

Abstract
We have generated a high-spin FeIII–OOH complex supported by tetramethylcyclam via protonation of its conjugate base and characterized it in detail by various spectroscopic methods. This FeIII–OOH species converts quantitatively to an FeIV=O complex via O–O bond cleavage, which represents the first example of such a conversion. This conversion is promoted by two factors: the strong FeIII–OOH bond that inhibits Fe–O bond lysis and the addition of protons that facilitate O–O bond cleavage. This example provides a synthetic precedent for how O–O bond cleavage of high-spin iron(III)-peroxo intermediates of nonheme iron enzymes may be promoted.
Cleavage of the O–O bond of iron(III)-hydroperoxo (FeIII–OOH) species is a key step in the O2 activation mechanisms of cytochrome P450,1 Rieske dioxygenases,2 and even methane monooxygenase (MMO),3 leading to high-valent iron-oxo species that effect organic substrate oxidation. On the other hand, Fe–O bond cleavage must occur to release H2O2 in the catalytic cycle of superoxide reductase (SOR)4 and for cytochrome P450 reactions that exhibit uncoupling.1 While protonation of the proximal O atom of the FeIII–OOH unit can be readily envisioned as the step needed to release H2O2, the mechanism for O–O bond cleavage is not as simple. For heme peroxidases and cytochrome P450, it is generally accepted that protonation of the distal O atom of the low-spin FeIII–OOH intermediate facilitates the heterolysis of the O–O bond.1 This notion should also apply to nonheme iron systems, but the likely high-spin state of iron-peroxo species in nonheme enzymes could raise the barrier for O–O bond lysis relative to that for low-spin counterparts in heme enzymes.5 The scarcity of experimental evidence further limits insights into O–O bond scission by nonheme enzymes. The only mechanistically relevant information available is for MMO, where the conversion of the peroxodiiron(III) intermediate to the diiron(IV) oxidant exhibits a pH dependence and a H/D solvent kinetic isotope effect,3 emphasizing the key role played by a proton in O–O bond cleavage. Among synthetic complexes, there are only a few nonheme FeIII–OOH complexes that are spectroscopically well characterized,6,7 but none of them has been directly observed to generate a high-valent iron-oxo intermediate, thus making it difficult to obtain mechanistic insights into this key step in iron-catalyzed oxygen activation.
To obtain the first example of a FeIII–OOH complex that can undergo O–O bond cleavage to generate a high-valent iron-oxo complex, we have focused on trapping [FeIII(TMC)(OOH)]2+ (2; see Scheme 1A for a structure of TMC), a yet elusive species invoked in the reactions of O2 with [FeII(TMC)(CH3CN)]2+ that afford [FeIV(O)(TMC)(CH3CN)]2+ (3) in 60–80% yield as the final product.8 Herein we report the high-yield generation of 2 by protonation of the previously reported [FeIII(TMC)(O2)]+ complex (1)9 at −40 °C, its detailed spectroscopic characterization, and kinetic studies that shed mechanistic light on the quantitative conversion of 2 to 3 (Scheme 1B).
Scheme 1.

(A) TMC ligand used in this study. (B) Conversion of 1 to 2 and then to 3 via a putative short-lived FeV=O intermediate.
Complex 1 was generated using the published procedure7 by treating 2.0 mM [FeII(TMC)(CH3CN)]2+ in CH3CN with 10 equiv. NEt3 followed by 20 equiv. H2O2 in CH3CN at −40 °C. Complex 1 exhibits a λmax of 835 nm (Figure 1 left panel) with an ε value of 650 M−1cm−1, established with the aid of Mössbauer data (vide infra). Upon addition of 20 equiv. HClO4 at −40 °C, 1 converted immediately to a short-lived (t1/2 ~ 1 min) maroon intermediate (2) with a shoulder-like absorption feature at ~500 nm (ε = 450 M− 1cm−1), which in turn decayed to give the signature absorption feature of 3 (Figure 1). The significant blue shift observed in the conversion of 1 to 2 has been noted previously in the protonation of other nonheme FeIII(η2-O2) species, consistent with the weaker basicity of the hydroperoxo monoanion compared to the peroxo dianion.6a–b The conversion of 1 to 2 is reversible. Addition of excess NEt3 to the solution of 2 instantly results in the near-quantitative regeneration of 1, as shown by its characteristic absorption band. This cycle can be repeated several times (see Figure S1). These results further underscore that 2 is the conjugate acid of 1 and can be formulated as [FeIII(TMC)(OOH)]2+.
