

Abstract
Paxillin acts as an adaptor protein in integrin signaling. We have shown that paxillin exists in a relatively large cytoplasmic pool, including perinuclear areas, in addition to focal complexes formed at the cell periphery and focal adhesions formed underneath the cell. Several ADP-ribosylation factor (ARF) GTPase-activating proteins (GAPs; ARFGAPs) have been shown to associate with paxillin. We report here that Git2-short/KIAA0148 exhibits properties of a paxillin-associated ARFGAP and appears to be colocalized with paxillin, primarily at perinuclear areas. A fraction of Git2-short was also localized to actin-rich structures at the cell periphery. Unlike paxillin, however, Git2-short did not accumulate at focal adhesions underneath the cell. Git2-short is a short isoform of Git2, which is highly homologous to p95PKL, another paxillin-binding protein, and showed a weaker binding affinity toward paxillin than that of Git2. The ARFGAP activities of Git2 and Git2-short have been previously demonstrated in vitro, and we provided evidence that at least one ARF isoform, ARF1, is an intracellular substrate for the GAP activity of Git2-short. We also showed that Git2-short could antagonize several known ARF1-mediated phenotypes: overexpression of Git2-short, but not its GAP-inactive mutant, caused the redistribution of Golgi protein β-COP and reduced the amounts of paxillin-containing focal adhesions and actin stress fibers. Perinuclear localization of paxillin, which was sensitive to ARF inactivation, was also affected by Git2-short overexpression. On the other hand, paxillin localization to focal complexes at the cell periphery was unaffected or even augmented by Git2-short overexpression. Therefore, an ARFGAP protein weakly interacting with paxillin, Git2-short, exhibits pleiotropic functions involving the regulation of Golgi organization, actin cytoskeletal organization, and subcellular localization of paxillin, all of which need to be coordinately regulated during integrin-mediated cell adhesion and intracellular signaling.
INTRODUCTION
Integrins play an essential role in a number of dynamic aspects of cell regulation, including adhesion and migration. A number of different cytoplasmic proteins, with scaffolding as well as signaling properties, must assemble on the cytoplasmic tails of integrins for proper integrin functioning (Hynes, 1992; Clark and Brugge, 1995; Burridge and Chrzanowska-Wodnicka, 1996). Formation of integrin-mediated adhesive contacts is dynamically regulated during cell adhesion and migration. It is believed that there must be mechanisms that orchestrate and coordinate protein recruitment at the cytoplasmic tails of integrins, but the molecular processes largely remain to be established (Burridge and Chrzanowska-Wodnicka, 1996; Norman et al., 1998; Kondo et al., 2000).
It has been well documented that Rho-family GTPases Rho, Rac, and Cdc42 regulate focal adhesion formation and actin cytoskeletal organization (Nobes and Hall, 1995; reviewed by Hall, 1998). It has been shown recently, however, that another family of the small GTP-binding proteins, ADP-ribosylation factors (ARFs), is also involved in actin-based cytoskeletal organization, as well as focal adhesion formation (Norman et al., 1998; Song et al., 1998; Radhakrishna et al., 1999). ARF-family proteins have been primarily implicated in the membrane and vesicle traffic in mammalian cells (for reviews see Moss and Vaughan, 1998; Roth, 1999). The family includes six isoforms of ARF and the ARF-like proteins. The six ARF isoforms are highly homologous to one another and assigned to class I, II, or III based on sequence similarity (reviewed by Nuoffer and Balch, 1994; Moss and Vaughan, 1995). Class I includes ARF1, 2, and 3; class II includes ARF4 and 5; and class III includes ARF6. Among them, ARF1 has been most thoroughly studied (reviewed by Roth, 1999) and has been shown to regulate intracellular traffic at multiple sites within the cell. ARF1 can be colocalized with Golgi-associated proteins and acts primarily at the Golgi (Stearns et al., 1990; Serafini et al., 1991; Donaldson et al., 1992; Kahn et al., 1992). However, ARF1 has also been shown to function in endoplasmic reticulum (ER)-to-Golgi transport (Balch et al., 1992; Dascher and Balch, 1994), trans-Golgi network (Stamnes and Rothman, 1993), endosome-endosome fusion (Lenhard et al., 1992; West et al., 1997; Ooi et al., 1998), protein secretion and fluid-phase endocytosis (Zhang et al., 1994), and phospholipase D activation (reviewed by Kahn et al., 1993; Cockcroft, 1996). The GTP-bound form of ARF1 recruits protein coats to membranes and initiates budding of the membrane vesicles (reviewed by Donaldson and Klausner, 1994; Boman and Kahn, 1995; Schekman and Orci, 1996; Springer et al., 1999). Subsequent hydrolysis of GTP to GDP by ARF1 may trigger disassembly of the coat from the vesicle, which is necessary for the vesicle to fuse to target membranes. Unlike other small GTP-binding family proteins, such as Ras-family and Rho-family proteins, it is noteworthy that the intrinsic GTPase activity of ARF proteins is almost undetectable in vitro (Kahn and Gilman, 1986); thus, involvement of a GTPase-activating protein (GAP) seems to be crucial for the regulation of ARF activity.
Paxillin, one of the integrin-assembly proteins, localizes to focal contacts and acts as a scaffolding adaptor protein in integrin signaling by binding to several other integrin-assembly proteins, including focal adhesion kinase, Pyk2, c-Src, and vinculin (reviewed by Clark and Brugge, 1995; Turner, 1998). Paxillin is also highly tyrosine phosphorylated upon integrin activation (Burridge et al., 1992) and thus creates binding sites for several src homology 2-containing proteins such as Crk-I, Crk-II, Crk-L and Csk (reviewed by Turner, 1998). The importance of paxillin in protein assembly and signaling has also been suggested by the lack of paxillin tyrosine phosphorylation in neutrophils isolated from a patient with a leukocyte adhesion deficiency (Graham et al., 1994). Moreover, paxillin-binding activity toward different types of papilloma virus E6 proteins correlates with the degree of disruption of the actin cytoskeletal architecture induced by infection with each type of papilloma virus (Tong and Howley, 1997; Tong et al., 1997). Miyamoto et al. have demonstrated that paxillin is not constitutively associated with integrins but is recruited to cell surface integrins only after the integrins are activated (Miyamoto et al., 1995).
We have shown that a relatively large cytoplasmic pool of paxillin exists, largely overlapped with the Golgi apparatus (Mazaki et al., 1998). The perinuclear localization of paxillin may have some relationship to the intracellular dynamics of paxillin, including its recruitment to focal contacts to be engaged in integrin signaling. We found that several unidentified proteins seem to be associated with paxillin (Mazaki et al., 1997). We analyzed these novel paxillin-interacting proteins and found that most of them contain ARFGAP motifs and named these proteins PAGs (paxillin-associated protein with ARFGAP activity; Kondo et al., 2000). We show in this paper that a significant fraction of paxillin is indeed localized to the Golgi-like structure in cultured fibroblasts, and we report that one of the PAG proteins (which we originally named PAG1α) seems to be colocalized with paxillin primarily at the perinuclear areas and is involved in the subcellular localization of paxillin. This protein corresponds to KIAA0148, previously isolated by Nagase et al. (1995), and has turned out to be identical to Git2-short, a short isoform of Git2 (Premont et al., 2000; Vitale et al., 2000). Git2 is closely homologous to another paxillin-binding protein p95PKL (Turner et al., 1999). We show that Git2-short exhibits much weaker binding affinity toward paxillin α than that of Git2. Git2 has been shown to stimulate hydrolysis of GTP bound to all classes of ARF proteins in vitro (Vitale et al., 2000), and our results indicated that Git2-short acts as a GAP at least for ARF1 in vivo. We also show that Git2-short appears to be involved in several distinct aspects of intracellular regulation, such as the Golgi organization, subcellular localization of paxillin, and actin cytoskeletal organization, all of which should be coordinately regulated during integrin-mediated cell adhesion and signaling. Based on our analysis, a possible functional difference between Git2-short and Git2/p95PKL will also be discussed.
MATERIALS AND METHODS
Cells and Antibodies
293T, HeLa, and COS-7 cells were grown in DMEM (with 4.5 g glucose/l) (Life Technologies, Grand Island, NY) supplemented with 10% fetal calf serum (Hyclone, Logan, UT). 3Y1 and NIH3T3 cells were grown in DMEM with 5% fetal calf serum.
Anti-paxillin antibody (Ab199-217) was described previously (Mazaki et al., 1997). Mouse polyclonal anti-Git2-short antiserum was raised against glutathione S-transferase (GST)-fusion forms of Git2-short protein (see below). Other antibodies were purchased from commercial sources: anti-paxillin (mouse monoclonal antibody clone 349; Transduction Laboratories, Lexington, KY), anti-influenza hemagglutinin (HA; mouse monoclonal antibody clone 16B12 and polyclonal antibodies; Berkeley Antibody, Richmond, CA), anti-myc (mouse monoclonal antibody clone 9E10, and polyclonal antibodies; Berkeley Antibody), anti-green fluorescent protein (GFP) (mouse monoclonal antibody clone B34, Berkeley Antibody), anti-GST (mouse monoclonal antibody clone 4C10, Berkeley Antibody), anti-fibroblast growth factor receptor 1 (mouse monoclonal antibody clone 19B2, Upstate Biotechnology, Lake Placid, NY), anti-ARF (mouse monoclonal antibody clone 1D9, Affinity BioReagents, Neshanic Station, NJ), anti-CrkII (Santa Cruz Biotechnology, Santa Cruz, CA), anti-Erk1/2 (Upstate Biotechnology), and anti-phosphotyrosine (mouse monoclonal antibody 4G10, Upstate Biotechnology). Polyclonal anti-β-COP antibody was a gift from M. Tagaya (Tokyo Pharmaceutical College, Tokyo, Japan; Yamaguchi et al., 1998). Donkey antibodies to rabbit or mouse IgG conjugated with horseradish peroxidase, Cy2, Cy3, or Cy5 were from Jackson ImmunoResearch Laboratories (West Grove, PA).
Protein Purification and Sequencing Analysis
Approximately 2 × 109 HeLa cells (150-mm culture dishes) were solubilized in 20 ml of 1% Nonidet P-40 (NP-40) buffer (1% NP-40, 150 mM NaCl, 20 mM Tris-HCl pH 7.4, 5 mM EDTA, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1% aprotinin, 2 μg/ml leupeptin, and 3 μg/ml pepstatin A; Sabe et al., 1994). After the supernatant was centrifuged at 16,000 × g for 30 min at 4°C, it was first passed through 1 ml of glutathione-Sepharose 4B beads (Pharmacia, Piscataway, NJ) coupled to GST (5 μg, produced in Escherichia coli from pGEX2T) and then applied onto 0.1 ml of glutathione beads coupled with GST-Paxillin (N), which contained the NH2-terminal one half of paxillin α (amino acids 1–324) produced in the baculovirus system (Kondo et al., 2000). After the proteins were extensively washed with the same buffer (10 ml) followed by a washing with the same buffer without aprotinin, leupeptin, and pepstatin A (10 ml), those that were retained on the beads were eluted with 1 ml of 0.1 N NH4OH and then lyophilized in vacuo. Proteins were then separated by SDS-PAGE (8%), blotted to a polyvinylidene difluoride membrane (Applied Biosystems, Foster City, CA), and identified by staining with Ponceau S. Each protein band immobilized on polyvinylidene difluoride membrane was reduced, S-carboxymethylated, and digested in situ with Achromobacter protease I. Digested peptides were chromatographed by reverse-phase high-performance liquid chromatography using a Wakosil-II AR C18 300 Å column (Wako Pure Chemical, Osaka, Japan), and amino acid sequencing was performed with a gas-phase sequencer (model PPSQ-10; Shimadzu, Kyoto, Japan), as described previously (Iwamatsu, 1992).
