

Using large scale experimental datasets, the authors show how modular protein interaction domains such as PDZ, SH3 or WW domains, frequently display unexpected multiple binding specificity. The observed multiple specificity leads to new structural insights and accurately predicts new protein interactions.
Keywords: binding specificity, peptide recognition domains, PDZ, phage display, residue correlations
Abstract
Modular protein interaction domains form the building blocks of eukaryotic signaling pathways. Many of them, known as peptide recognition domains, mediate protein interactions by recognizing short, linear amino acid stretches on the surface of their cognate partners with high specificity. Residues in these stretches are usually assumed to contribute independently to binding, which has led to a simplified understanding of protein interactions. Conversely, we observe in large binding peptide data sets that different residue positions display highly significant correlations for many domains in three distinct families (PDZ, SH3 and WW). These correlation patterns reveal a widespread occurrence of multiple binding specificities and give novel structural insights into protein interactions. For example, we predict a new binding mode of PDZ domains and structurally rationalize it for DLG1 PDZ1. We show that multiple specificity more accurately predicts protein interactions and experimentally validate some of the predictions for the human proteins DLG1 and SCRIB. Overall, our results reveal a rich specificity landscape in peptide recognition domains, suggesting new ways of encoding specificity in protein interaction networks.
Introduction
Modular peptide recognition domains are a widespread class of protein domains that mediate important protein interactions in cell signaling pathways and are involved in the assembly of many protein complexes (Pawson and Nash, 2003). Some of the largest families of these domains, including PDZ (Doyle et al, 1996; Harris and Lim, 2001), WW (Hu et al, 2004), SH3 (Mayer, 2001) and kinases (Hutti et al, 2004; Miller et al, 2008), bind selectively to short linear motifs (Gould et al, 2010) often found in disordered regions on the surface of proteins. These interactions are usually sufficiently specific so that a detailed knowledge of the binding preferences of a given domain allows for accurate prediction of its interactions (Tonikian et al, 2009). Many high-throughput experimental techniques have been developed to characterize the binding specificity of modular peptide recognition domains. Microarrays (Stiffler et al, 2007) and synthetic peptide array technology (SPOT; Wiedemann et al, 2004) have been used to measure the binding of hundreds of selected peptides with different domains. Kinase specificity has been studied using different methods, such as oriented peptide libraries (Hutti et al, 2004) or quantitative phosphoproteomics (Olsen et al, 2010). Phage display provides an accurate and unbiased way of studying in vitro the specificity of modular peptide recognition domains (Tong et al, 2002; Tonikian et al, 2008). This technology uses bacteriophage to express libraries of up to 10 billion random peptides as genetic fusions to phage coat proteins (Tonikian et al, 2007). After repeatedly incubating the phage particles with a domain, and washing away non-interacting phage, a specificity profile consisting of a set of strongly interacting peptides can be retrieved by sequencing the phage-encapsulated DNA.
For protein domains interacting with unstructured peptides found on the surface of proteins, it is often assumed that each residue contributes independently to the binding affinity. In other words, the presence of a given residue at some position does not significantly influence the amino acid preference at another position along the interacting peptide. This assumption of uncorrelated positions is underlying several computational models of binding specificity (Chen et al, 2008; Tonikian et al, 2008). One such popular model is the position weight matrix (PWM, sometimes called position-specific scoring matrix; Obenauer et al, 2003), which can be visualized as a sequence logo. Disregarding correlations when modeling specificity implicitly assumes that domains are characterized by a single class of binding peptides, all following the same binding mode.
Using large data sets derived from phage display experiments for human and worm PDZ domains (Tonikian et al, 2008), yeast SH3 domains (Tonikian et al, 2009) and human WW domains, we show that highly significant positional correlations are found for almost half of the domains analyzed here. Moreover, we observe that most correlation patterns can be captured by clustering the peptides into a small number of clusters. This result prompted us to represent domain-binding specificity with a mixture model that makes use of multiple PWMs, instead of a single one, and our results reveal a widespread occurrence of multiple specificity. Other machine learning algorithms, such as hidden Markov models (HMMs; Noguchi et al, 2002), artificial neural networks (ANNs; Brusic et al, 1998; Blom et al, 1999; Nielsen et al, 1999; Emanuelsson et al, 2000; Miller et al, 2008) or support vector machines (Hui and Bader, 2010; Shao et al, 2010) have been used previously in different contexts to account for positional correlations. Our work suggests that the full complexity and nonlinearity of these models may not be required to accurately model the specificity of protein domains binding to short linear peptides. Moreover, thanks to simple visualization (which, mathematically speaking, can be related to linear approximations of HMMs or ANNs) and direct interpretation, the multiple specificity model gives new structural insights into binding modes of modular peptide recognition domains and predicts new protein interactions within signaling pathways mediated by these domains.
Results
Positional correlations are widespread in known specificity profiles
Positional correlations reflect the influence of an amino acid at one position in a set of interacting peptides over the amino acid preferences at other positions. For instance, in peptides binding to the first DLG1 PDZ domain, I and L are both observed seven times at position −1 (Figure 1A). However, W at position 0 is always found together with I at position −1 and never with L. To measure correlations among pairs of residue positions, we used mutual information. Taking a P-value threshold of 0.001 to define significant correlations (see Materials and methods), we observe that roughly a third of all tested domains have at least one pair of significantly correlated positions in their specificity profile. Specifically, 24 out of 82 PDZ domains, 13 out of 24 SH3 domains and the 3 WW domains display positional correlations. For instance, peptides interacting with DLG1 PDZ1 display many correlated positions, and significant P-values are observed among most of the last five residues (red edges in Figure 1A). The profiles displaying strong positional correlations do not conform to the assumptions of positional independence of a single PWM model and hence cannot be accurately modeled in this way.
Figure 1.

