

Abstract
The aggregation of polypeptides into amyloid fibrils is associated with a number of human diseases. Because these fibrils – or intermediates on the aggregation pathway – play important roles in the etiology of disease, considerable effort has been expended to understand which features of the amino acid sequence promote aggregation. One feature suspected to direct aggregation is the π-stacking of aromatic residues. Such π-stacking interactions have also been proposed as the targets for various aromatic compounds that are known to inhibit aggregation. In the case of Alzheimer’s disease, the aromatic side chains Phe19 and Phe20 in the wild-type amyloid beta (Aβ) peptide have been implicated. To explicitly test whether the aromaticity of these side chains plays a role in aggregation, we replaced these two phenylalanine side chains with leucines or isoleucines. These residues have similar sizes and hydrophobicities as Phe, but are not capable of π-stacking. Thioflavin-T fluorescence and electron microscopy demonstrate that replacement of residues 19 and 20 by Leu or Ile did not prevent aggregation, but rather enhanced amyloid formation. Further experiments showed that aromatic inhibitors of aggregation are as effective against Ile- and Leu-substituted versions of Aβ42 as they are against wild type Aβ. These results suggest that aromatic π-stacking interactions are not critical for Aβ aggregation or for the inhibition of Aβ aggregation.
Alzheimer’s disease, Huntington’s disease, and type II diabetes are among the human diseases where the self-assembly of polypeptides into amyloid structures has been associated with significant physiological impairments (1–3). Recognizing the need for effective therapeutics to combat these debilitating conditions, much attention has been focused on understanding what drives particular peptide sequences to aggregate. Though the sequences of the peptides associated with these diseases differ considerably from one another, the similarity of their cross-β sheet fibrillar structures suggests a common underlying molecular organization (4). Probing the primary sequences of these amyloidogenic polypeptides for features that are critical to fibril formation may reveal specific interactions that must be disrupted to prevent aggregation. This knowledge can then be used to direct the design of therapeutics that move beyond symptom management and towards treatments targeted at the misfolding and aggregation pathways that underlie amyloid disorders.
In Alzheimer’s disease (AD), the accumulation and aggregation of the amyloid-beta peptide (Aβ) is posited as triggering a cascade of events that ultimately results in memory loss, dementia, and other neurocognitive deficits. This “amyloid cascade hypothesis” proposes that the path toward disease begins with the release of Aβ from the amyloid precursor protein (APP) (5–9). Variable cleavage by secretase enzymes produces Aβ isoforms of various lengths with the 40-residue Aβ40 and the more toxic and highly aggregating 42-residue Aβ42 variants being most prevalent (10, 11). Once released, the Aβ peptide self-associates into oligomers, which ultimately assemble into the fibrils that comprise the extracellular plaque first observed by Alois Alzheimer over a century ago (12). It is now believed that early oligomeric intermediates – rather than the later fibrils and plaques – are the toxic species in AD (13, 14). Therefore, disrupting the aggregation pathway at its earliest stages may represent an effective route toward the development of therapeutics. In order to design targeted inhibitors of aggregation, it is important to understand which features of the primary sequence lead peptides to aggregate in AD and other amyloid disorders.
The π-stacking of aromatic residues has been suggested as a key feature promoting the assembly of polypeptides into amyloid structures. While relatively infrequent in proteins in general, aromatic residues occur frequently in amyloid sequences – a fact suggestive of their potential involvement in the aggregation process (15). Furthermore, examination of shorter variants of amyloid-forming polypeptides showed that the minimal fragments necessary for aggregation almost always contain aromatic residues. Well-ordered fibrils have been generated from short penta- and tetra-peptides containing phenylalanine (16, 17), and even a Phe-Phe dipeptide assembles into tubular structures with some similarities to amyloid fibrils (18).
The putative role of aromatic-aromatic interactions in aggregation was also suggested by an examination of compounds that inhibit fibril formation. Aromatic groups are a frequent feature in these inhibitors. For example, various polyphenols inhibit aggregation in vitro, and in some cases, provide protective effects in vivo in animal models of AD and other amyloid diseases (19–22). Gazit and coworkers suggested that aromatic groups in these inhibitors prevent amyloid formation by interfering with π-stacking interactions between aromatic side chains (21, 22). They posited that such stacking interactions provide both energetic and directional contributions toward peptide self-assembly and that disrupting these interactions provides a mechanism for inhibition (15).
Initial evidence from scanning mutagenesis experiments supported the premise that aromatic side chains promote amyloidogenesis. Replacement of phenylalanine with alanine diminished fibril formation in fragments of islet amyloid polypeptide (IAPP) and calcitonin, which are both associated with amyloid-based human diseases, (17, 23). However, the Phe→Ala mutations in those studies not only change the phenylalanine to an aliphatic side chain but also considerably alter the hydrophobicity, size, and β-sheet propensity of the side chains (Table 1).
Table 1.
Side-chain properties of Ala, Phe, Ile, and Leu residuesa
| Amino Acid Residue | Van der Waals Volume (Å) | Hydrophobicity (Fauchere-Pliska Index) | β-Sheet, ΔΔG (kcal/mol) |
|---|---|---|---|
| Ala | 67 | 0.31 | 0 |
| Phe | 135 | 1.79 | −1.08 |
| Ile | 124 | 1.80 | −1.25 |
| Leu | 124 | 1.70 | − 0.45 |
Recent mutagenesis studies using more conservative substitutions suggest that aromatic π-stacking interactions may not be critical for fibril formation. For example, Tracz et al. showed that in IAPP fragments, the replacement of Phe residues with Leu, a residue of similar size and hydrophobicity, did not prevent aggregation into amyloid fibrils (28). Furthermore, Marek et al. reported that fibrillization remained possible even when all three aryl residues in IAPP were mutated to Leu in a F15L/F23L/Y37L triple mutant (29).
These findings suggest that π-stacking may not be critical for amyloid formation, and point to other driving forces for aggregation. Indeed, the smaller size and lower hydrophobicity of alanine may account for the reduced aggregation of the Phe→Ala mutants of IAPP relative to the wild-type and Phe→Leu peptides. Thus hydrophobic burial, rather than aromaticity, may be the driving force in amyloidogenesis. Consistent with this suggestion, we have previously shown that random mutations of nonpolar side chains in Aβ42 to other nonpolar residues do not prevent aggregation, thereby demonstrating that “generic” hydrophobic interactions may suffice for amyloidogeneis (30). Our observation that the aggregation rates and morphologies of nonpolar→nonpolar Aβ42 mutants differed from the wild-type peptide, however, suggested that specific steric interactions may guide the precise aggregation pathway (30).