Plasmids and Recombinant Proteins
All procedures for nucleic acid manipulation were done according to standard methods (Sambrook et al., 1989). For construction of the HA-tagged Git2-short expression vector (pcDNA3/HA-Git2-s), an HA-Git2-short cDNA fragment was amplified by polymerase chain reaction (PCR) from KIAA0148 plasmid (a gift from Dr. T. Nagase) using primers 5′-GGGGTACCGCCATGTCGAAACGGCTCCGGAG-3′ and 5′-GCTCTAGATTAAGCGTAGTCTGGGACGTCGTATG-GGTAATTAGCATCTTTTCCAGCA-3′, digested with KpnI and XbaI, and ligated into the KpnI-XbaI site of pcDNA3 (Invitrogen, San Diego, CA) to be transcribed under the control of the cytomegalovirus promoter. HA-Git2-short retrovirus vector (pBabePuro/HA-Git2-s) was constructed by amplifying the HA-Git2-short cDNA fragment using primers 5′-GGTACGTAATGTACCCATACGACGTCCCAGAC-TACCGTTCGAAACGGCTCCGGAGCAG-3′ and 5′-GCTACGTATTAATTAGCATCTTTTCCAA-3′, digested with SnaBI, and ligated into the SnaBI site of pBabePuro (Morgenstern and Land, 1990) to be transcribed under the control of the murine retrovirus long terminal repeat promoter. GST-Git2-short baculovirus vector was constructed by amplifying the Git2-short cDNA from KIAA0148 plasmid using 5′- and 3′-primers with an SmaI site (5′-TCCCCCGGGAATGTCGAAACGGCTCCGGAGCAG-3′ and 5′-TCCCCCGGGTTAATTAGCATCTTTTCCAA-3′) and cloned into pAcG2T (PharMingen, San Diego, CA) to be fused in-frame to the COOH terminus of GST (pAcG2T/Git2-s). Plasmids for mutant forms of GST-Git2-short proteins were constructed using primers 5′-TCCCCCGGGAACACCGTGGCCTCCAACACT-3′ and 5′-TCCCCCGGGTTAATTAGCATCTTTTCCAA-3′ for the M1 mutant encompassing amino acids 53–471 of Git2-short, 5′-TCCCCCGGGAGACGATAGTGTGACTGCCAA-3′ and 5′-TCCCCCG-GGTTAATTAGCATCTTTTCCAA-3′ for the M2 mutant (amino acids 125–471), 5′-TCCCCCGGGAATGTCGAAACGGCTCCGGAGCAG-3′ and 5′-TCCCCCGGGTTATTCAGGATGAAAGAAGTT GG-3′ for the M3 mutant (amino acids 1–165), 5′-TCCCCCGGGAATGTCGAAACGGCTCCGGAGCAG-3′ and 5′-TCCCCCGGGTTAGCTAAGATCTTTGGCAGTCA-3′ for the M4 mutant (amino acids 1–134), 5′-TCCCCCGGGAAAAGGAAACACCCCACTCCA-3′ and 5′-TCCCCCGG-GTTAATTAGCATCTTTTCC-AA-3′ for the M5 mutant (amino acids 166–471), and 5′-TCCCCCGGGAAGACTAGCCTTCTATCTCTG-3′ and 5′-TCCCCCGGGTTAATTAGCATCTTTTCCAA-3′ for the M6 mutant (amino acids 229–471). Each resulting fragment was digested with SmaI and ligated into pAcG2T vector in-frame with the COOH terminus of GST. A plasmid for the C11A mutant of Git2-short (the CA mutant; pAcG2T/Git2-s CA), in which the critical cysteine residue for the GAP activity, cysteine 11, was mutated to alanine to diminish the GAP activity (Cukierman et al., 1995), was constructed by removing the BamHI fragment from pAcG2T/Git2-s, and a synthetic double-strand DNA fragment containing the mutation 5′-GATCCCCGGGAATGTCGAAACGGCTCCG-GAGCAGCGAGGTGGCCGCTGACTGCAGC-GGGCCG-3′ and 5′-GATCCGGCCCGCTGCAGTCAGCGGCCACCTCGCTGCTCCGGA-GCCGTTTCGACATTCCCGGG-3′ was ligated into the resulting vector. Recombinant proteins were produced in the baculovirus system according to the manufacturer's instructions (PharMingen, San Diego, CA). His-Git2-short and the CA mutant were constructed by isolating each SmaI fragment from pAcG2T/Git2-s or pAcG2T/Git2-s CA and ligating into the SmaI site of pQE-30 (QIAGEN, Valencia, CA). Recombinant proteins were produced in E. coli according to the manufacturer's instructions (QIAGEN). pcDNA3/HA-Git2-s CA was constructed from pAcG2T/Git2-s CA using primers 5′-GGGGTACCGCCATGTCGAAACGGCTCCGGAG-3′ and 5′-TCCCCCGGGTTATTCAGGATGAAAGAAGTTGG-3′; the resultant PCR-fragment was digested with KpnI and EcoRI and replaced with the KpnI-EcoRI fragment in the pcDNA/Git2-s. Tetracycline-regulated expression vectors of HA-Git2-short (pTet-Splice/HA-Git2-s) and the CA mutant (pTet-Splice/HA-Git2-s CA) were constructed by isolating each KpnI-XbaI fragment from pcDNA3/HA-Git2-s or pcDNA3/HA-Git2-s CA, blunt ending, and ligating into the EcoRV site of pTet-Splice (Life Technologies). For construction of the GST-paxillin α mammalian expression vector (pEBG/paxillin α), a synthetic double-strand oligonucleotide with a NotI site 5′-AAT TCG CGG CCG CG-3′ was ligated into the EcoRI site of pGEX2T/paxillin α (Mazaki et al., 1997). A BamHI-NotI fragment was then isolated and ligated into the BamHI-NotI site of pEBG (Mayer et al., 1995) to be fused in-frame to the COOH terminus of GST. For construction of the enhanced green fluorescent protein (EGFP)-tagged Git2 expression vector (pEGFP-C1/Git2), Git2 cDNA was isolated by PCR amplification of the first-strand cDNAs prepared from mRNA of 12-O-tetradecanoyl-phorbol acetate-stimulated U937 cells using primers 5′-ATGAATTCAATGTCGAAACGGCTCAGGAGC-3′ and 5′-ATGTCGACTCAGTTGTTGTTCTCTTTGGTTGTG-3′. The resulting cDNA fragment was digested with EcoRI and SalI and ligated into the EcoRI-SalI site of pEGFP-C1 (Clontech, Palo Alto, CA).
cDNAs in pcDNA3 vectors each encoding myc-ARF1, myc-ARF5, myc-ARF6, HA-ARF1, HA-ARF6, myc-ARF1Q71L, HA-ARF1N126I, and HA-ARF6N122I were gifts from Dr. K. Nakayama. myc-ARF1, myc-ARF5, and myc-ARF6 cDNAs in pBabePuro vectors were constructed by isolating each HindIII-XbaI fragment from pcDNA3/myc-ARF1, pcDNA3/myc-ARF5, or pcDNA3/myc-ARF6, blunt ending, and ligating into the SnaBI site of pBabePuro. Nucleotide sequences were confirmed with all the plasmids after construction.
Recombinant paxillin α, β, and γ, as well as paxillin (N) (amino acids 1–324 of paxillin α) and paxillin (LIM) (amino acids 325–557 of paxillin α) produced in the baculovirus system were described previously (Kondo et al., 2000). Recombinant nonmyristoylated ARF1 was a gift from Dr. K. Nakayama.
Protein Expressions in Cultured Cells
For stable transfectants, the pBabePuro/HA-Git2-s retrovirus was packaged using BOSC23 cells (Pear et al., 1993) as described previously (Mazaki et al., 1998). Virus titers were in the range of 105-106 infectious units/ml. Cells were then infected with these viruses and selected 2 d later with puromycin (1.5 μg/ml for 3Y1, 2.0 μg/ml for NIH3T3; Sigma, St. Louis, MO) for 1 wk more. Levels of exogenous proteins expressed by the BOSC-derived retroviruses were similar to or only two- to threefold higher than those of each corresponding endogenous protein. For transient protein expression, 0.5–1 × 105 cells in a 35-mm culture dish were transfected with 4 μg of plasmid DNAs by the calcium phosphate precipitation method (Bonifacino et al., 1989) or with 1 μg of plasmid DNAs using FuGENE 6 (Boheringer Mannheim, Indianapolis, IN) and incubated for 48 h before fixation. For protein production in 293T cells, 2 × 106 cells in a 90-mm culture dish were transfected with 20 μg of plasmid DNAs by the calcium phosphate precipitation method and incubated for 48 h before preparation of the cell lysates. For transient overexpression, plasmid DNAs with pcDNA3 vector were used, which utilizes the cytomegarovirus promoter for the expression of inserted cDNAs.
pTet-Splice/HA-Git2-s or pTet-Splice/HA-Git2-s CA NIH3T3 cell clones were established by transfection of cells with pTet-tTAK Hygro (Life Technologies) and selected in 0.2 mg/ml hygromycin B. Cell clones thus obtained were further transfected with pTet-Splice/HA-Git2-s or pTet-Splice/HA-Git2-s CA and a 1:10 M ratio of pBabePuro DNA and selected in 2.0 μg/ml puromycin. Each cell clone obtained was then tested for the tetracycline-regulated expression of the HA-Git2-s proteins. These cell clones were normally cultured in the presence of 0.5 μg/ml tetracycline.
Protein-binding Analysis
Protein-binding analysis was performed as described previously (Sabe et al., 1994). Briefly, 500 μg of cell lysate prepared in 1% NP-40 buffer was mixed with 5 μg of GST-fusion protein bound to glutathione beads, incubated for 2 h at 4°C, and then washed four times with 1% NP-40 buffer. Proteins retained on the beads were subjected to immunoblotting analysis after separation by SDS-PAGE (10%) and visualized by an enzyme-linked chemiluminescence method. Protein concentrations were determined using a Dc protein assay kit (Bio-Rad, Hercules, CA) with bovine serum albumin (BSA; Sigma) as a standard. Each figure showed representative results from at least three independent experiments.
Subcellular Fractionation of Git2-short
Subcellular fractionation was performed according to a method previously described (Abe et al., 1999) with a slight modification. In brief, cells were washed in ice-cold phosphate-buffered saline (PBS) and lysed in a buffer (2 mM Tris-HCl, pH 8.0, 140 mM NaCl, 0.25 M sucrose, 1 mM EDTA, 1 mM Na3VO4, 10 μM Na3MoO4, 1 mM phenylmethylsulfonyl fluoride, 20 μg/ml leupeptin, 1% aprotinin, 3 μg/ml pepstatin A) with a Dounce-type glass homogenizer. After nuclei and undisrupted cells were removed by centrifugation at 1000 × g for 10 min at 4°C, lysate was centrifuged at 105 × g for 1 h at 4°C to be fractionated into soluble and particulate fractions. The ratio of protein amounts recovered in the soluble and the particulate fractions was ∼2:1 by weight.
Immunoelectron Microscopy
Immunoelectron microscopy was carried out using the silver-enhancement technique as described previously (Mizoguchi et al., 1994; Yamamoto et al., 1997). Briefly, cells were fixed in 4% paraformaldehyde in PBS, rinsed with PBS, and permeabilized with 5% BSA and 0.02% saponin in PBS. The permeabilized cells were incubated with anti-paxillin monoclonal antibody (1:100) or anti-HA monoclonal antibody (1:100) in PBS containing 0.005% saponin, followed by a wash with 0.005% saponin in PBS. The cells were incubated with anti-mouse antibody coupled with 1.4-nm gold particles (Nanoprobes, Stony Brook, NY). After the sample was washed, gold particles were enhanced by incubation with silver developer (HQ silver, Nanoprobes). The sections were postfixed with 0.5% OsO4, dehydrated with ethanol, and embedded in Epon. Ultrathin sections were then made and analyzed using electron microscope (JEM-1200; JEOL, Tokyo, Japan).
