

Abstract
Penetrating traumatic insult during pregnancy is a leading cause of human fetal demise; in particular trauma to the brain may lead to devastating long-term cognitive sequelae. Perinatal brain injury involves glial precursors, but the neural mechanisms controlling astrocyte ontogeny after injury remain incompletely understood, partly due to a lack of appropriate markers and animal models. We analyzed astrocyte precursor response to injury at the beginning (E11) and peak (E15) of gliogenesis in an avian tectal model of penetrating embryonic brain trauma, without confounding maternal and sibling effects. At both ages, lateral ventricular dilatation, necrotic foci, periventricular cysts and intraventricular hemorrhages were observed distal to stab wounds two days after a unilateral stab injury to optic tecta. Neuronal (TUBB3) and oligodendrocyte precursor (PLP) markers were down-regulated, even far-removed from the wound site. In contrast, the mature astrocyte marker, GFAP, was up-regulated at the wound site, around necrotic areas and cysts, plus in usual areas of GFAP expression. Increased inflammatory response and apoptotic cell death were also confirmed in the injured tecta. Increased expression of NFIA, SOX9 and GLAST at the wound site and in the ventricular zone (VZ) of the injured tecta indicated an astroglial precursor response. However, cell division increased in the VZ only in early (E11) injury, but not later (E15), indicating that in late injury the astrogliogenesis occurring after acute injury is predominantly due to precursor differentiation rather than precursor proliferation. The inability to replenish the glial precursor pool during the critical period of vulnerability to injury may be an important cause of subsequent developmental abnormalities.
Keywords: astrocyte precursors, gliogenesis, brain stab-wound injury, ventricular zone, optic tecta
1. INTRODUCTION
Perinatal brain injury in humans following trauma, intrauterine infections, hypoxia, and/or ischemia, as well as the consequence of pre-maturity and low birth weight, often leads to devastating neurological consequences such as epilepsy, cerebral palsy, and behavioral and cognitive problems. Periventricular white matter injury (PWMI), the most frequently observed lesion in the neonatal brain, is strongly associated with several of these adverse outcomes (Dambska et al., 1989; Folkerth, 2005; Rees et al., 2008), but the etiology of this neuropathology is not fully understood (Johnston et al., 2002; Rezaie and Dean, 2002) and the mechanisms leading to PWMI have not been identified. From the clinical prospective, traumatic brain injury (TBI) elicits more severe responses during perinatal periods than in adults (Adelson and Kochanek, 1998; Levin et al., 1982). However, the pattern of neonatal brain injury differs significantly from that seen in the adult nervous system, and there are broad differences in regional vulnerability and plasticity (Chapman et al., 2003; Lawson and Perry, 1995; Milligan et al., 1991; Moses and Stiles, 2002; Stiles, 2000). Importantly, the susceptibility period for the most common forms of developmental brain injuries coincides with oligodendrocyte and astrocyte differentiation and maturation (Fig. 1). Deleterious effects on oligodendrocyte and astrocyte precursors together with functional vascular wall immaturity have been suggested to be vulnerability factors (McClure et al., 2008). If this hypothesis is valid, several types of injuries might be expected to produce similar phenotypes if they are inflicted during the susceptibility period.
Figure 1. Sequence of neuronal and macroglial development phases in avian brain compared to selected mammalian counterparts.

Comparison of chick, mouse/rat and human brain developmental timelines for the generation of the three cell types, based on the following reports: (Clancy et al., 2001; Clancy et al., 2007; Domowicz et al., 2008; Kim et al., 2006; Rice and Barone, 2000; Sauvageot and Stiles, 2002; Wiggins, 1986). The black bar in the human timeline represents the critical period of vulnerability in humans (Rice and Barone, 2000). E: embryonic age; P: post-natal day; M: month of gestation.
The literature describes a variety of animal models for the study of injury outcomes, ranging from hypoxia-ischemia to induction of the inflammatory response, that exhibit several of the phenotypic characteristics observed in individuals with PWMI (Back et al., 2007; Rees et al., 2008). Other types of brain injury such as traumatic or stab-wound injury have been less-studied in embryonic animal models, even though traumatic insult during pregnancy, in particular from penetrating trauma, has recently been reported to be the leading cause of fetal demise in humans (Petrone et al., 2010). Some information is available from the few animal models that have been described in the study of traumatic brain injury during development. Concussive head trauma in infant rats causes extensive neuronal cell death at the trauma site followed by degeneration of other, distant neurons (Bayly et al., 2006; Bittigau et al., 1999; Ikonomidou et al., 1996; Pohl et al., 1999). As brain development progresses, there is decreased susceptibility to apoptosis induced by traumatic injury, possibly related to the developmental expression of members of the caspase-3 pathway (Yakovlev et al., 2001) and to glutamate homeostasis (Lea and Faden, 2001). However, analyses of the responses of astrocytes and their precursors after stab-wound brain injury in the developing embryo are lacking, even though these cells may be expected to be key players in controlling glutamate metabolism after injury. In fact, astrocytic response towards reactive astrogliosis has been deemed both harmful and beneficial to neural repair after adult brain injury (Sofroniew, 2005; Sofroniew, 2009) and the response of glial precursors has only recently been assessed in a few adult animal injury models, where indications of oligodendrocyte precursor cell death and temporal shifts in progenitor fates have been reported (Gotts and Chesselet, 2005; Hampton et al., 2004; Sellers et al., 2009). In contrast to adult injury, during perinatal injury the brain lacks sufficient mature astrocytes to respond to injury; thus either progenitors need to divide to increase the pool of glial precursors or the existing precursors will need to accelerate their maturation to the astrocytic phenotype and then to reactive astrocytes. However, these aspects of the glial precursor response to embryonic injury have not been adequately examined, largely due to the limited number of in vivo markers, especially for astrocyte precursors.
Although the origin of astrocyte precursors during development is poorly defined, it is thought to occur in at least two phases. Astrocytes can be generated in early development by migration and differentiation of radial glial cells (Luskin et al., 1988; Price and Thurlow, 1988; Seo et al., 2008) and at later stages from migratory progenitors that emerge from the dorsal sub-ventricular zone (Levison and Goldman, 1993). Other than early glial markers such as brain lipid binding protein (BLBP) (Feng et al., 1994; Kurtz et al., 1994), glial Na+-dependent glutamate/aspartate transporter (GLAST) (Shibata et al., 1997) and nestin (Frederiksen and McKay, 1988), a number of transcription factors such as Sox9 (Stolt et al., 2003; Wegner and Stolt, 2005) and nuclear factor I/A and B (NFIA, NFIB) (Deneen et al., 2006), and proteoglycans such as aggrecan (Domowicz et al., 2008) and brevican (Jaworski et al., 1995), have been reported to be expressed by these precursors. In humans and in several animal models, oligodendrocyte loss and astrogliosis response after perinatal brain injury have been well documented (Marin-Padilla, 1997; Marin-Padilla, 1999; Robinson et al., 2005), but significant questions such as whether glial precursors are recruited after injury to become hypertrophic astrocytes remain unanswered.