More detailed evidence that specific van der Waals packing of nonpolar residues promotes the formation of amyloid structures was provided by the crystallographic studies of Eisenberg and coworkers, who solved the structures of dozens of short peptides derived from various amyloid-forming polypeptides, including Aβ (31, 32). These peptides all formed “steric zipper” structures of tightly interdigitated β-sheets with closely packed side chains. The precise fit required for steric zipper formation may explain the lag-dependent kinetics of fibril formation: the side chains must adopt the proper rotamers for interdigitation and pack together tightly to exclude solvent. The accompanying decrease in entropy is balanced by enthalpically favorable packing interactions (32).
In the current study, we investigate the relative contributions of π-stacking interactions versus generic hydrophobic packing in the aggregation of Aβ by examining variants in which the Phe side chains at positions 19 and 20 are replaced by Ile and Leu residues. Our results show that both the F19L/F20L and F19I/F20I mutants form amyloid fibrils, and do so at higher levels than the wild-type peptide. These findings provide clear evidence that aromatic interactions are not required for the aggregation of Aβ. Distinctions between the Leu and Ile mutants in both their aggregation pathways and fibril morphologies further implicate steric packing as a driving force for amyloidogenesis. Finally, we show that aromatic compounds inhibit not only wild-type Aβ aggregation but also, with similar effectiveness, the aggregation of non-aromatic mutant peptides. Together, these results suggest that steric packing of hydrophobic residues – rather than aromatic π-stacking interactions – promote Aβ aggregation.
EXPERIMENTAL PROCEDURES
Peptides
Crude peptides synthesized via FMOC solid-phase synthesis were purchased from the Keck Institute at Yale University, and purified using a C4 reverse-phase column. Solvent gradients were run at 60°C using a two-buffer system, where solvent A consisted of 95% water, 5% acetonitrile, and 0.1% trifluoroacetic acid (TFA), and solvent B consisted of 50% acetonitrile, 50% water, and 0.1% TFA (v/v). Purified peptides were disaggregated via sequential treatment with TFA and 1,1,1,3,3,3-hexafluoro-2-isopropanol (33) and stored as a lyophilized film at −20°C.
Thioflavin-T Assays
20 μM peptide solutions were prepared by dissolving 0.5 mg of purified peptide in 300 μL DMSO and diluting the solution with 5 mL of 8 mM NaOH and 300 μL 20x PBS buffer (final concentration of 50 mM NaH2PO4, 100 mM NaCl and 5% (v/v) DMSO, pH 7.2–7.3). Samples were incubated at 37°C under quiescent conditions for the indicated time periods. At various time points, triplicate 100 μL aliquots of peptide were mixed with 100 μL of a solution of thioflavin-T (7 μM ThT, 50 mM glycine-NaOH, pH 7.1). Fluorescence was measured on a Varioskan plate reader (Thermo-Scientific) at an excitation wavelength of 450 nm and emission wavelength of 483 nm.
SDS-PAGE Electrophoresis
At the indicated time points, 60 μL aliquots were removed from the peptide samples described above, and centrifuged at 13000 rpm for 30 minutes at 4°C. The top 20 μL of the supernatant was removed, combined with 2x SDS sample buffer, and boiled ~7 minutes at 100°C. These samples were loaded onto a 10–20% Tris-HCl gel (Bio-rad, Hercules, CA), run at 100 V, and silver-stained.
Electron Microscopy
Formvar carbon-coated Cu grids were floated on a drop of 20 μM peptide. The grids were washed twice with distilled water, stained for two minutes with 1% uranyl acetate (J.T. Baker), and imaged with a Zeiss 912ab Electron Microscope.
Aβ-GFP Fusion Assay
Sequences encoding the Aβ peptides were inserted into the GFP folding reporter vector as described previously (34). The F19I/F20I and F19L/F20L mutations were then introduced via site-directed mutagenesis using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA) using primers obtained from Integrated DNA Technologies (Coralville, IA). Sequences were verified and transformed into BL21/DE3 E. coli cells for screening. Cells were grown at 37°C in LB medium containing 35 μg/mL kanamycin until they reached an O.D. of ~0.6. Expression was induced with 1 mM IPTG and cells were grown for an additional 3 hours. A Thermo Varioskan plate reader was used to measure the fluorescence of GFP. The optical density of cells at 600 nm was used to normalize fluorescence measurements. Expression and solubility of the Aβ-GFP fusion proteins were also assessed by gel electrophoresis as described previously (34).
Thioflavin-T Assays in the Presence of Aggregation Inhibitors
20 μM solutions of peptide were prepared as described above and 1 μL of 10 mM of the indicated compounds in DMSO was added to 100 μL aliquots of the peptide solutions (final inhibitor concentration of 100 μM). Samples were then incubated at 37°C under quiescent conditions for seven days. 40 μL of the ThT solution described above were added, and fluorescence was measured as described above.
RESULTS
The Aβ sequence contains four aromatic residues: phenylalanines at positions 4, 19, and 20, and tyrosine at position 10 (Figure 1). Because previous studies showed that hydrophobic stretches in the central and C-terminal parts of Aβ42 are critical for aggregation (35, 36), we focused our studies on the aromatic residues at positions 19 and 20 in the central hydrophobic cluster (Leu17-Val18-Phe19-Phe20-Ala21). Proline scanning mutagenesis showed that these residues are particularly sensitive to replacement (36), and this region has been targeted in the design of peptide inhibitors of aggregation (37–39). Furthermore, solid-state NMR studies by Tycko and coworkers have generated models of Aβ fibrils that place the central hydrophobic cluster within the core β-sheet of the fibrillar structure (40, 41). In particular, their model suggests that Phe19 makes contacts with C-terminal hydrophobic residues (Ile32, Leu34, Val36) on the interior of the β-hairpin (40). In contrast to residues in the central hydrophobic cluster, the N-terminal residues of Aβ, including Phe4 and Tyr10, are flexible and not involved in the ordered fibrillar structure (36, 42). If π-stacking interactions play a critical role in the aggregation of Aβ, then removing aromatic functionality at positions 19 and 20 should diminish aggregation. Thus, replacing Phe19 and Phe20 with Leu or Ile, which are similar to Phe in size and hydrophobicity (Table 1), but are not aromatic, provides a stringent test of the importance of π-stacking in Aβ aggregation.