Figure 1.

Left: UV-visible absorption spectra of [FeIII(TMC)(O2)]+ (1) (blue dashed line) and [FeIII(TMC)(OOH)]2+ (2) (red solid line) in CH3CN. Right: UV-visible absorption spectra during the conversion of 2 (red solid line) to [FeIV(O)(TMC)(CH3CN)]2+ (3) (brown dashed line) via the addition of ca. 20 equiv. HClO4 in CH3CN at −40 °C. b = 1 cm. Inset: Close-up view to show the isosbestic point at ~695 nm.
The Mössbauer samples of 1, 2 and 3 contain large fractions of the designated complexes. In the spectrum of 1 shown in Figure 2A, about 90% of total Fe belongs to 1, while 80% of the Fe in Figure 2B can be assigned to 2 with 10% of the iron belonging to FeIV=O complex 3 (vide infra for analysis details). Analysis of a sample of 2 that was allowed to decay for 5 min after addition of HClO4 shows that 90% of the total Fe corresponds to 3 (Fig. S9). Thus, the overall conversion of 1 to 3 is essentially quantitative.
Figure 2.
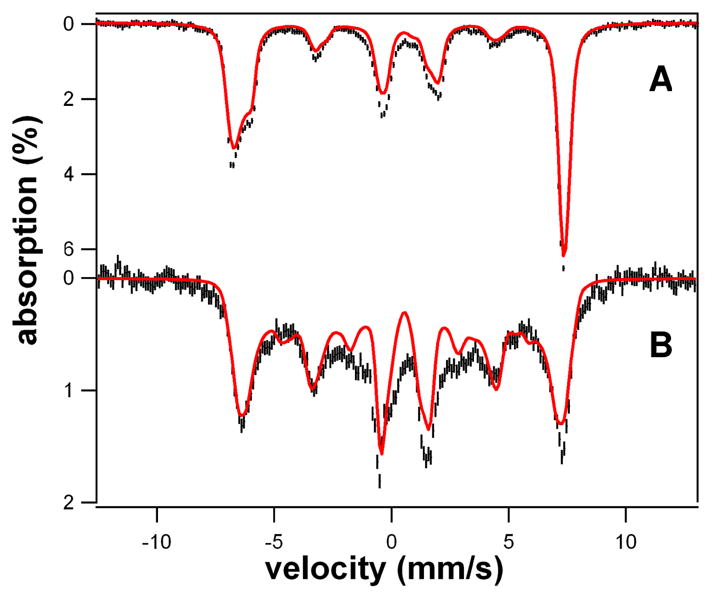
4.2 K Mössbauer spectra of 1 and 2 in 3:1 PrCN/MeCN (v/v) recorded in parallel applied fields (black lines) and simulations (red lines); simulation parameters and comments are given in Table S1 and Supporting Information. (A) 8.0 T spectrum of 1; same batch as used for EPR shown in Fig. 3A. (B) 1.2 T spectrum of 2. The red line is a spectral simulation, assuming slow relaxation of the electronic system. 10% of 3 was removed from the data. Approximately 80% of the Fe in the sample belongs to 2.