Confocal Immunofluorescence Microscopy
Immunolabeling and confocal immunofluorescence microscopic analysis was done as previously described (Kondo et al., 2000). Briefly, cells stably expressing exogenous proteins, or 20–24 h after DNA transfection, were replated onto glass chamber slides (Becton Dickinson, Franklin Lakes, NJ). Cells were fixed 20–24 h later in 4% paraformaldehyde (Sigma) in PBS. To activate GTP-binding proteins, cells were treated for 1 h with 30 mM NaF and 50 μM AlCl3 (aluminum fluoride [AlF]) at 37°C as previously described (Radhakrishna et al., 1996; Ooi et al., 1998), before fixation. Incubation for 1 h with AlF was chosen by our preliminary time-course study (10–120 min) as optimal for activation of transfected ARFs in COS-7 cells (Kondo et al., 2000). To inactivate ARFs, cells were treated with 5 μg/ml brefeldin A (BFA, Sigma) for 30 min before fixation. Cells were then subjected to immunolabeling analysis, and confocal images were acquired using a confocal laser scanning microscope (model 510; Carl Zeiss, Thornwood, NY). Each figure of microscopic analysis showed representative results that were observed in a majority (50–80%) of the transfected cells observed in three independent experiments (50–200 cells).
ARFGAP Assay
ARFGAP assay was performed as described previously (Vitale et al., 1997; Premont et al., 1998). Briefly, 0.5 μg of recombinant nonmyristoylated ARF1 was incubated for 30 min at 30°C in 20 mM Tris-HCl, pH 8.0, 10 mM dithiothreitol, 2.5 mM EDTA with 0.3 mg/ml BSA, 1 mg/ml cardiolipin, and 30 μg/ml phosphatidylserin and then for 40 min at 30°C in the same solution with 0.5 μM [α-32P]GTP and 10 mM MgCl2. Purified His-Git2-short or His-Git2-short CA were added and incubated for 5–20 min at 30°C. Unbound GTP was removed by vacuum filtration through nitrocellulose and washing six times. Bound nucleotides were eluted in 2 M formic acid and were separated by chromatography on polyethyleneimine-cellulose plates (Merck, Rahway, NJ) in 1 M formic acid/1 M LiCl.
Cell Adhesion, Migration Assays, and Haptotaxis Migration Assay
The cell adhesion assay was performed by plating 2 × 105 cells onto 35-mm culture dishes, precoated with fibronectin (10 μg/ml) and blocked with heat-inactivated BSA (inactivated at 70°C for 1 h), then incubated for 30 min at 37°C, and washed to remove the nonadhered cells. Adhered cells were then harvested with 0.125% trypsin in PBS and counted using a hemocytometer. Results are shown as means ± SEM from triplicate experiments.
Cell migration was traced using time-lapse video microscopy. Cells (2 × 105 in a 75-cm2 culture flask; NalgeNunc, Rochester, NY) were placed on a microscope stage (IX-70; Olympus) and maintained at 37°C using an air current incubator (IX-IBM, Olympus, Tokyo, Japan). Images were collected at one frame every 1 min for 24 h, using a three-charge–coupled device digital camera (MCD-350; Ikegami, Tokyo, Japan), and stored initially with an image-processing system (LVR-3000 AN/OL; SONY-Olympus, Tokyo, Japan). The stored images were then visualized using a scan converter (VSC-310–1; Tokyo Electronics, Tokyo, Japan) and a cathode-ray tube monitor (C/T-21B.J, Ikegami). The magnification of the entire system was such that the pixel diameter was 2.2 μm. Cell locomotion was then traced by marking the positions of the nuclei, and distances each cell traveled were measured using a digital curvimeter (with an accuracy of less than 3/1000 pulse; Uchida-Yoko, Tokyo, Japan). Velocities are shown as means ± SEM of distances (in micrometers) per hour (measured with 30 cells in each of three independent experiments).
Haptotaxis migration assay was performed using a modified Boyden chamber (tissue culture treated, 6.5-mm diameter, 10-μm thickness, 8-μm pores, Transwell; Costar, Cambridge, MA) as previously described (Kondo et al., 2000). In brief, only the underside of the polycarbonate membrane on upper chambers was coated with 10 μg/ml fibronectin (Sigma). Cells (1 × 105) were applied onto the upper migration chambers and allowed to migrate into the underside of the upper chamber for 3 h at 37°C with 5% CO2. After the nonmigrated cells on the upper membrane surface were removed with a cotton swab, cells that migrated to the underside of upper chamber were fixed with 4% paraformaldehyde in PBS, then stained with crystal violet solution (0.1% crystal violet, 0.1 M borate, pH 9.0, 2% ethanol), and counted. Results are shown as means ± SEM from triplicate experiments.
RESULTS
Localization of Paxillin to the Golgi-like Structure
We have shown that paxillin has a relatively large cytoplasmic pool, which seems to be largely overlapped with the Golgi apparatus (Mazaki et al., 1998). In an attempt to definitively determine whether paxillin can be localized to the Golgi, 3Y1 cells were examined by immunoelectron microscopy. As shown in Figure 1A, a significant fraction of paxillin localized to the Golgi-like structure. A fraction of paxillin also seemed to localize to the ER and several unidentified membrane structures in the perinuclear region. Focal adhesion localization of paxillin is also shown as a control (Figure 1B).
Figure 1.

Immunoelectron microscopic study of subcellular localization of paxillin. Anti-paxillin immunoelectron microscopic analysis was performed using 3Y1 cells. (A) Perinuclear area. (B) Focal adhesions. n, nucleus; er, ER; g, Golgi-like structure; FA, focal adhesion. The gold particles are shown as black dots. Bar, 500 nm.
Identification of Git2-short/KIAA0148 as a Paxillin-interacting Protein
Paxillin is not a Golgi resident protein. To explore the biological significance of the perinuclear localization of paxillin, we attempted to isolate the putative paxillin-interacting proteins that may be involved in the perinuclear localization of paxillin. We have shown that several unidentified proteins could be coprecipitated with paxillin (Mazaki et al., 1997). Using GST-fused paxillin (N), which contained the NH2-terminal half of paxillin α but not the COOH-terminal LIM domains, we purified paxillin-binding proteins from HeLa cell extracts and subjected them to amino acid sequence analysis. We used GST-paxillin (N), instead of the full-length paxillin, to reduce the size of the affinity probe. GST-paxillin (N) was found to precipitate most of the proteins detected with the full-length paxillin (Mazaki and Sabe, unpublished results). From one protein band thus purified, we obtained peptide sequences of KAEFIRAK (peptide 1), KQLHSSVRxGNLExCLRLLSLGAxA (x; uncertain amino acid residues; peptide 2), and KGNTPLHVASK (peptide 3), after Achromobacter protease I digestion (Figure 2A). These peptide sequences were found to be identical to those encoded by the KIAA0148 cDNA (Nagase et al., 1995). During our analysis, we found that the KIAA0148 cDNA was identical to Git2-short, which is thought to be a spliced variant of Git2 (Premont et al., 2000; Vitale et al., 2000) (Figure 2A). Git2 is highly homologous, but not identical, to p95PKL, which has also been identified as a paxillin-binding protein (Turner et al., 1999) (Figure 2A). The deduced amino acid sequence of both the KIAA0148/Git2-short and Git2 cDNAs contain a zinc finger motif highly homologous to that of ARF1GAP (Figure 2B), which has been shown to be localized to the Golgi membrane (Cukierman et al., 1995). KIAA0148/Git2-short and Git2 may thus also localize to the Golgi membrane and may thereby be involved in the perinuclear localization of paxillin. Therefore, these proteins were subjected to further analysis. We call these proteins Git2-short (KIAA0148) and Git2 in this paper and report here the function of Git2-short.
Figure 2.

Structure of Git2-short and its binding to paxillin. (A) Schematic diagram of Git2-short and the sequence comparison with Git2 and p95PKL. Peptide sequences obtained from the protein we purified are underlined. (B) Comparison of the Git2-short zinc finger domain (residues 7–80) with those of ARFGAP proteins, ARF1GAP (residues 18–91), GIT1 (residues 7–80; Premont et al., 1998), PIP3bp (residues 17–89; Tanaka et al., 1997), and Gcs1 (residues 22–95; Poon et al., 1996). The conserved cysteines are marked by dots, and the residue mutated in the CA mutant are marked by an asterisk. (C) Binding of Git2-short to paxillin α in vivo. 293T cells were transfected with 20 μg each of pEBG/paxillin α (lanes 1 and 4) or pcDNA3/HA-Git2-s (lanes 2 and 5) or cotransfected with each 10 μg of these plasmids (lanes 3 and 6) using a calcium phosphate method. GST-paxillin α was then pulled down from 500 μg of cell lysates using glutathione beads (lanes 4–6). Total cell lysates (20 μg) were also included (lanes 1–3). (D) Git2-short and Git2 binding to paxillin isoforms in vitro. Each 5 μg of GST-fusion forms of paxillin α (lanes 2 and 9), β (lanes 3 and 10), and γ (lanes 4 and 11), paxillin (N) (lane 5), paxillin (LIM) (lane 6), or GST alone (lanes 7 and 12), produced in baculovirus system and purified on glutathione beads, was incubated with 500 μg of 293T cell lysates expressing HA-Git2-short or EGFP-Git2 to test the binding (lanes 2–7 and 9–12). GST-paxillin was pulled down using glutathione beads. Total cell lysates (20 μg) of the 293T cells expressing HA-Git2-short (lane 1) or EGFP-Git2 (lane 8) were also included. (E) Dissection of Git2-short for paxillin binding. Each mutant Git2-short protein (5 μg; shown on the right), fused to GST and purified on glutathione beads, was incubated with recombinant paxillin α (each 20 μg of cell lysate) to test binding. All of the recombinant paxillin α and mutant Git2-short proteins were produced in the baculovirus system. wt, wild type. (F) Expression of Git2-short protein. Each cell lysate (20 μg) was separated by SDS-PAGE. (G) Coimmunoprecipitation of endogenous Git2-short with endogenous paxillin. U937 cell lysate (3 mg) prepared in 1% NP-40 buffer was immunoprecipitated with polyclonal anti-paxillin antibody Ab 199–217 coupled with protein G-Sepharose beads (lane 3). The control consisted of 3 mg of U937 cell lysate precipitated using preimmune rabbit serum coupled with protein G-Sepharose beads (lane 2). Total cell lysate (20 μg) of U937 cells was also included (lane 1). Immunoblotting of the same filter with anti-Erk1/2 antibody is also shown as a negative control. In C–G, immunoblots were done using anti-HA antibody (C, top; D, left), anti-GST antibody (C, bottom), anti-GFP antibody (D, right), monoclonal anti-paxillin antibody (E and G, middle), and anti-Git2-short antibody (F and G, top).
The potential binding of Git2-short to paxillin was then examined. HA-Git2-short was coexpressed with GST-paxillin α in 293T cells. As shown in Figure 2C, a pull-down of GST-paxillin α using glutathione beads precipitated a significant amount of HA-Git2-short. The relative affinities of Git2-short and Git2 toward paxillin were also compared (Figure 2D). We found that EGFP-Git2 exhibited a stronger binding affinity toward GST-paxillin α than did HA-Git2-short. There seemed to be an ∼10-fold difference in the binding affinities of Git2-short and Git2 toward paxillin α. Thus, Git2-short is a short isoform of Git2 interacting weakly with paxillin. Moreover, Git2-short exhibited only a marginal level of binding to the β isoform of paxillin, and no significant binding was detected to the γ isoform (Figure 2D). Similarly, Git2 showed strong affinity to the α isoform of paxillin, a weaker affinity to the β isoform, and no significant binding to the γ isoform (Figure 2D). Binding of Git2-short to the NH2-terminal half of paxillin α, but not to the COOH-terminal LIM domains, was also confirmed (Figure 2D). Coprecipitation of endogenous paxillin with endogenous Git2-short was also detected (Figure 2G), although the amount of coprecipitated Git2-short was very small, possibly reflecting its weak binding affinity toward paxillin. Git2-short did not show significant binding toward other focal adhesion proteins tested, such as vinculin, talin, Fak, Pyk2, and p130Cas (Mazaki and Sabe, unpublished results).