Figure 1.
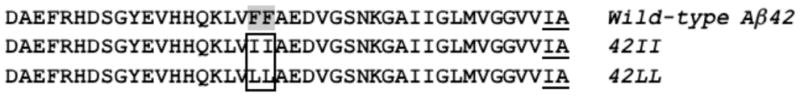
Amino acid sequences of wild-type Aβ42 and the F19I/F20I (42II) and F19L/F20L (42LL) mutants. Phe 19 and 20 are highlighted and the mutations are boxed. Ile41 and Ala42 are underlined. These C-terminal residues are not present in the 40-residue peptides.
Aggregation of Ile/Ile and Leu/Leu mutants of Aβ42
To probe the impact of introducing Leu or Ile at positions 19 and 20 of Aβ42, we first monitored thioflavin-T (ThT) fluorescence of the F19L/F20L and F19I/F20I mutants. ThT is a benzothiazole compound that fluoresces when bound to amyloid fibrils, and is used to detect amyloid (43). Interpretation of ThT fluorescence for mutant versus wild-type sequences can be complicated by possible differences in the number of dye binding sites or the quantum yield of modified sequences. Nonetheless, we used ThT binding, in conjunction with SDS-PAGE, to provide a semi-quantitative assessment of amyloid formation. Figure 2a displays the time course of ThT aggregation for wild-type (WT) Aβ42 and mutants lacking aromatic residues in the hydrophobic core: F19L/F20L and F19I/F20I, hereafter denoted as 42LL and 42II, respectively. A soluble double mutant (F19S/L34P) of Aβ42, known as GM6 (34) was used as a non-aggregating control. Figure 2a clearly shows that 42II and 42LL display increased ThT fluorescence relative to WT Aβ42, thereby demonstrating that aromatic residues in the central hydrophobic cluster are not required for aggregation.
Figure 2.

(a) Thioflavin-T fluorescence shows that the 42II and 42LL mutants display a higher aggregation propensity than wild-type Aβ42. 20 μM solutions of peptide were incubated at 37°C under quiescent conditions. At the indicated time points, aliquots were mixed with ThT and measured at an emission wavelength of 483 nm. Time points show an average of three measurements, and standard errors are indicated. (b) At 6 and 14 days, aliquots were centrifuged to remove insoluble aggregates. Soluble material was boiled in SDS sample buffer and loaded onto 10–20 % Tris-HCl gels. These gels confirm the ThT plots and show that 42II aggregates more quickly (leaving less soluble monomer) than WT Aβ42 or 42LL. After 14 days both non-aromatic peptides produce more insoluble material (leaving less soluble monomer) than the wild type peptide.
Peptide 42II exhibits rapid amyloid formation, with fluorescence increasing without the initial lag phase seen for WT Aβ42 or 42LL. The 42LL peptide has a slower initial rate, but ultimately produces considerably more ThT-staining amyloid than either 42II or WT Aβ42. At the final time-point at 21 days, ThT fluorescence for both 42II and 42LL exceeds WT Aβ42 with 42LL fluorescence markedly higher than the wild-type peptide. These results demonstrate that loss of aromaticity not only fails to prevent aggregation; it actually produces more ThT-reactive aggregates.
Gel electrophoresis confirmed the higher aggregation propensity of the Ile and Leu mutants (Figure 2b). After 6 and 14 days of quiescent incubation, samples were centrifuged to remove insoluble aggregates, and the remaining soluble material was quantified by removing the top third of the supernatant, boiling it in SDS sample buffer, and running it on a 10–20% Tris-HCl gel. These gels reveal that, after six days, 42II shows a reduced concentration of soluble peptide relative to WT Aβ42. After 14 days, the 4 kDa monomeric band in both 42II and 42LL was diminished relative to the wild-type peptide. Also notable is the higher concentration of soluble, SDS-resistant aggregates in the non-aromatic mutants. As indicated by the band at the interface of the stacking and gradient gels, the Leu double mutant, in particular, shows an increased amount of soluble aggregate at both 6 and 14 days.
Next, we used transmission electron microscopy (TEM) to monitor aggregation of the 42II and 42LL mutants, and to compare the morphologies of the aggregates. As shown in Figure 3, after 1 day of quiescent incubation, WT Aβ42 and 42LL both display small protofibrillar aggregates with the occasional appearance of short fibrils. Consistent with the lack of a lag phase seen in ThT fluorescence measurements, after only one day the 42II mutant had already formed larger aggregates comprising protofibrils and fibrils. By fourteen days, all three peptides exhibit considerable aggregation. Thus the TEM results, in agreement with ThT fluorescence, show that loss of aromaticity at residues 19 and 20 does not prevent amyloid formation.
Figure 3.

(a) TEM images after 1 day of quiescent incubation (top) show that 42II formed clumps of fibrils and protofibrils, while WT Aβ42 and 42LL form mostly dispersed small protofibrils with an occasional short fibril. After 14 days, all three peptides show significant aggregation with distinct fibril morphologies. Scale bars indicate 100 nm. (b) Individual fibrils show a lack of twist in any of the 42-residue peptides (i). However, lateral association of individual fibrils was observed, with 42II showing striated ribbons (ii). Scale bars indicate 40 nm in (i) and 80 nm in (ii). (c) The distribution of widths for 60 randomly chosen fibrils indicates that fibrils formed by 42LL are typically narrower than those formed by WT Aβ42 or 42LL. Average fibril widths were 9.00 ± 0.89 nm for WT Aβ42, 8.95 ± 1.06 nm for 42II, and 7.76 ± 1.10 nm for 42LL.
The morphologies of the fibrils differ for Aβ42, 42II, and 42LL. Though a range of morphologies could be found for all peptides, several features clearly distinguish the sequences from one another. As seen in Figure 3a, the mature 14-day fibrils of the 42II mutant are long and relatively straight, while WT Aβ42 and 42LL show a greater abundance of shorter kinked aggregates intermixed with longer fibrils. Supplemental Figure S1 shows these morphological distinctions at multiple magnifications. Though none of the mature 14-day fibrils displayed a discernible twist (3b, i), lateral association of fibrils was visible with a striated ribbon morphology most apparent in 42II (3b, ii). Finally, histograms plotting the distribution of fibril widths show that on average, fibrils formed by 42LL are narrower than those formed by wild-type or 42II (Fig. 3c). These morphological distinctions show that substitution with Ile or Leu—which differ only by branching at the beta versus gamma carbon of the side chain—have different impacts on fibrillization rate and structure.