Positional correlations are present in peptides binding to modular peptide recognition domains. (A) Phage peptides binding to the first PDZ domain of the human protein DLG1, aligned from the C terminus. The last five positions (red box) display positional correlations. Pairs of significantly correlated positions (P-value <0.001) are connected with a red edge, others with a black edge. An example of correlation can be found between the two last columns: W or L at position 0 always appears with I at −1, whereas V at position 0 is never found with I at −1. (B) Hierarchical clustering of the peptides shown in A. The heat map shows the similarity between the binding peptides based on correlated positions (see Materials and methods). The two main clusters (orange dashed line) are the ones identified by our method. Positional correlations are successfully removed within the two clusters (black edges). (C) Sequence logos for a single PWM (left) and the multiple PWMs (right).
Positional correlations originate from multiple specificity
To explore possible causes of these correlations and their relationship with the biophysical characteristics of protein interactions, we first applied a clustering algorithm on each set of binding peptides using the percentage of sequence identity as a similarity measure (see Figure 1B and Materials and methods). If correlations result from structural constraints, such as the presence of different binding modes, we expect to see clusters of related peptides with much less positional correlation within each cluster. Figure 1B shows how, in the case of DLG1 PDZ1, two clusters are sufficient to remove significant correlations for any position, suggesting two classes of specificity for this domain that can be accurately modeled with two PWMs (Figure 1C). Overall, we observe that a limited number of clusters (most often two or three, except for some WW domains) are necessary to significantly remove positional correlations. Figure 2 summarizes the number of domains with correlated positions before the clustering procedure (by construction, no domain displays correlated positions after clustering). The average number of clusters required to remove correlations is indicated in parenthesis for each domain family. Randomly grouping the peptides into clusters of the same size, as the ones identified by our algorithm, clearly leaves several correlated positions (blue bar in Figure 2, see Materials and methods), which highlights the relevance of the identified clusters. This observation led us to model the binding specificity of the domains with multiple PWMs rather than a single one. Toward this goal, we used the machine learning framework of mixture of PWMs (Bailey and Elkan, 1994; Barash et al, 2003; Hannenhalli and Wang, 2005) that provides a general and computationally efficient way of solving this problem (see Materials and methods and Supplementary information). The main idea of this approach is to fit K different PWMs to the aligned peptides, where K is chosen here as the number of clusters found to remove positional correlations. The parameters of the multiple PWMs, as well as their weights, are directly learned from the data using a maximum likelihood (ML) approach (Bailey and Elkan, 1994; Bishop, 2006). Within this model, the specificity of each domain can be visualized as K different sequence logos. For instance, Figure 1C shows both the single PWM and the multiple PWM results for DLG1 PDZ1. Importantly, this result shows that predictions based on the single PWM can be misleading; for instance, a peptide ending with ETIW appears to match fairly well with a single PWM, whereas it is clearly excluded in the multiple PWM model and it was never found in the phage data.
Figure 2.

Positional correlations are widespread in different domain families. Red bars indicate the number of domains displaying at least one pair of correlated positions in the set of interacting peptides before clustering. By construction, no domain displays correlations after clustering the peptides. Blue bars indicate the expected number of domains with positional correlations for randomized clusters. The numbers in parenthesis below the domain names indicate the average number of clusters necessary to remove correlations.
The results of the multiple PWM model for PDZ domains displaying multiple specificity are shown in Figure 3. As correlations are significantly reduced within each cluster, the multiple logos provide a more accurate description of the specificity of these domains. Interestingly, we observe that PWMs can be as drastically different as corresponding to different specificity classes (e.g., SCRIB#1). We note that multiple specificity in PDZ domains is found both in worms and humans, which are separated by over 800 million years of evolution, indicating that this is likely a general feature of the PDZ domain family. The results for the yeast SH3 domains and human WW domains are displayed in Supplementary Figure S1. It is also interesting to observe that clustering the peptides leads to a much enhanced specificity, with an average entropy over all positions and all domains of 0.52 before clustering and of 0.42 after clustering (P<10−4, see Supplementary information and Supplementary Figure S2). In particular, multiple PWMs reveal interesting specificities that tend to be smoothed out in a single PWM (see for instance MLLT4#1 or HTRA2#1 in Figure 3).
Figure 3.

The multiple specificity tree of PDZ domains. For all profiles displaying multiple specificities, the single PWMs and the total number of phage peptides are shown at the end of the brown branches. For each domain, the multiple PWMs are shown at the end of the green branches together with their weights in the multiple PWM model. Worm PDZ domains are highlighted in yellow. The red rectangles show PDZ domains whose multiple PWMs are most different from each other. For visualization purposes, the tree was built using average linkage hierarchical clustering with Euclidean distance between the single PWM of each domain. ‘#’ after the protein name indicates the domain number.
Multiple PWMs more accurately model binding specificity
To validate the multiple PWMs model, we first assessed its ability to predict protein interactions using 10-fold cross-validation. We compared the multiple PWMs with single PWMs using the method of Sharon et al (2008) (see Supplementary information). For 80% of the domains displaying correlations in their binding peptides, multiple PWMs give better performance than a single PWM (see Supplementary Figure S3). We then used receiver operating characteristic (ROC) curves to compare the multiple PWMs model with ANNs (Brusic et al, 1998; Blom et al, 1999; Nielsen et al, 1999; Miller et al, 2008) and HMMs (Noguchi et al, 2002), which are known to also accurately model positional correlations (see Supplementary information). Overall, the results of the different models are very good and quite similar, with an average area under ROC curve (AROC) of 0.98 for multiple PWMs and HMMs and 0.99 for ANNs (See Supplementary Table S1 for the full list of AROC).
We then benchmarked the multiple PWM model on several independent data sets (see Supplementary information). We first used a large interaction data set of 12 yeast SH3 domains and 2 human PDZ domains generated by the SPOT technique, which provides a measure of affinity between domains and peptides (Wiedemann et al, 2004; Tonikian et al, 2009). For all available domains, the correlation between the score of the multiple PWMs model and the SPOT signal is higher than that with a single PWM (see Supplementary Table S2). On average, multiple PWMs also give slightly better correlations than HMMs, although the trend is not the same for all domains (as ANNs are not probabilistic models, correlation values cannot be directly compared). We carried out another validation using yeast two-hybrid (Y2H) data (Tonikian et al, 2009). We again found that better predictions are obtained using the multiple PWMs compared with the single PWMs, while performance is similar with HMMs (see Supplementary Figure S4). In this case, ANNs did not perform as well (see Supplementary information). We finally retrieved all experimentally determined interactions from the PDZbase interaction database (Beuming et al, 2005) to build an independent benchmarking data set (see Supplementary Table S3). When tested on this data set, both multiple PWMs and HMMs give an average AROC of 0.86, whereas ANNs give an average AROC of 0.85 (see Supplementary Table S4 for the full list of AROC and P-values).