The electronic structures of 1 and 2 were established by EPR and Mössbauer spectroscopy. Both complexes are high-spin FeIII with quite different zero field splitting (ZFS) parameters and isomer shifts (Table S1). We have analyzed the data with the S = 5/2 spin Hamiltonian, eq 1,
where D and E are the axial and rhombic ZFS parameters, A is the 57Fe magnetic hyperfine tensor and
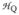 describes the nuclear quadrupole interactions.
describes the nuclear quadrupole interactions.
The X-band EPR spectrum of 1 exhibits, for the middle Kramers doublet, signals at geff = 4.58, 4.38, and ≈ 4.1 (Figures 3A and S4–7). Mössbauer analysis (details in Supporting Information) shows that D ≈ −0.9 cm−1 and E/D = 0.28(1). With E/D fixed, the explanation of the EPR features of 1 requires inclusion of substantial fourth-order ZFS parameters, eq 2.
Figure 3.
X-band EPR spectra of 1 and 2 in 3:1 PrCN/MeCN (v/v) shown in black lines (A) Complex 1. T = 15 K, microwave power, 0.02 mW, 1 mT modulation. The red line is a simulation using D = −0.91 cm−1, E/D = 0.28, fourth-order parameters F = −0.108 cm−1 and a = −0.017 cm−1 (see Supporting Information), g = (2.04, 1.98, 2.03) and distributed E/D with σE/D = 0.038. (B) Complex 2. T = 10 K, microwave power, 2.0 mW, 1 mT modulation. The red line is a simulation using D = 2.5 cm−1, E/D = 0.097, g = (2.00, 2.00, 2.00), and σE/D = 0.02. The sharp features at geff = 4.58 and 4.36 represent remaining 1, corresponding to (only) 1% of total Fe.
For the simulation of Figure 3A, we used F = −0.108 cm−1 and a = −0.017 cm−1. Large fourth-order parameters, namely a = 0.074 cm−1 and F = 0.043 cm−1, have been reported for Fe superoxide dismutase-azide,10 together with D = 0.46 cm−1 and E/D = 0.255. An 8.0 T Mössbauer spectrum of 1 together with a spectral simulation is shown in Figure 2A (see Figures S2 and S3 for additional spectra). The shoulder on the low energy feature (at ~ −6 mm/s Doppler velocity) and the fairly sharp high-energy line show that the 57Fe A-tensor is anisotropic. The spectrum shown gives a value for the isomer shift of 1, δ = 0.58 mm/s, similar to that of the side-on peroxo complex [FeIII(N4Py)(η2-O2)]+ (δ = 0.61 mm/s).6b See SI for additional details of the spectral analysis.
Analysis of the Mössbauer spectra of 2 shown in Figures 2B and S8 yielded D = +2.5 cm−1, E/D = 0.097(7) and δ = 0.51 mm/s. The EPR spectrum of 2 exhibits signals at geff = 8.00 (ground doublet), 5.71 (middle) and 3.4 (ground), consistent with the above D and E/D values (Figure 3B). The parameters used for the simulations of 1 and 2 are listed in Table S1.