Aggregation of Ile/Ile and Leu/Leu mutants of Aβ40
After showing that the Phe→Leu and Phe→Ile substitutions at residues 19 and 20 increase the aggregation of Aβ42, we were curious to see whether these substitutions exert a similar effect in the context of the shorter version of the Alzheimer’s peptide, Aβ40. Although produced in vivo in greater abundance than Aβ42, Aβ40 is less amyloidogenic, less toxic, and has a longer lag phase than the full length Aβ42 (44). As shown in Figure 4, introduction of the same F19I/F20I or F19L/F20L mutations into Aβ40 increased aggregation propensity, as they had for Aβ42. Ile at positions 19 and 20 shortens the lag phase – with a dramatic increase in ThT fluorescence occurring between 4 and 6 days – and leads to ThT fluorescence that is markedly greater than for wild-type Aβ40. In contrast, placing leucines at positions 19 and 20 produces a longer lag phase with a significant rise in ThT fluorescence occurring only after 10 days of quiescent incubation. This is similar to the time course for WT Aβ40. However, while the aggregation of WT Aβ40 levels off, the fluorescence of 40LL continues to increase. Consistent with the results presented above for Aβ42, these results indicate that introduction of the non-aromatic Ile or Leu mutations enhance the formation of ThT-reactive amyloid.
Figure 4.

(a) Time course of Thioflavin-T fluorescence showing increased amyloid formation by 40II and 40LL relative to WT-Aβ40. 20 μM solutions of peptide were incubated at 37°C under quiescent conditions. At the indicated time points, aliquots were mixed with ThT and measured at an emission wavelength of 483 nm. Time points are an average of three measurements and standard errors are indicated. (b) At 6 and 14 days, aliquots were centrifuged to remove insoluble aggregates, and boiled with SDS sample buffer before loading onto 10–20 % Tris-HCl gels. These gels confirm that 40II forms more insoluble aggregate (less monomer) than wild-type Aβ40. For 40LL, the quantity of monomer is comparable to WT Aβ40.
Aggregation was also assessed using gel electrophoresis to quantify the fractions of peptide that remained soluble (Figure 4b). For 40II, the electrophoresis results were consistent with the ThT results, showing an enhanced propensity to aggregate relative to WT Aβ40. Samples run after either six or fourteen days of incubation showed considerably less monomer remaining for 40II than for WT Aβ40. The behavior of 40LL is more subtle: the ThT assay showed its propensity to aggregate was slightly enhanced relative to WT Aβ40 (Figure 4a), however the gel assay was incapable of detecting this difference. This may be due either to the small magnitude of the difference or the possibility that some of the ThT active aggregates formed by 40LL are only marginally stable and become disaggregated by boiling in SDS.
We also assessed the aggregation of WT Aβ40, 40II, and 40LL by electron microscopy. Images were taken after seven and 21 days of quiescent incubation (Figure 5a). Consistent with the ThT fluorescence, after seven days, 40II displays considerable aggregation with a mixture of fibrils and large clumps of protofibrillar aggregates. In contrast, the TEM grid for WT Aβ40 was mostly blank with the occasional long fibril. 40LL contained some dispersed short fibrils. After 21 days, all three peptides show considerable fibril formation. Additional TEM images at varying magnifications are shown in Supplemental Figure S2.
Figure 5.
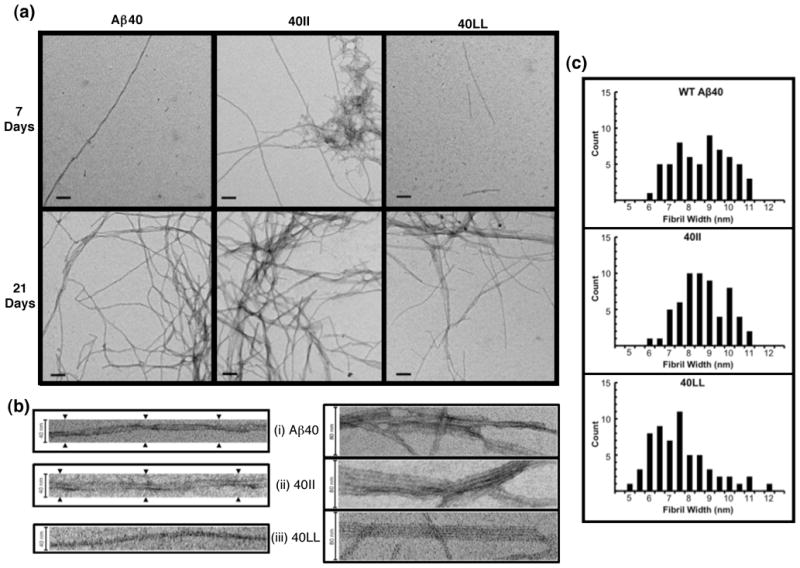
(a) TEM images taken after 7 days of quiescent incubation (top) indicate significant aggregation in 40II with a mixture of long fibrils and large clumps of protofibrillar species. Minimal aggregation was noted in WT40 with only an occasional, solitary long fibril, and in 40LL, which showed dispersed short fibrils. After 21 days (bottom), all peptides show considerable fibril formation. Scale bars indicate 100 nm. (b) Twisting was visualized in some WT-Aβ40 and 40II fibrils (i) while striated ribbons were apparent for 40II and 40LL (ii). Scale bars indicate 40 nm in (i) and 80 nm in (ii). (c) Histograms of the widths measured for 60 randomly chosen fibrils show a wide distribution in all mutants with the 40LL skewing towards narrower widths than and wild-type Aβ40 and 40II. Average fibril widths of 40LL, 40II, and WT were 7.31 nm (± 1.50), 8.44 nm (± 1.17), and 8.35 nm (± 1.32), respectively.
Morphological features of the fibrils are highlighted in Figure 5b. Both WT Aβ40 and 40II produced a twisted morphology, while no such fibrils were seen for 40LL (5b, i). Lateral association of fibrils was seen for all three peptides (5b, ii). Striated ribbons were most apparent for 40II and 40LL, while WT Aβ40 fibrils did not align in a regular pattern. Finally, as was observed for the full-length 42-residue peptides, fibrils formed by the LL mutant tended to be narrower than those formed by either the WT Aβ40 or 40II peptides (Figure 5c).