Taken together, the multiple PWMs outperform single PWMs when used to predict domain–peptide interactions. Comparing with more complex machine learning frameworks, such as HMMs or ANNs, we observe similar performance, as expected, as different but mathematically related methods trained on the same data, in general, provide similar results (Nielsen et al, 1999). Having the added advantage of intuitive interpretation and visualization, multiple PWMs provide a particularly suitable framework to study the specificity of modular peptide recognition domains.
Multiple specificity corresponds to known binding modes of SH3 domains
Having found that a limited number of PWMs accurately represent the binding specificity of modular peptide recognition domains, we then sought to gain structural insights from these multiple specificities. In particular, we assume that domains with clearly different PWMs may display interesting structural features in order to accommodate such diversity among their interacting peptides. To explore this issue, we first examined our results with SH3 domains. SH3 domains bind proline-rich regions, in particular, PxxP motifs. Early studies identified two specificity classes: class I domains bind to [R/K]xxPxxP motifs, whereas class II domains bind to PxxPx[R/K] motifs (Mayer, 2001). These two classes correspond to different orientations of the peptide in the binding pocket (Lim et al, 1994). Some SH3 domains have been found to display a dual specificity, accommodating both class I and class II ligands, as illustrated in the two structures of Figure 4A for the SRC SH3 domain (Feng et al, 1994). Three of the yeast SH3 domains (Rvs167p, Lsb1p and Pin3p) are particularly interesting in this regard. Our completely automated analysis reveals that the specificity of these domains is best modeled with two PWMs for Rvs167p and Lsb1p, and three PWMs for Pin3p (see Figure 4B). In all three cases, the Arg is positioned either on the left or on the right of the proline-rich region in the multiple PWM model, which is the hallmark of SH3 domains accommodating both class I and class II ligands. This result shows that the multiple PWMs can predict different binding modes of modular peptide recognition domains, even in the absence of crystal structures for a specific domain. For other SH3 domains with correlated residues (see Supplementary Figure S1), the interpretation of multiple specificity is not as clear as for the ones in Figure 4. Some of these cases may correspond to more detailed structural features of the molecular recognition events taking place at the SH3-binding site.
Figure 4.

Multiple specificity in SH3 domains. (A) Solution structures of the bi-specific chicken SRC SH3 domain in complex with class I (PDB: 1PRL) and class II (PDB: 1RLP) ligands. (B) Comparison between the single PWM (first column) and the multiple PWMs of three yeast SH3 domains. The total number of interacting peptides is indicated in parenthesis. The weight of each component in the multiple PWM model is indicated below the sequence logo. Here, the multiple PWMs reveal distinct binding modes of SH3 domains predicted to correspond to different binding orientations of the peptides on the surface of the domain, as illustrated in A.
Multiple specificity predicts new binding modes of PDZ domains
We next examine PDZ domains, which have fewer recognized binding modes than SH3 domains. Most PDZ domains bind to the C terminus of their ligands, with a binding site that contacts up to seven ligand residues (Doyle et al, 1996). A few PDZ domains have also been observed to act as internal binders (Brenman et al, 1996; Hillier et al, 1999; Penkert et al, 2004) or to display non-canonical binding modes in recent crystal structures (Elkins et al, 2010). The multiple specificity of DLG1 PDZ1 in Figure 1 provides an interesting example to analyze. DLG1 is part of a cluster of four close paralogs (DLG1–4), each containing three PDZ domains, and the binding site of the first PDZ domain of these proteins is 100% conserved. The first of the multiple PWMs (Figure 1C) corresponds to the canonical binding mode of PDZ domains with a hydrophobic residue (here Val0) at the C terminus. This well-known binding mode is illustrated in Figure 5A by a crystal structure of DLG3 PDZ1 in complex with a peptide EETSV (PDB: 2I1N; Elkins et al, 2007). To interpret the second specificity predicted by the multiple PWMs, we first notice that the two logos of Figure 1C align well if the second one is shifted by one position. This suggests the presence of another residue (Trp in the phage data) at the C terminus. The crystal structure of Figure 5A clearly shows that an additional residue cannot be accommodated without a significant displacement of the carboxylate-binding loop. Interestingly, the C-alpha atoms in this loop display much larger B-factors (between 35 and 40) than the ones found elsewhere in the PDZ-binding pocket (between 27 and 32), suggesting higher flexibility. A new crystal structure of DLG2 PDZ1 was recently released, in which the domain crystallized as a trimer with the C terminally extended sequences RRRPIL binding to the peptide-binding site of adjacent PDZ molecules (Figure 5B, PDB: 2WL7; Fiorentini et al, 2009). In this structure, Ile−1 is found at the same spatial position as Val0 in Figure 5A. Moreover, the binding is accompanied by a large displacement of the carboxylate-binding loop, with only minor changes elsewhere. This recent structure already confirms our interpretation, even if only the last three residues (PIL) are in contact with the PDZ-binding site. To go one step further, we used the Rosetta modeling software (Wang et al, 2007) to generate a model of the new structure bound to a peptide built according to the non-canonical specificity observed in phage (EETDIW). We then used the FoldX force field (Schymkowitz et al, 2005) to optimize the side-chain positions and compute the predicted binding energy of this peptide (see Materials and methods). Figure 5C shows the final result of the docking and side-chain remodeling. The binding energy predicted by FoldX for the extended ligand EETDIW (−12.7 kcal/mol) compares favorably with the one computed for the short ligand in the original DLG3 PDZ1 (2I1N) structure (−7.3 kcal/mol). The new position of the loop accommodates the additional Trp, preserving one of the two usual hydrogen bonds between the C terminal residue and the PDZ backbone. In the canonical binding mode of PDZ domains, the carboxyl group forms a salt bridge mediated by a water molecule to a conserved Arg/Lys (R229 in Figure 5; Doyle et al, 1996). In our case, the carboxyl group of Trp can directly form a salt bridge with this Arg. We then explored the amino acid preferences at each ligand position in silico (see Materials and methods). The results shown in Figure 5D agree well with experimental data. In particular, FoldX predicts a clear preference for Trp (as well as Phe or Tyr) at the C terminal position.