We next constructed GST-fusion forms of Git2-short and its mutants and tested their binding toward paxillin α (Figure 2E). The zinc finger domain containing the CX2C-X16-CX2C motif (deleted in the M1 or mutated in the CA) and the COOH-terminal half encompassing the second ankyrin repeat and thereafter (M5 and M6) were dispensable for the binding. On the other hand, amino acids 125–165 of Git2-short, present in the M1, M2, and M3 mutants but absent in the M4 mutant, seemed to be necessary for binding.
Git2-short/KIAA0148 mRNA and protein have been shown to be expressed ubiquitously (Nagase et al., 1995; Premont et al., 2000), although higher protein expression was observed in hematopoietic cell lines (Figure 2F).
Git2-short Is Localized to Perinuclear Areas and the Cell Periphery but Not Accumulates at Paxillin-containing Focal Adhesions underneath the Cell Body
We next examined the subcellular localization of Git2-short. Paxillin is localized to focal complexes, focal adhesions, and the cytoplasm, including perinuclear areas (Mazaki et al., 1998; Kondo et al., 2000; Nakamura et al., 2000). It should be noted that in this paper “focal complexes,” which are formed at the cell periphery, are distinguished from “focal adhesions,” which are formed underneath the cell body. This terminology has been used previously to describe these structures that are regulated by different intracellular machinery (see Manser et al., 1998, for example).
Perinuclear localization of paxillin is more readily observed in fibroblasts adhered to fibronectin (Mazaki et al., 1998) than in other types of cells, including epithelial cells, in which paxillin is more diffusely distributed in the cytoplasm (Kondo et al., 2000). Because antibodies raised against Git2-short are weak for immunostaining, we stably expressed HA-Git2-short cDNA in fibroblasts by BOSC-retrovirus infection. In these fibroblast cells, endogenous expression of Git2-short was low (Figure 2F), and HA-Git2-short protein was expressed at only slightly higher levels than the endogenous Git2-short (about two- to threefold; Mazaki and Sabe, unpublished results). In confocal immunofluorescence studies, when the focus was adjusted across the center of the cell body, Git2-short was detected at the perinuclear areas, largely, but not completely, overlapped with endogenous paxillin (Figure 3A). Perinuclear localization of Git2-short was also examined using the immunoelectron microscope and shown to be localized to the Golgi-like and other structures (Figure 3B). In contrast to paxillin, however, accumulation of HA-Git2-short to focal adhesions underneath the cell body was not evident (Figure 3A). On the other hand, a fraction of Git2-short was also localized to actin-rich structures at the cell periphery (Figure 3, A and B), where it seemed to be largely overlapped with paxillin (Figure 3A).
Figure 3.

Subcellular localization of Git2-short. 3Y1 cells stably expressing HA-Git2-short by BOSC23 retrovirus infection and plated onto fibronectin-coated (10 μg/ml) chamber slides are shown. Note that HA-Git2-short was not overexpressed but expressed at a level only about two- to threefold higher than that of the endogenous Git2-short (Mazaki and Sabe, unpublished results). (A) Comparison with paxillin. Cells were fixed and immunolabeled using mouse anti-HA antibody (a and d) and rabbit anti-paxillin antibody (b and e). Focuses were adjusted 3.0 μm above the surface of the glass chamber plates (a–c), across the center of the nucleus in the majority of cells, or 0.5 μm above the surface of the glass chamber plates (d–f), near the bottom layers of the cells. All confocal images were taken from the same single cell. Arrows in d–f indicate Git2-short localization to focal complexes at the cell periphery. Panels g and h are a higher magnification of the merged images shown in c and f (indicated by rectangles), respectively. (B) Immunoelectron microscopic study of Git2-short subcellular localization. Anti-HA antibody was used to detect HA-Git2-short. a, perinuclear area; b, cell periphery. n, nucleus; er, ER; g, Golgi-like structure. The gold particles are shown as black dots. Bar, 500 nm. In untransfected parental 3Y1 cells, no significant signals were detected. (C) Comparison with ARF isoforms. 3Y1/HA-Git2-short cells were further transfected with each of myc-tagged ARF cDNA (a–c, ARF1; d–f, ARF5; g–i, ARF6) in pBabe vector using the BOSC23 retrovirus infection method. After fixation, cells were immunolabeled with mouse anti-HA antibody (a, d, and g) and rabbit anti-myc antibody (b, e, and h). The size and morphology of cells expressing the ARFs were similar to the cells shown in A. Note that the cDNAs for these ARF proteins were driven bythe murine retrovirus promoter in the pBabe vector and hence not overexpressed. j, k, and l are a higher magnification of the merged images shown in c, f, and i (indicated by rectangles), respectively. (D) Comparison with β-COP. 3Y1/HA-Git2-short cells, untreated or treated with 5 μg/ml BFA for 30 min, were fixed and immunolabeled with mouse anti-HA antibody (a and d) and rabbit anti-β-COP antibody (b and e). β-COP is tightly localized to perinuclear areas (b), and the size and morphology of cells were almost unchanged by BFA treatment (d–f). g is a higher magnification of the merged image shown in c (indicated by a rectangle). In A, C, and D, each immunolabeling was visualized by further incubation with Cy2-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG. In C and D, focuses were adjusted 3.0 μm above the surface of the glass chamber plates, across the center of the nucleus in the majority of cells. The right columns represent the merging of the left and the middle images. Bars, 20 μm. Essentially the same results were obtained with NIH3T3 cells stably expressing HA-Git2-short (Mazaki and Sabe, unpublished results).
Localization of Git2-short at the perinuclear areas implies that Git2-short may be associated with an endomembrane structure. Consistent with this, biochemical fractionation showed that a significant fraction of endogenous Git2-short (∼30%) was associated with the particulate fraction (Figure 4). About 25% of total cellular paxillin was also recovered in the particulate fraction (Figure 4).
Figure 4.
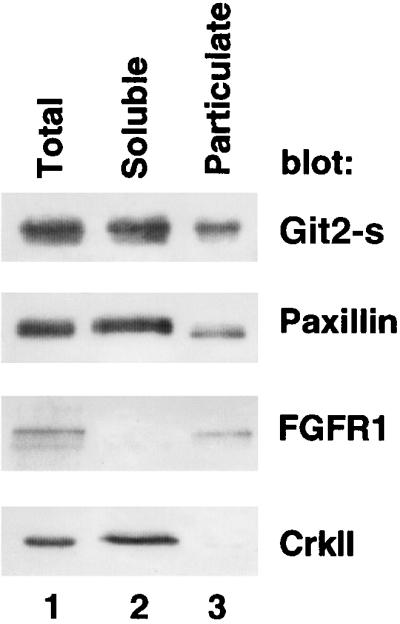
Subcellular fractionation of endogenous Git2-short. 3Y1 cells were subjected to subcellular fractionation as described in MATERIALS AND METHODS. Total, soluble, and particulate fractions (20 μg each) were separated by SDS-PAGE and immunobloted with anti-Git2-short, anti-paxillin, anti-fibroblast growth factor R1 (FGFR1), or anti-CrkII antibodies. Similar distributions of endogenous Git2-short in NIH3T3 and HA-Git2-short in 3Y1 or in NIH3T3 were also observed (Mazaki and Sabe, unpublished results).
Relationship between ARF Activities and Perinuclear Localization of Git2-short and Paxillin
The zinc finger motif found in ARF1GAP has been shown to be essential for the GAP activity (Cukierman et al., 1995). Because Git2-short contains a similar zinc finger motif (Figure 2B) and exhibits ARFGAP activity (Premont et al., 2000; Vitale et al., 2000; see below), possible colocalization of Git2-short with ARF proteins was then examined. As shown in Figure 3, Git2-short appeared to be well colocalized, but not completely, with ARF1 (class I ARF) at the perinuclear areas (Figure 3C). On the other hand, ARF5 (class II) and ARF6 (class III) have been shown to exhibit broader cytoplasmic distributions than ARF1 (Peters et al., 1995; Hosaka et al., 1996), and Git2-short exhibited a narrower cytoplasmic distribution than that of ARF5 and ARF6 (Figure 3C). Git2-short also seemed to be partially colocalized with a subunit of the COPI coat, β-COP (Figure 3D). COPI is recruited to the membrane after activation of ARF1 and is involved in the retrograde transport of recycling components during ER-to-Golgi transport and also in Golgi function (Letourneur et al., 1994; reviewed by Kreis et al., 1995). We used β-COP as a marker for several of the following experiments (see below).
Every class of ARF isoforms can affect the subcellular localization of paxillin (Norman et al., 1998; Kondo et al., 2000). We next explored a possible relationship between ARF activities and the perinuclear localization of Git2-short and paxillin. A fungous macrocyclic lactone, BFA, inhibits several ARF exchange factors and thus causes inactivation of several ARF isoforms (Zhang et al., 1998; reviewed by Chardin and McCormick, 1999). It has been well documented that treatment of cells with BFA thereby causes a rapid release of β-COP (reviewed by Chardin and McCormick, 1999) and the ARF1GAP protein (Cukierman et al., 1995) from the Golgi into the cytosol or into the ER. Upon BFA treatment, Git2-short became diffusely redistributed from their tight localization at perinuclear areas, together with β-COP (Figure 3D), as well as paxillin (Mazaki and Sabe, unpublished results). These results suggest that some ARF activity is involved in the perinuclear localization of paxillin and Git2-short. Interestingly, paxillin and Git2-short still seemed to colocalize well with each other in the BFA-treated cells (Mazaki and Sabe, unpublished results).
Evidence for the GAP Activity of Git2-short for ARF1 In Vivo
We next examined the specificity of the ARFGAP activity of Git2-short for the various ARF isoforms. As shown in Figure 5, we first confirmed the GAP activity of Git2-short in vitro, using a recombinant ARF1 protein preloaded with radiolabeled GTP as a substrate, as has been used recently by others (Premont et al., 2000; Vitale et al., 2000). A negligible activity, on the other hand, was observed with the CA mutant, in which a cysteine residue in the zinc finger domain critical for the GAP activity was mutated to alanine (Figure 5). The specificity of the GAP activity of Git2-short toward different ARF isoforms should then be determined. However, it has been shown that coatomer proteins directly participate in the GTPase reaction of ARF1GAP, accelerating GTP hydrolysis by an additional 1000-fold in vitro (Goldberg, 1999). Thus, the activities and specificities of ARFGAP proteins toward different ARF isoforms may not be adequately assessed in vitro, if the bona fide coatomer or effector proteins are not present. A number of different coatomers have been reported, and we do not know which may affect the GAP activity of Git2-short toward ARFs. We thus performed the following two different types of experiments to assess the ARFGAP activity of Git2-short toward ARF isoforms in vivo.
Figure 5.

In vitro GAP activity of Git2-short toward ARF1. ARF1 (0.5 μg) preloaded with [α-32P]GTP were incubated for 5 min at 30°C with buffer alone (lane 1), with 2.5 μg of Git2-short (lane 2), or the CA mutant (lane 3). The nucleotides were then extracted and separated on a polyethyleneimine-cellulose plate and subjected to autoradiography. Positions of GTP and GDP were identified using authentic nucleotides.
Like BFA treatment, it has been shown that overexpression of the dominant-negative form of ARF1 causes inactivation of ARF1 and thus also causes redistribution of β-COP into the cytosol or into the ER (Peters et al., 1995; Ooi et al., 1998) (Figure 6). On the other hand, overexpression of the dominant-negative form of ARF6, the ARF isoform most distantly related to ARF1, is incapable of causing such a redistribution of β-COP (Peters et al., 1995) (Figure 6). These effects of the dominant-negative forms of ARF proteins on β-COP redistribution have been readily observed in HeLa cells (Peters et al., 1995; Ooi et al., 1998). We thus used HeLa cells and found that transient overexpression of HA-Git2-short caused a redistribution of β-COP, whereas overexpression of the GAP-inactivated CA mutant did not (Figure 6). Redistribution of β-COP was observed in 70–80% of cells overexpressing HA-Git2-short. Moreover, the Git2-short-induced redistribution of β-COP could be suppressed by coexpression of a dominant-active ARF1 mutant, ARF1Q71L (Figure 6), in which the GTPase activity was negligible. These results are consistent with the notion that Git2-short acts as a GAP toward ARF1 rather than ARF6, and overexpression of Git2-short causes inactivation of ARF1.