Overall, the results with Aβ40 confirm what we observed with of Aβ42: Ile and Leu double mutants form fibrils despite loss of aromatic Phe residues. Moreover, the morphological differences indicate that specific steric packing arrangements may drive aggregation.
Aggregation of Aβ-GFP Fusions in E.coli
The biochemical and biophysical characterizations described above were performed in vitro using synthetic peptides. To assess the effects of mutations on Aβ aggregation in a biological milieu, we also studied the properties of Aβ42-GFP fusions expressed in E. coli. Previous work in our lab showed that wild type Aβ42 prevents the linked GFP from folding and fluorescing, while mutations in Aβ42 that prevent aggregation cause the entire fusion to remain soluble, thereby allowing the linked GFP to fold and fluoresce. Thus, the fluorescence of Aβ42-GFP fusions expressed in E. coli provides a rapid and accurate readout of the effect of amino acid replacements on the aggregation propensity of Aβ (30, 34, 45–47).
We compared the fluorescence of cells expressing GFP fusions to wild type Aβ42 with those expressing fusions to 42II and 42LL. A GFP fusion to the mutant GM6 was used as a soluble control (34). As shown in Figure 6, GFP fusions to WT Aβ42, 42II and 42LL all show low levels of fluorescence – less than 15% of the GM6 control. The GFP fusion to 42LL has a slightly higher fluorescence than the fusion to WT Aβ42, while the fusion to 42II produces lower fluorescence than the wild type fusion. Irrespective of these relatively small differences, it is clear that fusion to both 42II and 42LL diminishes GFP fluorescence far below the soluble GM6 mutant. These results indicate that the loss of aromatic residues in these peptides does not prevent aggregation.
Figure 6.
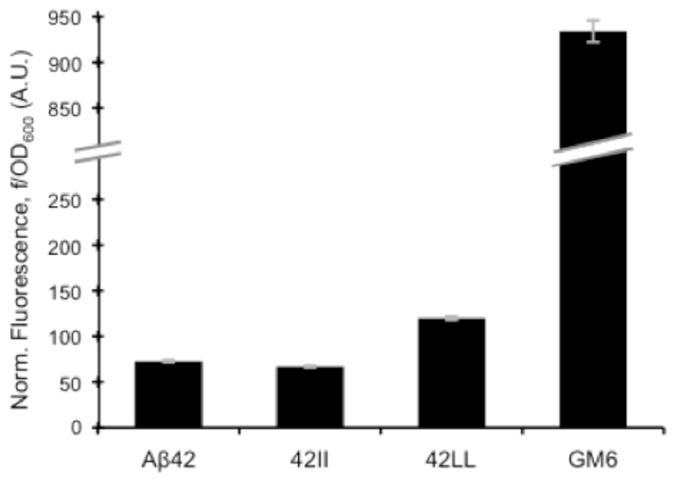
Fluorescence of Aβ42-GFP fusions. Fusions to WT Aβ42 (WT42) or to 42II or 42LL decreases the fluorescence of the fusion protein. In contrast, fusion to GM6, a soluble mutant of Aβ42 allows GFP to fold, remain soluble, and fluoresce.
The aggregation in vivo of the Aβ42-GFP fusions was also analyzed by gel electrophoresis (Figure 7). Although the various Aβ42-GFP fusions were expressed at similar levels (Fig. 7a), only the fusion to mutant GM6 was soluble (Fig. 7b). In contrast, mutants 42LL and 42II were similar to WT Aβ42 in causing the fusion protein to partition into the insoluble fraction. Both the fluorescence measurements shown in figure 6 and the gel analyses shown in Figure 7 are consistent with our results with synthetic peptides in demonstrating that aromatic residues are not required for the aggregation of Aβ.
Figure 7.

SDS-PAGE showing expression and solubility of the Aβ-GFP fusion proteins. (a) Whole cell lysates show that expression levels were similar for all fusions, with a slightly decreased band in the GM6 mutant. (b) After four cycles of freeze-thaw lysis, followed by centrifugation (34), the soluble fraction of the lysates was analyzed. Only the GM6-GFP control shows significant partitioning to the soluble fraction.
Response to aromatic and non-aromatic inhibitor compounds
The presence of aromatic groups in many documented aggregation inhibitors has been suggested as evidence that disrupting π-stacking interactions blocks amyloidogenesis (15). If the aromatic groups in inhibitors indeed function by disrupting π-stacking interactions, then one would expect these inhibitors to be less effective in reducing the aggregation of Aβ mutants in which Phe19 and Phe20 are replaced by non-aromatic side chains.
We tested the efficacy of three aromatic compounds previously shown to inhibit Aβ aggregation: curcumin, tannic acid, and triazine E2 (Figure 8). Curcumin and tannic acid are found in curry and tea, respectively, and both have been shown to prevent Aβ aggregation and fibrillization (48). E2, was discovered in our laboratory, using the Aβ42-GFP system as a high-throughput screen for inhibitors of aggregation (46). As a control, we also tested the non-aromatic inhibitor, myristyltrimethylammonium bromide (MTMA), a detergent-like compound, shown previously to reduce Aβ aggregation (49).
Figure 8.
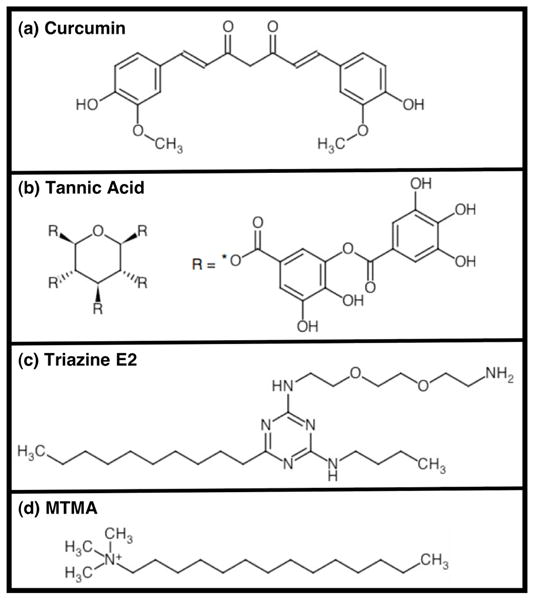
Compounds identified previously as inhibitors of Aβ aggregation were tested for their ability to prevent aggregation of 42II and 40II. Three aromatic compounds—(a) curcumin, (b) tannic acid, and (c) triazine E2 and one non-aromatic compound (d, MTMA) were tested.