The present study addresses how astrocyte precursors respond to embryonic injury by analyzing the expression and localization of a number of glial precursor markers using mRNA in situ hybridization in a novel avian model of penetrating tectal stab-wound injury. The chick has numerous advantages for such studies: i) the simple and well-studied laminar structure of the tectum (Gray and Sanes, 1991; LaVail and Cowan, 1971a; LaVail and Cowan, 1971b); ii) the opportunity to compare the ipsilateral (injured) and contralateral (uninjured) side tecta; iii) absence of confounding maternal or placental influences; and iv) the relatively advanced brain development at hatching in chicks (comparable to birth in humans), in contrast to that of small mammals like mouse and rat. In addition to these scientific advantages, chick embryos offer practical advantages as well: i) the use of eggs is economical, allowing large sample sizes; ii) ease of handling; iii) and ability to test at different developmental ages with accurate timing and reproducibility. Furthermore, timing of the generation of the three cell types within the chick brain correlates to comparable periods during mammalian development (Fig. 1), allowing direct temporal comparisons between the different species.
These significant advantages make embryonic chick brain an excellent model in which to assess changes in expression and localization of glial precursors. We have analyzed the consequences of stab-wound brain injury in this model at two critical times during astrocyte differentiation: in the early stages of gliogenesis (E11) and during the peak of gliogenesis (E15) (Fig. 1) (Domowicz et al., 2008). Interestingly, these ages coincide with the sensitivity period for impairment of memory after embryonic hypoxic injury in chicks (Rodricks et al., 2008) and the later period falls within the susceptibility periods described for human and rats (Fig. 1). We observed specific cell-type loss and pathology similar to that seen in human perinatal brain injuries, as well as activation of ventricular zone (VZ)-localized precursors. We also found increased expression of early glial markers at the wound site and in the VZ of the injured areas, supporting the hypothesis that glial precursors are recruited to astrocytic differentiation following injury. Furthermore, this recruitment appears independent of precursor proliferation when the injury is performed during the peak of gliogenesis (E15).
2. RESULTS
2.1 Embryonic Injury Model
Puncture injuries in the right optic tecta of chick embryos were made at E11 and E15 through windows cut in the egg shells. The optic tectum was the chosen target because it is a large and well-defined area of the chick brain with well-characterized cytoarchitecture comprising 15 laminae, where retinotectal projections have been thoroughly described, and for which the developmental formation process is known (Cepko et al., 1997; Yamagata et al., 2006). Eggs were re-sealed and the embryos were allowed to develop for up to 5 days after injury. We observed approximately 15% mortality among both injured and sham-control embryos, and in most cases death could be attributed to damage of the extra-embryonic vasculature during the handling of the embryo. Brains were collected, sectioned and analyzed with a battery of cell-type markers by in situ hybridization. Several examples of low magnification pictures of brain sections injured at E11 and E15 depicting the neuronal and astrocytic response to injury are shown in Supplementary Figure 1.
Besides the obvious puncture wounds in the right tecta (Fig. 2A, B), cavitating lesions (Fig. 2F), lateral ventricular dilatation (enlarged ventricles), periventricular cysts (Fig. 2G) and small hemorrhagic foci (Fig. 2C) were frequently observed in the injured midbrain either in the white matter or along the midbrain midline at E11. In a few cases, large lateral hemorrhages were observed (not shown), probably due to puncture of a major blood vessel during the injury.
Figure 2. E11 stab brain injury elicits early upregulation of GFAP mRNA in the ventricular zone and causes extensive neuronal damage.

Stab injuries were performed on E11 chick right tecta, and midbrains were collected two days later. Expression patterns of the neuronal marker Class III β-tubulin (TUBB3) (A,B,C, F) and the astrocytic marker glial fibrillary acidic protein (GFAP) (D, E & G) in the uninjured contralateral (Co) (A &D) and injured ipsilateral (Ip) (B,C,E and F) sides of the tecta detected by mRNA in situ hybridization are shown in midbrain transverse sections. Dotted line in B indicates needle track. Close-up of small hemorrhagic foci in the midbrain midline area (C), ventricular cavitating lesions (F), and peri-ventricular cysts (G) are shown. Arrows in B indicate reduced TUBB3 signal at a distance from the puncture site. Note that no hybridization signal for GFAP was detected in the contralateral side at this stage (D) compared to GFAP expression in the VZ of the ipsilateral side (E). Scale bar: 100 μm. Results depicted in this figure were consistently observed in multiple independent embryos (6 embryos)
Prenatal brain injury in humans results in gliosis as well as oligodendrocyte precursor and neuronal loss (Back et al., 2007; Rees et al., 2008; Rezaie and Dean, 2002; Volpe, 2001). In chick embryos, acute puncture injury at E11 led to extensive loss of the neuronal marker Class III β-tubulin (TUBB3), not only at the puncture site (Fig. 2B, dotted line) but at a distance from the injury site as well (Fig. 2B, arrows). This general loss of neurons starts to be evident 24 h after injury (not shown), in contrast to what has been described for adult brain injury models in which only localized neuronal loss is observed initially (Kazanis et al., 2003). Injury at the peak of gliogenesis (E15) yielded significant neuronal loss (Fig. 3A, B) and a very robust astroglial response, with strong increases in glial fibrillary acidic protein (GFAP) mRNA (Fig. 3C, D), not only in the puncture area (Fig. 3D, arrow) but also in the ventricular and intermediate zones (Fig. 3D, vertical line).
Figure 3. Increased GFAP mRNA expression on embryos injured at E15.
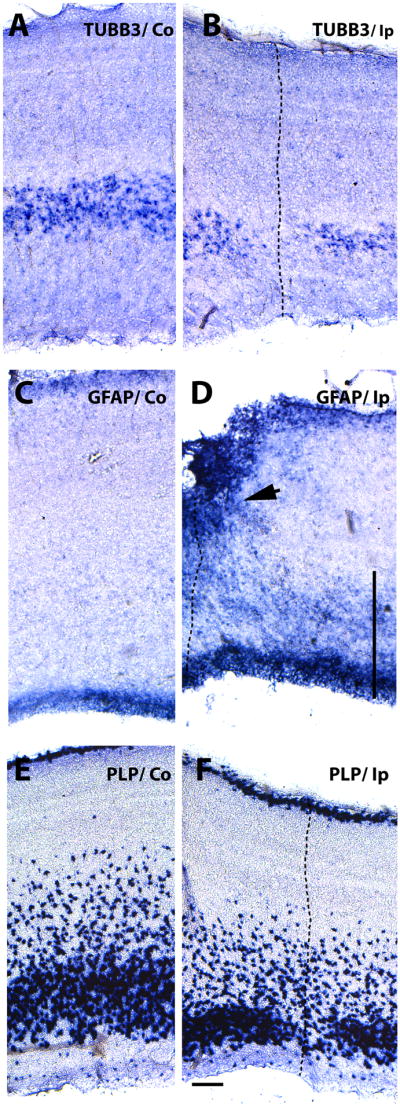
Expression patterns of post-mitotic neuron, astrocyte and oligodendrocyte precursor markers five days after stab-wound injury in E15 tectum. Serially adjoining cross-sections of the uninjured Co (contralateral) (A, C and E) and the injured Ip (ipsilateral) sides (B, D and F) of the tectal laminae probed for TUBB3 (neuronal marker) (A, B), GFAP (astrocyte marker) (C, D), and PLP (oligodendrocyte precursor marker) (E,F) expression by mRNA in situ hybridization. The needle track is marked with a dotted line. Note increased levels of GFAP in the VZ and around the injury site (arrow in D) and in the intermediate zone (vertical line in D), as well as the loss of neurons and oligodendrocyte precursors. Scale bar: 100μm. Results depicted in this figure were consistently observed in multiple independent embryos (6 embryos).