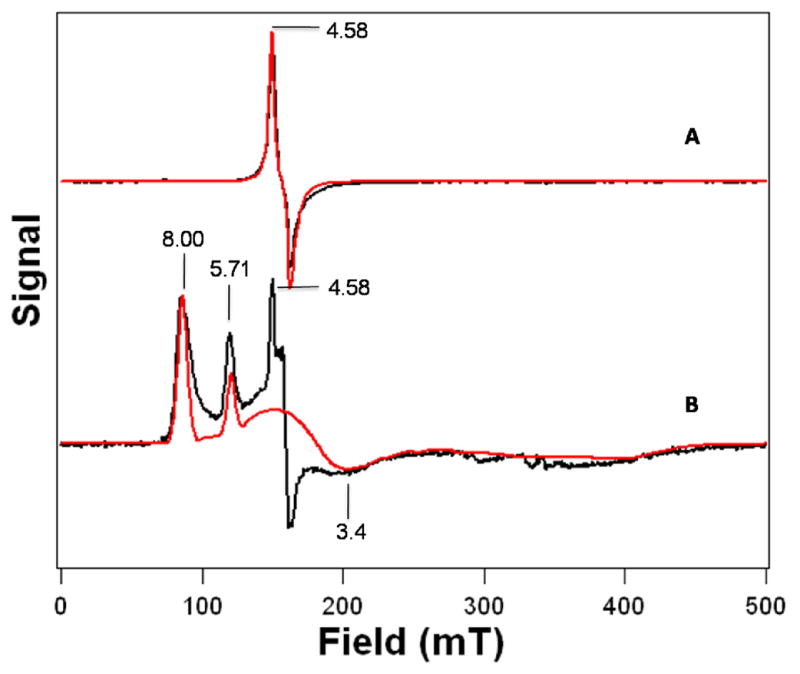
We also carried out Fe K-edge X-ray absorption spectroscopic (XAS) studies to obtain structural information and metric parameters for 1 and 2 (Figure 4, Tables S1–S3). The EXAFS spectrum of 1 is best fit by two N/O scatterers at 1.93 Å and four N/O scatterers at 2.20 Å, while the best fit for 2 consists of one N/O scatterer at 1.92 Å and four N/O scatterers at 2.15 Å. Interchanging the number of scatterers in the 1.9-Å sub-shells of 1 and 2, which arise from the peroxo ligands, significantly worsened the overall fit quality for both complexes with unacceptable Debye-Waller factors for this sub-shell in both cases (Tables S2 and S3). These results lead to the respective assignments of an η2-binding mode to the dianionic peroxo ligand in 1 and an η1-binding mode to the monoanionic hydroperoxo ligand of 2. This conclusion is supported by the observed 0.05-Å decrease in r(Fe-NTMC) from 1 to 2.11
Figure 4.
Top: Fourier transform of Fe K-edge EXAFS of 1 (dotted line) over a k-range of 2–15 Å−1 and a back-transformation range of 0.3–3.0 Å−1. Inset: Fourier-filtered k3χ(k) EXAFS data (dotted line). Solid lines represent fit 9 in Table S2. Bottom: Fourier transform of Fe K-edge EXAFS of 2 (dotted line) over a k-range of 2–14 Å−1 and a back-transformation range of 0.3–3.2 Å−1. Inset: Fourier-filtered k3χ(k) EXAFS data (dotted line). Solid lines represent fit 9 in Table S3.
Resonance Raman studies of 1 and 2 provide additional insight into how the difference in binding mode affects the two high-spin iron(III)-peroxo units. Resonance enhanced vibrations are found for 1 at 826 and 493 cm−1 and for 2 at 870 and 676 cm−1 (Figures 5 and S10). These features can be assigned respectively to ν(O–O) and ν(Fe–O) modes on the basis of downshifts observed upon 18O-labeling, which conform to predictions for diatomic harmonic oscillators by Hooke’s Law. The observed vibrational modes compare favorably to those reported for related complexes (Tables 1 and S4). The sole exception is the ν(Fe–O) of 2 at 676 cm−1, which lies above the range of ν(Fe–O) values (420–620 cm−1) found for other high-spin FeIII–OOH complexes studied thus far (Table 1). We speculate that the high ν(Fe–O) value for 2 may reflect the weaker electron donating ability of the presumed CH3CN ligand trans to the hydroperoxo unit relative to those of the other complexes in Table 1. The stronger Fe–O bond suggested by the high ν(Fe–O) value for 2 is also likely to be an important factor that contributes to the observed cleavage of its O–O bond. In contrast, the FeIII–OOH units of oxyhemerythrin12 and superoxide reductase (SOR),13 with ν(Fe–O) values that are at least 100 cm−1 smaller, undergo Fe–O bond cleavage in the course of their respective functions.
Figure 5.