We assayed the abilities of these compounds to reduce fibril formation for wild-type Aβ42 and the 42II and 42LL mutants. As shown in Figure 9a, ThT fluorescence for all three peptides was reduced by incubation with any of the four compounds. These findings were confirmed by TEM, with representative images shown in Figure 9b. Although a few fibrils were observed in some of the treated samples, fibril formation was significantly reduced relative to the dense fibril collections observed for untreated peptides. Most significantly, the aromatic compounds are not more effective at disrupting the aggregation of WT Aβ42 than of mutants lacking aromatic residues at positions 19 and 20. These results suggest that inhibition of aggregation by aromatic compounds is not mediated by the disruption of π-stacking interactions.
Figure 9.

Aromatic and non-aromatic inhibitors reduce fibril formation for 42II and 42LL to an extent similar to that observed for wild-type Aβ42. 20 μM peptide solutions were incubated at 37°C under quiescent conditions with inhibitor compounds present at 100 μM. (a) ThT fluorescence of the samples was measured after seven days at an emission wavelength of 483 nm. Scale bars represent an average of three measurements, and the standard error is shown. (b) Representative TEM images after seven days incubation confirm that the inhibitors reduced fibril formation in all three peptides.
The non-aromatic compound, MTMA, was also effective at inhibiting aggregation of Aβ42 and the 42II and 42LL mutants. As shown in Figure 9, MTMA reduces fluorescence in ThT assays and diminishes fibrillization in TEM images. This non-aromatic compound is equally effective against peptides with and without aromatic residues. These results indicate that aromatic groups in inhibitor compounds do not prevent aggregation by disrupting π-stacking interactions, and provides further evidence that aromatic moieties are not critical for Aβ aggregation.
DISCUSSION
If π-stacking interactions were a critical driving force for amyloidogenesis, one would expect that removal of aromatic functionality from the hydrophobic core of Aβ would significantly impair, if not entirely prevent, aggregation. Instead, our results show that Aβ peptides in which Phe19 and Phe20 are replaced by non-aromatic side-chains not only retain a propensity to form fibrils, but actually yield higher levels of thioflavin-T reactive material. Thus, aromaticity does not appear to play a critical role in driving aggregation.
Our results are consistent with those of de Groot et al. who systematically replaced Phe19 with other residues and found that aggregation decreased as the substitutions became less conservative of the side chain’s physicochemical properties (50). Particularly, they found that the aggregation propensity of Aβ could be correlated with the hydrophobicity and β-sheet propensity of residues at this position (50). Likewise, Dobson and co-workers have developed an algorithm which robustly predicts aggregation propensity based on consideration of only three physicochemical properties – charge, hydrophobicity, and β-sheet propensity (51). That aromaticity is not a critical factor for predicting aggregation is consistent with mutagenesis studies of IAPP (28, 29) and acylphosphatase (52), which demonstrated that aggregation of these other amyloidogenic sequences also does not require aromatic residues.
Beyond demonstrating that π-stacking interactions are not required for Aβ aggregation, the differential impact of introducing Ile or Leu substitutions into the Aβ peptide suggests the importance of specific packing in driving amyloid formation. While the F19I/F20I peptides rapidly formed amyloid aggregates with a significantly diminished lag phase, the F19L/F20L mutants were slower to nucleate. With identical volumes and hydrophobicities (Table 1), Ile and Leu differ only in the position of a methyl group. Thus, despite their similarities, Ile and Leu represent slightly dissimilar puzzle pieces and pack differently against other side chains. As noted by Eisenberg and coworkers, side chain interdigitation may account for the slow and entropically disfavored nucleation step of the aggregation process (32). Therefore, even small differences in the shape of a residue could influence steric zipper formation and aggregation rate. Such steric factors might be especially important for a side chain making contacts on the interior of the β-hairpin, as seen for residue 19 in the solid-state NMR structure of Aβ reported by Tycko and coworkers (40).
Variations in the precise molecular arrangements of specific side chains have been shown to produce different fibril morphologies for a range of amyloid sequences. Eisenberg and coworkers showed that different sequences form different steric zipper structures, and in some cases a single sequence can assemble into several different structures (31). In the case of Aβ, subtle changes in growth conditions are sufficient to produce different fibril morphologies. Solid state NMR shows that the different morphologies are due to variations in side chain packing that propagate into polymorphic fibrillar structures (41, 53, 54). Given these precedents showing the importance of steric packing in dictating amyloid structure, it is not surprising that introducing Ile or Leu at the core of Aβ produces different fibrillar morphologies (Figures 3 & 5).
Gazit and coworkers postulated that the frequency of aromatic residues in amyloidogenic sequences suggests a role for π-stacking in aggregation (15). Our results do not dispute the propensity of Phe to promote aggregation, but suggest that this propensity is due to hydrophobic packing, rather than π-stacking interactions. In agreement with this finding, an analysis of protein-protein interactions by Ma and Nussinov revealed that Phe is one of the most conserved residues (along with Trp, another aromatic side chain) at interaction interfaces. In contrast, Ile is among the least conserved (55). They suggest that nature may have avoided Ile in protein-protein interfaces precisely because it has a high propensity to form amyloid aggregates (55). Indeed, evolution may have selected aromatic side chain interfaces because of their ability to form π-stacking interactions in specific protein-protein interactions. Ile, on the other hand, is a more generic hydrophobic residue that does not participate in π-stacking interactions, but serves to simply drive sequences towards amyloid aggregation. Because nature selects for well-structured interfaces and against amyloid-like aggregation, Phe is used frequently, while Ile is used only sparingly on protein surfaces.
While our results indicate definitively that aromatic interactions are not critical for aggregation of the Aβ peptide, the implications of these findings for the development of aggregation inhibitors is somewhat less clear. In spite of our demonstration that π-stacking interactions are not essential for aggregation, the frequent presence of aromatic functionalities in previously identified inhibitors suggests a role for these groups in targeted drug design. As our results showed that phenolic compounds still act as potent inhibitors of aggregation for Aβ mutants in which the Phe-Phe aromatic motif was removed, it seems likely that these compounds are effective for reasons unrelated to the disruption of π-stacking. Thus, our results do not disqualify the incorporation of aromatic groups into aggregation inhibitors, but merely suggest an alternate explanation for the mechanism of inhibition. Perhaps, phenolic compounds are effective because their planar structure allows them to intercalate between monomer layers or cap growing fibrillar ends, thereby inhibiting aggregation and serving as platforms for the further development of more potent treatments for Alzheimer’s disease.