To facilitate study of oligodendrocyte precursor cells (OPC) in the chick brain, myelin proteolipid protein (PLP) mRNA levels can be used to detect late oligodendrocyte precursors. Since PLP mRNA was only detectable in the laminar tecta after E13, we evaluated the expression changes of this marker only following the later, E15 injuries. Expression of PLP mRNA was down-regulated in E15 injured tecta (Fig. 3F) and the affected areas correspond closely to the areas of neuronal marker loss (Fig. 3B). Interestingly, the superficial layers of the OPCs were less affected by injury. Previously, two populations of OPC have been distinguished in optic tecta on the bases of their origin and migration patterns: a population located in the superficial layers of the retinal nerve tract, and another population which originates in the ventricular zone of the mesencephalic neural tube and migrates first tangentially through the intermediate zone of the optic tectum and then radially towards the external layers (Kim et al., 2006). Our findings regarding the deeper tectal layers may indicate that this second population of OPCs is preferentially affected by injury.
2.2 Apoptosis
To explore whether the decreased neuronal and oligodendrocyte marker levels could be due to apoptotic cell death, levels of caspase-3/7 activity were measured in fresh tecta homogenates at different times after injury using a fluorogenic peptide substrate, Ac-DEVD-AMC (Fig. 4A). The lower levels of caspase-3/7 in the experiments performed at E15 (Fig. 4B) are expected, since most of the developmental neuronal cell death is over by E15 in the chick (Oppenheim, 1991). The results also indicate that, in tecta injured at either E11 or E15, increased caspase-3/7-specific activity peaks 24 h post-injury; however, no significant differences were observed between the uninjured and sham-control tecta. Furthermore, histological analysis of Nissl and DAPI fluorescent stained sections from the injured ipsilateral side (Fig. 4C and E) showed many pyknotic nuclei, in contrast with the uninjured contralateral side (Fig. 4D and F) 24h after E11 or E15 injuries. Thus, the loss of neurons and oligodendrocytes after injury may be attributable at least in part to apoptotic cell death, as has been described in other embryonic injury models (Bayly et al., 2006; Leviton and Gressens, 2007). Even though these findings do not identify the cell types affected by apoptosis they allow us to define the first 24–48 h after injury as a critical window for apoptotic damage.
Figure 4. Time course of neural death after stab-wound injury demonstrates a peak of apoptotic damage after 24h.

Levels of Caspase 3/7 were determined as a measure of apoptosis in the injured midbrains. Tecta were collected from embryos injured unilaterally on E11(A) or E15 (B) and sacrificed at the indicated times after injury. The ipsilateral and contralateral tecta were homogenized and caspase 3/7 catalytic activity was determined by measuring the proteolytic cleavage of the fluorogenic substrate Ac-DEVD-AMC (AnaSpec). Production of AMC was measured continuously in a Victor3 microplate fluorescent reader (PerkinElmer). Total protein content was normalized using the BCA protein assay (Pierce). As sham control, tecta from embryos in which eggs were windowed at E11 or E15 and collected at the indicated times were also analyzed. Initial maximum velocities are shown, expressed as relative fluorescent units per minute per mg of protein. Tukey HSD test was used after one-way ANOVA to generate the p values. **: p<0.01; *: p<0.05. Paraffin sections from embryos injured unilaterally on E11 (C, D) or E15 (E, F) and sacrificed 24h after injury were stained with green fluorescent Nissl stain (Invitrogen) and DAPI (Sigma). Panel C and E depict a representative image from the injured side and panel D and F from the uninjured contralateral side of the same section. All pictures correspond to the neuronal laminae i/j of the SGFS (stratum griseum et fibrosum superficial). Arrowhead indicates some of the pyknotic nuclei.
2.3 Inflammatory response
An inflammatory response to the stab-wound injuries was also observed, evidenced by up-regulation of cyclooxygenase-2 (COX-2) mRNA in the injured tecta as assayed by Northern blot (Fig. 5A). Interestingly, this up-regulation was evident in brains damaged at E11, even though the levels of COX-2 are relatively low at this embryonic stage since the immune system is not fully developed (Cuadros et al., 1997; Cuadros et al., 2000). Also, the levels of COX-2 in the uninjured-side tecta were higher than in age-matched sham-treated animals, indicating a remote response to injury in the acutely injured embryonic brains (Fig. 5B), a finding consistent with previous results in mammals (Caggiano et al., 1996). Since COX-2 has been reported to be expressed by all cell types in the brain (Laflamme et al., 1999; Minghetti, 2004; Samad et al., 2001; Vardeh et al., 2009), we analyzed expression of COX-2 by in situ hybridization in sections from midbrain injured at E15 and collected at E18 (Fig. 5C). We were unable to detect any signal for this probe in the tectum laminae except for the area immediately surrounding the needle track (Fig 5B, arrow), suggesting that the increased expression of COX-2 may be due at least in part to infiltrating activated microglia. We further analyzed expression of the transcription factor HIF-1, which has also been implicated in migration of microglia after hypoxic injury (Wang et al., 2008), and found a pattern of expression similar to that of COX-2 (Fig. 5C). Taken together these results support the idea that there is an inflammatory response after embryonic brain injury.
Figure 5. Embryonic brain stab injury triggers widespread pro-inflammatory changes.

A. COX-2 and GAPDH mRNA expression in optic tecta from embryos injured unilaterally on E11 and sacrificed on E14 (E11–13) or injured on E15 and sacrificed on E18 (E15–E18) determined by Northern blot analysis. As a sham control (S), tecta from embryos in which eggs were windowed at E11 or E15 and collected in E15 and E18, respectively, were used. A representative Northern blot is shown. B. COX-2 mRNA levels from injured I (ipsilateral) and uninjured C (contralateral) tecta were normalized with respect to GAPDH mRNA levels using Quantity One image analysis software (Bio-Rad). Data represent the average ± the standard deviation from three independent embryos. Tukey HSD test was used after one-way ANOVA to generate the p values. *: p<0.05. C. Expression of GFAP, COX-2, HIF-1 by in situ hybridization in three serially adjoining sections around the stab-wound area (arrow) of embryos injured unilaterally on E15 and sacrificed on E18.
2.4 Astrocyte precursor response
As mentioned, astrocyte precursors are thought to occur in at least two phases: generated by differentiation of radial glial cells in early development (Luskin et al., 1988; Price and Thurlow, 1988; Seo et al., 2008) and from migratory progenitors that emerge from the dorsal sub-VZ at later stages (Levison and Goldman, 1993). Intermediate filament proteins such as GFAP have been used in immunohistochemical studies to characterize the temporal spatial changes in reactive astrocytosis (Eng et al., 2000); this intermediate filament protein is also used in developmental studies as the major mature-astrocyte marker. We found GFAP mRNA levels to be up-regulated at the wound site and around necrotic areas and cysts, as well as in the VZ and intermediate zones in E15-punctured tecta (Fig. 3D & Fig. 7B & C), in multiple embryos confirming an astrogliosis response. Interestingly, increased GFAP mRNA expression in the VZ of the injured side of E11-punctured brains (Fig. 2E), before any GFAP-positive cells are detected in normal development, suggests that the gliogenesis process is accelerated by acute injury in the immature brain. To test this possibility, we analyzed the expression of a marker of astrocyte precursors at this stage, GLAST (Hartfuss et al., 2001; Shibata et al., 1997) and of two transcription factors, NFIA (Deneen et al., 2006) and SOX9 (Stolt et al., 2003), which recently have been proposed to regulate gliogenesis. Increased levels of all three markers were observed in the injured side of E11-punctured brains two days after injury (Fig. 6), particularly in the VZ but also in cells scattered throughout the tectal laminae (Fig. 6B, D, F). In brains injured at E15 these differences were more apparent and extended to all layers of the stratum central (Fig. 7E, H, K). Also, increased levels of GFAP around cyst and necrotic areas were accompanied by increased levels of these precursor markers (Fig. 7F, I, L). These data suggest that astrocyte precursors, derived from their normal sites of developmental generation, are responding to the acute injury. Also, since we observed improved needle-wound sealing following injuries at E11 compared to E15 (data not shown), perhaps the better healing capacity in the early injuries reflect a larger plasticity from earlier precursors.