Figure 5.

Structural modeling of the predicted non-canonical binding mode of DLG1 PDZ1. (A) Crystal structure of DLG3 PDZ1 in complex with the ligand EETSV (PDB: 2I1N; Elkins et al, 2007). (B) Recent crystal structure of DLG2 PDZ1 (PDB: 2WL7; Fiorentini et al, 2009). The peptide PIL in the binding site corresponds to the C terminus of the adjacent PDZ in the crystal structure. The carboxylate-binding loop displays a large conformational change on binding to this peptide. (C) Computational modeling of DLG1 PDZ1 in complex with an extended ligand EETDIW corresponding the new specificity observed in phage display-derived binding peptides. The orange carboxylate-binding loop corresponds to the canonical binding mode of the PDZ domain in A and the green one corresponds to the new binding mode predicted by our method and observed in the crystal structure shown in B. Dashed lines indicate important hydrogen bonds between the peptide and the domain. (D) Heat map view of FoldX results for the amino acid preferences in the model of extended ligand binding to DLG1 PDZ1. The amino acids of the initial ligand EETDIW are marked with black boxes.
Overall, both phage display data and the structural analysis predict that DLG1 PDZ1 has two distinct binding modes, one following the canonical C terminal PDZ-binding mode and another unexpected one allowing for an additional residue at the C terminus. As all residues involved in the non-canonical binding of DLG1 PDZ1 are exactly conserved in the other three DLG proteins (DLG2–4), we predict that this new binding mode applies to the first PDZ domain of all four DLG proteins. Interestingly, a similar multiple specificity is also observed for the tenth PDZ domain of MPDZ (see Figure 3), suggesting a similar binding mode. To gain insights into the generality of the carboxylate-binding loop remodeling observed for DLG1 PDZ1, we surveyed all PDZ domains from the PDB database and found five other crystal structures with a displaced loop (see Supplementary Figure S5). One pertains to Par-6 binding internally to Pals (Penkert et al, 2004), two come from the second PDZ domain of DLG1 (Haq et al, 2010) and DLG3, which are known to act as internal binders (Brenman et al, 1996), one from SYNJ2BP and one from MAGI1 PDZ4. The latter shares 63% identity and a very similar carboxylate-binding loop (ETGFG versus ESGFG) with MAGI3 PDZ4, which has phage display data available and exhibits multiple specificity (see Figure 3). Overall, it appears that remodeling of the carboxylate-binding loop is often associated with non-canonical binding modes of PDZ domains, some of which can be predicted by our analysis of phage display data, as shown in detail for DLG1 PDZ1.
The agreement between phage data and structural calculations for DLG1 PDZ1 prompted us to test whether some of the different binding specificities could be structurally predicted for other PDZ domains. We focused on DLG2 PDZ3, SCRIB PDZ1 and MPDZ PDZ10, which display interesting multiple specificity and for which structural data are available. Using FoldX, we scanned all residue positions of the ligand (see Supplementary information). The canonical specificity could be approximately retrieved, in agreement with previous work (Smith and Kortemme, 2010; see Supplementary Figure S6). In particular, the Thr/Ser at −2 is given a good score for the three domains, which is a hallmark of canonical PDZ binding. On the contrary, the structural analysis failed to predict the non-canonical specificities, most likely because the backbone conformation of the existing structures correspond to canonical ligands and does not accommodate other binding modes. As such, multiple PWMs provide an unbiased way of extracting new features from high-throughput data that are not easily predicted, unless different structures corresponding to distinct binding modes already exist.
Protein interaction predictions
For human PDZ domains exhibiting multiple specificity, we used the multiple PWM model to scan the human proteome and predict protein interactions. To test some of the predicted interactions, we manually chose conservative thresholds on multiple PWM scores, leading to a low false-positive rate. The resulting network is displayed in Supplementary Figure S7. Out of 31 predicted interactions, 8 are known from previous studies. To test whether some of the unknown interactions could be confirmed experimentally, we used the membrane Y2H system (Deribe et al, 2009; Snider et al, 2010) and tested three interactions of DLG1 and SCRIB, that were not previously described, with integral membrane proteins. These interactions could be confirmed experimentally: DLG1 was shown to bind to ANO9 and CYSLTR2, while SCRIB was shown to interact with SLC6A12 (see Figure 6). CYSLTR2 is a G-protein-coupled receptor. Binding of cysteinyl leukotrienes such as LTC4 to CYSLTR2 has been shown to activate chemokine production through induction of NF-κB and AP-1 transcription factors, although the molecular mechanism of this signaling is still poorly understood (Thompson et al, 2008). Our results suggest a role for DLG1 within this pathway, possibly acting as a scaffolding protein, as is often the case for multi-domain proteins. SLC6A12 is an integral membrane protein involved in GABA transport and linked to aspirin-intolerant asthma (Pasaje et al, 2010). Interestingly, a GABAergic system with a crucial role for mucus production in asthma has been recently found in airway epithelium (Xiang et al, 2007), which is consistent with the expression of SCRIB in epithelial cells.
Figure 6.

Validation of predicted protein–protein interactions by membrane yeast two-hybrid assay (see Materials and methods). Bait proteins ANO9 and CYSLTR2 interact with DLG1 and SLC6A12 interacts with SCRIB, as predicted, while control baits EPHA2 and SLC6A5 are negative. Results are representative of three independent experiments.