Supplementary Material
Acknowledgments
We thank Woojin Kim for initiating this project, Margaret Bisher for assistance with the EM, and Ehud Gazit for helpful comments on the manuscript.
Funding Information: This work was supported by Award R21-AG028462 from the National Institute on Aging, and Award IIRG-08-89944 from the Alzheimer’s Association.
ABBREVIATIONS
- Aβ
amyloid-β peptide
- Aβ40
40-residue wild-type Aβ
- Aβ42
42-residue wild-type Aβ
- 40II
40-residue Aβ with F19I/F20I mutations
- 40LL
40-residue Aβ with F19L/F20L mutations
- 42II
42-residue Aβ with F19I/F20I mutations
- 42LL
42-residue Aβ with F19L/F20L mutations
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- FMOC
9-fluorenylmethoxycarbonyl
- GM6
42-residue Aβ with F19S/L34P mutations
- IAPP
islet amyloid polypeptide
- MTMA
myristyltrimethylammonium bromide
- NMR
nuclear magnetic resonance
- PBS
phosphate-buffered saline
- TA
tannic acid
- TEM
transmission electron microscopy
- ThT
thioflavin-T
- TFA
trifluoroacetic acid
- v/v
volume-to-volume
- WT
wild-type
Footnotes
SUPPORTING INFORMATION AVAILABLE: Additional TEM images at lower and higher magnification. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Koo EH, Lansbury J, Peter T, Kelly JW. Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci USA. 1999;96:9989–9990. doi: 10.1073/pnas.96.18.9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McGowan DP, van Roon-Mom W, Holloway H, Bates GP, Mangiarini L, Cooper GJS, Faull RLM, Snell RG. Amyloid-like inclusions in Huntington’s disease. Neuroscience. 2000;100:677–680. doi: 10.1016/s0306-4522(00)00391-2. [DOI] [PubMed] [Google Scholar]
- 3.Hoppener JWM, Ahren B, Lips CJM. Islet amyloid and type 2 diabetes mellitus. N Engl J Med. 2000;343:411–419. doi: 10.1056/NEJM200008103430607. [DOI] [PubMed] [Google Scholar]
- 4.Sunde M, Serpell LC, Bartlam M, Fraser PE, Pepys MB, Blake CCF. Common core structure of amyloid fibrils by synchrotron X-ray diffraction. J Mol Biol. 1997;273:729–739. doi: 10.1006/jmbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- 5.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 6.Lesne S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-beta protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- 7.Head E, Lott IT. Down syndrome and beta-amyloid deposition. Curr Opin Neurol. 2004;17:95–100. doi: 10.1097/00019052-200404000-00003. [DOI] [PubMed] [Google Scholar]
- 8.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM. Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging. 2003;24:1063–1070. doi: 10.1016/j.neurobiolaging.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 9.De Jonghe C, Zehr C, Yager D, Prada CM, Younkin S, Hendriks L, Van Broeckhoven C, Eckman CB. Flemish and Dutch mutations in amyloid beta precursor protein have different effects on amyloid beta secretion. Neurobiol Dis. 1998;5:281–286. doi: 10.1006/nbdi.1998.0202. [DOI] [PubMed] [Google Scholar]
- 10.Aisen PS. The development of anti-amyloid therapy for Alzheimer’s disease : from secretase modulators to polymerisation inhibitors. CNS Drugs. 2005;19:989–996. doi: 10.2165/00023210-200519120-00002. [DOI] [PubMed] [Google Scholar]
- 11.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 12.Dahm R. Alzheimer’s discovery. Curr Biol. 2006;16:R906–R910. doi: 10.1016/j.cub.2006.09.056. [DOI] [PubMed] [Google Scholar]
- 13.Lue L, Kuo Y, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid-beta peptide concentration as a predictor of synaptic change in alzheimer’s disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. J Am Med Ass. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 15.Gazit E. A possible role for pi-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002;16:77–83. doi: 10.1096/fj.01-0442hyp. [DOI] [PubMed] [Google Scholar]
- 16.Levy M, Garmy N, Gazit E, Fantini J. The minimal amyloid-forming fragment of the islet amyloid polypeptide is a glycolipid-binding domain. FEBS J. 2006;273:5724–5735. doi: 10.1111/j.1742-4658.2006.05562.x. [DOI] [PubMed] [Google Scholar]
- 17.Reches M, Porat Y, Gazit E. Amyloid fibrils formation by penta- and tetrapeptide fragments of human calcitonin. J Biol Chem. 2002;277:35475–35480. doi: 10.1074/jbc.M206039200. [DOI] [PubMed] [Google Scholar]
- 18.Reches M, Gazit E. Casting metal nanowires within discrete self-assembled peptide nanotubes. Science. 2003;300:625–627. doi: 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- 19.Lim GP, Chu T, Yang F, Beech W, Frautschy SA, Cole GM. The Curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J Neurosci. 2001;21:8370–8377. doi: 10.1523/JNEUROSCI.21-21-08370.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ono K, Yoshiike Y, Takashima A, Hasegawa K, Naiki H, Yamada M. Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: implications for the prevention and therapeutics of Alzheimer’s disease. J Neurochem. 2003;87:172–181. doi: 10.1046/j.1471-4159.2003.01976.x. [DOI] [PubMed] [Google Scholar]
- 21.Porat Y, Abramowitz A, Gazit E. Inhibition of amyloid fibril formation by polyphenols: structural similarity and aromatic interactions as a common inhibition mechanism. Chem Biol Drug Des. 2006;67:27–37. doi: 10.1111/j.1747-0285.2005.00318.x. [DOI] [PubMed] [Google Scholar]
- 22.Scherzer-Attali R, Pellarin R, Convertino M, Frydman-Marom A, Egoz-Matia N, Peled S, Levy-Sakin M, Shalev DE, Caflisch A, Gazit E, Segal D. Complete phenotypic recovery of an Alzheimer’s disease model by a quinone-tryptophan hybrid aggregation inhibitor. PLoS ONE. 2010;5:e11101. doi: 10.1371/journal.pone.0011101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azriel R, Gazit E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. J Biol Chem. 2001;276:34156–34161. doi: 10.1074/jbc.M102883200. [DOI] [PubMed] [Google Scholar]
- 24.Creighton TE. Proteins: Structures and Molecular Properties. 2. W.H. Freeman and Company; New York: 1993. [Google Scholar]