Figure 7. Increased astrocytic precursor marker expression from the VZ to the parenchyma on embryos injured at E15.

Detection of NFIA (D, E, F), GLAST (G, H, I) and SOX9 (J, K, L) mRNAs by in situ hybridization in serially adjoining cross-sections of the tectal laminae three days after stab injuries performed at E15. Pattern of GFAP expression is shown for comparison (A–C). Increased expression levels of astrocyte precursor markers in the Ip (ipsilateral) tecta with respect to the Co (contralateral) side suggests activation of astrocyte differentiation in the VZ (B, E, H & K) (arrows) and in the parenchyma (vertical line) as well as surrounding the necrotic and cystic areas (C, F, I, L). Scale bar: 100μm. Results depicted in this figure were consistently observed in multiple independent embryos (3 embryos).
Figure 6. Increased expression of astrocytic precursor markers in the VZ of embryos injured at E11.

Detection of NFIA (A, B), GLAST (C, D) and SOX9 (E, F) mRNAs in serially adjoining cross-sections of the tectal laminae three days after stab injuries performed at E11. Increased expression levels of astrocyte precursor markers in Ip (ipsilateral) injured tecta (B, D and F) with respect to the Co (contralateral) side (A, C and D) in the VZ (arrows) and scattered throughout the tectal laminae (B arrowheads) suggests activation of astrocyte differentiation. Scale bar: 100μm. Results depicted in this figure were consistently observed in multiple independent embryos (3 embryos).
The question remained whether these changes following injury reflect an increased hypertrophic astrocytic response or an increased differentiation response. Since, at the embryonic stages studied, most of the tectal GFAP- positive astrocytes still have radial-glial morphology, we analyzed these cells for morphological changes after injury, using GFAP immunocytochemistry (Fig 8). Four days after stab injuries performed at E11, increased levels of GFAP immunoreactivity were observed in the ipsilateral side distal from the puncture site with respect to the contralateral side, in agreement with the up-regulation of GFAP mRNA (Fig 8A and B). The GFAP-positive cells in the injured side exhibited radial glial morphology, while astrocytes with reactive/hypertrophic morphology (Wilhelmsson et al., 2006) were identified in areas where pyknotic nuclei were observed (Fig 8C and D). Similar results were obtained in E15 punctured tecta (Fig. 8E and F), where astrocytes with hypertrophic morphology were found both at the puncture site (not shown) and around hemorrhagic foci (Fig 8G, I) and periventricular cysts (Fig 8H, J), while in the adjacent areas most of the GFAP-positive cells exhibit radial-glial morphology (Fig 8C, G and H). Thus, most of the transcriptional changes occurring after injury in the ventricular zone reflect precursor differentiation to the astrocytic fate rather than hypertrophy.
Figure 8. Morphology of GFAP-positive cells on embryos injured at E11 and E15.

A–D. Immunocytochemistry with GFAP antibody and confocal microscopy confirmed increased expression of GFAP protein in the ipsilateral side (B) of brains injured on E11 and sacrificed on E15 with respect to the contralateral (A) side of the same animal. Radial glial morphology characterized most cells distal from the stab wound (B) and damage areas. Cells with hypertrophic morphology were evident in areas with pyknotic nuclei (C). D depicts magnified confocal XY projections of the area marked with an arrowhead in C; the arrow in D indicates an astrocyte with hypertrophic morphology. E–J. GFAP-positive cells in brains injured at E15 and sacrificed on E20 maintained the radial cell morphology on the contralateral side and ipsilateral side distal from the injury (E and F, respectively). GFAP-positive cells around periventricular hemorrhagic foci (G, I) and periventricular cysts (H, J) exhibited a hypertrophic morphology. I and J depict magnified confocal XY projections of the area marked with arrowheads in G and H, respectively. Arrows in G and H indicate astrocytes with hypertrophic morphology. Scale bar represent 50 μm (A–C & E–H) and 10 μm D, I & J.
2.5 Proliferation
BrdU incorporation studies were performed to determine if the observed elevated levels of markers for astrocytes and their precursors can be attributed to increased proliferation in the VZ of the injured hemisphere. Analysis of brains injured at E11 and labeled with BrdU for 6 h at E13 revealed an increase in BrdU-positive cells in the VZ of the injured side (Fig. 9C, D), while in brains injured at E15, and labeled with BrdU no apparent increase in cell division was observed in any region of the tectum cortex at E18, including the VZ (Fig. 9G, H). These observations were confirmed by quantifying the number of proliferative cells two (E13) and four (E15) days after E11 injury, and three (E18) and five days (E20) after E15 injury (Fig 9I). A significant difference in the number of BrdU-positive cells per square mm was observed two days after E11 injury on the ipsilateral side with respect in the contralateral side. Four days after injury the same tendency was observed but the results were not statistically significant. In contrast, three or five days after brain injury at E15, no increase in proliferation was found in the injured compared to the uninjured side. These results suggest that increased precursor differentiation may be the primary mechanism driving the astrocytic response to injury at E15, and that the precursor population is not replenished, at least initially, by increased precursor mitosis.
Figure 9. Proliferation is activated after stab-wound brain injury at E11 but not at E15.

BrdU incorporation was used to assess cell proliferation. Embryos were injured at E11 (A–D) and E15 (E–H) and BrdU (1.6 mg/100 ml) was applied to the vitelline membrane 3h prior to tissue harvest at E13 and E18, respectively. Tissue was then fixed, embedded, sectioned and processed for immunocytochemistry using an anti-BrdU IgG (BD) primary antibody and an alkaline phosphatase conjugated anti-rabbit IgG (Roche) secondary antibody (C–D and G–H). Neighboring sections were processed for mRNA in situ hybridization with GFAP anti-sense probes. Left panels are representative sections from the Co (contralateral) side of the same injured Ip (ipsilateral) tectum side shown in the right panels. Arrows indicate the VZ and the dotted lines mark the needle tracks. Scale bar: 100μm. Bars in I represent BrdU-positive cells per square mm +/− standard error counted in sections similar to the ones shown above, from the ipsilateral (Ip) and contralateral (Co) sides in 3 independent animals, using Image J software. *: p<0.001 (One-way ANOVA)
3. DISCUSSION
Diverse animal injury models using conditions such as hypoxia-ischemia in rats and guinea pigs, umbilical cord occlusion in late ovine gestation, and exposure to endotoxin which mimics fetal infection and the associated inflammation response in sheep and rats, all replicate aspects of the pathology observed in humans, and therefore have been used to study different aspects of fetal brain injury (Back et al., 2007; Johnston et al., 2001; Rees and Harding, 2004; Rees et al., 2008; Riddle et al., 2006; Robinson et al., 2005; Svedin et al., 2005; Tan et al., 2005). Since many of the pathological sequelae of PWMI observed in human neonates closely resemble those observed after puncture injury in chick tectum, this injury model can be useful to test emerging hypotheses of PWMI pathogenesis: i) injury to oligodendrocyte progenitors may contribute to the pathogenesis of PWMI by arresting the maturation of oligodendrocytes (Segovia et al., 2008); ii) the vascular end zones in white matter may be particularly susceptible to ischemia, and several lesions of periventricular leukomalacia (PVL) may arise from severe or persistent ischemia in vascular end zones of long-penetration arteries (Nakamura et al., 1994; Volpe, 2001); iii) transient over-expression of activated microglia in cerebral white matter during the peak window of vulnerability for PVL could render this region susceptible to insults caused by prolonged microglial cell activation (Billiards et al., 2006). Thus far, the avian penetrating injury model has led to novel findings with respect to glial precursor response to embryonic injury.