Discussion
Efficient computational strategies are crucial to retrieve the most relevant information encoded in large protein interaction data sets, which leads to better understanding of the many signaling pathways mediated by participating proteins. Here, we have addressed at a large scale the issue of positional dependencies within short linear stretches of residues targeted by modular peptide recognition domains. For the domains analyzed in this work, we have found cases of positional correlations for more than 25% of PDZ domains, more than 50% of SH3 domains and all three WW domains. Moreover, we have shown that most correlations can be resolved by clustering the peptides into a few groups corresponding to different specificity. This clearly shows that multiple specificity is a common phenomenon. From a computational point of view, the multiple PWMs give similar performance as other machine learning algorithms. We suggests that, because of the structural constraints underlying short peptide-binding events, a simple decomposition into multiple PWMs is sufficient to handle correlations, while more complex problems, such as predicting subcellular localization from protein sequence (Emanuelsson et al, 2000), may require more advanced machine learning algorithms.
We can distinguish different categories of multiple specificity from our analysis. For some domains, the multiple PWMs are very different from each other (some examples are highlighted in red in Figure 3). In these cases, significant structural changes are likely to take place at the level of the domain–peptide interface, and our analysis of SH3 domains and of DLG1 PDZ1 confirm that multiple PWMs provide a very useful computational tool to guide structural analysis. For other domains, the differences between the PWMs are less dramatic and mostly depend on two or three positions located close to each other. In general, we do not expect such cases to correspond to large structural remodeling of the binding sites, but rather to side-chain–side-chain interactions within the peptide. One potential example is the PDZ domain of worm shn-1, which appears to prefer either R at −3 or K at −4, but not both together, suggesting that one single charged residue at these positions is more favorable for peptides binding to this domain. A possible way to automatically distinguish between the two kinds of multiple specificity is to compute the residue preference similarity between correlated positions. On the basis of our results, we suggest that if clear differences are found at three or fewer correlated positions, and all of them are within four residues, then the correlations most likely correspond to interactions between ligand residues. Conversely, if all correlated positions are different (e.g., DLG1#1) or if correlated positions are far away from each other (e.g., SH3 domains in Figure 4), multiple specificity more likely corresponds to distinct binding modes of the domain. Finally, not all domains appear to display positional correlations. This may be due to the availability of a limited number of binding peptides, which prevents us from observing slightly less favorable binding specificities, or some domains may be optimized to accommodate only one specific kind of peptide.
An obvious question to ask is whether multiple specificity can still be observed using other experimental data sets. To observe statistically significant correlated positions and automatically detect multiple specificity, at least a dozen interacting peptides are required (e.g., for two clusters of six identical peptides each, the mutual information P-value is ≈0.002), which partly explains why this feature has not often been observed in previous work. For instance, a recent study (Stiffler et al, 2007) used 217 peptides derived from Mouse natural C termini to probe the specificity of PDZ domains in a protein-chip experiment. This screen yielded <10 interacting peptides for most domains. Moreover, the set of initial peptides is highly biased toward canonical PDZ-binding motifs. As a result, we observed significant positional correlations for only one domain in this dataset. However, with future technological advances, this may change. High-throughput experimental techniques such as phosphoproteomic methods to study kinase specificity (Olsen et al, 2010) or ribosome display (Hanes and Pluckthun, 1997), are increasingly becoming available to generate large and unbiased sets of interacting peptides. Moreover, new sequencing technologies are currently revolutionizing phage display experiments by allowing rapid sequencing of hundred of thousands of peptides or proteins that have passed the selection runs (Ernst et al, 2010; Fowler et al, 2010). Hence, such data are likely to become available in the near future for many natural domains, offering new opportunities to enhance both our biophysical understanding of molecular recognition events and the accuracy of computational protein interaction predictions. In the field of protein–DNA interactions, very large data sets are available to map the specificity of transcription factors, and multiple specificity has also been recently observed (Badis et al, 2009). It is likely that similar approaches, as the one presented in this work, will yield new insights into modular peptide recognition domains or transcription factors studied with these other experimental techniques. For other kinds of protein interactions, such as those involving larger binding interface or non-peptide substrates, sequence-based approaches are more difficult to apply. As such, the multiple PWM model is especially suited for proteins interacting with small ligands made out of a limited number of building blocks (e.g., amino acids or nucleotides) and adopting a few different binding modes on their targets.
At a system-wide level, the multiple specificity observed in modular peptide recognition domains, such as PDZ, SH3 or WW, has several interesting consequences. First, it enables additional potential crosstalk in signaling pathways, where domains displaying multiple specificity could act as linkers between different pathways. Second, multiple specificity enables optimization of a domain to interact in a highly specific manner with a few very different ligands. This binding-site plasticity yields interesting evolutionary advantages: it may provide a greater repertoire to build on the pathway topologies required to sustain cell activity using only a limited number of components. In addition, it allows for the emergence of new specificities without necessarily altering the initial one. As such, the evolution of specificity does not necessarily need to follow a gradual process, but could rather consist in exploration of novel binding specificities that do not perturb the existing protein interactions and are retained only when conferring an advantage to the organism. Such neutral evolutionary pathways are critical to enable innovation in a system while preserving its robustness (Ciliberti et al, 2007), and our results suggest that multiple specificity may act as a key factor in this process.
Materials and methods
Phage display data
The experimental data sets used in this work come from large-scale phage display experiments (Tonikian et al, 2007). For PDZ domains, all phage display peptides found in Tonikian et al (2008) for wild-type domains in humans and worms were used in our analysis. For each domain, the peptides were aligned from the C terminus. Owing to the presence of STOP codons in the phage library, some peptides are shorter than seven amino acids (missing residues are labeled with X). For yeast SH3 domains, the phage peptides from Tonikian et al (2009) were automatically aligned with the MUSCLE alignment software (Edgar, 2004) using settings that prevented internal gaps. The new data for the three WW domains come from a recent phage display experiment run with similar protocols as for the PDZ and SH3 domains (see Supplementary Table S5 for the raw data). Because of the amplification step in phage display, the frequency of the peptides pulled out experimentally is difficult to interpret in terms of binding strength. For this reason, peptides retrieved multiple times were treated as unique throughout our analysis.