Top: resonance Raman spectra of 1 prepared with H2O2 (black line) or H2 18O2 (red line) and obtained with λexc = 647.1 nm. Bottom: resonance Raman spectra of 2 prepared with H2O2 (black line) or H2 18O2 (red line) and obtained with λexc = 514.5 nm.
Table 1.
Vibrational frequencies of high-spin FeIII–OOH species.
| Complexesa | ν(Fe–O), cm−1 (Δ18O) [Δ2H] | ν(O–O), cm−1 (Δ18O) [Δ2H] | ref |
|---|---|---|---|
| 2 | 676 (−24) [−1] | 870 (−50) [−1] | b |
| [Fe(H2bppa)(OOH)]2+ | 621 (−22) | 830 (−17) [−4] | 7a |
| E114A SOR | 567 (−4) | 838 (−23) | 13 |
| oxyhemerythrin | 503 (−24) [−3] | 844 (−48) [+4] | 12 |
| [Fe(cyclam-PrS)(OOH)]+ | 419 (−19) | 891 (−35) [c] | 7b |
H2bppa = bis(6-pivalamido-2-pyridyl-methyl)(2-pyridylmethyl)amine; SOR = superoxide reductase; cyclam-PrS = 1-(3′-mercaptopropyl)-1,4,8,11-tetraazacyclotetradecane anion.
This work;
Fermi doublet observed to collapse in deuterated solvent but no isotopic shift reported.
After its formation from 1, 2 quickly decayed and underwent O–O bond cleavage to form [FeIV(O)(TMC)(CH3CN)]2+ (3)14 in essentially quantitative yield (vide supra). The decay of 2 (monitored at 500 nm) occurred concomitantly with the appearance of 3 (monitored at 820 nm), with an isosbestic point at ~695 nm (Figure 1 right panel). The time courses of the absorbance changes at both 500 and 820 nm could be fit with a simple first-order kinetic model (Figure 6 left panel), affording rate constants (kobs) that were found to be identical within experimental error. The temperature dependence of the kobs values was determined between −40 °C to −20 °C (Figure S11) and gave rise to an Eyring plot with activation parameters of ΔH‡ = 44(2) kJ/mol and ΔS‡ = −90(10) J/mol·T. These parameters are quite distinct from those determined for the reaction of [FeII(TMC)(CH3CN)]2+ with H2O2 in CH3CN in the presence of 2,6-lutidine to form 3 (ΔH‡ = 29(2) kJ/mol and ΔS‡ = −144(10) J/mol·T), which involves a direct FeII/FeIV=O conversion.15 On the other hand, the intermediacy of the FeIII–OOH complex 2 has been postulated in the reactions of [FeII(TMC)(NCCH3)]2+ with O2 in the presence of an H-atom donor or its equivalent that afford 3 as the final product, but 2 has been elusive in these reactions.8 Our results thus provide the first direct evidence to support this hypothesis.
Figure 6.

Left: Plots of absorbance at 500 nm and 820 nm vs. time for the conversion of 2 to 3. Solid lines represent fits of reaction progress (absorbance at 500 nm and 820 nm) against time to a typical first-order rate equation. Experimental conditions: 1.5 mM 2, 34 mM HClO4, −40 °C, in CH3CN. Right: Plot of kobs(820) and kobs(500) vs. added [HClO4] for the conversion of 2 to 3. Experimental conditions: 1.5 mM 2, −40 °C, in CH3CN. The black line is a linear fit for kobs(820). See Supporting Information for additional experimental details.
This first unequivocal example of converting a high-spin FeIII–OOH species to an FeIV=O complex provides an opportunity to discern what factors promote O–O bond cleavage in such species. Importantly, the conversion of 2 to 3 is found to be proton-dependent. 16 As shown in Figure 6 right panel, the values for both kobs(500) and kobs(820) increase linearly with [HClO4] added, and a second-order rate constant k2 of 0.9(1) M−1s−1 at −40 °C can be extracted from the slope of this plot. Also of significance is the observation that the quantitative yield of 3 from 1 is not affected even at the highest amounts of acid added, indicating that the added protons do not lead to Fe–O bond cleavage in 2 to release H2O2. The observed proton dependence in the formation of 3 strongly suggests that a proton promotes O–O bond cleavage.