Compared to needle stab-wound injury in adult mice (Burns et al., 2009), the brain damage occurring after such injury in the developing chick embryo is more severe with respect to the extent of astrocyte activation, as well as in pathological consequences. Increased levels of GFAP expression extend beyond the damaged areas and affect precursor population locations such as those in the VZ. Since increased GFAP expression is also indicative of reactive astrocytes, we needed to distinguish the contributions from reactive astrogliosis and differentiating astrocytes. Therefore, to better define the astrocyte precursor response, the expression of transcription factors known to be involved in astrocyte differentiation such as NFIA and SOX9 (Deneen et al., 2006; Stolt et al., 2003; Wegner and Stolt, 2005) as well as the morphological changes of the astrocytic cell population following injury were assessed. At both ages E11 and E15, hypertrophic morphology (Wilhelmsson et al., 2006) is observed around damaged areas, but for the most part the GFAP-positive cells exhibit the radial glial morphology that characterizes glial precursors at this developmental stage. These data suggest that the transcriptional changes observed in the VZ precursors are not due to hypertrophy of the cells, but rather are associated with increased differentiation towards the astrocytic phenotype following injury.
NFIA has thus far never been reported to be associated with reactive astrocytes, and our study is the first to analyze its expression after embryonic injury. Increases in SOX9 expression have been observed after adult spinal cord injury (Gris et al., 2007; Meletis et al., 2008); genetic cell mapping of the cells in the glial scar after such injury indicated that the SOX9-positive cells were astrocyte-like cells derived from the ependymal cell precursors, and were a population distinct from the resident reactive astrocytes (Meletis et al., 2008). Thus, increased expression of mRNAs for the transcription factors SOX9 and NFIA observed after embryonic injury likely indicates that either existing precursors accelerated their differentiation to the astrocytic phenotype or that more precursors were recruited to this fate. The latter effect could be particularly damaging if it reduced the pool of OPCs in the brain. This finding together with the loss of OPCs to apoptosis after injury, suggest this population of cells is most affected by developmental injuries. In addition, it has been suggested that extracellular matrix changes stemming from the gliosis response may limit the differentiation abilities of OPCs (Riddle et al., 2006), therefore alterations in matrix composition and fate are also being investigated (unpublished observations).
Although the modes of insult/damage are diverse in nature they all lead to an inflammatory cascade, and it has been suggested that this response, together with vascular and glial immaturity, may comprise the common denominators in the dramatic and widespread outcomes in neonatal brain injury (Folkerth, 2005; Reddy et al., 1986; Rees et al., 2008). We also demonstrated increased post-injury levels of COX-2, which is consistent with the occurrence of an inflammatory response in our model, even though the embryonic immune system is under-developed. Although COX-2 is known to be up-regulated in neurons after adult injury, when it aids in developing a central component of inflammatory pain hypersensitivity (Vardeh et al., 2009), we were unable to associate the increased COX-2 expression to neurons after injury. We did detect increased expression of COX-2 and HIF1A in the wound area by in situ hybridization, probably associated with activated microglia. Additionally, the reduction in neuronal and oligodendrocyte precursor marker levels indicate significant cell loss occurs during acute injury. Since levels of caspase-3/7 were dramatically increased 24 h post-injury, we can conclude that this loss may be due in part to apoptotic cell death. Susceptibility of other glial precursors and astrocytes to cell death should not be overlooked, since several other embryonic models have exhibited rapid loss of these cell types as well (Sen and Levison, 2006).
Proliferation analyses showed increased BrdU incorporation when injury was inflicted early (E11) but not later (E15) in development, suggesting that the diminished ability of precursors to respond to injury with progression of development by activating their cell cycle, might be an additional detrimental factor affecting recovery from injury during the susceptibility period (E15). Furthermore, we can conclude that the changes in SOX9 and NFIA expression patterns after E15 injuries were not due to the generation of more astrocyte precursors by cell division following injury. As well, the acute gliosis response by astrocytes at the injury site or associated with cystic areas was initially due to nonproliferative hypertrophy. Commensurate with our studies, lower levels of cell proliferation were observed (Robinson et al., 2005) in embryonic brain models of hypoxia-ischemia, while in models of chronic hypoxia increases in BrdU incorporation in the SVZ only become evident one week after cessation of hypoxia (Fagel et al., 2006). Also, brain injury in adults has been reported to increase cell division in precursor niches, but the effect occurs several weeks after injury at the earliest.
In conclusion, the pathology of avian prenatal brain injury described here closely parallels the response-to-injury process observed in mammalian models. Importantly, it provides a means to analyze differentiation, apoptosis and proliferation of glial precursors after injury and during the inflammation response, in an economical, technically straight-forward manner free of confounding maternal and sibling influences. The studies, thus far, indicate that the astrogliogenesis response to acute injury results predominantly in precursor differentiation and recruitment. The subsequent pathophysiology is therefore most likely due to a diminished precursor pool during the period of greatest susceptibility to injury, making the sequelae of embryonic injury significantly different from that observed in adult acute injury, and may explained the more severe pathophysiology.
4. EXPERIMENTAL PROCEDURES
4.1 Embryonic Brain Injury
Eggs containing E11 or E15 chick embryos were windowed and the vitelline membranes were opened. Embryos were carefully (avoiding damaging the extra-embryonic blood vessels) exposed using a sterile glass hook to gain access to the head. The exposed skull was stabbed at the level of the right tectum using a sterile 16-gauge needle which is scored at 1mm to mark the deepness of the wound. Eggs were resealed with 3M vinyl tape and returned to incubation at 39°C for up to five days after injury. Control eggs were sham-treated, omitting the stabbing step.
All age embryos were euthanized by decapitation prior to dissection of the brain in agreement with recommendations of ILAR, the NIH intramural recommendations for rodent neonates, and the AVMA Panel on Euthanasia.
4.2 mRNA In Situ Hybridization
At different times after injury, brains were dissected, fixed in 4% paraformaldehyde, and processed for non-radioactive in situ hybridization as described previously (Domowicz et al., 2008). To prepare the digoxigenin (DIG)-labeled RNA probes used for in situ hybridizations, cDNA fragments from the GFAP, PLP, TUBB3, SOX9, COX-2 and GLAST/SLC1A3 genes were prepared as previously described (Domowicz et al., 2008; Domowicz et al., 2009) and cDNA fragments for NFIA (nt 1330–1864 of NM_205273) and HIF1A (nt 1562–2228 of NM_204297.1) were obtained by PCR from E8 chick brain cDNA using specific primers based on GenBank sequences. Riboprobes incorporating DIG-labeled nucleotides were synthesized from linearized plasmid templates with SP6 or T7 polymerase (Roche). After hybridization, DIG-labeled RNA duplexes were detected with an alkaline phosphatase-conjugated anti-DIG antibody (Roche). Alkaline phosphatase activity was detected using BCIP and NBT substrates (Roche). Results depicted in Figs. 2, 3, 6 and 7 were consistently observed in multiple independent embryos (3–6) and contralateral tecta patterns are comparable to sham controls (data not shown).