Identifying correlated positions in peptide alignments
To identify correlated positions in peptide alignments, we used mutual information for all possible position pairs. Mutual information is computed as:
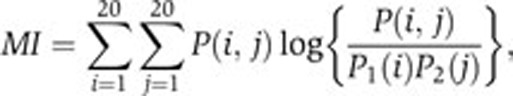 |
where P(i,j) stands for the probability of having amino acid i at one position together with amino acid j at the other position in a peptide. P1(i) is the probability of having amino acid i at one position and P2(j) the probability of having amino acid j at the other position. MI=0 corresponds to independence of the two positions, whereas larger values indicate that knowing which amino acids are found at one position gives some information about which ones are expected at the other position. One limitation of mutual information is that non-zero values are often expected to be present by chance, even for randomly generated peptides. We therefore used the mutual information P-value as a filter (other statistical measures such as the Z-scores could also be used). All P-values have been computed by randomly shuffling the amino acids within the alignment columns. A threshold of 0.001 has been used to define correlated positions.
Clustering domain-binding peptides
For each domain displaying correlated positions, the peptides were clustered using the average linkage hierarchical clustering algorithm implemented in R. The similarity measure between two peptides was computed as the ratio of identical amino acids, including only positions that are correlated with at least one other position. Although other measures of similarity, such as BLOSUM62 or biochemical similarity, may be used, we did not observe any improvement when using them. The final clusters were defined by following the branches of the dendrogram from its root and stopping whenever no more correlated positions are present within a cluster according to our mutual information P-value cutoff (see Figure 1B). Although larger P-values are always expected for smaller peptide sets, the absence of significant positional correlations within the clusters does not originate from this scaling effect, since random clusters of the same size do not remove correlations, as shown in Figure 2. Clusters containing less than two peptides most often correspond to false-positives in phage and were filtered out in subsequent analyses.
Mixture of PWMs
A mixture of K PWMs is described by the model parameters θlik, which correspond to the probability of having amino acid i and position l according to the kth PWM, and the mixing coefficients πk quantifying the weight of each PWM ∑k = 1k π k = 1 (Bailey and Elkan, 1994; Bishop, 2006). The score of a peptide X=(x1…xL) of length L is then given by the equation:
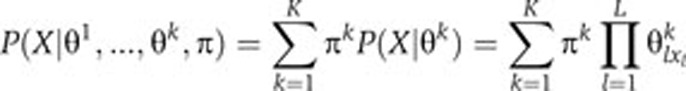 |
Given a data set of N interacting peptides, the different parameters of the mixture of PWMs are directly learned from the data using standard maximum likelihood algorithms (see Supplementary information). Choosing the best value for K (i.e., number of PWMs) is a difficult machine learning problem that does not have a general solution. In practice, one can either test different values and choose the most meaningful one or design heuristic strategies to estimate a reasonable value of K. Here, we chose K as the number of clusters found to remove all positional correlations. However, we stress that the mixture model provides a general framework that can be used with any other method of choosing K. In particular, the method can also be used as a fast exploration tool by probing different values of K (i.e., different number of PWMs) and manually identifying the most meaningful one, without performing the initial clustering step.
Molecular modeling
The Rosetta software (Wang et al, 2007) was used to dock the ligand EETDIW to DLG1 PDZ1, using the recent PDB structure 2WL7 for the PDZ domain. Ligand position was optimized with Rosetta2.3 allowing for backbone flexibility, and the highest scoring trajectory out of 100 optimization runs was used in our model. Binding energies and residue preferences in the peptides were analyzed with FoldX (Schymkowitz et al, 2005; see Supplementary information). All structures were visualized with Pymol (http://www.pymol.org).
Protein interaction predictions
The C-termini of all human proteins were scanned with the multiple PWM model and fairly stringent thresholds were used to generate the network of Supplementary Figure S7. As DLG1 and DLG2 share >80% sequence identity, the two proteins were merged in the network of predicted interactions. To experimentally test some of the predicted interactions, we filtered away the ones already known from literature and databases and the non-membrane proteins to comply with the requirements of the membrane Y2H system. We manually selected three proteins (ANO9 and CYSLTR2 predicted to bind to DLG1 and SLC6A12 predicted to bind to SCRIB) from the network in Supplementary Figure S7. All three proteins were tested with both DLG1 and SCRIB.
Experimental testing of protein interactions
Membrane Y2H constructs
Full-length human ANO9, CYSLTR2 and SLC6A12 cDNAs, as well as the controls EPHA2 and SLC6A5, were amplified by PCR and subcloned by homologous recombination in yeast into bait vectors pBT3-N and pTLB-1 (DualSystems Biotech) conferring the C terminal ubiquitin (Cub) moiety and LexA-VP16 transcription factor at the bait N terminus (except for CYSLTR2 and EPHA2 in which the Cub-LexA-VP16 tag was fused to the C terminus of the bait protein). Similarly, full-length human DLG1 and SCRIB cDNA were subcloned into prey vector pPR3N (DualSystems Biotech), which confers the N terminal ubiquitin (Nub) moiety to the N terminus of the prey protein.
Membrane Y2H assay
Yeast reporter strain THY.AP40 (MATa trp1 leu2 his3 LYS2::lexA-HIS3 URA3::lexA-lacZ) was transformed with the indicated LexA-VP16-Cub-BAIT constructs by the lithium acetate protocol. Self-activation and membrane localization were assessed by the Fur4-NubI/Fur4-NubG and Ost-NubI/Ost-NubG tests, as previously described (Deribe et al, 2009; Snider et al, 2010). On passing the NubG/I test, pPR3N-DLG1/SCRIB preys were transformed into bait-containing yeast and transformants were selected on SD-Trp-Leu. Three colonies for each transformation were spotted on selective media containing 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-GAL), which turns blue in the presence of β-galactosidase, indicating activation of the reporter system. Figure 6 displays the results for these three independent experiments for each interaction. The protein interactions from this publication have been submitted to the IMEx (http://imex.sf.net) consortium through IntAct (Aranda et al, 2010) and assigned the identifier IM-15347.
Supplementary Material
Supplementary Tables S1–5, Supplementary Figures S1–7
Acknowledgments
DG acknowledges the financial support of the Swiss National Science Foundation (SNSF) and the European Molecular Biology Organization (EMBO). This work was supported by the Canadian Institutes of Health Research (Grant MOP-84324). The Stagljar lab is supported by grants from the Canada Foundation for Innovation (CFI), the Canadian Institutes of Health Research (CIHR), the Canadian Cancer Society Research Institute (CCSRI), the Heart and Stroke Foundation, the Cystic Fibrosis Foundation, the Ontario Genomics Institute and Novartis. The Kim lab is funded by grants from the Natural Sciences and Engineering Research Council (NSERC), the Canada Foundation for Innovation (CFI) and the Ontario Research Fund (ORF).