Figure 6.

Overexpression of Git2-short, but not its GAP-inactive mutant, causes a redistribution of β-COP. HeLa cells were transiently transfected with 4 μg of pcDNA3 plasmids encoding HA-ARF1N126I (a and b), ARF6N122I (c and d), HA-Git2-short (e and f), the CA mutant (g and h), or 2 μg each of HA-Git2-short and myc-ARF1Q71L (i and j), using the calcium phosphate method. After 48 h, cells were fixed and HA-ARFs or HA-Git2-short proteins were visualized using mouse anti-HA antibody and Cy3 anti-mouse IgG (a, c, e, and g). Endogenous β-COP was visualized with rabbit anti-β-COP antibody and Cy2 anti-rabbit IgG (b, d, f, h, and j). In i, HA-Git2-short was visualized using biotin-labeled mouse monoclonal anti-HA antibody and Cy3 streptavidin (but shown in green), and myc-ARF1Q71L was visualized using monoclonal anti-myc antibody coupled with Cy5 anti-mouse IgG (shown in red). Each right column is of the same field as the left column, and arrows indicate cells overexpressing HA-ARFs (a–d), HA-Git2-short (e and f), or HA-Git2-short CA mutant (g and h), or both HA-Git2-short and myc-ARF1Q71L proteins (i and j). Focuses were adjusted 3.0 μm above the surface of the plates. Bar, 20 μm.
AlF is a G protein activator that can affect the behavior of ARFs, including ARF1 and ARF6 (Donaldson et al., 1991; Finazzi et al., 1994; Radhakrishna et al., 1996). It has been well documented that AlF treatment of ARF-overexpressing cells induces distinct cell phenotypes, depending on the ARF cDNAs transfected: it causes an appearance of a number of enlarged ARF1-containing punctate structures in the ARF1-transfected cells (Ooi et al., 1998) (Figure 7A), and induces ARF6-containing membrane protrusion in the ARF6-transfected cells (Radhakrishna et al., 1996) (Figure 7B). Because these phenotypes induced by the AlF treatment are conspicuous and easily recognized and could be observed within a relatively short time after the addition of AlF (10–60 min), we used this system to test whether overexpression of Git2-short could suppress the ARF-dependent phenotypes. We have shown with this experimental system that another PAG protein, PAG3/Papα/KIAA0400, acts as a GAP for ARF6 (Kondo et al., 2000). As shown in Figure 7, we found that overexpression of HA-Git2-short appeared to suppress the ARF1-induced phenotype. After AlF treatment for 1 h, an increase in the number and size of the ARF1-associated punctate structures was seen in 80–90% of cells expressing myc-ARF1 (Figure 7A; Mazaki and Sabe, unpublished results). However, when HA-Git2-short was co-overexpressed, the AlF-induced phenotype was suppressed in the majority of cells expressing myc-ARF1 (Figure 7A; we observed such suppression in 50–60% of cells expressing both myc-ARF1 and HA-Git2-short). Co-overexpression of the CA mutant of HA-Git2-short did not exert such an effect (Figure 7A), suggesting that the GAP activity is necessary for the suppression. On the other hand, unlike PAG3, we did not observe significant suppressive activity of Git2-short toward the AlF-activated ARF6-phenotype (Figure 7B). Overexpression of the HA-Git2-short CA mutant also did not suppress the ARF6-induced phenotype (Figure 7B).
Figure 7.

Assessment of the ARFGAP activity of Git2-short in vivo. (A) Overexpression of Git2-short, but not its GAP-inactive CA mutant, suppresses the AlF-induced phenotype in ARF1 transfected cells. COS-7 cells were transiently transfected with a plasmid encoding myc-ARF1 alone (a–c and g–i) or a plasmid encoding myc-ARF1 was cotransfected with a plasmid encoding HA-Git2-short (d–f and j–l) or with the CA mutant of HA-Git2-short (m–o), using FuGENE 6. (B) Overexpression of Git2-short does not suppress the AlF-induced phenotype in ARF6 transfected cells. All panels correspond to those in A, except that a plasmid encoding myc-ARF6 was used instead of a plasmid encoding myc-ARF1. In A and B, all cDNAs were constructed in the pcDNA3 vector. For cotransfection, 0.5 μg of each Git2-short or the CA mutants and 0.5 μg of ARF plasmids were used, as determined by our preliminary titration experiments. Cells were untreated (a–c and d–f) or treated with AlF mixture for 1 h at 37°C (g–i, j–l, and m–o), before fixation. HA-Git2-short proteins were visualized by rabbit anti-HA antibody and Cy3 anti-rabbit IgG (d, j, and m). myc-ARF proteins were visualized with mouse anti-myc antibody coupled with Cy2 anti-mouse IgG (b, e, h, k, and n). The right columns represent the merging of the left and the middle images. Focuses were adjusted 3.0 μm above the surface of the plates. Bars, 20 μm. In c and i, differential interference contrast images (gray) are also included to show the subcellular localization of myc-ARF1. It should be noted that ARF1 existed as condensed structures localized to one side of the nucleus in the untreated COS-7 cells, as shown previously (Kondo et al., 2000); this is in contrast to the localization of ARF1 in 3Y1 cells, shown in Figure 3C.
These two lines of evidence suggest that Git2-short acts as a GAP at least for ARF1, the class I ARF, in vivo.
Overexpression of Git2-short Reduces Amounts of Paxillin-containing Focal Adhesions and Actin Stress Fibers
Norman et al. (1998) demonstrated that ARF1 activity is involved in paxillin recruitment to focal adhesions, as well as in Rho-stimulated stress fiber formation in Swiss 3T3 fibroblasts. Because our results described above suggest that Git2-short may act as a GAP for ARF1, we next examined whether Git2-short could affect the various cellular events in which ARF1 activity is thought to be involved.
As shown in Figure 8, we found that overexpression of Git2-short, but not its CA mutant, in 3Y1 and NIH3T3 fibroblasts affected paxillin localization and actin stress fiber formation. In fibroblasts, in which HA-Git2-short seemed to be overexpressed 20 times higher than the endogenous Git2-short (Mazaki and Sabe, unpublished results), the number of paxillin-containing focal adhesion plaques was significantly reduced (Figure 8A). The number and the amount of actin stress fibers were also reduced (Figure 8A). Because the focal accumulation of other focal adhesion proteins, such as vinculin (Figure 8B), and tyrosine-phosphorylated proteins (Mazaki and Sabe, unpublished results) also became almost undetectable upon Git2-short overexpression, the reduction in paxillin-containing focal adhesion plaques may be due to the overall disruption or disassembly of focal adhesion plaque structure upon Git2-short overexpression. The ARFGAP activity of Git2-short was also found to be involved in the perinuclear localization of paxillin (Figure 8, A and B). We also scanned the fibroblast cell body vertically and found that a large fraction of paxillin was existed diffusely in the cytoplasm, overlapping with overexpressed HA-Git2-short (Figure 8C). In contrast, it should be noted that localization of paxillin and vinculin to focal complexes at the cell periphery was almost unchanged or even slightly increased in HA-Git2-short–overexpressing cells (Figure 8). Although a single cell positive for HA-Git2-short is shown in Figure 8, 50–100 HA-Git2-short–overexpressing cells were examined, and 70–80% of them exhibited similar phenotypes.
Figure 8.

Effects of Git2-short overexpression on subcellular localization of paxillin and vinculin and actin cytoskeletal organization. (A and B) Overexpressing Git2-short, but not its GAP-inactive mutant, affects the perinuclear localization of paxillin and reduces the number of paxillin-containing focal adhesions at the cell bottom and actin stress fibers, whereas paxillin localization to focal complexes formed at the cell periphery is unaffected or slightly augmented. 3Y1 cells were transiently transfected with pcDNA3 plasmids encoding HA-Git2-short (a–e) or the CA mutant (f–j). Fibronectin-coated (10 μg/ml) chamber slides were used. a–e and f–j are each of the same field in A and B. HA-Git2-short proteins were visualized with rabbit anti-HA antibody coupled with Cy2 anti-rabbit IgG (a and f in A) or with Cy3 anti-rabbit IgG (a and f in B). Paxillin was visualized with mouse anti-paxillin antibody and Cy5 anti-mouse IgG (b and g in A) or by expression of EGFP-tagged paxillin (Mazaki et al., 1998) (b and g in B). F-actin (c and h in A) was visualized with Texas Red phalloidin. Vinculin (c and h in B) was visualized with anti-vinculin antibody coupled with Cy5 anti-mouse IgG. Focuses were adjusted 0.5 μm above the surface of the plates (Cell Bottom, a–d and f–i), or 3.0 μm above the surface of the plates (Cell Body, e and j). d and i represent the merging of panels b and c and g and h, respectively, in each A and B. (C) Subcellular localization of endogenous paxillin in Git2-short–overexpressing cells. 3Y1 cells were transiently transfected with pcDNA3 plasmids encoding HA-Git2-short. Paxillin was detected with mouse anti-paxillin antibody and Cy5 anti-mouse IgG (a, d, and g), and HA-Git2-short protein was detected with rabbit anti-HA antibody and Cy2 anti-rabbit IgG (b, e, and h). c, f, and i represent the merging of each of the left and the middle panels. All panels represent the same field in different focal planes: 3.0 μm (Cell Body, a–c), 1.7 μm (d–f), and 0.5 μm (Cell Bottom, g–i) above the surface of the plates. j represents the vertical section of the cell, indicated by a thin line in c, f, and i. Note that a large fraction of paxillin in the Git2-short–overexpressing cells seemed to be colocalized with overexpressed HA-Git2-short and was diffusely distributed in the cytoplasm, accompanied by the substantial disappearance of its tight perinuclear localization and focal adhesion localization. Localization of paxillin to focal complexes at the cell periphery was again unaffected or even slightly augmented. In A–C, arrows indicate cells overexpressing HA-Git2-short or its CA mutant. Essentially the same results were obtained in NIH3T3 cells (Mazaki and Sabe, unpublished results). Bars, 20 μm.
We also established clonal NIH3T3 cell lines in which HA-Git2-short expression was tightly controlled by a tetracycline-regulated promoter. In these cell clones, HA-Git2-short expression was induced ∼20-fold higher than that of endogenous Git2-short when tetracycline was depleted from the culture medium (Figure 9). Using these cell clones, we confirmed essentially the same results as described above, which were drawn using a transient transfection system. We also confirmed that overexpression of HA-Git2-short did not affect the expression levels of paxillin, vinculin, actin, Rho A, and ARFs (Figure 9A).
Figure 9.

Effects of Git2-short overexpression assessed in a single cell clone. An NIH3T3 cell clone bearing pTet-tTAK and pTet-Splice/HA-Git2-s was used. (A) Expression of HA-Git2-short, paxillin, vinculin, actin, Rho A, and ARFs in cells cultured in the presence (0.5 μg/ml, +) or in the absence (−) of tetracycline for 48 h. Cell lysates (20 μg) prepared with RIPA buffer (Sabe et al., 1994) were separated by SDS-PAGE (8%) and subjected to immunoblot analysis using antibodies as indicated. (B) Reduction in the number of paxillin-containing focal adhesion plaques and actin stress fibers by overexpression of HA-Git2-short and their partial restoration by co-overexpression of myc-ARFQ71L. Cells were cultured in the presence (a–c) or in the absence (d–i) of tetracycline, as above. Fibronectin-coated (10 μg/ml) chamber slides were used. a–c, d–f,and g–i are each of the same field; paxillin and F-actin were visualized as in Figure 8A. Arrows in g–i indicate a cell also expressing myc-ARF1Q71L (shown in i as blue), as visualized using rabbit anti-myc antibody and Cy2 anti-rabbit IgG. Focuses were adjusted 0.5 μm above the surface of the plates. Note that paxillin localization to focal complexes formed at the cell periphery was again almost unaffected or slightly augmented when HA-Git2-short was induced to be overexpressed (see d). Paxillin localization at the cell periphery was also augmented when ARF1Q71L was overexpressed (see g). (C–E) Effects of HA-Git2-short overexpression on cell migration on culture dishes (C), haptotaxis migration toward fibronectin in modified Boyden chambers (D), and cell adhesion to fibronectin (E). Cells were cultured in the presence (+) or in the absence (−) of tetracycline as above, and cell adhesion and migration activity were measured as described in MATERIALS AND METHODS. Each bar represents the mean ± SEM of triplicate experiments. *p < 0.02. In A–C, essentially the same results were obtained with three independent cell clones, in which HA-Git2-short was induced to be expressed at levels 20-fold higher than that of endogenous Git2-short. We also tried to examine the CA mutant overexpression in the same tetracycline-regulated system, but no cells overexpressed the CA mutant at levels similar to that of the wild-type HA-Git2-short clone. The cell clones we obtained could express the CA mutant up to about threefold higher than the endogenous Git2-short, and no significant changes in the amounts of paxillin-containing focal adhesions and stress fibers were observed (Mazaki and Sabe, unpublished results).