4.3 Northern Blot Analysis
Total brain RNA was extracted using TRIzol reagent (Invitrogen). RNA (10 μg per lane) was electrophoresed in a denaturing 1.2% agarose-formaldehyde gel and transferred to Nytran SuperCharge (Schleicher and Schuell) membranes by TurboBlotter rapid downward transfer. Hybridization was performed using QuikHyb solution (Stratagene). Radiolabeled probes were prepared using the Rediprime system (Roche). Following hybridization, the nylon membranes were washed twice with 2X SSC/0.05% SDS for 30 min at room temperature and twice with 0.1X SSC/0.1% SDS for 30 min at 58 °C. Autoradiography was performed overnight at −70°C with Kodak MR2 film. Bands were quantified with Quantity One image analysis software (Bio-Rad). Blots were stripped by boiling in 0.1X SSC/0.1%SDS and re-probed with a GAPDH control probe. GAPDH mRNA level was used as the internal standard for normalizing loading differences.
4.4 Proliferation Assay
For proliferation assays, BrdU (1.6 mg/100μL) was applied to the vitelline membranes of eggs 3 h prior to tissue harvest. Tissue was then fixed, embedded, sectioned and processed for immunocytochemistry using an anti-BrdU IgG (BD Bioscience) primary antibody and alkaline phosphatase conjugated anti-rabbit IgG (Roche, Germany) secondary antibody (Domowicz et al., 2008). BrdU-positive cells were quantified using ImageJ software (NIH) for three embryos at each indicated embryonic age.
4.5 Caspase 3/7 Activity
At the indicated times after injury, the right and left tecta were dissected and homogenized in lysis buffer (AnaSpec, Ca). Caspase 3/7 catalytic activity was determined by measuring the proteolytic cleavage of the fluorogenic substrate Ac-DEVD-AMC (AnaSpec, Ca). Production of AMC was monitored continuously in a Victor3 fluorescence plate reader (PerkinElmer) (King et al., 2009). Total protein content was determined using the BCA protein assay (Pierce). Initial maximum velocities are represented (Fig. 4) and expressed as relative fluorescence units per minute per μg of protein. Data were analyzed for statistical significance using the Student’s t-test.
4.6 Immunocytochemistry and histology
Immunocytochemistry with anti-GFAP antibody (Clone GF5, Millipore) was performed as described previously (Domowicz et al., 2008). Fluorescence images were obtained with a Leica SP2 spectral confocal microscope in the BSD Digital Light Microscopy Core Facility of The University of Chicago Cancer Research Center.
Green fluorescent Nissl staining (Invitrogen) and DAPI (Sigma) staining were performed on paraffin sections from embryos injured unilaterally on E11 or E15 and sacrificed 24h after injury. Fluorescence images were obtained with an Olympus DSU spinning disk confocal microscope in the BSD Digital Light Microscopy Core Facility of The University of Chicago Cancer Research Center.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institute of Child Health and Human Disorders (HD 09402; P30 HD054275), the National Institute of General Medical Science (R25 GM066522-06), the National Institute of Neurological Disorders and Stroke (NS-19108), and the White Foundation.
Footnotes
Authors’ contributions. The initial study concept and design was a joint effort of MSD and NBS. MSD and JGH developed and performed the injury model protocol. MSD, JGH, NW, and AN performed the experiments. Analysis of the data was done by MSD and discussed with NBS and RPK. MSD, NBS and RPK wrote the paper. The final version contains contributions of all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelson PD, Kochanek PM. Head injury in children. J Child Neurol. 1998;13:2–15. doi: 10.1177/088307389801300102. [DOI] [PubMed] [Google Scholar]
- Back SA, Riddle A, McClure MM. Maturation-dependent vulnerability of perinatal white matter in premature birth. Stroke. 2007;38:724–30. doi: 10.1161/01.STR.0000254729.27386.05. [DOI] [PubMed] [Google Scholar]
- Bayly PV, Dikranian KT, Black EE, Young C, Qin YQ, Labruyere J, Olney JW. Spatiotemporal evolution of apoptotic neurodegeneration following traumatic injury to the developing rat brain. Brain Res. 2006;1107:70–81. doi: 10.1016/j.brainres.2006.05.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiards SS, Haynes RL, Folkerth RD, Trachtenberg FL, Liu LG, Volpe JJ, Kinney HC. Development of microglia in the cerebral white matter of the human fetus and infant. J Comp Neurol. 2006;497:199–208. doi: 10.1002/cne.20991. [DOI] [PubMed] [Google Scholar]
- Bittigau P, Sifringer M, Pohl D, Stadthaus D, Ishimaru M, Shimizu H, Ikeda M, Lang D, Speer A, Olney JW, Ikonomidou C. Apoptotic neurodegeneration following trauma is markedly enhanced in the immature brain. Ann Neurol. 1999;45:724–35. doi: 10.1002/1531-8249(199906)45:6<724::aid-ana6>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Burns KA, Murphy B, Danzer SC, Kuan CY. Developmental and post-injury cortical gliogenesis: a genetic fate-mapping study with Nestin-CreER mice. Glia. 2009;57:1115–29. doi: 10.1002/glia.20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caggiano AO, Breder CD, Kraig RP. Long-term elevation of cyclooxygenase-2, but not lipoxygenase, in regions synaptically distant from spreading depression. J Comp Neurol. 1996;376:447–62. doi: 10.1002/(SICI)1096-9861(19961216)376:3<447::AID-CNE7>3.0.CO;2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepko CL, Golden JA, Szele FG, Lin JC. Linage analysis in the vertebrate Central Nervous System. In: Cowan WM, Jessell TM, Zipursky SL, editors. Molecular and cellular approches to neural development. Oxford University Press; New York: 1997. pp. 391–439. [Google Scholar]
- Chapman SB, Max JE, Gamino JF, McGlothlin JH, Cliff SN. Discourse plasticity in children after stroke: age at injury and lesion effects. Pediatr Neurol. 2003;29:34–41. doi: 10.1016/s0887-8994(03)00012-2. [DOI] [PubMed] [Google Scholar]
- Clancy B, Darlington RB, Finlay BL. Translating developmental time across mammalian species. Neuroscience. 2001;105:7–17. doi: 10.1016/s0306-4522(01)00171-3. [DOI] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931–7. doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadros MA, Rodriguez-Ruiz J, Calvente R, Almendros A, Marin-Teva JL, Navascues J. Microglia development in the quail cerebellum. J Comp Neurol. 1997;389:390–401. [PubMed] [Google Scholar]
- Cuadros MA, Martin D, Perez-Mendoza D, Navascues J, Clarke PG. Response of macrophage/microglial cells to experimental neuronal degeneration in the avian isthmo-optic nucleus during development. J Comp Neurol. 2000;423:659–69. doi: 10.1002/1096-9861(20000807)423:4<659::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Dambska M, Laure-Kamionowska M, Schmidt-Sidor B. Early and late neuropathological changes in perinatal white matter damage. J Child Neurol. 1989;4:291–8. doi: 10.1177/088307388900400408. [DOI] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52:953–68. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Domowicz MS, Sanders TA, Ragsdale CW, Schwartz NB. Aggrecan is expressed by embryonic brain glia and regulates astrocyte development. Developmental Biology. 2008;315:114–124. doi: 10.1016/j.ydbio.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domowicz MS, Cortes M, Henry JG, Schwartz NB. Aggrecan modulation of growth plate morphogenesis. Dev Biol. 2009;329:242–57. doi: 10.1016/j.ydbio.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng LF, Ghirnikar RS, Lee YL. Glial fibrillary acidic protein: GFAP-thirty-one years (1969–2000) Neurochem Res. 2000;25:1439–51. doi: 10.1023/a:1007677003387. [DOI] [PubMed] [Google Scholar]