Proton-assisted O–O bond cleavage has generally been associated with O–O bond heterolysis,1,17,18 as protonation of the terminal oxygen atom of the Fe–OOH moiety converts hydroxide into a much better leaving group. Indeed this is the generally accepted mechanism for the generation of the high-valent iron-oxo intermediate Compound I in heme enzymes.1,17 Protons also promote the conversion of the peroxo intermediate of MMO into the corresponding diiron(IV) oxidant Q.3 In model systems, it has been demonstrated that acid facilitates O–O bond heterolysis for the conversion of acylperoxoiron(III) porphyrin complexes to oxoiron(IV) porphyrin cation radical species.18 Proton-assisted O–O bond heterolysis of FeIII–OOH intermediates to generate FeV=O oxidants is also proposed in the mechanisms of nonheme iron catalysts that use H2O2 as oxidant to carry out C–H hydroxylation, C=C epoxidation and cis-dihydroxylation, and aromatic ring hydroxylation.19 The fact that many of the oxidations are highly stereoselective argues against the involvement of HO· species that would be produced from O–O bond homolysis.19a Indeed an EPR signal assigned to the putative FeV oxidant has been reported.20 On the other hand, DFT calculations of Solomon and co-workers reveal a very significant barrier for O–O bond homolysis of high-spin FeIII–OOH(R) species.5 Taken together, the points presented above and our observed proton dependence for the conversion of 2 into 3 lead us to favor a heterolytic cleavage mechanism that would initially afford a formally FeV=O species. Unfortunately, our attempts to intercept the putative FeV=O species have not been successful. Owing to the neutral nature of TMC ligand, it is perhaps not surprising for the putative [FeV(O)(TMC)]3+ species to have such a short lifetime; it could be rapidly reduced by one of several possible reductants present in the reaction mixture such as H2O2, NEt3, or even the CH3CN solvent to afford 3 that is experimentally observed (Scheme 1B). Indeed the only well characterized FeV=O complex to date is supported by a tetraanionic macrocyclic ligand that significantly extends the lifetime of the FeV=O unit.21
In summary, we report here the first example of a synthetic high-spin FeIII–OOH complex that quantitatively converts to an oxoiron(IV) complex via O–O bond cleavage. This transformation is promoted by two factors: a) the strong Fe–O bond found for 2 as indicated by its high Raman frequency, which appears to prevent Fe–O bond scission even in the presence of 0.1 M HClO4, and b) the key role of protons. Irrespective of the precise nature of the cleavage mechanism, the conversion of 2 to 3 demonstrates that O–O bond cleavage can indeed occur readily at a high-spin iron(III) center, even at −40 °C. This example thus serves as a synthetic precedent for the proton-assisted conversion of high-spin FeIII–OOH intermediates to high-valent iron-oxo oxidants in the proposed mechanisms of dioxygen-activating nonheme enzymes such as the cis-dihydroxylating Rieske dioxygenases2 and bacterial multi-component monooxygenases like MMO3 and toluene monooxygenase.22
Supplementary Material
Acknowledgments
We are grateful for support provided by NIH grants GM-33162 and GM-38767 (L.Q.) and EB-001475 (E.M.), NIH postdoctoral fellowships ES017390 (M.A.C.) and GM093479 (K.V.H.), and a University of Minnesota Doctoral Dissertation Fellowship (F.L.). We are indebted to Dr. Erik R. Farquhar for helpful guidance and discussion on the XAS analysis of 1 and 2 and to Dr. Jason England for insightful discussions.