We also examined whether a dominant-active form of ARF1, ARF1Q71L, could suppress these Git2-short–induced phenotypes. As shown in Figure 9B, overexpression of ARF1Q71L caused partial restoration of paxillin-containing focal adhesion plaques and actin stress fibers. Perinuclear localization of paxillin was also significantly, but not completely, restored. However, the number of the adhesion plaques and stress fibers remained fewer compared with that in the control Tet (+) cells. Therefore, unlike in the case of the redistribution of β-COP (Figure 6), inactivation of ARF1 caused by the overexpression of Git2-short may not be the sole reason for the disappearance of paxillin-containing focal adhesions and stress fibers. It should be noted that paxillin localized to the cell periphery was altered in the ARF1Q71L-expressing cells compared with that in Tet (+) cells.
Finally, possible effects of Git2-short overexpression on cell adhesion and migratory activities were examined. After the induction of HA-Git2-short for 48 h, cell migration was traced over 24 h by a video recording under a relatively sparse cell culture. We found that overexpression of Git2-short could increase the cell migration speed, but only by ∼20% (Figure 9C). Haptotactic cell migration activity toward fibronectin, as assessed using modified Boyden chambers, was also enhanced, but only slightly (Figure 9D). On the other hand, cell adhesion activity onto fibronectin was not significantly affected by the overexpression of Git2-short (Figure 9E). Thus, the altered paxillin subcellular localization and the apparent decrease in amount of actin stress fibers caused by Git2-short overexpression do not hamper the cell adhesion activity significantly.
DISCUSSION
Integrins require the recruitment and assembly of a variety of proteins at their cytoplasmic tails for proper functioning. A number of integrin-mediated focal contacts are formed within a single cell during cell migration. Thus, for a cell to migrate in a certain direction, there must exist a mechanism that orchestrates the dynamic process of the recruitment and assembly of specific proteins at the cytoplasmic tails of each integrin molecule, as previously mentioned. To address this issue, we examined the subcellular localization and intracellular dynamics of paxillin (Mazaki et al., 1998; Kondo et al., 2000). We chose paxillin as a molecular tool because it can be tagged with GFP without affecting its biological properties (Mazaki et al., 1998), permitting analysis of its intracellular dynamics in situ in migrating cells (Nakamura et al., 2000; Yano et al., 2000; Kondo et al., 2000).
This study extends our previous inquires (Mazaki et al., 1998; Kondo et al., 2000). In this paper, we analyzed the perinuclear localization of paxillin using immunoelectron microscopy to provide the structural basis for our previous observation (Mazaki et al., 1998). Our next effort was to identify proteins that could interpret the intracellular dynamics of paxillin, including its Golgi localization. We had observed that several unknown proteins could be coprecipitated with paxillin (Mazaki et al., 1997), as others also reported recently (Turner et al., 1999). Given the assumption that, among paxillin-associated proteins, a protein(s) may exist that could regulate the subcellular localization of paxillin, we identified many paxillin-associated proteins and found that several of them contain zinc-finger motifs highly homologous to those of ARFGAP proteins, and we named them PAGs (Kondo et al., 2000). We have also shown that each class of ARF (we used ARF1, ARF5, and ARF6) can affect the subcellular localization of paxillin (Kondo et al., 2000), extending the previous report that ARF1 helps to regulate the focal adhesion recruitment of paxillin (Norman et al., 1998). We have therefore focused on analyzing a possible role for these PAG proteins in regulation of the subcellular localization of paxillin. We previously reported that one of the PAG proteins, PAG3, exhibits GAP activity for ARF6 and is colocalized with paxillin primarily at the cell periphery of epithelial and mature monocyte cells (Kondo et al., 2000). We showed here that, in contrast to PAG3, one of other PAGs, Git2-short, is primarily localized to perinuclear areas, exhibits a GAP activity for ARF1, and hence appears to be involved in the subcellular localization of paxillin and actin cytoskeletal organization. Our present study, however, is based primarily on deductive reasoning, and further study using inductive methods will be required to clarify whether Git2-short regulates ARF1 activity and hence regulates the subcellular localization of paxillin or whether paxillin regulates the subcellular localization of Git2-short and hence is involved in the regulation of ARF activities, or both.
Git2-short is a short isoform of Git2. The first 465 amino acids and the corresponding nucleotide sequences of Git2-short are identical to the corresponding region of Git2. However, the COOH-terminal amino acid sequence of Git2-short is different from that of Git2 (Figure 2A). Thus, Git2-short is not simply a truncated short isoform of Git2. We also showed that the binding affinity of Git2-short toward paxillin seems to be almost 10-fold weaker than that of Git2. Thus, Git2-short is an alternative spliced isoform of Git2 and interacts only weakly with paxillin. Such a weak interaction with paxillin has been observed, for example, with vinculin and talin (Mazaki et al., 1997), but the biological relevance of these weak interactions remains to be elucidated. Moreover, among paxillin isoforms, both Git2-short and Git2 exhibit selective binding to the α isoform. Similar selective binding of paxillin isoforms was observed toward focal adhesion kinase (Mazaki et al., 1997). The biological significance of such selective binding of paxillin isoforms also remains to be established but may indicate that the γ isoform acts as a dominant-negative form for the association of paxillin α with Git2-short and Git2. We have also tried to identify the amino acid sequence primarily responsible for the Golgi localization of Git2-short, to examine in more detail the biological significance of the perinuclear localization of Git2-short. However, so far we have been unable to obtain firm results on this point because of the instability of several of the mutant Git2-short proteins when expressed in mammalian cells.
Git2 is highly homologous to p95PKL (Turner et al., 1999) in both molecular size and amino acid sequence (89.5% homology in the amino acid sequence; Figure 2A), which has also been identified as a paxillin-binding protein. However, the peptide sequences we obtained do not correspond to those of p95PKL, because serine-175 in peptide KGNTPLHVASK (Figure 2A), encoded by the codon TCC, is an alanine in p95PKL, encoded by the codon GCT. Two potential paxillin-binding subdomains (PBS; Wood et al., 1994; Tachibana et al., 1995; Brown et al., 1998) have been identified within p95PKL (Turner et al., 1999). Amino acids 643–679 of p95PKL, which correspond to the second potential PBS sequence (PBS2), seem to make up the major paxillin-binding site (Turner et al., 1999) and are conserved in Git2. However, this region is not present in Git2-short. In Git2-short, we found that amino acid residues 125–165, which are also conserved in Git2 and p95PKL and correspond to the first potential PBS sequence (PBS1) of p95PKL, are primarily responsible for the direct binding to paxillin. Thus, Git2, and possibly also p95PKL, appears to have two distinct binding sites for paxillin; one (PBS2) is strong and the other (PBS1) is weak.
p95PKL binds to a guanine nucleotide exchanger, βPix (Turner et al., 1999). βPix also binds to the Pak kinase (Manser et al., 1998); p95PKL has therefore been proposed to function as a kinase linker connecting paxillin to Pak (Turner et al., 1999). We confirmed that Git2-short and Git2 also bind to βPix (Hashimoto and Sabe, unpublished results), as shown previously (Bagrodia et al., 1999). However, paxillin binding to Git2-short competes with βPix binding to Git2-short (Hashimoto and Sabe, unpublished results). Therefore, Git2-short does not seem to function to physically link paxillin to Pak.
We provided several lines of evidence supporting that Git2-short acts as a GAP for ARF1, rather than for ARF6, in vivo. During our analysis, however, Vitale et al. (2000) showed that Git2-short exhibits almost similar GTP-hydrolyzing activities against ARF1, 2, 3, 4, and 6 in vitro. As already mentioned, unlike GAP proteins for other small GTP-binding proteins such as Rho, coatomer proteins participate directly in the GTPase reaction of ARFGAP, and thus the activity and the specificity of ARFGAP may not be adequately assessed in vitro if the bona fide coatomer or effector protein is not present (Goldberg, 1999). Indeed, another PAG protein, PAG3/Papα/KIAA0400, exhibits much stronger GAP activity toward ARF1 and ARF5 than ARF6 in vitro (Andreev et al., 1999), but this protein turned out to exhibit significant GAP activity toward ARF6 in vivo in our assay system: we previously demonstrated that the overexpression of PAG3 suppresses the ARF6-induced phenotype of AlF-treated cells (Kondo et al., 2000). In contrast, here we have shown that the overexpression of Git2-short does not suppress the ARF6-induced phenotype but suppresses the ARF1-induced phenotype of AlF-treated cells. Moreover, we (Kondo et al., 2000) and others (Andreev et al., 1999) have shown that overexpression of PAG3/Papα/KIAA0400 does not affect the perinuclear localization of β-COP, unlike in the case of Git2-short overexpression. Furthermore, we have also confirmed the specificity of PAG3 and Git2-short using Fcγ receptor-mediated phagocytosis of macrophages (Uchida and Sabe, unpublished results), in which the class III ARF (ARF6), but not the other classes of ARFs, is involved (Zhang et al., 1998; Uchida and Sabe, unpublished results). However, different GAP proteins may be involved in the recruitment of different coatomer proteins to the same ARF (Springer et al., 1999). This may be the reason the subcellular localization of Git2-short is not completely overlapped with those of ARF1 and β-COP, and it also should be noted that Git2-short is not completely colocalized with ARF5 or ARF6 (Figure 3C). Moreover, the same ARFGAP may act on different ARF isoforms, and our results do not preclude the possibility that Git2-short may also act as a GAP for ARF6 in vivo in some other cellular contexts, as has been suggested by biochemical analysis (Vitale et al., 2000). Also, we have not yet examined a possible interaction of Git2-short with the class II ARFs, because the function of the class II ARF isoforms has not been well studied.
It is intriguing to suppose that ARF-regulated vesicle/membrane transport is involved in the focal adhesion recruitment of paxillin. Integrins are membrane proteins and are thus transported through intracellular vesicle/membrane trafficking. On the other hand, it has been demonstrated that paxillin is associated with integrins only after integrins are activated at cell surfaces (Miyamoto et al., 1995). Thus, paxillin may not associate with integrins within intracellular vesicles or the membrane. However, a recent model suggests that ARFGAP proteins may be constitutively associated with transporting vesicles (Springer et al., 1999). It is thus possible that the interaction of paxillin with several ARFGAP proteins, such as PAG3, Git-2, Git2-short, and p95PKL, can thereby allow paxillin to be transported along vesicle/membrane-trafficking pathways. However, this hypothesis remains to be explored. Moreover, in addition to paxillin, several other focal adhesion proteins, or proteins assembled as signaling complexes beneath the plasma membrane, may also be transported actively through some intracellular transporting pathways, because, for example, Pyk2 also binds to PAG3/Papα/KIAA0400 (Andreev et al., 1999) and vinculin also exhibits a perinuclear localization (Mazaki et al., 1998).