- Fagel DM, Ganat Y, Silbereis J, Ebbitt T, Stewart W, Zhang H, Ment LR, Vaccarino FM. Cortical neurogenesis enhanced by chronic perinatal hypoxia. Exp Neurol. 2006;199:77–91. doi: 10.1016/j.expneurol.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Feng L, Hatten ME, Heintz N. Brain lipid-binding protein (BLBP): A novel signaling system in the developing mammalian CNS. Neuron. 1994;12:895–908. doi: 10.1016/0896-6273(94)90341-7. [DOI] [PubMed] [Google Scholar]
- Folkerth RD. Neuropathologic substrate of cerebral palsy. J Child Neurol. 2005;20:940–9. doi: 10.1177/08830738050200120301. [DOI] [PubMed] [Google Scholar]
- Frederiksen K, McKay RD. Proliferation and differentiation of rat neuroepithelial precursor cells in vivo. J Neurosci. 1988;8:1144–51. doi: 10.1523/JNEUROSCI.08-04-01144.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotts JE, Chesselet MF. Migration and fate of newly born cells after focal cortical ischemia in adult rats. J Neurosci Res. 2005;80:160–71. doi: 10.1002/jnr.20434. [DOI] [PubMed] [Google Scholar]
- Gray GE, Sanes JR. Migratory paths and phenotypic choices of clonally related cells in the avian optic tectum. Neuron. 1991;6:211–225. doi: 10.1016/0896-6273(91)90357-6. [DOI] [PubMed] [Google Scholar]
- Gris P, Tighe A, Levin D, Sharma R, Brown A. Transcriptional regulation of scar gene expression in primary astrocytes. Glia. 2007;55:1145–55. doi: 10.1002/glia.20537. [DOI] [PubMed] [Google Scholar]
- Hampton DW, Rhodes KE, Zhao C, Franklin RJ, Fawcett JW. The responses of oligodendrocyte precursor cells, astrocytes and microglia to a cortical stab injury, in the brain. Neuroscience. 2004;127:813–20. doi: 10.1016/j.neuroscience.2004.05.028. [DOI] [PubMed] [Google Scholar]
- Hartfuss E, Galli R, Heins N, Gotz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol. 2001;229:15–30. doi: 10.1006/dbio.2000.9962. [DOI] [PubMed] [Google Scholar]
- Ikonomidou C, Qin Y, Labruyere J, Kirby C, Olney JW. Prevention of trauma-induced neurodegeneration in infant rat brain. Pediatr Res. 1996;39:1020–7. doi: 10.1203/00006450-199606000-00015. [DOI] [PubMed] [Google Scholar]
- Jaworski DM, Kelly GM, Hockfield S. The CNS-specific hyaluronan-binding protein BEHAB is expressed in ventricular zones coincident with gliogenesis. J Neurosci. 1995;15:1352–62. doi: 10.1523/JNEUROSCI.15-02-01352.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston MV, Trescher WH, Ishida A, Nakajima W. Neurobiology of hypoxic-ischemic injury in the developing brain. Pediatr Res. 2001;49:735–41. doi: 10.1203/00006450-200106000-00003. [DOI] [PubMed] [Google Scholar]
- Johnston MV, Nakajima W, Hagberg H. Mechanisms of hypoxic neurodegeneration in the developing brain. Neuroscientist. 2002;8:212–20. doi: 10.1177/1073858402008003007. [DOI] [PubMed] [Google Scholar]
- Kazanis I, Bozas E, Philippidis H, Stylianopoulou F. Neuroprotective effects of insulin-like growth factor-I (IGF-I) following a penetrating brain injury in rats. Brain Res. 2003;991:34–45. doi: 10.1016/s0006-8993(03)03525-x. [DOI] [PubMed] [Google Scholar]
- Kim DW, Park SW, Jeon GS, Seo JH, Golden JA, Cho SS. The multiple dorsoventral origins and migratory pathway of tectal oligodendrocytes in the developing chick. Brain Res. 2006;1076:16–24. doi: 10.1016/j.brainres.2006.01.010. [DOI] [PubMed] [Google Scholar]
- King LA, Schwartz NB, Domowicz MS. Glial migratory streams in the developing hindbrain: a slice culture approach. J Neurosci Methods. 2009;177:30–43. doi: 10.1016/j.jneumeth.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Zimmer A, Schnutgen F, Bruning G, Spener F, Muller T. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development. 1994;120:2637–49. doi: 10.1242/dev.120.9.2637. [DOI] [PubMed] [Google Scholar]
- Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1beta in mediating NF-kappaB activity and COX-2 transcription in cells of the blood-brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. 1999;19:10923–30. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail JH, Cowan WM. The development of the chick optic tectum. I. Normal morphology and cytoarchitectonic development. Brain Res. 1971a;28:391–419. doi: 10.1016/0006-8993(71)90053-9. [DOI] [PubMed] [Google Scholar]
- LaVail JH, Cowan WM. The development of the chick optic tectum. II. Autoradiographic studies. Brain Res. 1971b;28:421–41. [PubMed] [Google Scholar]
- Lawson LJ, Perry VH. The unique characteristics of inflammatory responses in mouse brain are acquired during postnatal development. Eur J Neurosci. 1995;7:1584–95. doi: 10.1111/j.1460-9568.1995.tb01154.x. [DOI] [PubMed] [Google Scholar]
- Lea PMt, Faden AI. Traumatic brain injury: developmental differences in glutamate receptor response and the impact on treatment. Ment Retard Dev Disabil Res Rev. 2001;7:235–48. doi: 10.1002/mrdd.1033. [DOI] [PubMed] [Google Scholar]
- Levin HS, Eisenberg HM, Wigg NR, Kobayashi K. Memory and intellectual ability after head injury in children and adolescents. Neurosurgery. 1982;11:668–73. doi: 10.1227/00006123-198211000-00009. [DOI] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–12. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Leviton A, Gressens P. Neuronal damage accompanies perinatal white-matter damage. Trends Neurosci. 2007;30:473–8. doi: 10.1016/j.tins.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Luskin MB, Pearlman AL, Sanes JR. Cell lineage in the cerebral cortex of the mouse studied in vivo and in vitro with a recombinant retrovirus. Neuron. 1988;1:635–47. doi: 10.1016/0896-6273(88)90163-8. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. II: white matter lesions of the neocortex. J Neuropathol Exp Neurol. 1997;56:219–35. doi: 10.1097/00005072-199703000-00001. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Developmental neuropathology and impact of perinatal brain damage. III: gray matter lesions of the neocortex. J Neuropathol Exp Neurol. 1999;58:407–29. doi: 10.1097/00005072-199905000-00001. [DOI] [PubMed] [Google Scholar]
- McClure MM, Riddle A, Manese M, Luo NL, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Vinecore K, Roberts CT, Hohimer AR, Back SA. Cerebral blood flow heterogeneity in preterm sheep: lack of physiologic support for vascular boundary zones in fetal cerebral white matter. J Cereb Blood Flow Metab. 2008;28:995–1008. doi: 10.1038/sj.jcbfm.9600597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletis K, Barnabe-Heider F, Carlen M, Evergren E, Tomilin N, Shupliakov O, Frisen J. Spinal cord injury reveals multilineage differentiation of ependymal cells. PLoS Biol. 2008;6:e182. doi: 10.1371/journal.pbio.0060182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CE, Levitt P, Cunningham TJ. Brain macrophages and microglia respond differently to lesions of the developing and adult visual system. J Comp Neurol. 1991;314:136–46. doi: 10.1002/cne.903140113. [DOI] [PubMed] [Google Scholar]