Footnotes
Supporting Information Available: Detailed experimental procedures and physical methods, a plot for the interconversion between 1 and 2, a more extensive discussion of the Mössbauer and EPR simulations of 1 and 2, details of the EXAFS analysis, and an Eyring plot for conversion of 2 to 3. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2278. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]; (b) Sligar SG, Makris TM, Denisov IG. Biochem Biophys Res Commun. 2005;338:346–354. doi: 10.1016/j.bbrc.2005.08.094. [DOI] [PubMed] [Google Scholar]; (c) Watanabe Y, Nakajima H, Ueno T. Acc Chem Res. 2007;40:554–562. doi: 10.1021/ar600046a. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kovaleva EG, Neibergall MB, Chakrabarty S, Lipscomb JD. Acc Chem Res. 2007;40:475–483. doi: 10.1021/ar700052v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neibergall MB, Stubna A, Mekmouche Y, Münck E, Lipscomb JD. Biochemistry. 2007;46:8004–8016. doi: 10.1021/bi700120j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423–4432. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]; (b) Tinberg CE, Lippard SJ. Biochemistry. 2009;48:2145–12158. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kurtz DM., Jr J Inorg Biochem. 2006;100:679–693. doi: 10.1016/j.jinorgbio.2005.12.017. [DOI] [PubMed] [Google Scholar]; (b) Kovacs JA, Brines LM. Acc Chem Res. 2007;40:501–509. doi: 10.1021/ar600059h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Solomon EI, Decker A, Lehnert N. Proc Nat Acad Sci USA. 2003;100:3589–3594. doi: 10.1073/pnas.0336792100. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lehnert N, Ho RYN, Que L, Jr, Solomon EI. J Am Chem Soc. 2001;123:12802–12816. doi: 10.1021/ja011450+. [DOI] [PubMed] [Google Scholar]
- 6.Low-spin FeIII–OOH complexes: Simaan AJ, Döpner S, Banse F, Bourcier S, Bouchoux G, Boussac A, Hildebrandt P, Girerd JJ. Eur J Inorg Chem. 2000:1627–1633.Roelfes G, Vrajmasu V, Chen K, Ho RYN, Rohde JU, Zondervan C, la Crois RM, Schudde EP, Lutz M, Spek AL, Hage R, Feringa BL, Münck E, Que L., Jr Inorg Chem. 2003;42:2639–2653. doi: 10.1021/ic034065p.Mairata i Payeras A, Ho RYN, Fujita M, Que L., Jr Chem–Eur J. 2004;10:4944–4953. doi: 10.1002/chem.200400480.Shearer J, Scarrow RC, Kovacs JA. J Am Chem Soc. 2002;124:11709–11717. doi: 10.1021/ja012722b.
- 7.High-spin FeIII–OOH complexes: Wada A, Ogo S, Nagatomo S, Kitagawa T, Watanabe Y, Jitsukawa K, Masuda H. Inorg Chem. 2002;41:616–618. doi: 10.1021/ic001058h.Kitagawa T, Dey A, Lugo-Mas P, Benedict JB, Kaminsky W, Solomon E, Kovacs JA. J Am Chem Soc. 2006;128:14448–14449. doi: 10.1021/ja064870d.
- 8.(a) Kim SO, Sastri CV, Seo MS, Kim J, Nam W. J Am Chem Soc. 2005;127:4178–4179. doi: 10.1021/ja043083i. [DOI] [PubMed] [Google Scholar]; (b) Thibon A, England J, Martinho M, Young VG, Frisch JR, Guillot R, Girerd J-J, Münck E, Que L, Jr, Banse F. Angew Chem, Int Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Hong S, Lee YM, Shin W, Fukuzumi S, Nam W. J Am Chem Soc. 2009;131:13910–13911. doi: 10.1021/ja905691f. [DOI] [PubMed] [Google Scholar]; (d) Lee YM, Hong S, Morimoto Y, Shin W, Fukuzumi S, Nam W. J Am Chem Soc. 2010;132:10668–10670. doi: 10.1021/ja103903c. [DOI] [PubMed] [Google Scholar]
- 9.Annaraj J, Suh Y, Seo MS, Kim SO, Nam W. Chem Commun. 2005:4529–4531. doi: 10.1039/b505562h. [DOI] [PubMed] [Google Scholar]
- 10.Schmidt M, Scherk C, Iakovleva O, Nolting HF, Meier B, Parak F. Inorg Chim Acta. 1998;275:65–72. [Google Scholar]
- 11.The high-spin state for 2 results from constraints imposed by the TMC macrocycle that prevent formation of shorter Fe–N bonds required for a low-spin iron(III) center.
