dixon DA, Sabri MI, Kim M-S, Spencer PS. J Am Chem Soc. 2002;124:2744. doi: 10.1021/ja0113394. [DOI] [PubMed] [Google Scholar]
- 50.Dixon DA, Feller D, Zhan C-G, Francisco JS. Int J Mass spectrom. 2003;227:421. [Google Scholar]
- 51.Lu H, Chen X, Zhan CG. J Phys Chem B. 2007;111:10599. doi: 10.1021/jp072917r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yano JK, Hsu MH, Griffin KJ, Stout CD, Johnson EF. Nat Struct Mol Biol. 2005;12:822. doi: 10.1038/nsmb971. [DOI] [PubMed] [Google Scholar]
- 53.Case DA, et al. University of California. San Francisco: 2004. [Google Scholar]
- 54.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P. J Comput Chem. 2003;24:1999. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 55.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 56.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926. [Google Scholar]
- 57.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639. [Google Scholar]
- 58.Ryckaert J-P, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327. [Google Scholar]
- 59.Kollman PA, Massova I, Reyes C, Kuhn B, Huo S, Chong L, Lee M, Lee T, Duan Y, Wang W, Donini O, Cieplak P, Srinivasan J, Case DA, Cheatham TE. Acc Chem Res. 2000;33:889. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- 60.Gilson MK, Sharp KA, Honig BH. J Comput Chem. 1988;9:327. [Google Scholar]
- 61.Jayaram B, Sharp KA, Honig B. Biopolymers. 1989;28:975. doi: 10.1002/bip.360280506. [DOI] [PubMed] [Google Scholar]
- 62.Sanner MF, Olson AJ, Spehner J-C. Biopolymers. 1996;38:305. doi: 10.1002/(SICI)1097-0282(199603)38:3%3C305::AID-BIP4%3E3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 63.Pan Y, Gao D, Zhan C-G. J Am Chem Soc. 2008;130:5140. doi: 10.1021/ja077972s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raha K, Merz KM. J Med Chem. 2005;48:4558. doi: 10.1021/jm048973n. [DOI] [PubMed] [Google Scholar]
- 65.Berkman CE, Park SB, Wrighton SA, Cashman JR. Biochem Pharmacol. 1995;50:565. doi: 10.1016/0006-2952(95)00168-y. [DOI] [PubMed] [Google Scholar]
- 66.Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, Kuroiwa Y. Drug Metab Dispos. 1996;24:1212. [PubMed] [Google Scholar]
- 67.Messina ES, Tyndale RF, Sellers EM. J Pharmacol Exp Ther. 1997;282:1608. [PubMed] [Google Scholar]
- 68.Yamazaki H, Inoue K, Hashimoto M, Shimada T. Arch Toxicol. 1999;73:65. doi: 10.1007/s002040050588. [DOI] [PubMed] [Google Scholar]
- 69.Le Gal A, Dreano Y, Lucas D, Berthou F. Toxicol Lett. 2003;144:77. doi: 10.1016/s0378-4274(03)00229-7. [DOI] [PubMed] [Google Scholar]
- 70.Inoue K, Yamazaki H, Shimada T. Arch Toxicol. 2000;73:532. doi: 10.1007/s002040050005. [DOI] [PubMed] [Google Scholar]
- 71.Liu H, Zhang Y, Yang W. J Am Chem Soc. 2000;122:6560. [Google Scholar]
- 72.Zhang Y, Kua J, McCammon JA. J Am Chem Soc. 2002;124:10572. doi: 10.1021/ja020243m. [DOI] [PubMed] [Google Scholar]
- 73.Cisneros GA, Liu H, Zhang Y, Yang W. J Am Chem Soc. 2003;125:10384. doi: 10.1021/ja029672a. [DOI] [PubMed] [Google Scholar]
- 74.Cheng Y, Zhang Y, McCammon JA. J Am Chem Soc. 2005;127:1553. doi: 10.1021/ja0464084. [DOI] [PubMed] [Google Scholar]
- 75.Corminboeuf C, Hu P, Tuckerman ME, Zhang Y. J Am Chem Soc. 2006;128:4530. doi: 10.1021/ja0600882. [DOI] [PubMed] [Google Scholar]
- 76.Hu P, Wang S, Zhang Y. J Am Chem Soc. 2008;130:3806. doi: 10.1021/ja075896n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zheng F, Yang W, Ko M-C, Liu J, Cho H, Gao D, Tong M, Tai H-H, Woods JH, Zhan C-G. J Am Chem Soc. 2008;130:12148. doi: 10.1021/ja803646t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu J, Hamza A, Zhan C-G. J Am Chem Soc. 2009;131:11964. doi: 10.1021/ja903990p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poyner RR, Larsen TM, Wong S-W, Reed GH. Arch Biochem Biophys. 2002;401:155. doi: 10.1016/S0003-9861(02)00024-3. [DOI] [PubMed] [Google Scholar]
- 80.Cisneros GA, Wang M, Silinski P, Fitzgerald MC, Yang W. Biochemistry (Mosc) 2004;43:6885. doi: 10.1021/bi049943p. [DOI] [PubMed] [Google Scholar]
- 81.Metanis N, Brik A, Dawson PE, Keinan E. J Am Chem Soc. 2004;126:12726. doi: 10.1021/ja0463841. [DOI] [PubMed] [Google Scholar]
- 82.Wachters AJH. J Chem Phys. 1970;52:1033. [Google Scholar]
- 83.Bauschlicher CW, Langhoff SR, Partridge H, Barnes LA. J Chem Phys. 1989;91:2399. [Google Scholar]
- 84.Zhang Y, Liu H, Yang W. In: Methods for Macromolecular Modeling. Schlick T, Gan HH, editors. Springer-Verlag; New York: 2002. p. 332. [Google Scholar]
- 85.Yano JK, Denton TT, Cerny MA, Zhang XD, Johnson EF, Cashman JR. J Med Chem. 2006;49:6987. doi: 10.1021/jm060519r. [DOI] [PubMed] [Google Scholar]
- 86.Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 87.Northrop DB. Biochemistry (Mosc) 1981;20:4056. doi: 10.1021/bi00517a017. [DOI] [PubMed] [Google Scholar]
- 88.Northrop DB. J Am Chem Soc. 1981;103:1208. [Google Scholar]
- 89.Northrop DB. J Chem Ed. 1998;75:1153. [Google Scholar]
- 90.Guengerich FP. Chem Res Toxicol. 2001;14:611. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 91.Guengerich FP. Biol Chem. 2002;383:1553. doi: 10.1515/BC.2002.175. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.