- Minghetti L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J Neuropathol Exp Neurol. 2004;63:901–10. doi: 10.1093/jnen/63.9.901. [DOI] [PubMed] [Google Scholar]
- Moses P, Stiles J. The lesion methodology: contrasting views from adult and child studies. Dev Psychobiol. 2002;40:266–77. doi: 10.1002/dev.10031. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Okudera T, Hashimoto T. Vascular architecture in white matter of neonates: its relationship to periventricular leukomalacia. J Neuropathol Exp Neurol. 1994;53:582–9. doi: 10.1097/00005072-199411000-00005. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Petrone P, Talving P, Browder T, Teixeira PG, Fisher O, Lozornio A, Chan LS. Abdominal injuries in pregnancy: a 155 month study at two level 1 trauma centers. Injury. 2010 doi: 10.1016/j.injury.2010.06.026. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Pohl D, Bittigau P, Ishimaru MJ, Stadthaus D, Hubner C, Olney JW, Turski L, Ikonomidou C. N-Methyl-D-aspartate antagonists and apoptotic cell death triggered by head trauma in developing rat brain. Proc Natl Acad Sci U S A. 1999;96:2508–13. doi: 10.1073/pnas.96.5.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J, Thurlow L. Cell lineage in the rat cerebral cortex: a study using retroviral-mediated gene transfer. Development. 1988;104:473–82. doi: 10.1242/dev.104.3.473. [DOI] [PubMed] [Google Scholar]
- Reddy P, Jacquier AC, Abovich N, Peterson G, Rosbash M. Cell. 1986;46:53–61. doi: 10.1016/0092-8674(86)90859-7. [DOI] [PubMed] [Google Scholar]
- Rees S, Harding R. Brain development during fetal life: influences of the intra-uterine environment. Neurosci Lett. 2004;361:111–4. doi: 10.1016/j.neulet.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Rees S, Harding R, Walker D. An adverse intrauterine environment: implications for injury and altered development of the brain. Int J Dev Neurosci. 2008;26:3–11. doi: 10.1016/j.ijdevneu.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Rezaie P, Dean A. Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology. 2002;22:106–32. doi: 10.1046/j.1440-1789.2002.00438.x. [DOI] [PubMed] [Google Scholar]
- Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle A, Luo NL, Manese M, Beardsley DJ, Green L, Rorvik DA, Kelly KA, Barlow CH, Kelly JJ, Hohimer AR, Back SA. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–55. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S, Petelenz K, Li Q, Cohen ML, Dechant A, Tabrizi N, Bucek M, Lust D, Miller RH. Developmental changes induced by graded prenatal systemic hypoxic-ischemic insults in rats. Neurobiol Dis. 2005;18:568–81. doi: 10.1016/j.nbd.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Rodricks CL, Gibbs ME, Jenkin G, Miller SL. The effect of hypoxia at different embryonic ages on impairment of memory ability in chicks. Int J Dev Neurosci. 2008;26:113–8. doi: 10.1016/j.ijdevneu.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ. Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–5. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- Sauvageot CM, Stiles CD. Molecular mechanisms controlling cortical gliogenesis. Current Opinion in Neurobiology. 2002;12:244–249. doi: 10.1016/s0959-4388(02)00322-7. [DOI] [PubMed] [Google Scholar]
- Segovia KN, McClure M, Moravec M, Luo NL, Wan Y, Gong X, Riddle A, Craig A, Struve J, Sherman LS, Back SA. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:520–30. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers DL, Maris DO, Horner PJ. Postinjury niches induce temporal shifts in progenitor fates to direct lesion repair after spinal cord injury. J Neurosci. 2009;29:6722–33. doi: 10.1523/JNEUROSCI.4538-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen E, Levison SW. Astrocytes and developmental white matter disorders. Ment Retard Dev Disabil Res Rev. 2006;12:97–104. doi: 10.1002/mrdd.20106. [DOI] [PubMed] [Google Scholar]
- Seo JH, Chang JH, Song SH, Lee HN, Jeon GS, Kim DW, Chung CK, Cho SS. Spatiotemporal Gradient of Astrocyte Development in the Chick Optic Tectum: Evidence for Multiple Origins and Migratory Paths of Astrocytes. Neurochem Res. 2008 doi: 10.1007/s11064-008-9590-3. [DOI] [PubMed] [Google Scholar]
- Shibata T, Yamada K, Watanabe M, Ikenaka K, Wada K, Tanaka K, Inoue Y. Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. J Neurosci. 1997;17:9212–9. doi: 10.1523/JNEUROSCI.17-23-09212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–7. doi: 10.1177/1073858405278321. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles J. Neural plasticity and cognitive development. Dev Neuropsychol. 2000;18:237–72. doi: 10.1207/S15326942DN1802_5. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Lommes P, Sock E, Chaboissier MC, Schedl A, Wegner M. The Sox9 transcription factor determines glial fate choice in the developing spinal cord. Genes Dev. 2003;17:1677–89. doi: 10.1101/gad.259003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedin P, Kjellmer I, Welin AK, Blad S, Mallard C. Maturational effects of lipopolysaccharide on white-matter injury in fetal sheep. J Child Neurol. 2005;20:960–4. doi: 10.1177/08830738050200120501. [DOI] [PubMed] [Google Scholar]
- Tan S, Drobyshevsky A, Jilling T, Ji X, Ullman LM, Englof I, Derrick M. Model of cerebral palsy in the perinatal rabbit. J Child Neurol. 2005;20:972–9. doi: 10.1177/08830738050200120801. [DOI] [PubMed] [Google Scholar]
- Vardeh D, Wang D, Costigan M, Lazarus M, Saper CB, Woolf CJ, Fitzgerald GA, Samad TA. COX2 in CNS neural cells mediates mechanical inflammatory pain hypersensitivity in mice. J Clin Invest. 2009;119:287–94. doi: 10.1172/JCI37098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatr Res. 2001;50:553–62. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Wang X, Li C, Chen Y, Hao Y, Zhou W, Chen C, Yu Z. Hypoxia enhances CXCR4 expression favoring microglia migration via HIF-1alpha activation. Biochem Biophys Res Commun. 2008;371:283–8. doi: 10.1016/j.bbrc.2008.04.055. [DOI] [PubMed] [Google Scholar]
- Wegner M, Stolt CC. From stem cells to neurons and glia: a Soxist’s view of neural development. Trends Neurosci. 2005;28:583–8. doi: 10.1016/j.tins.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Wiggins RC. Myelination: a critical stage in development. Neurotoxicology. 1986;7:103–20. [PubMed] [Google Scholar]
- Wilhelmsson U, Bushong EA, Price DL, Smarr BL, Phung V, Terada M, Ellisman MH, Pekny M. Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci U S A. 2006;103:17513–8. doi: 10.1073/pnas.0602841103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev AG, Ota K, Wang G, Movsesyan V, Bao WL, Yoshihara K, Faden AI. Differential expression of apoptotic protease-activating factor-1 and caspase-3 genes and susceptibility to apoptosis during brain development and after traumatic brain injury. J Neurosci. 2001;21:7439–46. doi: 10.1523/JNEUROSCI.21-19-07439.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata M, Weiner JA, Dulac C, Roth KA, Sanes JR. Labeled lines in the retinotectal system: markers for retinorecipient sublaminae and the retinal ganglion cell subsets that innervate them. Mol Cell Neurosci. 2006;33:296–310. doi: 10.1016/j.mcn.2006.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.