Git2-short–overexpressing cells showed a slight increase in migration speed (∼20%). Similarly, it has been shown that fibroblasts treated with Y-27632 to block activity of the Rho-effector p160ROCK are also characterized by increased cell migration speed (∼30%) (Nobes and Hall, 1999). Under both of these conditions, cells contain decreased amounts of actin stress fibers and focal adhesions (Figure 9; Nobes and Hall, 1999). It has been proposed that ARF1 can potentiate Rho A-stimulated stress fiber formation and ARF1 and Rho A activate complementary pathways that together lead to the formation of paxillin-rich focal adhesions (Norman et al., 1998). Therefore, our results again are not inconsistent with the notion that Git2-short acts as a GAP for ARF1: overexpression of Git2-short may affect an intracellular process(es) involving the activity of ARF1 together with Rho A and may thereby lead to a slight increment in cell migratory activity, which is accompanied by the reduction of actin stress fibers and focal adhesions (also see below).
Git2-short also binds to βPix, a guanine nucleotide exchange factor for Rac1 (Manser et al., 1998). Therefore, Git2-short appears to be versatile, possibly involved in regulation of different subfamilies of the small GTP-binding proteins by interacting with ARF1 and βPix. Moreover, βPix is enriched in focal complexes at the cell periphery colocalized with paxillin, but it does not accumulate at focal adhesions underneath the cell body (Manser et al., 1998). This subcellular localization of βPix is similar to that of Git2-short. We have shown that Git2-short overexpression seems to cause the overall disruption of focal adhesions rather than simply blocking the localization of paxillin to focal adhesions, as well as a slight increase in the amounts of paxillin and vinculin at the cell periphery. These phenotypes are again similar to those caused by the overexpression of βPix and by the activation of Rac1 (Nobes and Hall, 1995; Manser et al., 1998). We showed that a dominant-active form of ARF1 was incapable of completely suppressing the Git2-short–induced reduction in amounts of paxillin-containing focal adhesions and actin stress fibers (Figure 9B), whereas the dominant-active form of ARF1 could suppress the Git2-short–induced redistribution of β-COP at perinuclear areas (Figure 6). Thus, the pleiotropic functions of Git2-short appear to be involved in the formation of both paxillin-containing focal adhesions and actin stress fibers, whereas Git2-short seems to regulate Golgi organization only by its ARF1GAP activity. Norman et al. (1998) have shown that ARF1 is involved in the focal adhesion recruitment of paxillin but not vinculin. The versatility of Git2-short, in addition to its simple ARF1GAP activity, may be the reason for the apparent differences in phenotypes caused by Git2-short overexpression from those mediated by ARF1 (Norman et al., 1998).
A number of problems remain to be answered. First, more detailed analyses of the biological relevance of the weak interaction of paxillin with Git2-short should be conducted, as was discussed earlier; together with the functional relationship between Git2-short and Git2, the latter is the strong binding partner of paxillin. This may also provide further clues regarding the understanding of the mechanism of coordination of actin cytoskeletal reorganization and membrane/vesicle/receptor trafficking that occurs during cell migration. The precise mechanism for the intracellular association and dissociation of paxillin with Git2 isoforms (especially with Git2) should also be explored. This process may be related to the changes of the GTPases regulating actin-based cytoskeletal organization from Cdc42 to Rac, and then to Rho, which perhaps take place near the cell periphery during cell migration. Finally, during the revision process of our manuscript, an excellent paper (Cesare et al., 2000) was published that dealt with p95-APP1, another paxillin-binding ARFGAP, perhaps for ARF6, and addressed the cell motile machinery in a way similarly to ours (Mazaki et al., 1998; Kondo et al., 2000; this paper).
ACKNOWLEDGMENTS
We are grateful to T. Nagase (Kazusa DNA Research Institute, Chiba, Japan) for KIAA0148 cDNA, K. Nakayama (Tsukuba University, Ibaraki, Japan) for ARF cDNAs and useful discussions, and M. Tagaya (Tokyo University of Pharmacy and Life Science, Tokyo, Japan) for β-COP antibody. We also thank Mihoko Sato and Manami Hiraishi for their technical assistance, Mayumi Yoneda for her secretary work, and Heidi Greulich and Helena Akiko Popiel for their critical reading of the manuscript. This work was supported in part by Japan Science and Technology Corporation, grant in-aid from Ministry of Education, Science, Sports and Culture of Japan, grants from Takeda Medical Foundation, Mitsubishi Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and Novartis Foundation for the Promotion of Science.
Abbreviations used:
- AlF
aluminum fluoride
- ARF
ADP-ribosylation factor
- BFA
brefeldin A
- BSA
bovine serum albumin
- COP
coat protein
- ECM
extracellular matrix
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- GAP
GTPase-activating protein
- GFP
green fluorescent protein
- Git2
G protein-coupled receptor kinase interactor 2
- GST
glutathione S-transferase
- HA
influenza hemagglutinin
- NP-40
Nonidet P-40
- PAG
paxillin-associated protein with ARFGAP activity
- PBS
phosphate- buffered saline
- PCR
polymerase chain reaction
REFERENCES
- Abe K, Whitehead IP, O'Bryan JP, Der CJ. Involvement of NH2-terminal sequences in the negative regulation of Vav signaling and transforming activity. J Biol Chem. 1999;274:30410–30418. doi: 10.1074/jbc.274.43.30410. [DOI] [PubMed] [Google Scholar]
- Andreev J, Simon JP, Sabatini DD, Kam J, Plowman G, Randazzo PA, Schlessinger J. Identification of a new Pyk2 target protein with Arf-GAP activity. Mol Cell Biol. 1999;19:2338–2350. doi: 10.1128/mcb.19.3.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagrodia S, Bailey D, Lenard Z, Hart M, Guan JL, Premont RT, Taylor SJ, Cerione RA. A tyrosine-phosphorylated protein that binds to an important regulatory region on the Cool family of p21-activated kinase-binding proteins. J Biol Chem. 1999;274:22393–22400. doi: 10.1074/jbc.274.32.22393. [DOI] [PubMed] [Google Scholar]
- Balch WE, Kahn RA, Schwaninger R. ADP-ribosylation factor is required for vesicular trafficking between the endoplasmic reticulum and the cis-Golgi compartment. J Biol Chem. 1992;267:13053–13061. [PubMed] [Google Scholar]
- Boman AL, Kahn RA. Arf proteins: the membrane traffic police? Trends Biochem Sci. 1995;20:147–150. doi: 10.1016/s0968-0004(00)88991-4. [DOI] [PubMed] [Google Scholar]
- Bonifacino JS, Suzuki CK, Lippincott-Schwartz J, Weissman AM, Klausner RD. Pre-Golgi degradation of newly synthesized T-cell antigen receptor chains: intrinsic sensitivity and the role of subunit assembly. J Cell Biol. 1989;109:73–83. doi: 10.1083/jcb.109.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Curtis MS, Turner CE. Paxillin LD. motifs may define a new family of protein recognition domains. Nat Struct Biol. 1998;5:677–678. doi: 10.1038/1370. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Burridge K, Turner CE, Romer LH. Tyrosine phosphorylation of paxillin and pp125FAK accompanies cell adhesion to extracellular matrix: a role in cytoskeletal assembly. J Cell Biol. 1992;119:893–903. doi: 10.1083/jcb.119.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare AD, Paris S, Albertinazzi C, Dariozzi S, Andersen J, Mann M, Longhi R, de Curtis I. p95-APP1 links membrane transport to Rac-mediated reorganization of actin. Nat Cell Biol. 2000;2:521–530. doi: 10.1038/35019561. [DOI] [PubMed] [Google Scholar]
- Chardin P, McCormick F. Brefeldin A: the advantage of being incompetitive. Cell. 1999;97:153–155. doi: 10.1016/s0092-8674(00)80724-2. [DOI] [PubMed] [Google Scholar]
- Clark EA, Brugge JS. Integrins and signal transduction pathways: the road taken. Science. 1995;268:233–239. doi: 10.1126/science.7716514. [DOI] [PubMed] [Google Scholar]
- Cockcroft S. ARF-regulated phospholipase D, a potential role in membrane traffic. Chem Phys Lipids. 1996;80:59–80. doi: 10.1016/0009-3084(96)02546-7. [DOI] [PubMed] [Google Scholar]
- Cukierman E, Huber I, Rothman M, Cassel D. The ARF1 GTPase-activating protein: zinc finger motif and Golgi complex localization. Science. 1995;270:1999–2002. doi: 10.1126/science.270.5244.1999. [DOI] [PubMed] [Google Scholar]
- Dascher C, Balch WE. Dominant inhibitory mutants of ARF1 block endoplasmic reticulum to Golgi transport and trigger disassembly of the Golgi apparatus. J Biol Chem. 1994;269:1437–1448. [PubMed] [Google Scholar]
- Donaldson JG, Cassel D, Kahn RA, Klausner RD. ADP-ribosylation factor, a small GTP-binding protein, is required for binding of the coatomer protein beta-COP to Golgi membranes. Proc Natl Acad Sci USA. 1992;89:6408–6412. doi: 10.1073/pnas.89.14.6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson JG, Kahn RA, Lippincott-Schwartz J, Klausner RD. Binding of ARF and β-COP to Golgi membranes: possible regulation by a trimeric G proteins. Science. 1991;254:1197–1199. doi: 10.1126/science.1957170. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Klausner RD. ARF: a key regulatory switch in membrane traffic and organelle structure. Curr Opin Cell Biol. 1994;6:527–532. doi: 10.1016/0955-0674(94)90072-8. [DOI] [PubMed] [Google Scholar]
- Finazzi D, Cassel D, Donaldson JG, Klausner RD. Aluminum fluoride acts on the reversibility of ARF1-dependent coat protein binding to Golgi membranes. J Biol Chem. 1994;269:13325–13330. [PubMed] [Google Scholar]
- Goldberg J. Structural and functional analysis of the ARF1-ARFGAP complex reveals a role for coatomer in GTP hydrolysis. Cell. 1999;96:893–902. doi: 10.1016/s0092-8674(00)80598-x. [DOI] [PubMed] [Google Scholar]
- Graham IL, Anderson DC, Holers VM, Brown E. Complement receptor 3 (CR3, Mac-1, integrin alpha M beta 2, CD11b/CD18) is required for tyrosine phosphorylation of paxillin in adherent and nonadherent neutrophils. J Cell Biol. 1994;127:1139–1147. doi: 10.1083/jcb.127.4.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Toda K, Takatsu H, Torii S, Murakami K, Nakayama K. Structure and intracellular localization of mouse ADP-ribosylation factors type 1 to type 6 (ARF1-ARF6) J Biochem. 1996;120:813–819. doi: 10.1093/oxfordjournals.jbchem.a021484. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 1992;69:11–25. doi: 10.1016/0092-8674(92)90115-s. [DOI] [PubMed] [Google Scholar]
- Iwamatsu A. S-carboxymethylation of proteins transferred onto polyvinylidene difluoride membranes followed by in situ protease digestion and amino acid microsequencing. Electrophoresis. 1992;13:142–147. doi: 10.1002/elps.1150130129. [DOI] [PubMed] [Google Scholar]
- Kahn RA, Gilman AG. The protein cofactor necessary for ADP-ribosylation of Gs by cholera toxin is itself a GTP binding protein. J Biol Chem. 1986;261:7906–7911. [PubMed] [Google Scholar]
- Kahn RA, Randazzo P, Serafini T, Weiss O, Rulka C, Clark J, Amherdt M, Roller P, Orci L, Rothman JE. The amino terminus of ADP-ribosylation factor (ARF) is a critical determinant of ARF activities and is a potent and specific inhibitor of protein transport. J Biol Chem. 1992;267:13039–13046. [PubMed] [Google Scholar]