- 12.(a) Brunold TC, Solomon EI. J Am Chem Soc. 1999;121:8277–8287. [Google Scholar]; (b) Shiemke AJ, Loehr TM, Sanders-Loehr J. J Am Chem Soc. 1984;106:4951–4956. [Google Scholar]
- 13.Katona G, Carpentier P, Nivière V, Amara P, Adam V, Ohana J, Tsanov N, Bourgeois D. Science. 2007;316:449–453. doi: 10.1126/science.1138885. [DOI] [PubMed] [Google Scholar]
- 14.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 15.Li F, England J, Que LJ., Jr J Am Chem Soc. 2010;132:2134–2135. doi: 10.1021/ja9101908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.On the other hand, neither changing the concentration of H2O2 nor switching solvent from CH3CN to CD3CN affected the kobs(500) and kobs(820) values.
- 17.(a) Aikens J, Sligar SG. J Am Chem Soc. 1994;116:1143–1144. [Google Scholar]; (b) Vidakovic M, Sligar SG, Li H, Poulos TL. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. [DOI] [PubMed] [Google Scholar]; (c) Dunford HB, Hewson WD, Steiner H. Can J Chem. 1978;56:2844–2852. [Google Scholar]
- 18.Groves JT, Watanabe Y. J Am Chem Soc. 1988;110:8443–8452. [Google Scholar]
- 19.(a) Chen K, Costas M, Kim J, Tipton AK, Que L., Jr J Am Chem Soc. 2002;124:3026–3035. doi: 10.1021/ja0120025. [DOI] [PubMed] [Google Scholar]; (b) Mas-Ballesté R, Que L., Jr J Am Chem Soc. 2007;129:15964–15972. doi: 10.1021/ja075115i. [DOI] [PubMed] [Google Scholar]; (c) Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]; (d) Makhlynets OV, Das P, Taktak S, Flook M, Mas-Ballesté R, Rybak-Akimova EV, Que L., Jr Chem–Eur J. 2009;15:13171–13180. doi: 10.1002/chem.200901296. [DOI] [PubMed] [Google Scholar]; (e) Joon Y, Wilson SA, Jang YK, Seo MS, Nehru K, Hedman B, Hodgson KO, Bill E, Solomon EI, Nam W. Angew Chem Int Ed. 2009;48:1257–1260. doi: 10.1002/anie.200802672. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen MS, White MC. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]; (g) Das P, Que L., Jr Inorg Chem. 2010;49:9479–9485. doi: 10.1021/ic101144s. [DOI] [PubMed] [Google Scholar]; (h) Makhlynets OV, Rybak-Akimova EV. Chem–Eur J. 2010;16:13995–14006. doi: 10.1002/chem.201002577. [DOI] [PubMed] [Google Scholar]
- 20.Lyakin OY, Bryliakov KP, Britovsek GJP, Talsi EP. J Am Chem Soc. 2009;131:10798–10799. doi: 10.1021/ja902659c. [DOI] [PubMed] [Google Scholar]
- 21.Tiago de Oliveira F, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Jr, Bominaar EL, Münck E, Collins TJ. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]
- 22.Song WJ, McCormick MS, Behan RK, Sazinsky MH, Jiang W, Lin J, Krebs C, Lippard SJ. J Am Chem Soc. 2010;132:13582–13585. doi: 10.1021/ja1063795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.