- 25.Smith CK, Withka JM, Regan L. A thermodynamic scale for the beta-sheet forming tendencies of the amino acids. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 26.Minor DL, Kim PS. Measurement of the beta-sheet-forming propensities of amino acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 27.Fauchere J, Pliska V. Hydrophobic parameters of amino-acid side chains from the partitioning of N-acetyl-amino-acid amides. Eur J Med Chem. 1983;18:369–375. [Google Scholar]
- 28.Tracz SM, Abedini A, Driscoll M, Raleigh DP. Role of aromatic interactions in amyloid formation by peptides derived from human amylin. Biochemistry. 2004;43:15901–15908. doi: 10.1021/bi048812l. [DOI] [PubMed] [Google Scholar]
- 29.Marek P, Abedini A, Song B, Kanungo M, Johnson ME, Gupta R, Zaman W, Wong SS, Raleigh DP. Aromatic interactions are not required for amyloid fibril formation by islet amyloid polypeptide but do influence the rate of fibril formation and fibril morphology. Biochemistry. 2007;46:3255–3261. doi: 10.1021/bi0621967. [DOI] [PubMed] [Google Scholar]
- 30.Kim W, Hecht MH. Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer’s Abeta42 peptide. Proc Natl Acad Sci U S A. 2006;103:15824–15829. doi: 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, Thompson MJ, Balbirnie M, Wiltzius JJW, McFarlane HT, Madsen AO, Riekel C, Eisenberg D. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–457. doi: 10.1038/nature05695. [DOI] [PubMed] [Google Scholar]
- 32.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jao SC, Ma K, Talafous J, Orlando R, Zagorski MG. Trifluoroacetic acid pretreatment reproducibly disaggregates the amyloid-beta peptide. Amyloid. 1997;4:240–252. [Google Scholar]
- 34.Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer’s Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–1290. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 35.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural features of the A-beta amyloid fibril elucidated by limited proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 36.Williams AD, Portelius E, Kheterpal I, Guo J-t, Cook KD, Xu Y, Wetzel R. Mapping abeta amyloid fibril secondary structure using scanning proline mutagenesis. J Mol Biol. 2004;335:833–842. doi: 10.1016/j.jmb.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 37.Tjernberg LO, Näslund J, Lindqvist F, Johansson J, Karlström AR, Thyberg J, Terenius L, Nordstedt C. Arrest of beta-amyloid fibril formation by a pentapeptide Ligand. J Biol Chem. 1996;271:8545–8548. doi: 10.1074/jbc.271.15.8545. [DOI] [PubMed] [Google Scholar]
- 38.Soto C, Sigurdsson EM, Morelli L, Asok Kumar R, Castano EM, Frangione B. Beta-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: Implications for Alzheimer’s therapy. Nat Med. 1998;4:822–826. doi: 10.1038/nm0798-822. [DOI] [PubMed] [Google Scholar]
- 39.Findeis MA, Musso GM, Arico-Muendel CC, Benjamin HW, Hundal AM, Lee JJ, Chin J, Kelley M, Wakefield J, Hayward NJ, Molineaux SM. Modified-peptide inhibitors of amyloid-beta peptide polymerization. Biochemistry. 1999;38:6791–6800. doi: 10.1021/bi982824n. [DOI] [PubMed] [Google Scholar]
- 40.Petkova AT, Yau WM, Tycko R. Experimental constraints on quaternary structure in Alzheimer’s beta-amyloid fibrils. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petkova AT, Ishii Y, Balbach JJ, Antzutkin ON, Leapman RD, Delaglio F, Tycko R. A structural model for Alzheimer’s beta-amyloid fibrils based on experimental constraints from solid state NMR. Proc Natl Acad Sci USA. 2002;99:16742–16747. doi: 10.1073/pnas.262663499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naiki H, Higuchi K, Hosokawa M, Takeda T. Fluorometric determination of amyloid fibrils in vitro using the fluorescent dye thioflavin T. Anal Biochem. 1989;177:244–249. doi: 10.1016/0003-2697(89)90046-8. [DOI] [PubMed] [Google Scholar]
- 44.Jarrett JT, Berger EP, Lansbury PT., Jr The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 1993;32:4963–4967. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 45.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 46.Kim W, Kim Y, Min J, Kim DJ, Chang YT, Hecht MH. A high-throughput screen for compounds that inhibit aggregation of the Alzheimer’s peptide. ACS Chem Biol. 2006;1:461–469. doi: 10.1021/cb600135w. [DOI] [PubMed] [Google Scholar]
- 47.Kim W, Hecht MH. Sequence determinants of enhanced amyloidogenicity of Alzheimer Abeta42 peptide relative to Abeta40. J Biol Chem. 2005;280:35069–35076. doi: 10.1074/jbc.M505763200. [DOI] [PubMed] [Google Scholar]
- 48.Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- 49.Wood SJ, MacKenzie L, Maleeff B, Hurle M, Wetzel R. Selection inhibition of abeta fibril formation. J Biol Chem. 1996;271:4086–4092. doi: 10.1074/jbc.271.8.4086. [DOI] [PubMed] [Google Scholar]
- 50.de Groot NS, Aviles FX, Vendrell J, Ventura S. Mutagenesis of the central hydrophobic cluster in Abeta42 Alzheimer’s peptide. Side-chain properties correlate with aggregation propensities. FEBS J. 2006;273:658–668. doi: 10.1111/j.1742-4658.2005.05102.x. [DOI] [PubMed] [Google Scholar]
- 51.Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- 52.Bemporad F, Taddei N, Stefani M, Chiti F. Assessing the role of aromatic residues in the amyloid aggregation of human muscle acylphosphatase. Prot Sci. 2006;15:862–870. doi: 10.1110/ps.051915806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Self-propagating, molecular-Level polymorphism in Alzheimer’s beta-amyloid fibrils. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 54.Paravastu AK, Petkova AT, Tycko R. Polymorphic fibril formation by residues 10–40 of the Alzheimer’s beta-amyloid peptide. Biophys J. 2006;90:4618–4629. doi: 10.1529/biophysj.105.076927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma B, Nussinov R. Trp/Met/Phe hot spots in protein-protein interactions: Potential targets in drug design. Curr Top Med Chem. 2007;7:999–1005. doi: 10.2174/156802607780906717. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.