- Kahn RA, Yucel JK, Malhotra V. ARF signaling: a potential role for phospholipase D in membrane traffic. Cell. 1993;75:1045–1048. doi: 10.1016/0092-8674(93)90314-g. [DOI] [PubMed] [Google Scholar]
- Kondo A, Hashimoto S, Yano H, Nagayama K, Mazaki Y, Sabe H. A new paxillin-binding protein, PAG3/Papα/KIAA0400, bearing an Arf GTPase-activating protein activity is involved in paxillin recruitment to focal adhesions and cell migration. Mol Biol Cell. 2000;11:1315–1327. doi: 10.1091/mbc.11.4.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis TE, Lowe M, Pepperkok R. COPs regulating membrane traffic. Annu Rev Cell Dev Biol. 1995;11:677–706. doi: 10.1146/annurev.cb.11.110195.003333. [DOI] [PubMed] [Google Scholar]
- Lenhard JM, Kahn RA, Stahl PD. Evidence for ADP-ribosylation factor (ARF) as a regulator of in vitro endosome-endosome fusion. J Biol Chem. 1992;267:13047–13052. [PubMed] [Google Scholar]
- Letourneur F, Gaynor EC, Hennecke S, Demolliere C, Duden R, Emr SD, Riezman H, Cosson P. Coatomer is essential for retrieval of dilysine-tagged proteins to the endoplasmic reticulum. Cell. 1994;79:1199–1207. doi: 10.1016/0092-8674(94)90011-6. [DOI] [PubMed] [Google Scholar]
- Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cells. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- Mayer BJ, Hirai H, Sakai R. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 1995;5:296–305. doi: 10.1016/s0960-9822(95)00060-1. [DOI] [PubMed] [Google Scholar]
- Mazaki Y, Hashimoto S, Sabe H. Monocyte cells and cancer cells express novel paxillin isoforms with different binding properties to focal adhesion proteins. J Biol Chem. 1997;272:7437–7444. doi: 10.1074/jbc.272.11.7437. [DOI] [PubMed] [Google Scholar]
- Mazaki Y, Uchida H, Hino O, Hashimoto S, Sabe H. Paxillin isoforms in mouse: lack of the gamma isoform and developmentally specific beta isoform expression. J Biol Chem. 1998;273:22435–22441. doi: 10.1074/jbc.273.35.22435. [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Teramoto H, Coso OA, Gutkind JS, Burbelo PD, Akiyama SK, Yamada KM. Integrin function: molecular hierarchies of cytoskeletal and signaling molecules. J Cell Biol. 1995;131:791–805. doi: 10.1083/jcb.131.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi A, Yano Y, Hamaguchi H, Yanagida H, Ide C, Zahraoui A, Shirataki H, Sasaki T, Takai Y. Localization of rabphilin-3A on the synaptic vesicle. Biochem Biophys Res Commun. 1994;202:1235–1243. doi: 10.1006/bbrc.1994.2063. [DOI] [PubMed] [Google Scholar]
- Morgenstern JP, Land H. Advanced mammalian gene transfer: high titer retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990;18:3587–3596. doi: 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss J, Vaughan M. Structure, and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem. 1995;270:12327–12330. doi: 10.1074/jbc.270.21.12327. [DOI] [PubMed] [Google Scholar]
- Moss J, Vaughan M. Molecules in the ARF orbit. J Biol Chem. 1998;273:21431–21434. doi: 10.1074/jbc.273.34.21431. [DOI] [PubMed] [Google Scholar]
- Nagase T, Seki N, Tanaka A, Ishikawa K, Nomura N. Prediction of the coding sequences of unidentified human genes. IV. The coding sequences of 40 new genes (KIAA0121-KIAA0160) deduced by analysis of cDNA clones from human cell line KG-1. DNA Res. 1995;2:167–174. doi: 10.1093/dnares/2.4.167. [DOI] [PubMed] [Google Scholar]
- Nakamura K, Yano H, Uchida H, Hashimoto S, Schaefer E, Sabe H. Tyrosine phosphorylation of paxillin is involved in temporospatial regulation of paxillin-containing focal adhesion formation and F-actin organization in motile cells. J Biol Chem. 2000;275:27155–27164. doi: 10.1074/jbc.M000679200. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases, control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JC, Jones D, Barry ST, Holt MR, Cockcroft S, Critchley DR. ARF1 mediates paxillin recruitment to focal adhesions and potentiates Rho-stimulated stress fiber formation in intact and permeabilized Swiss 3T3 fibroblasts. J Cell Biol. 1998;143:1981–1995. doi: 10.1083/jcb.143.7.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuoffer C, Balch WE. GTPases: multifunctional molecular switches regulating vesicular traffic. Annu Rev Biochem. 1994;63:949–990. doi: 10.1146/annurev.bi.63.070194.004505. [DOI] [PubMed] [Google Scholar]
- Ooi CE, Dell'Angelica EC, Bonifacino JS. ADP-ribosylation factor 1 (ARF1) regulates recruitment of the AP-3 adaptor complex to membranes. J Cell Biol. 1998;142:391–402. doi: 10.1083/jcb.142.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters PJ, Hsu VW, Ooi CE, Finazzi DB, Teal S, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon PP, Wang X, Rotman M, Huber I, Cukierman E, Cassel D, Singer RA, Johnston GC. Saccharomyces cerevisiae Gcs1 is an ADP-ribosylation factor GTPase-activating protein. Proc Natl Acad Sci USA. 1996;93:10074–10077. doi: 10.1073/pnas.93.19.10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Claing A, Vitale N, Freeman JL, Pitcher JA, Patton WA, Moss J, Vaughan M, Lefkowitz RJ. beta2-Adrenergic receptor regulation by GIT1, a G protein-coupled receptor kinase-associated ADP ribosylation factor GTPase-activating protein. Proc Natl Acad Sci USA. 1998;95:14082–14087. doi: 10.1073/pnas.95.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Premont RT, Claing A, Vitale N, Perry SJ, Lefkowitz RJ. The GIT family of ADP-ribosylation factor GTPase-activating proteins. J Biol Chem. 2000;275:22373–22380. doi: 10.1074/jbc.275.29.22373. [DOI] [PubMed] [Google Scholar]
- Radhakrishna H, Al-Awar O, Khachikian Z, Donaldson JG. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J Cell Sci. 1999;112:855–866. doi: 10.1242/jcs.112.6.855. [DOI] [PubMed] [Google Scholar]
- Radhakrishna H, Klausner RD, Donaldson JG. Aluminum fluoride stimulates surface protrusions in cells overexpressing the ARF6 GTPase. J Cell Biol. 1996;134:935–947. doi: 10.1083/jcb.134.4.935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth MG. Snapshots of ARF1: implications for mechanisms of activation and inactivation. Cell. 1999;97:149–152. doi: 10.1016/s0092-8674(00)80723-0. [DOI] [PubMed] [Google Scholar]
- Sabe H, Hata A, Okada M, Nakagawa H, Hanafusa H. Analysis of the binding of the Src homology 2 domain of Csk to tyrosine-phosphorylated proteins in the suppression and mitotic activation of c-Src. Proc Natl Acad Sci USA. 1994;91:3984–3988. doi: 10.1073/pnas.91.9.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schekman R, Orci L. Coat proteins and vesicle budding. Science. 1996;271:1526–1533. doi: 10.1126/science.271.5255.1526. [DOI] [PubMed] [Google Scholar]
- Serafini T, Orci L, Amherdt M, Brunner M, Kahn RA, Rothman JE. ADP-ribosylation factor is a subunit of the coat of Golgi-derived COP-coated vesicles: a novel role for a GTP-binding protein. Cell. 1991;67:239–253. doi: 10.1016/0092-8674(91)90176-y. [DOI] [PubMed] [Google Scholar]
- Song J, Khachikian Z, Radhakrishna H, Donaldson JG. Localization of endogenous ARF6 to sites of cortical actin rearrangement and involvement of ARF6 in cell spreading. J Cell Sci. 1998;111:2257–2267. doi: 10.1242/jcs.111.15.2257. [DOI] [PubMed] [Google Scholar]
- Springer S, Spang A, Schekman R. A primer on vesicle budding. Cell. 1999;97:145–148. doi: 10.1016/s0092-8674(00)80722-9. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell. 1993;73:999–1005. doi: 10.1016/0092-8674(93)90277-w. [DOI] [PubMed] [Google Scholar]
- Stearns T, Willingham MC, Botstein D, Kahn RA. ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc Natl Acad Sci USA. 1990;87:1238–1242. doi: 10.1073/pnas.87.3.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana K, Sato T, D'Airro N, Moromoto C. Direct association of pp125FAK with paxillin, the focal adhesion-targeting mechanism of pp125FAK. J Exp Med. 1995;182:1089–1100. doi: 10.1084/jem.182.4.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Imajoh-Ohmi S, Sawada T, Shirai R, Hashimoto Y, Iwasaki S, Kaibuchi K, Kanaho Y, Shirai T, Terada Y, Kimura K, Nagata S, Fukui Y. A target of phosphatidylinositol 3,4,5-trisphosphate with a zinc finger motif similar to that of the ADP-ribosylation-factor GTPase-activating protein and two pleckstrin homology domains. Eur J Biochem. 1997;245:512–519. doi: 10.1111/j.1432-1033.1997.00512.x. [DOI] [PubMed] [Google Scholar]
- Tong X, Howley PM. The bovine papillomavirus E6 oncoprotein interacts with paxillin and disrupts the actin cytoskeleton. Proc Natl Acad Sci USA. 1997;94:4412–4417. doi: 10.1073/pnas.94.9.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Salgia R, Li J-L, Griffin JD, Howley PM. The bovine papillomavirus E6 protein binds to the LD motif repeats of paxillin and blocks its interaction with vinculin and the focal adhesion kinase. J Biol Chem. 1997;272:33373–33376. doi: 10.1074/jbc.272.52.33373. [DOI] [PubMed] [Google Scholar]
- Turner CE. Paxillin. Int J Biochem Cell Biol. 1998;30:955–959. doi: 10.1016/s1357-2725(98)00062-4. [DOI] [PubMed] [Google Scholar]
- Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale N, Moss J, Vaughan M. Interaction of GTP-binding and GTPase-activating domains of ARD1 involves the effector region of the ADP-ribosylation factor domain. J Biol Chem. 1997;272:3897–3904. doi: 10.1074/jbc.272.7.3897. [DOI] [PubMed] [Google Scholar]
- Vitale N, Patton WA, Moss J, Vaughan M, Lefkowitz RJ, Premont RT. GIT proteins, a novel family of phosphatidylinositol 3,4,5-triphosphate-stimulated GTPase-activating proteins for ARF6. J Biol Chem. 2000;275:13901–13906. doi: 10.1074/jbc.275.18.13901. [DOI] [PubMed] [Google Scholar]
- West MA, Bright NA, Robinson MS. The role of ADP-ribosylation factor and phospholipase D in adaptor recruitment. J Cell Biol. 1997;138:1239–1254. doi: 10.1083/jcb.138.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood CK, Turner CE, Jackson P, Critchley DR. Characterization of the paxillin-binding site and the C-terminal focal adhesion targeting sequence in vinculin. J Cell Sci. 1994;197:709–717. [PubMed] [Google Scholar]
- Yamaguchi T, Nakayama K, Hatsuzawa K, Tani K, Himeno M, Tagaya M. ADP-ribosylation factor-1 is sensitive to N-ethylmaleimide. J Biochem. 1998;124:1229–1236. doi: 10.1093/oxfordjournals.jbchem.a022242. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Harada N, Kano K, Taya S, Canaani E, Matsuura Y, Mizoguchi A, Ide C, Kaibuchi K. The Ras target AF-6 interacts with ZO-1 and serves as a peripheral component of tight junction in epithelial cell. J Cell Biol. 1997;139:785–795. doi: 10.1083/jcb.139.3.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano H, Uchida H, Iwasaki T, Mukai M, Akedo H, Nakamura K, Hashimoto S, Sabe H. Paxillin α and Crk-associated substrate exert opposing effects on cell migration and contact inhibition of growth through tyrosine phosphorylation. Proc Natl Acad Sci USA. 2000;97:9076–9081. doi: 10.1073/pnas.97.16.9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C-J, Rosenwald AG, Willingham MC, Skuntz S, Clark J, Kahn RA. Expression of a dominant allele of human ARF1 inhibits membrane traffic in vivo. J Cell Biol. 1994;124:289–300. doi: 10.1083/jcb.124.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang O, Cox D, Tseng C-C, Donaldson JG, Greenberg S. A requirement of ARF6 in Fcγ receptor-mediated phagocytosis in macrophages. J Biol Chem. 1998;273:19977–19981. doi: 10.1074/jbc.273.32.19977. [DOI] [PubMed] [Google Scholar]