Author contributions: DG, GDB and PMK designed research; DG, FB, EV, PV, HH, ND performed research; DG, AE, LS, IS, SSS, GDB and PMK analyzed data; DG, GDB and PMK wrote the paper.
Footnotes
The authors declare that they have no conflict of interest.
References
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J, Kerssemakers J, Leroy C, Menden M, Michaut M, Montecchi-Palazzi L, Neuhauser SN, Orchard S, Perreau V, Roechert B, van Eijk K et al. (2010) The IntAct molecular interaction database in 2010. Nucleic Acids Res 38: D525–D531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, Kuznetsov H, Wang CF, Coburn D, Newburger DE, Morris Q, Hughes TR, Bulyk ML (2009) Diversity and complexity in DNA recognition by transcription factors. Science 324: 1720–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36 [PubMed] [Google Scholar]
- Barash Y, Elidan G, Friedman N, Kaplan T (2003) Modeling dependencies in protein-DNA binding sites. In RECOMB '03: Proceedings of the Seventh Annual International Conference on Research In Computational Molecular Biology. Berlin, Germany: ACM [Google Scholar]
- Beuming T, Skrabanek L, Niv MY, Mukherjee P, Weinstein H (2005) PDZBase: a protein-protein interaction database for PDZ-domains. Bioinformatics 21: 827–828 [DOI] [PubMed] [Google Scholar]
- Bishop CM (2006) Pattern Recognition and Machine Learning, Vol. 1 Singapore: Springer [Google Scholar]
- Blom N, Gammeltoft S, Brunak S (1999) Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol 294: 1351–1362 [DOI] [PubMed] [Google Scholar]
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS (1996) Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 84: 757–767 [DOI] [PubMed] [Google Scholar]
- Brusic V, Rudy G, Honeyman G, Hammer J, Harrison L (1998) Prediction of MHC class II-binding peptides using an evolutionary algorithm and artificial neural network. Bioinformatics 14: 121–130 [DOI] [PubMed] [Google Scholar]
- Chen JR, Chang BH, Allen JE, Stiffler MA, MacBeath G (2008) Predicting PDZ domain-peptide interactions from primary sequences. Nat Biotechnol 26: 1041–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciliberti S, Martin OC, Wagner A (2007) Innovation and robustness in complex regulatory gene networks. Proc Natl Acad Sci USA 104: 13591–13596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deribe YL, Wild P, Chandrashaker A, Curak J, Schmidt MH, Kalaidzidis Y, Milutinovic N, Kratchmarova I, Buerkle L, Fetchko MJ, Schmidt P, Kittanakom S, Brown KR, Jurisica I, Blagoev B, Zerial M, Stagljar I, Dikic I (2009) Regulation of epidermal growth factor receptor trafficking by lysine deacetylase HDAC6. Sci Signal 2: ra84. [DOI] [PubMed] [Google Scholar]
- Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R (1996) Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell 85: 1067–1076 [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Gileadi C, Shrestha L, Phillips C, Wang J, Muniz JR, Doyle DA (2010) Unusual binding interactions in PDZ domain crystal structures help explain binding mechanisms. Protein Sci 19: 731–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins JM, Papagrigoriou E, Berridge G, Yang X, Phillips C, Gileadi C, Savitsky P, Doyle DA (2007) Structure of PICK1 and other PDZ domains obtained with the help of self-binding C-terminal extensions. Protein Sci 16: 683–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson O, Nielsen H, Brunak S, von Heijne G (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300: 1005–1016 [DOI] [PubMed] [Google Scholar]
- Ernst A, Gfeller D, Kan Z, Seshagiri S, Kim PM, Bader GD, Sidhu SS (2010) Coevolution of PDZ domain-ligand interactions analyzed by high-throughput phage display and deep sequencing. Mol Biosyst 6: 1782–1790 [DOI] [PubMed] [Google Scholar]
- Feng S, Chen JK, Yu H, Simon JA, Schreiber SL (1994) Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science 18: 1241–1247 [DOI] [PubMed] [Google Scholar]
- Fiorentini M, Nielsen AK, Kristensen O, Kastrup JS, Gajhede M (2009) Structure of the first PDZ domain of human PSD-93. Acta Crystallogr Sect F Struct Biol Cryst Commun 65: 1254–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Araya CL, Fleishman SJ, Kellogg EH, Stephany JJ, Baker D, Fields S (2010) High-resolution mapping of protein sequence-function relationships. Nat Methods 7: 741–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould CM, Diella F, Via A, Puntervoll P, Gemund C, Chabanis-Davidson S, Michael S, Sayadi A, Bryne JC, Chica C, Seiler M, Davey NE, Haslam N, Weatheritt RJ, Budd A, Hughes T, Pas J, Rychlewski L, Trave G, Aasland R et al. (2010) ELM: the status of the 2010 eukaryotic linear motif resource. Nucleic Acids Res 38: D167–D180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes J, Pluckthun A (1997) In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci USA 94: 4937–4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannenhalli S, Wang LS (2005) Enhanced position weight matrices using mixture models. Bioinformatics 21(Suppl 1): i204–i212 [DOI] [PubMed] [Google Scholar]
- Haq SR, Jurgens MC, Chi CN, Koh CS, Elfstrom L, Selmer M, Gianni S, Jemth P (2010) The plastic energy landscape of protein folding: a triangular folding mechanism with an equilibrium intermediate for a small protein domain. J Biol Chem 285: 18051–18059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris BZ, Lim WA (2001) Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci 114: 3219–3231 [DOI] [PubMed] [Google Scholar]
- Hillier BJ, Christopherson KS, Prehoda KE, Bredt DS, Lim WA (1999) Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 284: 812–815 [PubMed] [Google Scholar]
- Hu H, Columbus J, Zhang Y, Wu D, Lian L, Yang S, Goodwin J, Luczak C, Carter M, Chen L, James M, Davis R, Sudol M, Rodwell J, Herrero JJ (2004) A map of WW domain family interactions. Proteomics 4: 643–655 [DOI] [PubMed] [Google Scholar]
- Hui S, Bader GD (2010) Proteome scanning to predict PDZ domain interactions using support vector machines. BMC Bioinformatics 11: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE (2004) A rapid method for determining protein kinase phosphorylation specificity. Nat Methods 1: 27–29 [DOI] [PubMed] [Google Scholar]
- Lim WA, Richards FM, Fox RO (1994) Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature 372: 375–379 [DOI] [PubMed] [Google Scholar]
- Mayer BJ (2001) SH3 domains: complexity in moderation. J Cell Sci 114: 1253–1263 [DOI] [PubMed] [Google Scholar]
- Miller ML, Jensen LJ, Diella F, Jorgensen C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten T, Olhovsky M, Pasculescu A, Alexander J, Knapp S, Blom N, Bork P, Li S, Cesareni G, Pawson T, Turk BE et al. (2008) Linear motif atlas for phosphorylation-dependent signaling. Sci Signal 1: ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Brunak S, von Heijne G (1999) Machine learning approaches for the prediction of signal peptides and other protein sorting signals. Protein Eng 12: 3–9 [DOI] [PubMed] [Google Scholar]
- Noguchi H, Kato R, Hanai T, Matsubara Y, Honda H, Brusic V, Kobayashi T (2002) Hidden Markov model-based prediction of antigenic peptides that interact with MHC class II molecules. J Biosci Bioeng 94: 264–270 [DOI] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB (2003) Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res 31: 3635–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal 3: ra3. [DOI] [PubMed] [Google Scholar]
- Pasaje CF, Kim JH, Park BL, Cheong HS, Chun JY, Park TJ, Lee JS, Kim Y, Bae JS, Park JS, Yoon SH, Uh ST, Choi JS, Kim YH, Kim MK, Choi IS, Cho SH, Choi BW, Park CS, Shin HD (2010) Association of SLC6A12 variants with aspirin-intolerant asthma in a Korean population. Ann Hum Genet 74: 326–334 [DOI] [PubMed] [Google Scholar]
- Pawson T, Nash P (2003) Assembly of cell regulatory systems through protein interaction domains. Science 300: 445–452 [DOI] [PubMed] [Google Scholar]
- Penkert RR, DiVittorio HM, Prehoda KE (2004) Internal recognition through PDZ domain plasticity in the Par-6-Pals1 complex. Nat Struct Mol Biol 11: 1122–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L (2005) The FoldX web server: an online force field. Nucleic Acids Res 33: W382–W388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Tan CS, Voss C, Li SS, Deng N, Bader GD (2010) A regression framework incorporating quantitative and negative interaction data improves quantitative prediction of PDZ domain-peptide interaction from primary sequence. Bioinformatics 27: 383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Lubliner S, Segal E (2008) A feature-based approach to modeling protein-DNA interactions. PLoS Comput Biol 4: e1000154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Kortemme T (2010) Structure-based prediction of the peptide sequence space recognized by natural and synthetic PDZ domains. J Mol Biol 402: 460–474 [DOI] [PubMed] [Google Scholar]
- Snider J, Kittanakom S, Damjanovic D, Curak J, Wong V, Stagljar I (2010) Detecting interactions with membrane proteins using a membrane two-hybrid assay in yeast. Nat Protoc 5: 1281–1293 [DOI] [PubMed] [Google Scholar]
- Stiffler MA, Chen JR, Grantcharova VP, Lei Y, Fuchs D, Allen JE, Zaslavskaia LA, MacBeath G (2007) PDZ domain binding selectivity is optimized across the mouse proteome. Science 317: 364–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C, Cloutier A, Bosse Y, Poisson C, Larivee P, McDonald PP, Stankova J, Rola-Pleszczynski M (2008) Signaling by the cysteinyl-leukotriene receptor 2. Involvement in chemokine gene transcription. J Biol Chem 283: 1974–1984 [DOI] [PubMed] [Google Scholar]
- Tong AH, Drees B, Nardelli G, Bader GD, Brannetti B, Castagnoli L, Evangelista M, Ferracuti S, Nelson B, Paoluzi S, Quondam M, Zucconi A, Hogue CW, Fields S, Boone C, Cesareni G (2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules. Science 295: 321–324 [DOI] [PubMed] [Google Scholar]
- Tonikian R, Xin X, Toret CP, Gfeller D, Landgraf C, Panni S, Paoluzi S, Castagnoli L, Currell B, Seshagiri S, Yu H, Winsor B, Vidal M, Gerstein MB, Bader GD, Volkmer R, Cesareni G, Drubin DG, Kim PM, Sidhu SS et al. (2009) Bayesian modeling of the yeast SH3 domain interactome predicts spatiotemporal dynamics of endocytosis proteins. PLoS Biol 7: e1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Boone C, Sidhu SS (2007) Identifying specificity profiles for peptide recognition modules from phage-displayed peptide libraries. Nat Protoc 2: 1368–1386 [DOI] [PubMed] [Google Scholar]
- Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, Sidhu SS (2008) A specificity map for the PDZ domain family. PLoS Biol 6: e239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Bradley P, Baker D (2007) Protein-protein docking with backbone flexibility. J Mol Biol 373: 503–519 [DOI] [PubMed] [Google Scholar]
- Wiedemann U, Boisguerin P, Leben R, Leitner D, Krause G, Moelling K, Volkmer-Engert R, Oschkinat H (2004) Quantification of PDZ domain specificity, prediction of ligand affinity and rational design of super-binding peptides. J Mol Biol 343: 703–718 [DOI] [PubMed] [Google Scholar]
- Xiang YY, Wang S, Liu M, Hirota JA, Li J, Ju W, Fan Y, Kelly MM, Ye B, Orser B, O′Byrne PM, Inman MD, Yang X, Lu WY (2007) A GABAergic system in airway epithelium is essential for mucus overproduction in asthma. Nat Med 13: 862–867 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables S1–5, Supplementary Figures S1–7